Decreased UDP-GlcNAc levels abrogate proliferation control in EMeg32-deficient cells (original) (raw)
Abstract
The hexosamine pathway provides UDP-_N_-acetylhexosamine donor substrates used in cytosolic and Golgi-mediated glycosylation of proteins and for formation of glycosylphosphatidylinositol (GPI) anchors, which tether proteins to the outer plasma membrane. We have recently identified the murine glucosamine-6-phosphate (GlcN6P) acetyltransferase, EMeg32, as a developmentally regulated enzyme on the route to UDP-_N_-acetylglucosamine (UDP-GlcNAc). Here we describe embryos and cells that have the EMeg32 gene inactivated by homologous recombination. Homozygous mutant embryos die at around embryonic day (E) 7.5 with a general proliferative delay of development. In vitro differentiated EMeg32–/– ES cells show reduced proliferation. Mouse embryonic fibroblasts (MEFs) deficient for EMeg32 exhibit defects in proliferation and adhesiveness, which could be complemented by stable re-expression of EMeg32 or by nutritional restoration of intracellular UDP-GlcNAc levels. Reduced UDP-GlcNAc levels predominantly translated into decreased _O_-GlcNAc modifications of cytosolic and nuclear proteins. Interestingly, growth-impaired EMeg32–/– MEFs withstand a number of apoptotic stimuli and express activated PKB/AKT. Thus, EMeg32-dependent UDP-GlcNAc levels influence cell cycle progression and susceptibility to apoptotic stimuli.
Keywords: actin dynamics/glucosamine-6-phosphate acetyltransferase/glycosylation/phosphatidylinositol/PKB
Introduction
Nucleotide sugars in eukaryotic cells are donor substrates for glycosylation of proteins and lipids. The central module, UDP-_N_-acetylglucosamine (UDP-GlcNAc), is synthesized de novo from glucose by the hexosamine synthesis pathway (HXNP), but can also be produced via salvage pathways (Schachter, 1978). Two GlcNAc units are the first monosaccharides of a dolichol-pyrophosphate oligosaccharide, which is transferred en bloc to nascent proteins in the endoplasmic reticulum (ER) during asparagine (N)-linked glycosylation of secreted or membrane proteins. Glycoprotein maturation requires the action of α-glucosidases, α-mannosidases and Golgi-resident glycosyltransferases, which obtain nucleotide sugar substrates from the cytoplasm through specialized membrane transporters (Hirschberg et al., 1998). Golgi-routed O-linked glycosylation emanates from serine or threonine with GalNAc as the first residue, and employs sequential addition of monosaccharides rather than en bloc transfer of pre-assembled oligosaccharide. Synthesis of glycosaminoglycans such as heparan sulfate, keratan sulfate and hyaluronic acid also requires Golgi-routed UDP-GlcNAc. Distinct from Golgi protein glycosylation, many cytosolic and nuclear proteins are O-glycosylated on Ser/Thr with a single GlcNAc residue and this is a reversible modification (for reviews see Van den Steen et al., 1998; Comer and Hart, 1999). Moreover, UDP-GlcNAc is central in the generation of glycosylphosphatidylinositol (GPI) anchors, as the key synthesis step requires the addition of GlcNAc to phosphatidylinositol (PtdIns) (for review see Udenfriend and Kodukula, 1995).
We have recently characterized the murine glucosamine-6-phosphate (GlcN6P) acetyltransferase, EMeg32, as an important enzyme on the route to UDP-GlcNAc (Boehmelt et al., 2000). The 184 amino acid (aa) EMeg32 protein is associated with the cytoplasmic leaflet of Golgi and endosomal membranes and accumulates at the G2–M phase of the cell cycle. Further, p97/VCP, the homolog of the cell cycle regulator cdc48, copurified with recombinant EMeg32 in insect cells (Boehmelt et al., 2000). p97/VCP is an ATPase involved in homotypic membrane fusion events at mitosis (Acharya et al., 1995; Rabouille et al., 1995).
Inactivation of the EMeg32 homolog, PAT1/GNA1, in Saccharomyces cerevisiae is lethal and PAT1 was shown to be required for multiple steps in the cell cycle such as exit from the G0 phase and mitosis (Lin et al., 1996; Mio et al., 1999). A screen to suppress the defects of a temperature-sensitive PAT1p revealed mutated SAC1, originally identified as a suppressor of actin mutations (Cleves et al., 1989). Recently, the ER/Golgi-resident SAC1 was demonstrated to convert the phosphatidylinositols PtdIns(3)P, PtdIns(4)P and PtdIns(3,5)P2 into PtdIns and this activity might explain many of the phenotypes described for yeast SAC1 mutants, like impaired secretion, actin cytoskeletal reorganization or deficiencies in ATP transport activities in the ER (Whitters et al., 1993; Guo et al., 1999; Kochendörfer et al., 1999).
It is not clear whether the inactivation of GlcN6P acetyltransferase would affect intracellular UDP-GlcNAc levels and downstream routes, or how inactivation might influence cell cycle control. To address these questions, we inactivated the EMeg32 gene in mice by homologous recombination. EMeg32 appears to be indispensable for embryonic development beyond E7.5. EMeg32-deficient mouse embryonic fibroblasts (MEFs) showed reduced proliferation and substratum adhesiveness, which correlated with the extent of intracellular UDP-GlcNAc reduction. GlcNAc feeding or re-expression of EMeg32 normalized UDP-GlcNAc levels in EMeg32–/– MEFs and rescued their biological defects. Downstream routes emanating from UDP-GlcNAc were differentially affected with predominant aberrations in cytosolic O-glycosylation. Furthermore, mutant MEFs are impaired in depolymerizing actin bundles, are resistant to a number of apoptotic stimuli and express activated PKB/AKT, indicating that cellular proliferation and apoptosis signaling are influenced by the amount of intracellular UDP-GlcNAc.
Results
Generation of a null mutation of EMeg32
To inactivate the EMeg32 gene, we used a targeting vector designed to replace 1.4 kb of genomic DNA by an antisense neomycin resistance cassette, thereby removing the first two coding exons of the EMeg32 gene (Figure 1A). Homologous recombination of the targeting vector was confirmed by Southern blot analysis on two embryonic stem (ES) cell clones (M2R4H and M7R11C; Figure 1B). M7R11C ES cells were further subjected to a higher G418 concentration to generate EMeg32 homozygous mutant ES cell clones (Figure 1D). Western blot analysis of a number of these clones revealed absence of p21 EMeg32 protein using EMeg32-specific antibodies (Boehmelt et al., 2000; Figure 1E).


Fig. 1. Gene targeting of the murine EMeg32 locus. (A) Schematic representation of the targeting vector and mouse EMeg32 wild-type and mutant loci. Open boxes represent exons (ex). The large arrow indicates transcriptional direction of the neomycin resistance cassette (neo). Small arrows mark positions of PCR primers used to identify wild-type and mutant alleles. Bar, 1 kb of sequence; B, _Bst_1107I; C, _Cla_I; E, _Eco_RI; N, _Nco_I; No, _Not_I; S, _Sac_I; V, _Eco_RV. (B) Southern blot analysis of ES cell genomic DNA from wild-type (E14K) and +/– (M2R4H, M7R11C) cell lines after digestion with _Eco_RI plus _Cla_I. The 5′ flanking probe [_Eco_RV–_Nco_I fragment shown in (A)] was used. DNA fragments derived from the 9.4 kb wild-type (wt) and 6.4 kb mutant (mut) allele are indicated. (C) Lysates from E7.5 embryos were analysed by PCR using the primers specific for the wild-type (wt, 750 bp fragment, arrow) or mutant (mut, 880 bp fragment) allele, respectively. EMeg32 genotypes (+/+, +/–, –/–) are indicated. A background smear in the wild-type PCR reaction is unspecific. Control, no DNA added. (D) Southern blot analysis of EMeg32 ES cell clones surviving increased G418 concentration. Procedure and labeling as described in (B). (E) Western blot analysis demonstrating loss of EMeg32 protein (arrow) in four –/– ES clones (C3-21, C3-56, C3-94 and C3-87). An unspecific band (open triangle) usually detected by EMeg32 antibody #2087 and the positions of protein standards (M) are indicated. Control, heterozygous M7R11C ES cells.
Homozygosity for the EMeg32 mutation leads to embryonic lethality by E7.5
Both heterozygous ES cell clones contributed to germline transmission following blastocyst injection and generation of chimeric mice. Analysis of neonates obtained from M2R4H- (n = 155) or M7R11C-derived (n = 114) heterozygous mutant intercrosses revealed no homozygous mutant (EMeg32–/–) mice. We therefore determined the time point of developmental failure during gestation. At E6.5, 26.3% of the 19 sampled embryos were typed as –/–. At E7.5, 18.6% of the 43 typed embryos were abnormal (Figure 2A and E) and EMeg32–/– (Figure 1C and not shown). At E8.5 or E9.5, only empty yolk sacs, occasionally with a remnant of allantois, could be typed as EMeg32–/–; at later stages (E10.5–E12.5), embryos were either wild type or heterozygous. These results show that homozygosity for the EMeg32 mutation causes lethality at E7.5. Morphologies and time points of developmental failure were identical in embryos derived from the two targeted ES cell clones.

Fig. 2. Impaired development of mutant EMeg32 embryos. Morphology of wild-type (A) or mutant (E) embryos at E7.5. Bars, 280 µm. Hematoxylin and eosin (H+E) staining of E7.5 wild-type embryo (B) showing organized development of mesoderm (m), headfold (hf) structures, allantois (al), amnion (a), exocoel (ex) and chorion (ch). (F) E7.5 mutant embryos resemble wild-type embryos developmentally delayed by 1.0–1.5 days (see text for details). Bars, 450 µm. In situ hybridization on sections of wild type (C and D) or mutant (G and H) EMeg32 embryos using a Brachyury (C, G) or an _Otx2_-specific (D, H) antisense probe, respectively. Embryos were at E6.5 (C, G) or E7.5 (D, H). Abbreviations are as in (B); ep, epiblast; ee, embryonic ectoderm. Bars, 70 µm (G), 140 µm (C), 210 µm (D, H).
EMeg32 mutant embryos exhibit delayed development
Serial sections of E6.5–E7.5 embryos obtained from heterozygous intercrosses were classified morphologically as wild type or mutant. At E6.5, wild-type embryos showed a well organized ectoderm, endoderm and developing mesoderm, whereas mutant embryos were less organized, resembling E5.5 wild-type embryos. About 50% of the E6.5 mutant embryos displayed a small ectoplacental cone (EPC). At E7.5, wild-type embryos showed well organized mesoderm and headfold structures in addition to proper development of allantois, amnion, chorion and exocoel. These structures were absent or corrupted in EMeg32–/– embryos (Figure 2B and F). Expression of transcription factor Hnf-4 RNA, a visceral endoderm marker, and Mash-2 RNA, a trophoblast marker, was normal in EMeg32–/– embryos. In contrast, expression of Brachyury, an early marker of mesoderm formation (Kispert and Hermann, 1993), was mislocalized and less restricted in E6.5 EMeg32–/– embryos (Figure 2C and G), resembling Brachyury localization in wild-type E6.0 embryos. Expression of the Otx2 gene at E7.5 was not restricted to anterior ectoderm but was widespread in EMeg32 mutant epiblasts, thereby resembling Otx2 expression at earlier developmental stages (Figure 2D and H; Simeone et al., 1993). In summary, these results establish that inactivation of EMeg32 causes a growth and developmental delay that cannot be compensated for beyond E7.5.
Since this delay could be achieved by either increased apoptosis or a decrease in cellular proliferation, we examined embryos for: (i) fragmented DNA using the TUNEL assay; and (ii) the ability to incorporate 5-bromo-2′-deoxyuridine (BrdU), indicative of ongoing cellular DNA synthesis. EMeg32–/– embryos contained more TUNEL-positive apoptotic cells than wild-type embryos (Figure 3A–C). In addition, we found a reduction in proliferative capacity in all mutant embryos analyzed. EMeg32–/– embryos generally exhibited 20–25% fewer BrdU-positive cells than wild-type controls (Figure 3D).


Fig. 3. Increased apoptosis and decreased proliferation in EMeg32 mutant embryos. (A–C) TUNEL staining on sections of wild-type (A) and mutant (B, C) EMeg32 embryos at E6.5. Note the increased number of apoptotic cells (bright green) in the epiblast (ep) of mutant embryos. (D) BrdU incorporation into wild-type (open symbols) or mutant (filled symbols) EMeg32 embryos at E6.5 (triangles) or E7.5 (diamonds), respectively. The percentage of BrdU-positive nuclei was determined in fields of at least 150 or 230 cells for E6.5 or E7.5 embryos, respectively. Only embryonic tissue was evaluated.
Impaired proliferation in EMeg32–/– cells and resistance to apoptotic stimuli
When cultured in the presence of leukemia inhibitory factor (LIF), EMeg32 mutant and wild-type ES cells showed similar growth properties (Figure 4A). Furthermore, the number of wild-type and mutant ES cells able to form embryoid bodies (EBs) in the absence of LIF was comparable (Table I). In the absence of LIF, EMeg32–/– EBs reproducibly exhibited only 24–79% of the cellularity observed for wild-type EBs, depending on the ES cell line used, while the percentage of dead cells was similar (Table I, data not shown). Since EMeg32 is differentially expressed during adult hematopoiesis (Boehmelt et al., 2000), we tested the capacity of EBs to form hematopoietic colonies in vitro after trypsinization and replating. While wild-type and EMeg32+/– EBs formed hematopoietic colonies at the expected frequency, colonies from EMeg32–/– EBs were drastically reduced in number and size (Table I). The latter usually contained <50 cells, while wild-type colonies of the same type contained from 200 to >1000 cells (depending on the colony type). We interpret these findings as an impairment of EBs in proliferative rather than differentiative capacity.

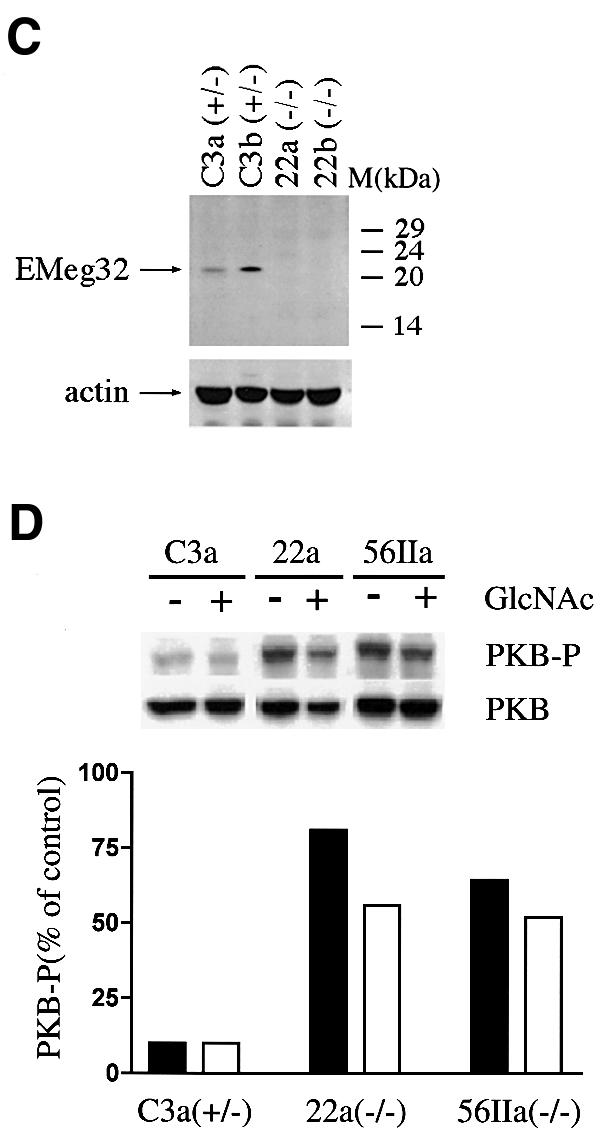
Fig. 4. Growth properties of EMeg32-deficient ES cells and MEFs. (A) Cumulative cell numbers from ES cells grown in the presence of LIF on gelatinized dishes. E14K (wild-type), square; C3 (+/–), diamond; C3-22 (–/–), circle; C3-56 (–/–), triangle. (B) Cumulative cell numbers of established MEF lines early after crisis. C3a (+/–), square; C3b (+/–), open triangle; 22a (–/–), circle; 22b (–/–), closed triangle. Average values of duplicate samples were plotted in (A) and (B). (C) Western blot demonstrating loss of EMeg32 protein (EMeg32) in –/– MEF lines (22a and 22b). +/– MEF lines (C3a and C3b) were loaded as controls. Anti-actin immunoblotting (actin) was used as loading control. (D) Constitutively activated PKB/AKT in EMeg32–/– cells. Western blot analysis of lysates from EMeg32–/– MEFs and heterozygous controls in the presence (+) or absence (–) of 10 mM GlcNAc. Antibodies used were anti-phospho-Ser473-PKB (PKB-P), anti-PKB raised against the unphosphorylated peptide (PKB) and anti-phospho-MAP kinase (control, not shown). Bands were quantified using a densitometer. Levels of phospho-PKB were plotted as a percentage of control. Black or white bars represent absence or presence of 10 mM GlcNAc, respectively.
Table I. Hematopoietic in vitro differentiation of ES cells.
| ES cell line | Embryoid bodies/1000 ES cells | Cells/embryoid bodya | Colonies in secondary culture | |
|---|---|---|---|---|
| Erythroidb | Non-erythroidb,c | |||
| E14K (+/+) | 91.5 (± 1.5)d | 714.7 | 10 (± 0) | 39 (± 0) |
| C3 (+/–) | 102 (± 1) | 710.4 | 14 (± 3) | 27 (± 5) |
| C3-22 (–/–) | 97.5 (± 0.5) | 160.1 | 0 (± 0) | 19.5 (± 3.5)e |
| C3-56 (–/–) | 89 (± 6) | 553.6 | 3 (± 3)e | 4.5 (± 2.5)e |
Next, we generated EMeg32+/– and EMeg32–/– MEFs after harvest of chimeric E12.5 embryos and G418 selection. Primary MEFs were split according to a 3T3 protocol until they reached crisis (with senescent, growth-arrested morphology). Heterozygous and homozygous mutant MEFs went into crisis at about the same passage number, but EMeg32–/– MEFs spontaneously immortalized much later (if at all) than control MEFs (Table II). Established EMeg32–/– MEF line cultures grew more slowly than heterozygous controls (Figure 4B) This was true for early passage and late passage cells (not shown), indicating that the difference in growth properties was a stable feature inherent to the culture. Absence of EMeg32 protein expression in these MEF lines was confirmed by western blotting (Figure 4C). Based on reactivity of the monoclonal antibody PAb240, which, under non-denaturing conditions, recognizes only mutant p53 (Gannon et al., 1990), we concentrated in the subsequent analysis on the MEF lines positive for mutated p53 (not shown). From this set of MEF lines, 22b–/– cells grew more quickly than other –/– lines, yet still more slowly than heterozygous controls.
Table II. Establishment of EMeg32–/– MEF lines.
| Line | Passage until crisis (3T3) | Days until establishmenta |
|---|---|---|
| C3a (+/–) | 5 | 15 |
| C3b (+/–) | 6 | 13 |
| C3-IIa (+/–) | 6 | 16 |
| C3-IIb (+/–) | 5 | 22 |
| C322a (–/–) | 6 | 27 |
| C322b (–/–) | 6 | 29 |
| C3-56a (–/–) | 6 | 72 |
| C3-56b (–/–) | 7 | 42 |
| C3-56IIa (–/–) | 5 | 77 |
| C3-56IIb (–/–) | 6 | >101 |
We also tested whether there was a greater rate of apoptosis in EMeg32–/– MEFs, since this might contribute to the increased proliferation doubling times measured. Using a variety of apoptosis-inducing agents, e.g. anisomycin, cycloheximide, tumor necrosis factor α (TNFα), staurosporine, UV irradiation or sorbitol, we determined via annexin V/propidium iodide staining and fluorescence-activated cell sorting (FACS) analysis the rate of early and late apoptotic cells after 8, 24 and 48 h of treatment. Untreated EMeg32–/– MEF lines showed no increase in apoptosis over heterozygous controls, and treated EMeg32–/– MEFs withstood apoptotic stimuli better than +/– MEFs (Table III). These findings rule out cell death as the cause of the observed decrease in proliferation of –/– MEFs. Interestingly, the latter expressed higher levels of active PKB/AKT than control cells as detected by anti-phospho-Ser473 AKT antibody (Figure 4D). The constitutive expression of phospho-PKB/AKT in –/– MEFs was also maintained in the presence of stress (high cell density) or apoptosis-inducing conditions (sorbitol treatment). Application of 10 mM GlcNAc partially decreased phospho-PKB/AKT levels in EMeg32–/– MEFs (Figure 4D). Activation of PKB/AKT is thought to counteract proapoptotic pathways (Datta et al., 1999). The decreased rate of apoptosis occurring in EMeg32–/– MEFs appears to contradict the findings obtained with mutant embryos, but might relate to the wild-type p53 state of the latter (also see Discussion).
Table III. Reduced apoptosis in EMeg32–/– MEFs.
| Treatmenta | EMeg32 genotypeb | Apoptosis, % (SD)c |
|---|---|---|
| Untreatedd | +/– | 1.54 (0.89) |
| –/– | 1.75 (0.99) | |
| Anisomycin (50 µM) | +/– | 38.20 (1.41) |
| –/– | 14.05 (0.98) | |
| Staurosporine (2 µM) | +/– | 90.56 (5.32) |
| –/– | 75.98 (1.10) | |
| UV (40 mJ/cm2) | +/– | 14.07 (0.39) |
| –/– | 7.23 (0.39) | |
| Sorbitol (400 mM) | +/– | 51.11 (4.44) |
| –/– | 20.97 (0.97) | |
| Cycloheximide (10 µg/ml) | +/– | 15.54 (1.01) |
| –/– | 2.86 (0.74) | |
| Cycloheximide (1 µg/ml) | +/– | 3.62 (0.53) |
| –/– | 0.37 (0.12) | |
| TNFα (10 ng/ml) | +/– | 4.10 (1.49) |
| –/– | 1.15 (0.46) | |
| Cycloheximide (1 µg/ml) | +/– | 76.68 (2.71) |
| + TNFα (10 ng/ml) | –/– | 9.99 (1.44) |
Restoration of EMeg32 function rescues growth and adhesiveness defects
To test whether the growth defect observed in various EMeg32–/– cell types is mediated directly by lack of EMeg32 protein and activity, we stably expressed EMeg32 cDNA in heterozygous and homozygous mutant MEFs using the recently described murine stem cell virus (MSCV)-EMeg32 retrovirus (Boehmelt et al., 2000). EMeg32 protein was only detected in MSCV-EMeg32 infected (ME) –/– MEFs and was absent in MSCV-neo infected (MN) –/– MEFs, as expected (Figure 5A). Expression levels achieved for EMeg32 protein in –/– MEFs were stable, but three to five times lower than in +/– control cells. Ectopic expression of EMeg32 cDNA in +/– cells did not lead to a significant increase of EMeg32 protein levels (Figure 5A).

Fig. 5. Restoration of EMeg32 function rescues the defects. (A) Western blots of +/– (C3IIb, C3a) or –/– (22a, 56IIa) MEF lines stably expressing MSCV-neo (neo) or MSCV-EMeg32 retroviruses (E) were incubated with anti-EMeg32 #2087 or anti-actin antibodies. Arrows indicate the respective specific signals. (B) Cumulative cell numbers were determined as in Figure 4 of MEF lines stably expressing MSCV-neo (open symbols) or MSCV-EMeg32 (filled symbols). Genotypes of parental cells are indicated on the right. (C) Cumulative cell numbers of C3a (+/–) and 22a (–/–) MEFs in the absence or presence of 10 mM GlcNAc (/GlcNAc) were determined as in Figure 4. (D) MEFs (250 000) were plated for 20 min and the percentage of adherent cells determined. Error bars, SEM. Samples were preincubated for 48 h with (gray) or without (white) 10 mM GlcNAc. C3a (+/–), 22a (–/–) and 56IIa (–/–) MEFs were analyzed.
Population doubling times for ME –/– MEFs were shorter than those for the respective MSCV-neo cells (Figure 5B). While MSCV-neo cells behaved indistinguishably from uninfected cells, all ME –/– MEFs showed normalization of growth rates (Figure 5B and data not shown). However, ME –/– MEFs did not quite reach the growth rates of heterozygous cells, possibly due to the lower EMeg32 protein levels. Addition of 10 mM GlcNAc to EMeg32–/– MEFs also rescued the growth defect (Figure 5C). EMeg32–/– MEFs also had a drastic defect in substratum adhesiveness, which could be complemented by re-expression of EMeg32 or by administration with GlcNAc (Figure 5D; data not shown).
UDP-GlcNAc levels are decreased in EMeg32–/– cells
Loss of GlcN6P acetyltransferase activity in EMeg32–/– cells is expected to result in decreased cytosolic UDP-GlcNAc levels, which could lead to multiple defects in pathways that usually employ this nucleotide sugar (Figure 6A). We therefore analyzed: (i) levels of nucleotide sugars and their precursors in EMeg32 wild-type, +/– and –/– EBs and MEFs using HPLC analysis; (ii) the status of Golgi and cytosolic glycosylation; and (iii) GPI-linker formation in EMeg32–/– cells.


Fig. 6. Decrease and replenishment of UDP-GlcNAc levels in EMeg32–/– cells. (A) Scheme depicting the synthesis of nucleotide sugars (boxed) with the pathway relevant for EMeg32 activity in bold. Main routes for further use of UDP-GlcNAc are indicated. Doubleheaded arrows represent several enzymatic steps not depicted in detail. Adapted from Schachter (1978). (B) EMeg32+/– (dashed line) or EMeg32–/– (solid line) EBs were labeled for 90 min with [3H]GlcN and analyzed by HPLC. Positions of GlcN-6P, GlcNAc-6P and UDP-GlcNAc standards are marked by arrows. (C) HPLC elution profile at _A_254 nm. Cells used were C3a+/–, 56IIa–/– or 56IIa–/– MEFs supplemented with 10 mM GlcNAc for 24 h (–/– GlcNAc). Arrows indicate elution positions of UDP-GlcNAc and UDP-Gal; the peak between is UDP-GalNAc. (D) Titration of GlcNAc into the culture medium for 24 h and determination of UDP-HexNAc:UDP-Hex ratio by HPLC at A254 nm. EMeg32–/– MEFs (56IIa, open square; 22a, open circle); +/– MEFs (C3a, filled triangle). 56IIa or 22a MEFs re-expressing EMeg32 (filled square or circle, respectively) were also analyzed. The UDP-Hex levels remained unchanged.
Heterozygous EMeg32 EBs incubated for 90 min with 3H-labeled GlcN incorporated four to five times more label into the UDP-_N_-acetylhexosamine (UDP-HexNAc) pool than EMeg32–/– EBs (Figure 6B). Labeled material eluting in the position of GlcN-P accumulated in EMeg32–/– EBs, while a labeled peak coincident with the GlcNAc-P marker was smaller than that from EMeg32+/– EBs (Figure 6B). This indicates that the GlcN6P acetyltransferase activity is impaired in EMeg32–/– cells, but it also shows that GlcN can be converted to GlcNAc-P and UDP-GlcNAc at a reduced efficacy in EMeg32–/– cells via an alternative route (Figure 6A).
Analysis of UDP-GlcNAc steady-state levels in 22b, 22a and 56IIa EMeg32–/– MEF lines revealed 33, 7 and 2% of that measured in C3a EMeg32+/– cells, respectively, suggesting that the EMeg32-dependent pathway is the major source of UDP-GlcNAc in MEFs (Figures 6C and 7D). The addition of 10 mM GlcNAc to fibroblasts for 24 h rescued the UDP-GlcNAc pools in both 22a and 56IIa EMeg32-deficient lines, without altering the UDP-hexose pool (Figure 6C). We observed a linear accumulation of UDP-GlcNAc in response to increasing amounts of GlcNAc or glucosamine (GlcN) in mutant and wild-type cells (Figure 6D, not shown). Rescue of UDP-GlcNAc pools to wild-type levels in EMeg32–/– fibroblasts was also accomplished with GalNAc, whereas supplements of d-mannose or d-glucose to the medium did not rescue UDP-GlcNAc pools in the mutant cells (Figure 6A, data not shown). Importantly, the UDP-GlcNAc pool was also normalized to wild-type levels in ME –/– MEFs, but not in control MN –/– MEFs (Figure 6D).




Fig. 7. Differential impairment of downstream routes of UDP-GlcNAc in EMeg32–/– cells. (A) Normalized lysates of 8 day differentiated EMeg32+/+ (E14K), EMeg32+/– (C3) and EMeg32–/– (C3-22, C3-56) EBs were blotted and analyzed using biotinylated lectins WGA, l-PHA or ConA in the presence (ConA/competitor) or absence (ConA) of 1 M 1-α-methyl-d-glucose. (B) Western blot of EB lysates [genotypes as in (A)] analyzed for E-cadherin expression (arrow). (C) LAMP-1 expression and glycosylation in EMeg32+/– and EMeg32–/– MEFs expressing MSCV-neo (neo) or MSCV-EMeg32 (E). LAMP-1 forms are indicated by a bracket; protein standards are in kDa at the right. (D) β-1,4-galactosyltransferase assay (open bars) on EMeg32+/– (C3a, C3IIb) and EMeg32–/– (22a, 56IIa, 22b) MEFs in relation to their UDP-GlcNAc:UDP-Gal ratio (filled bars) done by duplicate HPLC analysis as described in Figure 6C. (E) β-1,4-galactosyltransferase-mediated [3H]galactose labeling of cytosolic extracts prepared from C3a EMeg32+/– (filled triangle) or 56IIa EMeg32–/– MEFs grown in the absence (open square) or presence (open circle) of 10 mM GlcNAc. (F–H) GPI-linker formation as detected by proaerolysin sandwich-western of lysates from EBs (F), MEFs (G) and MEFs expressing MSCV-neo or MSCV-EMeg32 (G).
Glycosylation and GPI linker formation in EMeg32 mutant cells
Next, we analysed whether ER/Golgi-mediated glycosylation is impaired in EMeg32–/– cells. Extracts of EMeg32–/– EBs cultured for 8 days in the absence of LIF together with controls were subjected to immunoblot analysis using biotinylated lectins concanavalin A (ConA), wheatgerm agglutinin (WGA) and l-phytohemagglutinin (l-PHA). ConA binds to branched oligomannose chains on N-linked glycoproteins, WGA recognizes sialic acid and GlcNAc on N- and O-linked oligosaccharides and l-PHA recognizes the trisaccharide Galβ(1,4)GlcNAc-β(1,6)Man found specifically only on complex N-linked glycans.
No difference between EMeg32–/– and control EBs in the overall amount of glycoproteins was detected for the three lectins used (Figure 7A). Specificity of WGA staining was demonstrated by successful competition with GlcNAc (not shown), and ConA binding was abolished in the presence of 1-α-methyl-d-glucose (Figure 7A). A similar result was obtained upon analysis of extracts from undifferentiated ES cells cultured in the presence of LIF (data not shown). We found no difference between heterozygous and homozygous mutant EMeg32 MEFs in the overall pattern of glycosylated proteins in lectin blots, which was true for growing cultures and cultures deprived of serum for 18 h (not shown).
The _N_-glycosylation pattern of individual proteins was also analysed. Using anti-E-cadherin immunoblotting, we detected only fully glycosylated E-cadherin, migrating as a 120 kDa glycoprotein in sodium dodecyl sulfate (SDS)–polyacrylamide gels, in EMeg32–/– and control EBs (Figure 7B). Furthermore, lysosome-associated membrane protein 1 (LAMP-1), a glycoprotein migrating at 110 kDa in SDS–polyacrylamide gels when glycosylated with its 18 N-linked chains, appeared glycosylated in the EMeg32–/– MEF lines 22a and 22b and their respective MSCV-neo derivatives (Figure 7C). 22a/MSCV-EMeg32 MEFs showed slightly more slowly migrating forms of LAMP-1 than 22a/MSCV-neo cells. In EMeg32–/– 56IIa cells, only a faint LAMP-1 signal was obtained. Re-expression of EMeg32 or supplementing the cultures with 10 mM GlcNAc fully restored LAMP-1 expression, while MSCV-neo expression did not (Figure 7C, data not shown). In summary, these findings indicate that removal of EMeg32’s enzyme activity does not lead to generally impaired substitution of Asn-X-Ser/Thr sequences with oligosaccharides in the ER, but rather to a modest effect on Golgi processing.
To assess the extent of cytosolic O-linked GlcNAc modifications of proteins in EMeg32–/– MEFs, we labeled cytosolic extracts with UDP-[3H]Gal using β-1,4- galactosyltransferase. This enzyme specifically transfers Gal residues to unsubstituted GlcNAc residues on proteins, the majority of which are O-linked GlcNAc (Torres and Hart, 1984). The levels of cytosolic O-linked GlcNAc were much lower in lysates prepared from EMeg32–/– cells than in those from EMeg32+/– cells, except for 22b MEFs (Figure 7D). The defect in O-linked GlcNAc was rescued in the presence of 10 mM GlcNAc for both 22a and 56IIa cells (Figure 7E; data not shown).
To test for the presence of GPI-linked proteins, we made use of the pore-forming toxin proaerolysin, which has been reported to bind to the GPI moiety of most GPI-linked proteins with high affinity (Nelson et al., 1997; Diep et al., 1998). Cell lysates were subjected to proaerolysin sandwich-western analysis. Eight day differentiated EBs showed two predominant, GPI-linked proteins of 18 and 35 kDa and a characteristic pattern of several less abundant GPI-linked proteins in all samples, irrespective of EMeg32 genotype (Figure 7F). In the EMeg32–/– MEF lines 22a and 56IIa there was dramatic accumulation of four GPI-linked proteins between 11 and 31 kDa as compared with +/– controls or 22b cells (Figure 7G). The same pattern was observed in 22a and 56IIa cells expressing MSCV-neo. However, in 22a and 56IIa ME–/–MEFs, the smaller three proteins were absent (Figure 7H). In summary, GPI-linked proteins are produced in EMeg32–/– cells and their reduced UDP-GlcNAc levels are apparently not limiting for this important post-translational modification (also see Discussion).
Impaired actin depolymerization in EMeg32–/– MEFs
The defects of EMeg32–/– cells in substratum adhesiveness point towards problems in cytoskeletal dynamics. Furthermore, GNA1/PAT1 yeast mutants can be complemented by mutated SAC1 (Lin et al., 1996), which is implicated in regulation of actin dynamics (Cleves et al., 1989). We therefore analyzed whether actin dynamics are altered in EMeg32–/– cells. To this end, we made use of latrunculin A, which binds actin monomers (Ayscough, 1998) and hence prevents polymerization of actin. Treatment of cells with latrunculin A therefore allows assessment of depolymerization of exisiting actin bundles.
Using immunofluorescence analysis with fluorescein isothiocyanate (FITC)-conjugated phalloidin, we observed a drastic reduction in the number of actin bundles in control MEFs after a 30 min latrunculin A treatment. More than 80% of +/– cells had rounded up as a consequence of ongoing actin depolymerization and blocked polymerization of actin (Table IV). This effect was fully reversed within 1 h after removal of the drug. EMeg32–/– MEFs showed a dramatic resistance to latrunculin A, which manifested in the presence of clearly visible actin bundles in >40% of the cells (Table IV). The impairment in actin depolymerization was observed in several –/– MEFs, including 22b cells, to a comparable extent (Table IV). We also subjected the retrovirus-expressing MEFs to an actin cytoskeleton analysis using latrunculin A. Re-expression of EMeg32, but not plain MSCV-neo, rescued the defect in actin depolymerization to the levels seen in +/– cells (Figure 8A–H). As expected, additional expression of EMeg32 in +/– cells had no effect (Figure 8A–H). Impaired actin turnover could contribute to the growth suppression observed in EMeg32–/– cells.
Table IV. The defect in actin depolymerization is EMeg32-dependent.
| Latrunculin A treatment (min)a | MEF line | % actin bundle-positiveb |
|---|---|---|
| 120 | C3a (+/–) | 12.1 |
| 22a (–/–) | 44.4 | |
| 22b (–/–) | 44.8 | |
| 30 | C3IIb (+/–) | 17.9 |
| 22b (–/–) | 55.7 | |
| C3a (+/–)-neo | 9.8 | |
| C3a (+/–)-EMeg32 | 11.4 | |
| 56IIa (–/–)-neo | 48.6 | |
| 56IIa (–/–)-EMeg32 | 10.5 |

Fig. 8. EMeg32-dependent defect in actin depolymerization. (A–H) MEFs were incubated with 500 nM latrunculin A (B, D, F and H) or solvent (A, C, E and G) for 30 min prior to fixing and phalloidin–FITC and DAPI staining to visualize actin and nuclei, respectively. Cell types analyzed were (A, B) C3a/MSCV-neo, (C, D) C3a/MSCV-EMeg32, (E, F) 56IIa/MSCV-neo, (G, H) 56IIa/MSCV-EMeg32 MEFs. Genotypes are indicated.
Discussion
We demonstrate that genetic inactivation of the mouse glucosamine 6-phosphate acetyltransferase, EMeg32, is essential for embryonic development, and that such targeting of the HXNP in EMeg32–/– cells generates resistance to apoptotic stimuli and defects in proliferation. Most defects can be ascribed to an impairment in the synthesis of the nucleotide sugar UDP-GlcNAc, which manisfests in the lack of cytosolic O-glycosylation and lesser perturbation of Golgi-driven glycosylation. The impairment in actin depolymerization is surprising and may be connected to altered PtdIns metabolism in EMeg32–/– cells (see below).
Golgi-mediated glycosylation, GPI-linker formation and redundancy of UDP-GlcNAc synthesis
One major route that uses UDP-GlcNAc is the ER/Golgi-mediated N- and O-glycosylation of secreted and membrane glycoproteins. In murine embryonic development, disruption of _N_-glycosylation by inactivation of enzymes early in the pathway has more severe phenotypes than inactivation of enzymes acting later in the pathway (Ioffe and Stanley, 1994; Metzler et al., 1994; Asano et al., 1997; Priatel et al., 1997; Granovsky et al., 2000). Compared with the tunicamycin-induced lethality at the blastocyst stage (Iwakura and Nozaki, 1985), the lethality by E7.5 in EMeg32–/– embryos occurs late. This argues against a general impairment of ER/Golgi-mediated glycosylation in these embryos. We corroborated this interpretation by ConA- and WGA-staining of embryos (data not shown) and by analysis of various EMeg32–/– cell types.
UDP-GlcNAc concentrations are reduced in EMeg32–/– ES cells, EBs and MEFs. The salvage of GlcN and GlcNAc from serum and metabolic processes may be responsible for the cellular UDP-GlcNAc concentration in these EMeg32-deficient cells being 2–33% of the normal concentration. Apparently, UDP-GlcNAc levels in MEFs of ∼5% of normal suffice to provide UDP-GlcNAc for N-linked glycosylation in the ER and significant maturation by Golgi pathways. The alterations in LAMP-1 expression in 22a and 56IIa mutant MEFs might be explained through the suboptimal efficiency of Golgi nucleotide–sugar transporters and insufficient substrate delivery for medial or _trans_-Golgi GlcNAc transferases.
The order of depletion of UDP-GlcNAc in MEF lines, 56IIa > 22a > 22b, correlated with the impairment of growth, the degree of glycosylation defects and the accumulation of additional GPI-linked protein species. Since only one GPI anchor can be attached to a single polypeptide chain, it is possible that the three small proteins in Figure 7 constitute: (i) individual GPI-linked entities with increased expression in –/– MEFs; (ii) accumulated processing intermediates of a single polypeptide that, in addition to a GPI-linker is, for example, also modified by _N_-glycosylation at several sites; or (iii) degradation products of larger GPI-linked proteins. Accumulation in the latter two cases could be achieved through impaired routing of transport vesicles to or from the plasma membrane. EMeg32 has been found associated with Golgi and endosomal membranes and copurifies with p97/VCP (Boehmelt et al., 2000), which is also involved in ubiquitin-dependent protein degradation (Dai et al., 1998). Further studies are required to address whether the activity of p97/VCP is altered in EMeg32–/– cells.
Failure in cytosolic O-glycosylation
The most severe defect in downstream UDP-GlcNAc routes of EMeg32–/– cells is the apparent lack of cytosolic O-linked GlcNAc modifications as measured by the β-1,4-galactosyltransferase assay. Cytoplasmic and nuclear proteins are reversibly modified by single GlcNAc residues on Ser/Thr residues through the action of highly conserved cytosolic _O_-GlcNAc transferases (Kreppel et al., 1997; Lubas et al., 1997) and _N_-acetyl-β-d-glucosaminidases (Dong and Hart, 1994). Such _O_-GlcNAc modification is believed to compete in some instances with the action of Ser/Thr kinases (for reviews see Van den Steen et al., 1998; Comer and Hart, 1999). The nuclear proteins likely to be affected by the lack of O-glycosylation in EMeg32–/– cells are c-Myc, SP1, estrogen receptor, serum response factor, p53 and the C-terminal domain of the large subunit of RNA polymerase II. Amidst cytoplasmic proteins, the actin-binding protein talin, keratin 8, 13 and 18, or the microtubule-associated proteins MAP-2, MAP-4 and Tau are reported to carry _O_-GlcNAc. Removal of _O_-GlcNAc modification makes eukaryotic initiation factor 2-associated protein p67 and transcription factor SP1 susceptible to degradation, whereas in the case of p53, and c-Myc, transcriptional activities are affected (also see below). Thus, the regulatory influence of _O_-GlcNAc modification on proteins and their lack in EMeg32–/– cells could account for the observed impairments. An even more severe phenotype could be expected if nuclear and cytoplasmic O-glycosylation was abolished completely. Indeed, inactivation of the X-linked gene for _O_-GlcNAc transferase was shown to be lethal for male ES cells (Shafi et al., 2000).
Apoptosis versus decreased proliferative capacity
The HXNP is also associated with the development of insulin resistance. Increased levels of UDP-GlcNAc as induced with GlcN, uridine, free fatty acids or high glucose, correlate with inhibition of phosphatidylinositol 3-kinase (PI-3K) and glycogen synthase activities. Conversely, elevated insulin concentrations reduce the UDP-GlcNAc concentration and activate PI-3K and PKB/AKT in fat and muscle cells in vivo (Hresko et al., 1998; Wang et al., 1998; Heart et al., 2000). It is not clear at present whether altered UDP-GlcNAc concentrations control a signaling pathway for cell survival, or simply represent a downstream consequence of ongoing survival signaling. We observed increased levels of phospho-PKB/AKT in EMeg32–/– MEFs (Figure 4D), which might relate to a signaling event mediated by decreased UDP-HexNAc:UDP-Hex ratio. Since activated PKB/AKT has been reported to block apoptotic signaling via inactivation of proteins like BAD or forkhead transcription factor(s) (for review see Datta et al., 1999), the resistance of EMeg32–/– MEFs towards a number of apoptotic stimuli could be based on constitutive PKB/AKT activation.
The resistance to apoptotic stimuli of our MEF lines is probably not contributed by an event downstream of p53, because mutated p53 protein was equally expressed in the EMeg32 mutants and controls analyzed. However, EMeg32–/– embryos should express wild-type p53. Since the DNA-binding activity of p53 can be enhanced by O-linked glycosylation of the C-terminal regulatory domain (Shaw et al., 1996), it is possible that more p53 molecules are transcriptionally inactive in EMeg32–/– embryos than in wild-type controls. G1 arrest-inducing p53 target gene products, like p21/WAF, would be less prominent and suitable stimuli could possibly induce apoptosis rather than G1 arrest in mutant embryos (Zhang et al., 1999).
The increase in activated PKB/AKT does not lead to accelerated proliferation of EMeg32–/– cells, although it has been suggested that this kinase also induces cell cycle progression (Brennan et al., 1997). Possibly, downstream pathways usually mediating this function of PKB/AKT are no longer operative because of decreased cytosolic O-glycosylation in EMeg32–/– cells. The accumulation of EMeg32 protein at G2–M (Boehmelt et al., 2000) could provide high UDP-GlcNAc levels at this phase of the cell cycle. Since Golgi-mediated protein modifications are halted at this stage, while cytosolic O-glycosylation should still be functional, reduced UDP-GlcNAc levels in EMeg32–/– cells could affect cell cycle-relevant protein modifications. In cells, the proliferative defect was rescued by restoring the reduced UDP-HexNAc:UDP-Hex ratio. Attempts to rescue the developmental defect by feeding GlcNAc to pregnant mice have not succeeded so far (data not shown).
Actin dynamics and PtdIns metabolism
The defect in actin depolymerization was shared by all mutant MEF lines analyzed; of the attributes analyzed, it was the most sensitive to decreases in intracellular UDP-GlcNAc levels. Since the cell cycle defects evoked by the inactivation of PAT1/GNA1 could be complemented by mutated SAC1 (Lin et al., 1996), it is possible that altered PtdIns metabolism could mediate some of the effects observed in PAT1/GNA1-null or EMeg32–/– cells.
Using phosphate labeling and thin layer chromatography, we found increased PtdIns-bisphosphate (PIP2) levels in –/– MEFs, which probably represents the PtdIns(4,5)P2 isoform, since this is the most abundant (data not shown). PIP2 regulates several actin-binding proteins, including profilin, gelsolin, α-actinin and actin depolymerization factor (ADF, cofilin). ADF/cofilin can sever F-actin and binding of PtdIns(4,5)P2 inhibits ADF/cofilin binding to actin (for review see Bamburg et al., 1999). Thus, increased levels of PIP2 in EMeg32–/– MEFs could account for an inhibition of ADF/cofilin function and stabilize existing actin bundles. It will be interesting to test whether the increased PIP2 levels in EMeg32–/– MEFs relate to an altered activity of a murine SAC1 homolog.
Finally, impairments in the HXNP could influence PtdIns metabolism. Mediators of PI-3K activation, like insulin receptor substrate (IRS) 1 and 2 are modified by _O_-GlcNAc, and increased UDP-GlcNAc levels block PI-3K activity through decreased tyrosine phosphorylation of IRS-1 and -2 (Patti et al., 1999). Perhaps these PI-3K activators are more active in the unglycosylated form and lead to the increased PKB/AKT activity observed in EMeg32–/– cells. Further studies are clearly required to assess how the end products of the HXNP interfere with cellular signaling and the levels of phosphatidylinositols in particular. The EMeg32–/– cell system might prove to be an important tool to study these relationships.
Materials and methods
For histological and BrdU analysis, TUNEL assay, in situ hybridization, ES cell differentiation, apoptosis assays, retroviral expression, adhesiveness tests and protein detection procedures, see Supplementary data (available at The EMBO Journal Online).
Constructs and generation of EMeg32-deficient ES cells
A 129/J mouse genomic library was screened with an EMeg32 cDNA probe (nt 56–695; Boehmelt et al., 2000). An EMeg32 pseudogene was identified. From two other phage clones, a 9.5 kb _Eco_RI fragment was sequenced. It covered coding exons 1 and 2 of EMeg32. Downstream genomic sequences were identified by PCR. A 525 bp short arm was generated by PCR, introducing a _Not_I and a _Sac_I site at the ends (Figure 1). This _Not_I–_Sac_I fragment and a 4.68 kb _Bst_1107I fragment were used to generate the targeting vector (Figure 1). Electroporations of target vector and G418 selection of ES cells were as described (Sirard et al., 1998). Two ES clones (M2R4H and M7R11C) showed correct homologous recombination (determined by Southern blotting with a 5′ flanking _Nco_I–_Eco_RV and a _neo_-specific probe) and transmitted the mutation to the germline. To generate EMeg32–/– ES cells, M7R11C and M2R4H ES cells were selected in a high concentration of G418 (7 mg/ml). Positive clones were identified by PCR and Southern blotting. Only derivatives of M7R11C were used in this study.
Cell culture, generation of EMeg32 mutant mice and determination of genotype
Culture of E14K ES cells and MEFs was as described (Sirard et al., 1998). Chimeric mice were generated by microinjection of EMeg32+/– ES cells into E3.5 C57BL6/J blastocysts and transfer to CD1 foster mothers. Further handling was as described (Sirard et al., 1998). Primers routinely used for detection of the mutated allele were AS_neo_1476p (5′-GAGACTAGTGAGACGTGCTACTTC-3′) and SA3403m (5′-CTGTGATATCGGGTACGACTGATTC-3′). Primers specific for the wild-type allele were GEN6475p (5′-TACTGGAAAGATGAAACCCGATGAA-3′) and GEN7225m (5′-ACTTGGGAGGCAGAGACAGGTAGA-3′). F2 and F3 offspring from heterozygous intercrosses were used for most of the analyses in this study. Backcrossing into inbred C57BL6/J and outbred CD1 mice to the fourth and third generation, respectively, produced similar results in heterozygous intercrossings. Yolk sacs and embryos were analyzed by PCR as described (Sirard et al., 1998).
Generation of EMeg32-deficient MEFs
Heterozygous or EMeg32–/– ES cells were microinjected into E3.5 C57BL6/J blastocysts, which were transferred to CD1 foster mothers. On day 12.5 of pregnancy, MEFs were prepared from chimeric embryos and subjected to G418 selection (0.7 mg/ml). Cultures were split 1:3 every 3 days on to 10 cm tissue culture dishes. When cells reached crisis, medium was replaced every 3 days. Every 6 days, cells were trypsinized to be fully reseeded on to fresh dishes. G418 was maintained in the cultures until heterozygous MEFs left crisis. Two independently generated sets of EMeg32+/– and EMeg32–/– MEFs were used for this study.
Analysis of nucleotide sugars by HPLC
MEFs were grown in αMEM (Life Technologies) plus 10% fetal calf serum (MS). Nucleotide–sugars were extracted, separated on a Partisil SAX anion exchange high-performance liquid chromatography (HPLC) column (4.6 × 250 mm) and quantified by UV absorption at 254 nm as described (Robinson et al., 1995). To label nucleotide–sugar pools, trypsinized cells were washed once with medium, then 2 × 106 cells in 200 µl MS were incubated with 6 µCi of [6-3H]_N_-glucosamine (22.7 Ci/mmol, NEN, Boston) for 90 min. Cells were washed three times with 10 ml ice-cold phosphate-buffered saline (PBS) and nucleotide–sugars were extracted (Robinson et al., 1995).
β-1,4-galactosyltransferase assay
Cell lysates were prepared in PBS containing 1% Triton X-100 and centrifuged at 10 000 g for 10 min (4°C). Aliquots of lysate were incubated at 37°C for 20 min in 50 µl of assay mixture containing buffer (10 mM HEPES pH 7.0, 5 mM MnCl2 and 10 mM galactose), 0.01 mM UDP-Gal, 10 mU of bovine galactosyltransferase (Sigma) and 0.5 µCi UDP-[1-3H]Gal (NEN). Reactions were stopped by addition of 5 µl 100 mM EDTA. Protein reaction products were isolated in the exclusion volume from Sepharose G50 columns according to Holt and Hart (1986).
Latrunculin A treatment and actin staining
Seventy per cent confluent MEF cultures were treated with 500 nM latrunculin A (Molecular Probes) or vehicle (dimethylsulfoxide) for 30 min, washed with PBS, fixed in 4% PBS-buffered paraformaldehyde (for 15 min at room temperature) directly on the dish, permeabilized in PBS, 1% Trition X-100 (for 15 min at room temperature), blocked (for 30 min at room temperature) in PBS, 0.5% bovine serum albumin, 4% FCS and incubated (for 1 h at 37°C) with FITC-conjugated phalloidin (Sigma). Samples were washed, mounted in Vectashield (Vector Laboratories) containing DAPI (Sigma) (0.5 µg/ml) and evaluated using an immunofluorescence microscope coupled to a digital camera (Leica).
Supplementary data
Supplementary data for this paper are available at The EMBO Journal Online.
Acknowledgments
Acknowledgements
We cordially thank Charles E.Warren for biotinylated l-PHA, ConA and WGA, Gordon Keller for the kind gift of pretested PDS, Cybil Smith for sequencing, Martin Steegmaier for critical reading of the manuscript and Deborah Hyam for excellent technical assistance. This work was supported by NSERC, MRC and NCIC grants to J.W.D. and a special fellowship to G.B. from the Leukemia Society of America.
References
- Acharya U., Jacobs,R., Peters,J.M., Watson,N., Farquhar,M.G. and Malhotra,V. (1995) The formation of Golgi stacks from vesiculated Golgi membranes requires two distinct fusion events. Cell, 82, 895–904. [DOI] [PubMed] [Google Scholar]
- Asano M., Furukawa,K., Kido,M., Matsumoto,S., Umesaki,Y., Kochibe,N. and Iwakura,Y. (1997) Growth retardation and early death of β-1,4-galactosyltransferase knockout mice with augmented proliferation and abnormal differentiation of epithelial cells. EMBO J., 16, 1850–1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayscough K. (1998) Use of latrunculin-A, an actin monomer-binding drug. Methods Enzymol., 298, 18–25. [DOI] [PubMed] [Google Scholar]
- Bamburg J.R., McGough,A. and Ono,S. (1999) Putting a new twist on actin: ADF/cofilins modulate actin dynamics. Trends Cell Biol., 9, 364–370. [DOI] [PubMed] [Google Scholar]
- Boehmelt G., Fialka,I., Brothers,G., McGinley,M.D., Patterson,S.D., Mo,R., Hui,C.C., Huber,L.A., Mak,T.W. and Iscove,N.N. (2000) Cloning and characterization of the murine glucosamine-6-phosphate acetyltransferase EMeg32. J. Biol. Chem., 275, 12821–12832. [DOI] [PubMed] [Google Scholar]
- Brennan P., Babbage,J.W., Burgering,B.M., Groner,B., Reif,K. and Cantrell,D.A. (1997) Phosphatidylinositol 3-kinase couples the interleukin-2 receptor to the cell cycle regulator E2F. Immunity, 7, 679–689. [DOI] [PubMed] [Google Scholar]
- Cleves A.E., Novick,P.J. and Bankaitis,V.A. (1989) Mutations in the SAC1 gene suppress defects in yeast Golgi and yeast actin function. J. Cell Biol., 109, 2939–2950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comer F.I. and Hart,G.W. (1999) O-GlcNAc and the control of gene expression. Biochim. Biophys. Acta, 1473, 161–171. [DOI] [PubMed] [Google Scholar]
- Dai R.M., Chen,E., Longo,D.L., Gorbea,C.M. and Li,C.C. (1998) Involvement of valosin-containing protein, an ATPase co-purified with IκBα and 26S proteasome, in ubiquitin-proteasome-mediated degradation of IκBα. J. Biol. Chem., 273, 3562–3573. [DOI] [PubMed] [Google Scholar]
- Datta S.R., Brunet,A. and Greenberg,M.E. (1999) Cellular survival: a play in three Akts. Genes Dev., 13, 2905–2927. [DOI] [PubMed] [Google Scholar]
- Diep D.B., Nelson,K.L., Raja,S.M., Pleshak,E.N. and Buckley,J.T. (1998) Glycosylphosphatidylinositol anchors of membrane glycoproteins are binding determinants for the channel-forming toxin aerolysin. J. Biol. Chem., 273, 2355–2360. [DOI] [PubMed] [Google Scholar]
- Dong D.L. and Hart,G.W. (1994) Purification and characterization of an O-GlcNAc selective _N_-acetyl-β-d-glucosaminidase from rat spleen cytosol. J. Biol. Chem., 269, 19321–19330. [PubMed] [Google Scholar]
- Gannon J.V., Greaves,R., Iggo,R. and Lane,D.P. (1990) Activating mutations in p53 produce a common conformational effect. A monoclonal antibody specific for the mutant form. EMBO J., 9, 1595–1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granovsky M., Fata,J., Pawling,J., Muller,W.J., Khokha,R. and Dennis,J.W. (2000) Suppression of tumor growth and metastasis in Mgat5-deficient mice. Nature Med., 6, 306–312. [DOI] [PubMed] [Google Scholar]
- Guo S., Stolz,L.E., Lemrow,S.M. and York,J.D. (1999) SAC1-like domains of yeast SAC1, INP52, and INP53 and of human synaptojanin encode polyphosphoinositide phosphatases. J. Biol. Chem., 274, 12990–12995. [DOI] [PubMed] [Google Scholar]
- Heart E., Choi,W.S. and Sung,C.K. (2000) Glucosamine-induced insulin resistance in 3T3-L1 adipocytes. Am. J. Physiol. Endocrinol. Metab., 278, E103–E112. [DOI] [PubMed] [Google Scholar]
- Hirschberg C.B., Robbins,P.W. and Abeijon,C. (1998) Transporters of nucleotide sugars, ATP, and nucleotide sulfate in the endoplasmic reticulum and Golgi apparatus. Annu. Rev. Biochem., 67, 49–69. [DOI] [PubMed] [Google Scholar]
- Holt G.D. and Hart,G.W. (1986) The subcellular distribution of terminal _N_-acetylglucosamine moieties. Localization of a novel protein–saccharide linkage, O-linked GlcNAc. J. Biol. Chem., 261, 8049–8057. [PubMed] [Google Scholar]
- Hresko R.C., Heimberg,H., Chi,M.M. and Mueckler,M. (1998) Glucosamine-induced insulin resistance in 3T3-L1 adipocytes is caused by depletion of intracellular ATP. J. Biol. Chem., 273, 20658–20668. [DOI] [PubMed] [Google Scholar]
- Ioffe E. and Stanley,P. (1994) Mice lacking _N_-acetylglucosaminyltransferase I activity die at mid-gestation, revealing an essential role for complex or hybrid N-linked carbohydrates. Proc. Natl Acad. Sci. USA, 91, 728–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwakura Y. and Nozaki,M. (1985) Effects of tunicamycin on preimplantation mouse embryos: prevention of molecular differentiation during blastocyst formation. Dev. Biol., 112, 135–144. [DOI] [PubMed] [Google Scholar]
- Kispert A. and Hermann,B.G. (1993) The Brachyury gene encodes a novel DNA binding protein. EMBO J., 12, 4898–4899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kochendörfer K.U., Then,A.R., Kearns,B.G., Bankaitis,V.A. and Mayinger,P. (1999) Sac1p plays a crucial role in microsomal ATP transport, which is distinct from its function in Golgi phospholipid metabolism. EMBO J., 18, 1506–1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreppel L.K., Blomberg,M.A. and Hart,G.W. (1997) Dynamic glycosylation of nuclear and cytosolic proteins. Cloning and characterization of a unique O-GlcNAc transferase with multiple tetratricopeptide repeats. J. Biol. Chem., 272, 9308–9315. [DOI] [PubMed] [Google Scholar]
- Lin R., Allis,C.D. and Elledge,S.J. (1996) PAT1, an evolutionarily conserved acetyltransferase homologue, is required for multiple steps in the cell cycle. Genes Cells, 1, 923–942. [DOI] [PubMed] [Google Scholar]
- Lubas W.A., Frank,D.W., Krause,M. and Hanover,J.A. (1997) O-Linked GlcNAc transferase is a conserved nucleocytoplasmic protein containing tetratricopeptide repeats. J. Biol. Chem., 272, 9316–9324. [DOI] [PubMed] [Google Scholar]
- Metzler M., Gertz,A., Sarkar,M., Schachter,H., Schrader,J.W. and Marth,J.D. (1994) Complex asparagine-linked oligosaccharides are required for morphogenic events during post-implantation development. EMBO J., 13, 2056–2065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mio T., Yamada-Okabe,T., Arisawa,M. and Yamada-Okabe,H. (1999) _Saccharomyces cerevisiae GN_A1, an essential gene encoding a novel acetyltransferase involved in UDP-_N_-acetylglucosamine synthesis. J. Biol. Chem., 274, 424–429. [DOI] [PubMed] [Google Scholar]
- Nelson K.L., Raja,S.M. and Buckley,J.T. (1997) The glycosylphosphatidylinositol-anchored surface glycoprotein Thy-1 is a receptor for the channel-forming toxin aerolysin. J. Biol. Chem., 272, 12170–12174. [DOI] [PubMed] [Google Scholar]
- Patti M.E., Virkamaki,A., Landaker,E.J., Kahn,C.R. and Yki-Jarvinen,H. (1999) Activation of the hexosamine pathway by glucosamine in vivo induces insulin resistance of early postreceptor insulin signaling events in skeletal muscle. Diabetes, 48, 1562–1571. [DOI] [PubMed] [Google Scholar]
- Priatel J.J., Sarkar,M., Schachter,H. and Marth,J.D. (1997) Isolation, characterization and inactivation of the mouse Mgat3 gene: the bisecting _N_-acetylglucosamine in asparagine-linked oligosaccharides appears dispensable for viability and reproduction. Glycobiology, 7, 45–56. [DOI] [PubMed] [Google Scholar]
- Rabouille C., Levine,T.P., Peters,J.M. and Warren,G. (1995) An NSF-like ATPase, p97, and NSF mediate cisternal regrowth from mitotic Golgi fragments. Cell, 82, 905–914. [DOI] [PubMed] [Google Scholar]
- Robinson K.A., Weinstein,M.L., Lindenmayer,G.E. and Buse,M.G. (1995) Effects of diabetes and hyperglycemia on the hexosamine synthesis pathway in rat muscle and liver. Diabetes, 44, 1438–1446. [DOI] [PubMed] [Google Scholar]
- Schachter H. (1978) Glycoprotein biosynthesis. In Pigman,D. and Horowitz,M.I. (eds), The Glycoconjugates. Academic Press, New York, NY, Vol. II, pp. 87–181. [Google Scholar]
- Shafi R., Iyer,S.P., Ellies,L.G., O’Donnell,N., Marek,K.W., Chui,D., Hart,G.W. and Marth,J.D. (2000) The O-GlcNAc transferase gene resides on the X chromosome and is essential for embryonic stem cell viability and mouse ontogeny. Proc. Natl Acad. Sci. USA, 97, 5735–5739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw P., Freeman,J., Bovey,R. and Iggo,R. (1996) Regulation of specific DNA binding by p53: evidence for a role for O-glycosylation and charged residues at the carboxy-terminus. Oncogene, 12, 921–930. [PubMed] [Google Scholar]
- Simeone A., Acampora,D., Mallamaci,A., Stornaiuolo,A., D’Apice,M.R., Nigro,V. and Boncinelli,E. (1993) A vertebrate gene related to orthodenticle contains a homeodomain of the bicoid class and demarcates anterior neuroectoderm in the gastrulating mouse embryo. EMBO J., 12, 2735–2747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirard C. et al. (1998) The tumor suppressor gene Smad4/Dpc4 is required for gastrulation and later for anterior development of the mouse embryo. Genes Dev., 12, 107–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres C.R. and Hart,G.W. (1984) Topography and polypeptide distribution of terminal _N_-acetylglucosamine residues on the surfaces of intact lymphocytes. Evidence for O-linked GlcNAc. J. Biol. Chem., 259, 3308–3317. [PubMed] [Google Scholar]
- Udenfriend S. and Kodukula,K. (1995) How glycosylphosphatidylinositol-anchored membrane proteins are made. Annu. Rev. Biochem., 64, 563–591. [DOI] [PubMed] [Google Scholar]
- Van den Steen P., Rudd,P.M., Dwek,R.A. and Opdenakker,G. (1998) Concepts and principles of O-linked glycosylation. Crit. Rev. Biochem. Mol. Biol., 33, 151–208. [DOI] [PubMed] [Google Scholar]
- Wang J., Liu,R., Hawkins,M., Barzilai,N. and Rossetti,L. (1998) A nutrient-sensing pathway regulates leptin gene expression in muscle and fat. Nature, 393, 684–688. [DOI] [PubMed] [Google Scholar]
- Whitters E.A., Cleves,A.E., McGee,T.P., Skinner,H.B. and Bankaitis,V.A. (1993) SAC1p is an integral membrane protein that influences the cellular requirement for phospholipid transfer protein function and inositol in yeast. J. Cell Biol., 122, 79–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y., Fujita,N. and Tsuruo,T. (1999) Caspase-mediated cleavage of p21Waf1/Cip1 converts cancer cells from growth arrest to undergoing apoptosis. Oncogene, 18, 1131–1138. [DOI] [PubMed] [Google Scholar]