Identification of Macrodomain Proteins as Novel O-Acetyl-ADP-ribose Deacetylases (original) (raw)
Abstract
Sirtuins are a family of protein lysine deacetylases, which regulate gene silencing, metabolism, life span, and chromatin structure. Sirtuins utilize NAD+ to deacetylate proteins, yielding_O_-acetyl-ADP-ribose (_O_AADPr) as a reaction product. The macrodomain is a ubiquitous protein module known to bind ADP-ribose derivatives, which diverged through evolution to support many different protein functions and pathways. The observation that some sirtuins and macrodomains are physically linked as fusion proteins or genetically coupled through the same operon, provided a clue that their functions might be connected. Indeed, here we demonstrate that the product of the sirtuin reaction_O_AADPr is a substrate for several related macrodomain proteins: human MacroD1, human MacroD2, _Escherichia coli_YmdB, and the sirtuin-linked MacroD-like protein from Staphylococcus aureus. In addition, we show that the cell extracts derived from MacroD-deficient Neurospora crassa strain exhibit a major reduction in the ability to hydrolyze _O_AADPr. Our data support a novel function of macrodomains as _O_AADPr deacetylases and potential in vivo regulators of cellular_O_AADPr produced by NAD+-dependent deacetylation.
Keywords: Enzyme Catalysis, Enzyme Mechanisms, Protein Acylation, Protein Structure, Sirtuins
Introduction
Macrodomains are evolutionarily conserved structural domains found in proteins with diverse cellular functions (1, 2). Prior evidence suggested that macrodomains function as binding modules of NAD+ metabolites, including ADP-ribose/poly(ADP-ribose) (3, 4, 5, 6, 7) and_O_-acetyl-ADP-ribose (_O_AADPr)5 (8,9)._O_AADPr is produced in reactions catalyzed by NAD+-dependent protein/histone deacetylases (10, 11), which regulate gene silencing, metabolic enzymes, life span, and many other cellular processes (12, 13, 14)._O_AADPr has been implicated as a signaling molecule, modulating cellular processes affected by NAD+-dependent protein/histone deacetylation (15, 16, 17). The binding of_O_AADPr and other NAD+ metabolites to macrodomains such as the histone variant macroH2A1.1 (1, 5, 8, 9) suggests a possible connection between metabolic regulation, gene activity, and chromatin structure. The mechanism by which cells regulate and utilize _O_AADPr is not well understood. In yeast, the NUDIX ADP-ribose pyrophosphatase Ysa1 modulates the cellular levels of both ADPr and _O_AADPr, converting each to AMP and the corresponding ribose-phosphate (17). Cells lacking ysa1 exhibit an increased resistance to oxidative insults and produce lower levels of endogenous reactive oxygen species. In vitro, the poly (ADP-ribose) glycohydrolase ARH3 was capable of removing the acetyl group from_O_AADPr (18), though the activity was orders of magnitude slower than that observed for the NUDIX family (19). In eukaryotic cell extracts at least two different cellular activities were shown to contribute to _O_AADPr deacetylation (19), but the identities of these factors remain unknown.
Here we report a direct functional connection between sirtuins and a family of macrodomain proteins. We demonstrate that proteins belonging to this distinct branch of macrodomains are _O_AADPr deacetylases that efficiently catalyze the hydrolysis of_O_AADPr to produce ADP-ribose and free acetate. This group comprises eukaryotic MacroD proteins (orthologues of human MacroD1 and MacroD2), bacterial YmdB proteins and sirtuin-linked macrodomain proteins from certain pathogenic bacteria and fungi. Sirtuin-linked macrodomains constitute a macrodomain subfamily that are either fusions with sirtuin proteins or are genetically coupled through the same operon.
EXPERIMENTAL PROCEDURES
Materials
3H-Labeled acetyl-CoA was purchased from Moravek Biochemicals (Brea, CA). Synthetic peptide corresponding to the 11 amino acids surrounding lysine-14 of the histone H3, H2N-KSTGGKAPRKQ-CONH2(11-mer H3 peptide), was purchased from the University of Wisconsin-Madison Biotechnology Center Peptide Synthesis Facility. [3H]acetyl H3 peptide, H2N-KSTGGK(3H-acetyl)APRKQ-CONH2, was synthesized enzymatically from the 11-mer H3 peptide and purified as previously described (20). All other chemicals used were of the highest purity commercially available and were purchased from Sigma, Aldrich, or Fisher Scientific.
Constructs and Cloning
Human MacroD1, MacroD2, and GDAP2 were amplified from a human HeLa cDNA library. MacroD-like protein SAV0325 was cloned from S. aureus genomic DNA (ATCC). All genes were cloned into the pDONR221 entry vector. Specific point mutations were introduced using the QuickChange II site-directed mutagenesis kit (Stratagene). The two MacroD1 truncation mutants were generated in order to exclude the first 77 amino acids and the first 135 amino acids, respectively.
Protein Expression and Purification for Enzyme Assays
Expression and purification of HST2 (11, 21) and nicotinamidase from Salmonella enterica fused to maltose-binding protein (MBP-PncA) (22) were performed as described previously. Overexpression of the His-tagged macrodomain proteins was achieved by transforming _E. coli_BL21(DE3) cells with plasmids containing the macrodomain gene inserts (pDEST17-MacroD1, pDEST17-MacroD2, pET41a-YmdB, or pET28-SAV0325) and induction of mid-log phase cells (_A_600 ∼0.7) with 0.1 mmisopropyl-1-thio-β-d-galactopyranoside at 23 °C. All protein purification and cell extract preparation were performed at 4 °C or on ice. Cells from two 1-liter cultures are harvested by centrifugation, re-suspended in lysis buffer (50 mm Tris-HCl at pH 8.0, 250 mm NaCl, 1 mm 2-mercaptoethanol, 0.2 mm phenylmethylsulfonyl fluoride), and lysed by double passages of the suspended cells through a French press cell at 10,000–12,000 psi. Cell debris, large ribosomal protein and RNA were removed by ultracentrifugation for 45 min at 46,000 rpm (174,000 × g) using a Beckman 55.2Ti rotor. Supernatant was mixed with 8 ml bed volume of Ni-NTA resin equilibrated with wash buffer (50 mm Tris-HCl at pH 8.0, 250 mm NaCl, 1 mm 2-mercaptoethanol, 10 mm imidazole) and incubated for 1 h with gentle shaking. The protein-resin sample was centrifuged at 4000 × g for 5 min. The pellet was resuspended in ∼30 ml wash buffer and loaded onto a 1.5 × 5 cm column. After being washed with 200 ml of wash buffer, proteins were eluted with a 150 ml of linear gradient formed with wash buffer and elution buffer (50 mm Tris-HCl at pH 8.0, 250 mm NaCl, 1 mm 2-mercaptoethanol, 250 mm imidazole). Fractions containing the macrodomain protein, as visualized through Coomassie Brilliant Blue-stained SDS-PAGE gel, were pooled and concentrated using Amicon Ultra centrifugal filters. The concentrated proteins were dialyzed against two changes of 2-liter dialysis buffer (50 mm Tris-HCl at pH 8.0, 25 mm NaCl, 10% glycerol, and 3 mm DTT) or were passed through a 2.5 × 50 cm Sephadex G-75 column equilibrated with dialysis buffer. The concentration of the purified proteins was determined by the method of Bradford using bovine serum albumin as the standard (23). The purified protein aliquots were re-concentrated, flash-frozen in liquid nitrogen, and stored at −80 °C.
Synthesis of [3H]OAADPr
O-[3H]acetyl-ADP-ribose ([3H]_O_AADPr) was synthesized enzymatically using yeast deacetylase HST2 following procedures described previously (11) with the modification that 5 μm MBP-PncA was included in the synthetic reaction to increase [3H]_O_AADPr yield. Briefly, the reaction consisted of 20 μm HST2, 5 μm MBP-PncA, 1.0 mm NAD+, and 1.5 mm [3H]acetyl-H3 peptide in 50 mm Tris-HCl at pH 7.3. The reaction was carried out at 23 °C and monitored enzymatically for ammonium formation using glutamate dehydrogenase. Trifluoroacetic acid (TFA) was added to 1% (v/v) upon reaction completion. [3H]_O_AADPr was purified by HPLC using a 10 × 250 mm small pore Vydac C18 column as described. The purified [3H]_O_AADPr is lyophilized and re-dissolved in de-ionized water to make ∼10 μm solution with specific activity of ∼2.0 × 105 CPM/μl. Identity and purity were confirmed by comparison with authentic_O_AADPr using analytic HPLC. The [3H]_O_AADPr solution was aliquoted into single-use units, flash-frozen in liquid nitrogen, and stored at −80 °C.
Enzymatic Assays
Enzyme assays for _O_AADPr deacetylation to ADPr and acetate were performed by radioactive as well as HPLC methods. In the radioactive assays, the reactions contained from 0 to 1,600 μm[3H]_O_AADPr and 0 to 2.0 μm macrodomain enzymes in 50 to 200 mm Tris-HCl or 50 mm phosphate buffer at pH 7.3 in a total reaction volume of 250 μl. The reactions were initiated by the addition of macrodomain enzymes at 23 °C. A 40-μl aliquot was taken at the desired time points and mixed with 60 μl of charcoal slurry to terminate the reactions. Charcoal slurry was prepared by mixing 1 volume of activated charcoal with 1.5 volumes of 100 mm Tris-HCl or phosphate at pH 7.3. Activated charcoal effectively terminates the enzymatic reaction independent of the pH and temperature of the reactions (21). Under these conditions, ADPr, _O_AADPr, and proteins bind to the charcoal, leaving small molecular weight buffer component and acetate in solution. ADPr,_O_AADPr, and proteins bound to the charcoal are removed by centrifugation at 20,000 × g. After re-centrifugation to remove the residual charcoal, the supernatant was analyzed by scintillation counting to determine the radioactivity released from [3H]_O_AADPr to the solution. To assay enzyme activity in cell extracts, cells were lysed by sonication and cell debris was removed by centrifugation at 20,000 × g. Protein concentrations were normalized before being mixed with [3H]_O_AADPr substrates in the absence or presence of 1.25 mmEDTA in 50 mm Tris-HCl or phosphate buffer at pH 7.3 to start the reaction.
In the HPLC assays, the same reaction conditions are used except that the reactions are terminated by addition of TFA to 1% (v/v). Precipitated protein was removed by centrifugation. The supernatants were loaded onto a 4.6 × 250 mm analytical C18 column (Vydac) and analyzed by Shimadzu HPLC (LC 2010) under conditions described previously (17). HPLC elution profiles were monitored at 214 and 260 nm. Authentic acetate, ADPr, and_O_AADPr were used as standards under the same chromatographic conditions. Effluent fractions monitored at 260 nm were collected and characterized by mass spectrometry. The amount of ADPr formed was calculated from integration of the confirmed ADPr and _O_AADPr peaks and from the initial_O_AADPr concentration in the reactions. When [3H]_O_AADPr was used as substrate, effluent fractions monitored at 214 nm and 260 nm were collected and analyzed by scintillation counting.
LC-MS/MS Analyses of Reaction Products
Reactions of _O_AADPr deacetylation were stopped at desired time points by addition of TFA to 1% (v/v). The reaction mixtures were then centrifuged and the supernatants were mixed with acetonitrile to 50% (v/v). The solutions were centrifuged again to remove precipitated proteins. The supernatants were analyzed at the University of Wisconsin-Madison Biotechnology Center using an ABI 3200 Q-trap mass spectrometer coupled to an Agilent 1100 HPLC with an HILIC column (Nest Group, 300 Å, 5 μm, polyhydroxyethyl A, 4.6 × 200 mm). Samples were eluted into the mass spectrometer using conditions as described (24). The Q-trap mass spectrometer parameters were optimized using direct infusions of 1.0 μm _O_AADPr and 1.0 μm ADPr in 50% acetonitrile with a flow of 80 μl/min. The mass spectrometer was operated in negative SRM mode and monitored the SRM transitions 600.3/346.0 and 558.3/346.0 for _O_AADPr and ADPr, respectively.
Product Inhibitions
Product inhibition reactions were performed in 250 μl reaction volumes containing from 20 to 1,600 μm[3H]_O_AADPr, 0.5 μm macrodomain enzyme, and ADPr concentrations ranging from 0 to 400 μm in 50 mm Tris (pH 7.3) at 23 °C. Reactions were initiated by addition of enzyme and terminated by mixing an aliquot of the reaction mixtures with activated charcoal at desired time points. Inhibition was analyzed by measuring the initial forward rate of [3H]acetate formation as described above. The initial velocity data were fit and displayed using Kaleidagraph (Synergy Software, Reading, PA).
Protein Production and Crystallization
To obtain crystals of MacroD1, several truncated versions of the coding sequence were amplified by PCR and inserted into the expression vector pNH-TrxT using ligation-independent cloning (25). The clone used for crystallization encompassed aa 58–325 of MacroD1 downstream of a fusion tag, which consisted of a hexahistidine followed by the E. coli thioredoxin gene and a TEV-protease cleavage site. During the process of purification and crystallization, the protein was proteolyzed further, leading to loss of residues 58–81, and possibly an internal segment (see below), as verified by mass spectrometry of protein recovered from crystals. This unintentional degradation may have enabled us to achieve highly ordered crystals diffracting to 1.7 Å resolution. The structure includes residues 91–136 and 143–325; residues 81–90 and 137–142 are not visible in the electron density map.
For expression, the plasmids were transformed into BL21(DE3)-R3-pRARE2, a phage-resistant derivative of Rosetta2 (Merck). A glycerol stock was used to inoculate an overnight starter culture (37 °C) in LB supplemented with kanamycin (50 μg/ml) and chloramphenicol (34 μg/ml). The starter culture was then diluted 1.5:100 into 4 liters of Terrific Broth (TB) containing kanamycin but no chloramphenicol and grown in Ultrayield flasks at 37 °C to an_A_600 of 1. The cultures were transferred to 19 °C, and after 45 min, IPTG was added to 0.75 mm. The growth was continued overnight at 19 °C. The cells were harvested by centrifugation, suspended in 105 ml of lysis buffer (50 mm HEPES, pH 7.4, 500 mm NaCl, 5% glycerol, 5 mm imidazole pH 7.4, 0.5 mm TCEP) supplemented with Calbiochem Protease inhibitor set VII (1:5000 dilution) and stored at −80 °C. The suspended cells were lysed by sonication. Polyethyleneimine (PEI) was added to a final concentration of 0.15% from a 5% (w/v, pH 7.5) stock, followed by stirring for 15 min, then centrifugation for 45 min at 25,000 × g to remove cell debris and nucleic acids.
The protein was purified by a succession of Ni-affinity chromatography, gel filtration, tag removal with TEV protease, a second Ni-affinity column to remove the tag and other impurities, and a final gel filtration. The initial lysate was loaded on a 5-ml HisTrap column (GE Healthcare) in lysis buffer. The column was washed with lysis buffer and lysis buffer containing 30 mm imidazole, and the bound protein was eluted with lysis buffer containing 300 mm imidazole. The eluted fraction was collected and injected on a Superdex 75 HR60/10 column equilibrated with GF buffer (20 mmHEPES, pH 7.5, 250 mm NaCl, 5% glycerol, 1.0 mm TCEP) and fractionated at 1.2 ml/min. Fractions containing MacroD1 identified by SDS-PAGE and mass spectrometry were combined and digested with histidine-tagged TEV protease (1:30 w/w) overnight at 4 °C. The cleaved tag, the protease and other contaminants were removed by passing the lysate over a 5-ml column of Ni-NTA (Qiagen); the flow-through fraction was concentrated to 3 ml using a 10-kDa cutoff spin concentrator (Millipore). The fraction was loaded on a Superdex 75 HR60/10 column and fractionated as before; the purified protein was concentrated to 76 mg/ml and frozen in liquid nitrogen. The mass of the protein following cleavage of the tag by TEV protease was lower than expected. Mass spectrometry indicated that the protein was lacking 25 aa downstream of the TEV protease cleavage site, resulting in a protein encompassing aa 82–325 of MacroD1.
For crystallization, the protein was diluted to 20 mg/ml in GF buffer. Crystals were grown in sitting drops using vapor diffusion, with a reservoir solution consisting of 0.2m(NH4)2SO4, 0.1 m HEPES pH 7.5, 25% PEG 3350 at 4 °C. Protein and reservoir solutions were mixed at a 1:2 ratio. Crystals emerged after 3 weeks.
Structure Determination
A crystal was cryoprotected with a mixture of 1 μl of reservoir solution and 1 μl of 100% PEG 300, which was added to the drop containing the crystal. This corresponds to a PEG 300 concentration of 50%. The crystal was flash-frozen in liquid nitrogen and all measurements were carried out at 100 K. Data collection was performed at Diamond Beamline 103 at a wavelength of 0.9795 Å. The data set was processed with iMOSFLM (26) and scaled and merged with Scala (27). The structure with the PDB ID 1SPV was used as a model for phasing in molecular replacement using Phaser (28). After initial phasing the model was further improved by an autobuild cycle with Buccaneer (29) followed by subsequent cycles of manual building in Coot (30) and refinement with Refmac (31). The final structure was refined to 1.7 Å resolution and_R_work/_R_free(%) of 16.4/20.6.
N. crassa Strains, Gene Knock-out, and Protein Extraction
General conditions for growth and manipulation of_N. crassa_ are described elsewhere (32). Strains were maintained on minimal medium. Knock-out of_NCU07925_, herein referred to as_macrod_, was carried out according to the published procedure (33), which entails gene replacement with a hygromycin phosphotransferase (hph) cassette by homologous recombination. Conidia harvested from 15-day cultures of the mus-52, his-3 Fungal Genetics Stock Centre (FGSC) strain 9720 of N. crassa were transformed with an hph cassette targeting the NCU07925 locus. The_hph_ cassette consisted of the hygromycin phosphotransferase (hph) gene flanked with 1.1–1.2 kb regions of genomic DNA found directly upstream and downstream of NCU07925. The 5′ flanking fragment was amplified with the primers 5′-GTAACGCCAGGGTTTTCCCAGTCACGACGGTAGACGTCGTTGAGGAAGC-3′ and 5′-ATCCACTTAACGTTACTGAAATCTCCAACACACAGATACGATACCGAGG-3′, while the 3′ flanking fragment was amplified with the primers 5′-CTCCTTCAATATCATCTTCTGTCTCCGACCAAGTTCAGTGTCAGAGACG-3′ and 5′-GCGGATAACAATTTCACACAGGAAACAGCCGGAATGTAGATGCTAGAGG-3′. The transformants were backcrossed to the wild-type strain FGSC 2489 to obtain homokaryotic strains. Progeny from crosses were screened for hygromycin resistance on minimal medium containing 200 μg/ml of hygromycin B (Duchefa). Hygromycin-resistant progeny were tested for_macrod_ deletion by Southern blotting and PCR. For protein extraction, conidia from 5–10-day wild-type and knock-out strains were resuspended in water, filtered through glass wool, and inoculated in 100 ml of minimal medium to a final concentration of 1.5 × 107 conidia/ml. Flasks were shaken on a rotary shaker at 125 rpm in constant light at 30 °C for 48 h, harvested by vacuum filtration onto filter paper, and frozen in liquid nitrogen. Protein extracts from the wild-type and knock-out strains were obtained as previously reported (34). The extraction buffer consisted of 50 mm Tris-Cl pH 7.5, 50 mm NaCl, 5 mm EDTA, 1 mm DTT, and protease inhibitors (Roche).
Poly(ADP-ribose) Hydrolysis Activity Assay
32P-labeled automodified PARP1 was used as a substrate (35) to analyze poly(ADP-ribose) hydrolysis ability of recombinant proteins. 1 μmmacrodomain proteins or 10 nm human PARG (Trevigen) were added to poly(ADP)-ribosylated PARP1 substrate in the buffer containing 50 mm Tris, pH 7.5, 50 mm NaCl, and 5 mm MgCl2 at room temperature. The reaction products were analyzed by SDS-PAGE and visualized by autoradiography.
RESULTS
MacroD-like Proteins
Macrodomains are found in numerous proteins from all kingdoms of life, either as a self-standing domain or appended to other functional domains. In humans, there are at least 10 different macrodomain proteins (7) (Fig. 1A), however their function is largely unknown. The two human macrodomain proteins MacroD1 and MacroD2 appear to be highly related to each other. Sequence comparisons with phylogenetic analysis place them close to bacterial YmdB proteins and to sirtuin-linked proteins from pathogenic bacteria and fungi (e.g. SAV0325 from_Staphylococcus aureus_ and_Candida_ MacroD-like protein) (Fig. 1, A and_B_) (8). The observation that some sirtuins and macrodomains co-exist as either fusion proteins or are genetically coupled through the same operon provides a compelling rationale that there is an important link between their cellular functions. To explore the molecular and biological functions of this subgroup of macrodomain proteins (MacroD-like proteins), we performed detailed biochemical analyses and solved the crystal structure of human MacroD1.
FIGURE 1.

MacroD-like proteins as a distinct subfamily of macrodomain proteins that includes sirtuin-linked factors.A, phylogenetic tree illustrating relationships between different macrodomain proteins. Branch of MacroD-like proteins is shown in_red_. Schematic representation of the genome arrangements of sirtuin-linked macrodomain proteins is shown in the_box_, and it relates to MacroD protein from_C. albicans_ and SAV0325 protein from S. aureus shown in the tree. B, structure-based alignment of macrodomain proteins. The protein chains were aligned according to three-dimensional structures. A sequence-based alignment (ClustalW) was then manually adjusted to align spatially equivalent residues. The boxed regions are the N-terminal segments common to human MacroD1 and MacroD2 but absent from other macrodomain proteins; the_red lettering_ above the sequences indicate residues which, when mutated, resulted in reduced enzymatic activity. The_cylinders_ and arrows above the sequences represent, respectively, helices and β-strands in the structure of MacroD1. The proteins are: MacroD1 (PDB code 2X47, this study); MacroD2 (sequence only), E. coli YmdB (PDB code 1SPV); Yeast YMX7 (1TXZ)(37); _Thermus thermophilus_hypothetical protein (2DX6); SARS virus (2FAV)(4); Human histone H2A1.1 (3IIF)(36), human PARP15 macrodomain (3KH6), and human GDAP2 (sequence only).
Overall Structure of Human MacroD1
The MacroD1 crystal structure (Fig. 2A and supplemental Table S1) includes residues 91–325 (residues 137–142 are not visible in the electron density map) and constitutes a macrodomain and an N-terminal region (residues 91–136, depicted in orange) rich in basic residues (Fig. 2B). This N-terminal region, which is common to other vertebrate MacroD1 and MacroD2 proteins, is arranged as an elongated chain of helical segments and a short β-strand. The macrodomain region of human MacroD1 encompasses amino acids 151–322 and exhibits the canonical macrodomain fold (Fig. 2C). This core fold consists of a three-layered α-β-α sandwich with a central six-stranded β-sheet. Shown in green,Fig. 2A depicts the arrangement of the macrodomain core. Structure-based sequence alignments show that human MacroD1 is highly related to the YmdB protein from_E. coli_ (whose structure was deposited as PDB ID 1SPV), suggesting that MacroD1, MacroD2, and bacterial YmdB proteins are likely to be structural and functional homologues (Fig. 2C).
FIGURE 2.
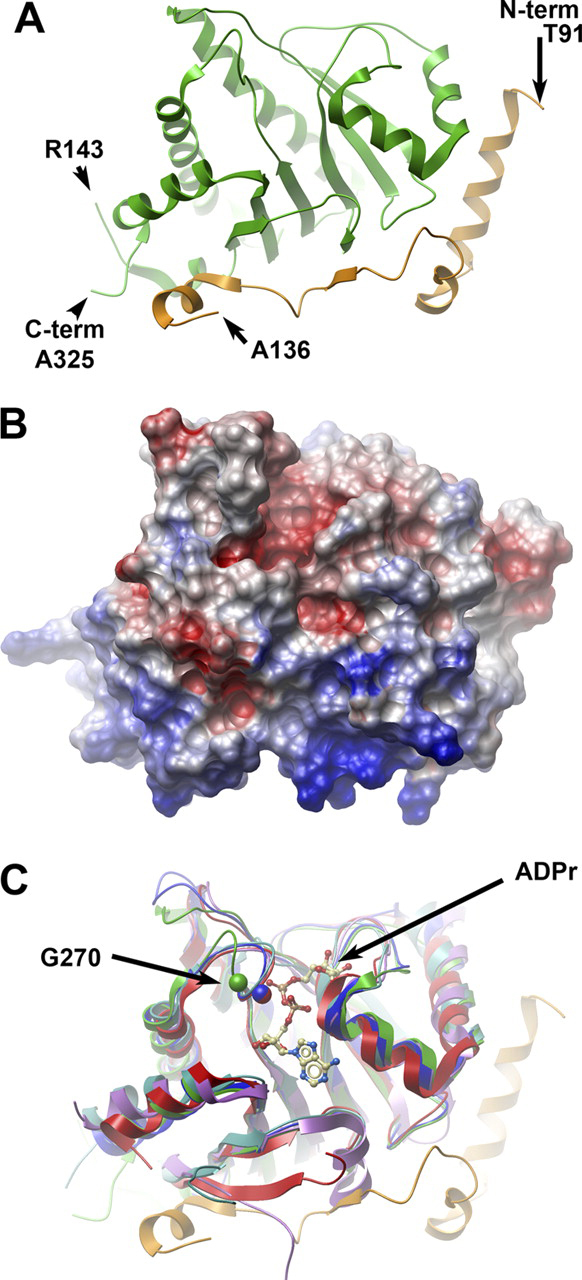
The crystal structure of human MacroD1.A, ribbon representation of the secondary structure of MacroD1. The conserved macrodomain is depicted in_green_, and the divergent N-terminal (aa 91–136) is in_orange_. The amino acids at the two termini of the ordered structure are marked, as well as the breakpoints of an internally missing fragment. B, surface potential diagram of MacroD1 shown in the same orientation as Fig. 2A. The N-terminal region (bottom) is highly positive.C, overlay of MacroD1 (green/orange) on the structures of other macrodomains: Feline sarcoma virus (PDB code 3JZT;purple), E. coli YmdB (PDB code 1SPV;blue), human PARP15 (PDB code 3KH6;cyan), and histone macroH2A1.1 (PDB code3IID;red). An ADPr molecule associated with the Feline sarcoma virus protein is shown as an atomic model. The Cα atom of a conserved glycine (Gly-270 of MacroD1) is marked in a green sphere; the homologous glycine of YmdB and MacroH2A1.1 are marked with_blue_ and red spheres, respectively. The shift of position of the loop containing Gly-270 (green) in MacroD1 relative to all other macrodomain structures is most likely the result of a close contact with a neighboring molecule in the crystal involving residues Val-271 and Phe-272 (not shown).
Identification of MacroD-Like Proteins as OAADPr Deacetylases
Based on the observation that some MacroD-like proteins are genetically and physically linked to sirtuins, we investigated whether these proteins might harbor catalytic activity toward _O_AADPr, the direct product of the NAD+-dependent deacetylation reaction of sirtuins. We initiated our investigation by incubating radiolabeled_O_-[3H]acetyl-ADPr with purified proteins and analyzing the incubation mixture using three different assays: charcoal-binding fractionation assays, HPLC, and mass spectrometry. In the charcoal-binding fractionation assay, enzymatic reactions are terminated by addition of activated charcoal at desired time points. Upon reaction termination, proteins,_O_AADPr, and ADPr bind to the charcoal leaving only small molecular weight acetate in solution (11). With _O_-[3H]acetyl-ADPr used as potential substrate, radioactivity released from substrate to solution would indicate the removal of the [3H]acetate moiety, which can be quantified by scintillation counting.
Using these assays we demonstrate that human MacroD1 catalyzes [3H]acetate removal from_O_-[3H]acetyl-ADPr. The time-dependent formation of [3H]acetate requires the presence of MacroD1 and the initial velocity of [3H]acetate formation increases with increasing MacroD1 concentration (Fig. 3A). Similar protein-dependent deacetylation activity was observed with truncated human MacroD1, human MacroD2, YmdB from E. coli and sirtuin-linked SAV0325 from S. aureus (seeTable 1). In contrast, the deacetylation activity was not observed with the human GDAP2 protein, the representative of the closest macrodomain branch to MacroD-like proteins (Fig. 1A, Table 1). To confirm that the deacetylation activity by MacroD-like proteins is dependent specifically on the macrodomain, we mutated the absolutely conserved glycine residue to a glutamate (residues 270 of human MacroD1, 124 of E. coli YmdB, and 214 of S. aureus SAV0325). This glycine residue is located on the P-loop interacting with the pyrophosphate moiety of the bound nucleotide. Mutation of this residue is predicted to block the ADP-ribose binding site in macrodomain proteins (36). The resulting MacroD mutants lacked _O_AADPr deacetylation activity under the same assay conditions (Table 2). The loss of catalytic activity of the glycine mutants provides convincing evidence that the MacroD-like domains are responsible for the catalytic activity on _O_AADPr.
FIGURE 3.

Deacetylation of_O_-acetyl-ADPr catalyzed by macroD-like proteins.A, [3H]acetate release from hydrolysis of _O_-[3H]acetyl-ADPr catalyzed by human MacroD1. _O_AADPr hydrolysis was monitored by measuring the radioactivity release as count per minute (CPM) due to [3H]acetate formation from_O_-[3H]acetyl-ADPr in the presence of 0 to 2.0 μm human MacroD1 and 800 μm_O_AADPr at pH 7.3. The data show the time dependent increase in free [3H]acetate formation and the rate of [3H]acetate formation increases with Macro D1 (E) concentration. B, HPLC assay of _O_AADPr hydrolysis catalyzed by human MacroD1. HPLC chromatograms monitored at 260 nm show the formation of ADPr from_O_AADPr in human MacroD1 catalyzed reaction. The chromatograms labeled 1–5 are reactions stopped by addition of TFA at 0, 3, 6, 10, and 16 min, respectively. The formation of ADPr from_O_AADPr depends on the presence of enzyme.Inset shows the time-dependent formation of ADPr from_O_AADPr in the presence of 0.5 μm MacroD1. C, LC-MS/MS analysis of human MacroD1 reaction. The precursor ions of 600.0 (_O_AADPr) and 558.0 (ADPr) and the major product ion of 346.0 shared by _O_AADPr and ADPr were selected. The SRM chromatogram shows the ion intensity versus time for_O_AADPr (dashed line) and ADPr (solid line) formed from the MacroD1 reaction stopped at 15 min. The inset shows the formation of ADPr increases with reaction time. The concentration of ADPr formed from each time point reaction was determined from external standards. D, steady-state kinetic analysis of human MacroD1. The deacetylation reaction of_O_AADPr catalyzed by MacroD1 follows saturation kinetics. The steady-state kinetic parameters are determined by radioactive assays for acetate formation and HPLC assays for ADP-ribose formation. An apparent Km was measured to be 375 ± 55 μm. The_V_max was measured to be 0.20 ± 0.04 s−1. The reaction mixtures contain 0.5 μm MacroD1. E, inhibition of ADPr to human MacroD1 activity. The deacetylation reactions were carried out in 50 mm Tris-HCl of pH 7.3 containing 0.5 μm MacroD1. The reactions were initiated in the presence of 0 μm(closed squares), 100 μm (open squares), 200 μm (open diamonds), and 400 μm (open circles) ADPr, respectively. The Km_′ values measured are 375.8, 590.9, 986.6, and 1350.3 μmin the presence of 0, 100, 200, and 400 μminitial ADPr, respectively. The V_maxvalues are 0.202, 0.204, 0.201, and 0.196 s−1. The_Ki was determined to be 145 ± 27 μm. The inhibition pattern is consistent with reversible competitive inhibition. F, kinetic comparison of wild-type MacroD1 and its variants. Conserved asparagine and aspartate residues located in vicinity to the 2′- and 3′-hydroxyl groups of the bound ADPr in the MacroD1 structure are mutated to alanine. The kinetic comparison of the wild type MacroD1 (closed squares) and its variants show that these residues are important to catalysis. The N171A (open squares), N174A (open diamonds), D184A (open circles), and N174A/D184A (open triangles), mainly affect the_V_max. The_Km values are only minimally affected.
TABLE 1.
Steady-state kinetic parameters of human MacroD1, MacroD2, and MacroD orthologs from E. coli and_S. aureus_
| Macro protein | Km | _k_cat | _k_cat/Km |
|---|---|---|---|
| μ_m_ | s_−_1 | m_−_1 s_−_1 | |
| Human MacroD1 (full-length) | 375 ± 55 | 0.20 ± 0.04 | (5.33 ± 1.23) × 102 |
| Human MacroD1 (ΔN58aa truncated) | 273 ± 43 | 0.21 ± 0.10 | (7.70 ± 3.86) × 102 |
| Human MacroD2 | 107 ± 38 | 0.12 ± 0.03 | (1.10 ± 0.47) × 103 |
| E. coli YmdB | 270 ± 76 | 0.48 + 0.14 | (1.78 ± 0.72) × 103 |
| S. aureus SAV0325 | 2000 ± 800 | 3.67 ± 1.22 | (1.84 ± 0.95) × 103 |
| Human GDAP2 | – | ∼0.001 | – |
TABLE 2.
_O_AADPr deacetylation activity of selected wild-type and mutant MacroD-like proteins
The activity below 0.005 s−1 is considered background or inactive under the assay conditions.
| Proteins | Organism | Mutations | Catalytic activity,kcat |
|---|---|---|---|
| s_−_1 | |||
| MacroD1 | Human | Wild type | 0.198 (100%) |
| MacroD1 | Human | D184A | 0.078 (∼39.3% of wt activity) |
| MacroD1 | Human | H188A | 0.114 (∼57.6% of wt activity) |
| MacroD1 | Human | D167A | 0.136 (∼68.7% of wt activity) |
| MacroD1 | Human | N171A | 0.082 (∼41.3% of wt activity) |
| MacroD1 | Human | N174A | 0.113 (∼57.3% of wt activity) |
| MacroD1 | Human | S176A | 0.195 (∼98.4% of wt activity) |
| MacroD1 | Human | C199A | 0.179 (∼90.4% of wt activity) |
| MacroD1 | Human | T226A | 0.204 (∼102.9% of wt activity) |
| MacroD1 | Human | D160A | 0.114 (∼57% of wt activity) |
| MacroD1 | Human | S268A | 0.153 (∼77% of wt activity) |
| MacroD1 | Human | N174A + D184A | 0.014 (∼7% of wt activity) |
| MacroD1 | Human | G270E | 0.002 (∼1% of wt activity) |
| YmdB | E. coli | Wild type | 0.48 (100%) |
| YmdB | E. coli | G124E | 0.005 (∼1.0% of wt activity) |
| SAV0325 | S. aureus | Wild type | 2.77 (100%) |
| SAV0325 | S. aureus | G214E | 0.007 (∼0.2% of wt activity) |
| SAV0325 | S. aureus | D122A | 0.679 (∼24.5% wt activity) |
To determine the identities of the reaction products from _O_AADPr hydrolysis, the MacroD-catalyzed reactions were analyzed by HPLC using authentic acetate, ADPr and _O_AADPr as standards. In analyzing the MacroD1-catalyzed reaction, the HPLC chromatograms monitored at 260 nm show the time-dependent increase of a peak at 9.7 min, which correlated with the decrease of peaks at 17.4/18.3 min (Fig. 3B). The retention times of these peaks match those of authentic ADPr (9.7 min) and_O_AADPr (17.4/18.3 min), indicating the formation of ADPr from _O_AADPr. The doublet of the _O_AADPr peak is due to the isomerization of the 2′-_O_-acetyl- and 3′-_O_-acetyl-isomers (11) under the HPLC conditions. The rapid formation of ADPr from_O_AADPr absolutely depends on the level of macrodomain enzymes used in the assay. When [3H]acetyl-ADPr was used as substrate, the radioactivity was associated with the _O_AADPr peak and the acetate peak, which elutes at 4.2 min when monitored at 214 nm. Radioactivity is not associated with the ADPr peak, indicating hydrolysis of the acetyl moiety from_O_AADPr. Analysis of the 9.7-min peak by electrospray ionization mass spectrometry (ESI-MS) operated in negative ion mode identified ion species of m/z 557.9 (−1), which matches the mass of the ADPr (559.3). Thus, the second reaction product of MacroD1-catalyzed _O_AADPr hydrolysis was consistent with ADPr.
The MacroD1-catalyzed reaction of_O_AADPr was also analyzed by liquid chromatography coupled to tandem mass spectrometry (LC-MS/MS). The ion intensities of _O_AADPr and ADPr from MacroD1 reactions were measured by selecting the precursor ions of 600.0 (O_AADPr), 558.0 (ADPr), and the common fragment ion of 346.0 (Fig. 3C). The concentration of ADPr formed from each time point reaction was determined utilizing external standards. The inset in Fig. 3C shows the time dependent increase of ADPr formation. The LC-MS/MS analyses of the MacroD1 reaction further confirmed ADPr as the second reaction product, which are consistent with the observations from radioactive and HPLC assays (Fig. 3, A and_B). These results indicate that human MacroD1 is a novel enzyme catalyzing the deacetylation of_O_AADPr to produce ADPr and acetate (supplemental scheme). To confirm the ability of this family of macrodomains to perform this deacetylation reaction, similar HPLC and MS assays were performed with human MacroD2, E. coli YmdB, S. aureus SAV0325, and human ganglioside-induced differentiation-associated protein 2 (GDAP2). Again, all evaluated MacroD-like proteins, with the exception of human GDAP2, exhibited _O_AADPr deacetylation activity (Table 1).
Substrate-Saturation Kinetics of MacroD-Catalyzed OAADPr Hydrolysis
To provide supporting evidence that MacroD-like proteins function as substrate-saturable enzymes, steady-state kinetic analyses were conducted with selected MacroD-like enzymes. Indeed, the deacetylation rates catalyzed by human MacroD1 follows saturation kinetics as the concentrations of_O_AADPr increased. An apparent_Km_ of 375 ± 55 μm and_V_max of 0.20 ± 0.04 s−1 at 23 °C were determined for the full-length MacroD1 (Fig. 3D). Steady-state kinetic parameters for O_AADPr deacetylation were also determined with other MacroD-like proteins and the results are summarized in Table 1. In all cases where deacetylation activity was observed, the macrodomains displayed the appropriate substrate saturation expected for an enzyme-catalyzed reaction with obligate formation of a specific Michaelis ES complex. As shown inTable 1, human MacroD2, truncated MacroD1, E. coli YmdB, and SAV0325 from S. aureus display similar saturation kinetic properties with Km values ranging from 100 to 400 μm, except for SAV0325 from S. aureus, which displays a much larger_Km of ∼2 mm. Nevertheless, all catalytic macrodomains have similar_V_max/_Km_values, ranging from 5.3 × 102 to 1.8 × 103 m−1s−1.
Product Inhibition of MacroD-like Enzymes
The fact that MacroD-like proteins display saturation kinetics with _O_AADPr suggests that these proteins harbor a specific binding site, which serves as the active site for catalysis. Previous x-ray crystallography and isothermal titration calorimetry studies revealed that human macroH2A1.1 functions as a binding domain of_O_AADPr and analogs (8, 9). Isothermal titration calorimetry also showed that human MacroD1 and MacroD2 bind ADP-ribose with high affinity (5). To provide direct evidence that these MacroD-like proteins contain a specific ADPr-binding site that is identical to the active site for _O_AADPr hydrolysis, we examined the ability of ADPr to serve as a simple competitive inhibitor of the _O_AADPr reaction. In this experiment, the deacetylation of_O_AADPr was initiated in the presence of ADPr ranging from 0 to 400 μm and the steady-state rate of deacetylation was measured. As shown inFig. 3E, increasing ADPr concentration resulted in an increase of the apparent Michaelis constant, _Km_′, while the maximum catalytic rates were unaffected. The data are consistent with reversible competitive inhibition of ADPr of MacroD1. An inhibition constant Ki of 145 ± 27 μm was determined at 23 °C. The results suggest that these MacroD-like proteins contain a specific ADPr-binding site, which is identical to the active site for _O_AADPr hydrolysis. The inhibition of_O_AADPr deacetylation by ADPr suggests that ADPr could act as an in vivo inhibitor of macrodomain-dependent _O_AADPr deacetylation and provide feedback regulation of cellular_O_AADPr concentration.
Modeling of ADPr into MacroD1 Structure
To identify residues important for_O_AADPr binding and hydrolysis, we modeled ADPr into the structure of human MacroD1 protein. Fig. 2C shows a structural alignment of MacroD1 with E. coli YmdB protein, as well as a number of distantly related macrodomain proteins from humans, coronaviruses and yeast, some of which were crystallized with bound ADPr (4, 37). All helices and β-strands of the core macrodomains are well-aligned. Based on the close structural alignment, we attempted to model a molecule of ADPr in the putative binding site on MacroD1, shown in a ball and stick depiction in Fig. 2C. The conformation and location of ADPr in the crystal structures of the viral and yeast macrodomain proteins is similar. In contrast, the ADPr molecule in the macrodomain of human PARP15 protein presents a different orientation of the distal ribose. The two conformations of ADPr are modeled on the surface of MacroD1 in supplemental Fig. S1A. It is most likely that ADPr (as well as O_AADPr) is bound in a conformation similar to that in the viral, yeast and_Archaeoglobus proteins. It should be noted that in this model of ADPr binding to MacroD1 a steric clash occurs between the adenine ring and the side chain of Phe-306, marked with an asterisk in supplemental Fig. S1A. However, as seen in supplemental Fig. S1B, the homologous residues in other macrodomain proteins (_e.g._Tyr-159 of E. coli YmdB) are rotated to a position that relieves the steric clash and provides a favorable stacking interaction with the adenine ring. It is likely that Phe-306 of MacroD1 assumes a similar conformation in the ADPr-bound state. Val-183 completes the other side of a hydrophobic adenine-binding pocket, and is highly conserved in macrodomain proteins. It is interesting to note that this model indicates that ADPr may be tightly bound to MacroD1, in line with observations on product inhibition of this enzyme.
Amino Acid Residues Important for Catalysis
To assess which of these amino acids are important for catalytic activity, a number of site-specific mutants of human MacroD1 were generated based on amino acid conservation and the structure of human MacroD1 with modeled ADP-ribose (Figs. 1,B and C, and4A). All the mutants were then tested for their ability to deacetylate _O_AADPr. As shown in Table 2, activity assays of macrodomain variants revealed varied effects on deacetylase activity. Mutation of the conserved residues Ser-176, Ser-268, Cys-199, and Thr-226 has minimal effect on activity. These residues can be classified as unimportant for catalysis and protein structure integrity. Mutation of His-188, Asp-167, Asp-160, Asn-171, Asn-174, Asp-184, and Gly-270 showed significant effect on activity. These residues can be classified as important either for direct catalysis or for maintaining protein structure integrity. Of particular interest were several highly conserved asparagine, aspartate, and histidine residues, which through structure modeling studies (Fig. 4A) are predicted to locate near the 2′- and 3′-hydroxyl groups of the bound ADPr. Residues Asn-171 and Asn-174 are widely conserved across multiple macrodomain classes. In contrast, aspartate and histidine at positions equivalent to Asp-184 and His-188 are seen in MacroD-like proteins (and partially in the yeast protein YMX7)(37) but are not conserved in other macrodomain proteins. Results of our mutational analysis indicate an important role for Asn-171, Asn-174, Asp-184, and His-188 in catalysis. Based on these observations (and on a speculative model of activity of the yeast YMX7), we propose a tentative model for the hydrolysis of O_AADPr, depicted in Fig. 4B. In this model, Asp-184 (as part of a catalytic triad including His-188 and, possibly, Thr-196) acts as a base that deprotonates a water molecule, which then attacks the carbonyl group, leading to hydrolysis of the acetyl group. Another plausible mechanism may involve a direct reaction of Asp-184 with the carbonyl, leading to an anhydride intermediate. Mutating any of the putative catalytic residues individually results in only a partial loss of activity although the double mutant D184A/N174A is severely impaired (Table 2). These residues do not seem to contribute significantly to the binding of substrate (see below), so the binding energy for weakening the ester bond for deacetylation may not be impacted. It is possible that other residues (or the ribose 3′-hydroxyl) partially compensate for the single mutations in D184A or N174A. Nevertheless, on the basis of the structure modeling and mutagenesis studies, Asn-171, Asn-174, and Asp-184 are most promising to function as important residues for catalysis or to interact directly with the acetyl moiety of the bound substrate. As displayed in Fig. 3F, these mutations significantly reduce the catalytic efficiency and lower the_V_max without affecting_Km significantly, indicating that Asn-171, Asn-174, and Asp-184 are important for catalysis.
FIGURE 4.
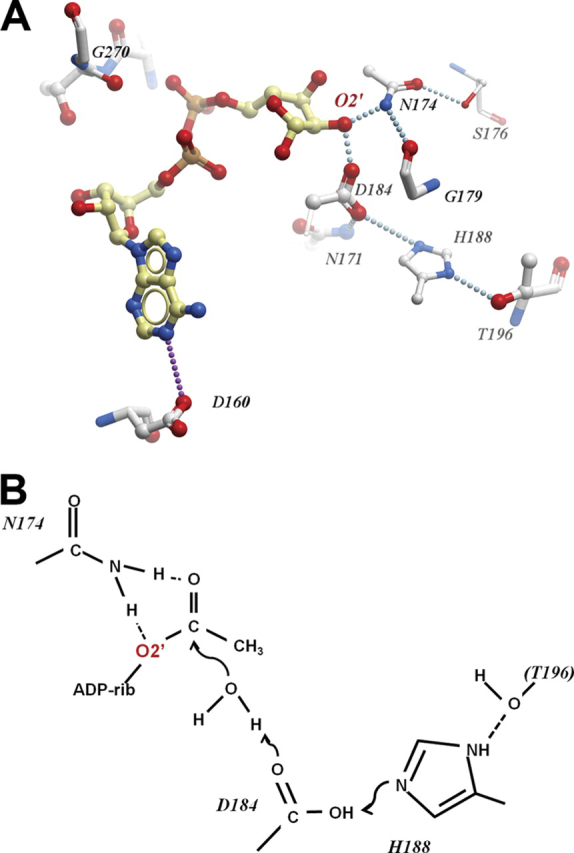
Possible arrangement of the MacroD1 catalytic site.A, residues of MacroD1 surrounding the modeled ADPr binding site. Dotted lines indicate potential hydrogen bonds either direct or water-mediated. B, hypothetical roles of catalytic residues in the hydrolysis of_O_AADPr.
Endogenous MacroD Functions as a OAADPr Deacetylase
Eukaryotic cell extracts are known to harbor at least two different cellular activities that contribute to_O_AADPr deacetylation (19), though their identity has not been determined. Unlike higher organisms, filamentous fungi like Neurospora crassa_possess only one MacroD homologue. For this reason we decided to use_N. crassa as a model to assess the physiological importance of MacroD proteins. We replaced the gene for MacroD protein (NCU07925) in N. crassa_with an antibiotic resistance gene (33). To demonstrate that_Neurospora MacroD indeed influences the ability to hydrolyze O_AADPr, we compared_N. crassa cell extracts prepared from wild type cells and from cells with the MacroD gene knocked out._O_AADPr deacetylase activities in these cell extracts were measured using the radioactive charcoal fractionation method. Deletion of the MacroD gene in N. crassa reduced the _O_AADPr deacetylation activities in cell extract by ∼30% (Fig. 5) and the measured activity was unaffected by the presence of EDTA. These results indicate that Neurospora MacroD contributes significantly to _O_AADPr deacetylation activities in vivo, as predicted from our detailed _in vitro_enzymatic analysis of MacroD-like proteins. The binding and deacetylation of _O_AADPr can therefore be proposed to be the general in vivo biological function of MacroD-like proteins.
FIGURE 5.

OAADPr deacetylation activity of cell extracts from wild type and MacroD knock-out (Δ_macrod_)neurospora.Neurospora cell extracts were assayed using radioactive method in the absence or presence of 1.25 mmEDTA. Protein concentrations in cell extracts were normalized before activity assays. MacroD1 knock-out in Neurospora reduced the_O_AADPr deacetylation activities in cell extracts for ∼30% independent of EDTA. The error bars represent standard deviations calculated from the specific activities of Neurospora extracts in five separate experiments.
DISCUSSION
_O_AADPr is a sirtuin reaction product generated from the NAD+-dependent protein deacetylation reactions and has been implicated as a signaling molecule (15, 16). Consequently, intracellular levels of _O_AADPr might be tightly regulated. However, the detailed mechanisms by which cells regulate the levels of _O_AADPr are poorly understood. The observation that some macrodomains and sirtuins are physically linked provides an important clue to their possible functional connections. In this study we demonstrate that the product of the sirtuin reaction is a substrate for the macrodomain. A subfamily of macrodomain proteins, which we have named MacroD-like proteins, is capable of efficient deacetylation of _O_AADPr to yield free acetate and ADPr. Our mutagenic analysis and structural studies suggest that they evolved a catalytic mechanism that is distinct from other macrodomain protein subfamilies.
Previously, only the 39 kDa human poly (ADP-ribose) glycohydrolase ARH3 had been reported to catalyze the deacetylation reaction of_O_AADPr in vitro (18). Unlike the deacetylation reaction catalyzed by MacroD-like enzymes, ARH3 requires the presence of magnesium ion and catalyzes _O_AADPr hydrolysis more than an order of magnitude slower than MacroD-like enzymes (18). Moreover, the action of ARH3 appears to be less specific, as this protein can also hydrolyze poly(ADP-ribose) (18). In contrast, MacroD-like enzymes are incapable of hydrolyzing poly(ADP-ribose) (supplemental Fig. S2). ARH3 is present only in vertebrates and cannot be responsible for hydrolysis of _O_AADPr in fungi (such as N. crassa), bacteria and archaea, which all encode sirtuins. On the other hand, MacroD-like proteins are highly conserved through all kingdoms of life. Within eukaryotic cells, sirtuins (and therefore _O_AADPr) are found in all cellular compartments. In this respect, it is interesting to note that highly related human MacroD1 and MacroD2 enzymes have quite distinct intracellular localization: MacroD1 is present in mitochondria and nucleus, whereas MacroD2 is mostly cytoplasmic (5). Therefore, together MacroD1 and MacroD2 co-exist in all cellular compartments where_O_AADPr is generated in human cells.
Cellular _O_AADPr levels can be also regulated by NUDIX hydrolyzes. NUDIX hydrolase Ysa1 from S. cerevisiae was shown to hydrolyze ADPr/_O_AADPr to produce AMP and ribose phosphate and acetyl-ribose phosphate, respectively (19). In yeast, Ysa1 modulated the levels of AMP and ADPr/_O_AADPr (17). The yeast Ysa1 enzyme hydrolyzes ADPr and_O_AADPr at similar efficiency in a Mg2+-dependent reaction. However, the human mitochondrial ortholog NudT9, prefers ADPr over _O_AADPr as substrate by 500-fold. To date, a NUDIX hydrolase specific for only_O_AADPr remains to be identified in higher eukaryotes.
Enzymatic activities that consume_O_AADPr have been reported in mammalian cells (19). Although the identities of the responsible enzymes were not determined, cytoplasmic fractions of HeLa cell protein extracts contain an O_AADPr hydrolase that produced acetate and ADPr. Nuclear fractionation enriched an enzymatic activity that removed the acetyl group from_O_AADPr to form ADPr, but the acetyl moiety was transferred to an unknown acceptor molecule (19). Both activities appeared to be independent of Mg2+, as reactions were performed in the presence of EDTA. Whether the enzymatic activity in HeLa cell extracts can be attributed to MacroD1/2 enzymes remain to be investigated. This possibility is supported by the fact that we were able to detect only Mg-independent hydrolysis in extracts of the lower eukaryote N. crassa and a significant fraction of this activity is due to a MacroD homologue in this organism (Fig. 5). Thus, it is plausible to suggest that MacroD-like proteins function as_O_AADPr-consuming enzymes in vivo. This role is further supported by genomic evidence, as MacroD-like proteins are found linked to sirtuin in many groups of pathogenic bacteria and fungi (S. aureus,Streptococci, Mycoplasma,C. albicans, and several_Aspergillus species).
The MacroD-like protein group is represented by numerous proteins from bacteria and eukaryotes, and we propose that dysregulation of MacroD-like protein function would disrupt _O_AADPr homeostasis and sirtuin pathways. The available literature suggests that the dysregulation of both human MacroD1 and MacroD2 levels in cells might lead to human disease. MacroD1 overexpression was linked to breast cancer progression, whereas deficiency in MacroD2 function has been linked to a developmental disorder called Kabuki syndrome (38, 39, 40). Clearly, more investigation is needed to clarify the proposed links between macrodomain dysregulation and these diseases.
In summary, this work identifies _O_AADPr deacetylation activities in MacroD-like proteins from bacteria and eukaryotes. Our data demonstrate that MacroD-like proteins regulate a variety of biochemical pathways by acting as potential in vivo regulators of cellular _O_AADPr produced by NAD+-dependent deacetylation.
Acknowledgments
The Structural Genomics Consortium (SGC) is a registered charity (number 1097737) that receives funds from the Canadian Institutes for Health Research, the Canadian Foundation for Innovation, Genome Canada through the Ontario Genomics Institute, GlaxoSmithKline, Karolinska Institutet, the Knut and Alice Wallenberg Foundation, the Ontario Innovation Trust, the Ontario Ministry for Research and Innovation, Merck & Co., Inc., the Novartis Research Foundation, the Swedish Agency for Innovation Systems, the Swedish Foundation for Strategic Research, and the Wellcome Trust.
Footnotes
The atomic coordinates and structure factors (code2X47) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).
5
The abbreviations used are:_O_AADPr,_O_-acetyl-ADP-ribose; PEI, polyethyleneimine; aa, amino acids; PDB, Protein Data Bank.
Contributor Information
John M. Denu, Email: jmdenu@wisc.edu.
Ivan Ahel, Email: IAhel@picr.man.ac.uk.
Supplementary Material
REFERENCES
- 1.Till S., Ladurner A.G. Front. Biosci. 2009;14:3246–3258. doi: 10.2741/3448. [DOI] [PubMed] [Google Scholar]
- 2.Ladurner A.G. Mol. Cell. 2003;12:1–3. doi: 10.1016/s1097-2765(03)00284-3. [DOI] [PubMed] [Google Scholar]
- 3.Karras G.I., Kustatscher G., Buhecha H.R., Allen M.D., Pugieux C., Sait F., Bycroft M., Ladurner A.G. EMBO J. 2005;24:1911–1920. doi: 10.1038/sj.emboj.7600664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Egloff M.P., Malet H., Putics A., Heinonen M., Dutartre H., Frangeul A., Gruez A., Campanacci V., Cambillau C., Ziebuhr J., Ahola T., Canard B. J. Virol. 2006;80:8493–8502. doi: 10.1128/JVI.00713-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Neuvonen M., Ahola T. J. Mol. Biol. 2009;385:212–225. doi: 10.1016/j.jmb.2008.10.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ahel D., Horejsí Z., Wiechens N., Polo S.E., Garcia-Wilson E., Ahel I., Flynn H., Skehel M., West S.C., Jackson S.P., Owen-Hughes T., Boulton S.J. Science. 2009;325:1240–1243. doi: 10.1126/science.1177321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kraus W.L. Nat. Struct. Mol. Biol. 2009;16:904–907. doi: 10.1038/nsmb0909-904. [DOI] [PubMed] [Google Scholar]
- 8.Kustatscher G., Hothorn M., Pugieux C., Scheffzek K., Ladurner A.G. Nat. Struct. Mol. Biol. 2005;12:624–625. doi: 10.1038/nsmb956. [DOI] [PubMed] [Google Scholar]
- 9.Comstock L.R., Denu J.M. Org. Biomol. Chem. 2007;5:3087–3091. doi: 10.1039/b710231c. [DOI] [PubMed] [Google Scholar]
- 10.Tanner K.G., Landry J., Sternglanz R., Denu J.M. Proc. Natl. Acad. Sci. U.S.A. 2000;97:14178–14182. doi: 10.1073/pnas.250422697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jackson M.D., Denu J.M. J. Biol. Chem. 2002;277:18535–18544. doi: 10.1074/jbc.M200671200. [DOI] [PubMed] [Google Scholar]
- 12.Smith B.C., Hallows W.C., Denu J.M. Chem. Biol. 2008;15:1002–1013. doi: 10.1016/j.chembiol.2008.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Oberdoerffer P., Michan S., McVay M., Mostoslavsky R., Vann J., Park S.K., Hartlerode A., Stegmuller J., Hafner A., Loerch P., Wright S.M., Mills K.D., Bonni A., Yankner B.A., Scully R., Prolla T.A., Alt F.W., Sinclair D.A. Cell. 2008;135:907–918. doi: 10.1016/j.cell.2008.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hallows W.C., Smith B.C., Lee S., Denu J.M. Cell. 2009;137:404–406. doi: 10.1016/j.cell.2009.04.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hoff K.G., Wolberger C. Nat. Struct. Mol. Biol. 2005;12:560–561. doi: 10.1038/nsmb0705-560. [DOI] [PubMed] [Google Scholar]
- 16.Tong L., Denu J.M. Biochim. Biophys. Acta. 2010;1804:1617–1625. doi: 10.1016/j.bbapap.2010.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tong L., Lee S., Denu J.M. J. Biol. Chem. 2009;284:11256–11266. doi: 10.1074/jbc.M809790200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ono T., Kasamatsu A., Oka S., Moss J. Proc. Natl. Acad. Sci. U.S.A. 2006;103:16687–16691. doi: 10.1073/pnas.0607911103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rafty L.A., Schmidt M.T., Perraud A.L., Scharenberg A.M., Denu J.M. J. Biol. Chem. 2002;277:47114–47122. doi: 10.1074/jbc.M208997200. [DOI] [PubMed] [Google Scholar]
- 20.Borra M.T., O'Neill F.J., Jackson M.D., Marshall B., Verdin E., Foltz K.R., Denu J.M. J. Biol. Chem. 2002;277:12632–12641. doi: 10.1074/jbc.M111830200. [DOI] [PubMed] [Google Scholar]
- 21.Jackson M.D., Schmidt M.T., Oppenheimer N.J., Denu J.M. J. Biol. Chem. 2003;278:50985–50998. doi: 10.1074/jbc.M306552200. [DOI] [PubMed] [Google Scholar]
- 22.Garrity J., Gardner J.G., Hawse W., Wolberger C., Escalante-Semerena J.C. J. Biol. Chem. 2007;282:30239–30245. doi: 10.1074/jbc.M704409200. [DOI] [PubMed] [Google Scholar]
- 23.Bradford M.M. Anal. Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 24.Lee S., Tong L., Denu J.M. Anal. Biochem. 2008;383:174–179. doi: 10.1016/j.ab.2008.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Savitsky P., Bray J., Cooper C.D., Marsden B.D., Mahajan P., Burgess-Brown N.A., Gileadi O. J. Struct. Biol. 2010;172:3–13. doi: 10.1016/j.jsb.2010.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Leslie A.G. Joint CCP4 + ESF-EAMCB Newsletter on Protein Crystallography. 1992 No. 26, BBSRC-CCP4. [Google Scholar]
- 27.Collaborative Computational Project N. Acta Crystallographica Section D. 1994;50:760–763. [Google Scholar]
- 28.McCoy A.J., Grosse-Kunstleve R.W., Adams P.D., Winn M.D., Storoni L.C., Read R.J. J. Appl. Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cowtan K. Acta Crystallogr D Biol Crystallogr. 2006;62:1002–1011. doi: 10.1107/S0907444906022116. [DOI] [PubMed] [Google Scholar]
- 30.Emsley P., Lohkamp B., Scott W.G., Cowtan K. Acta Crystallogr. D Biol. Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Murshudov G.N., Vagin A.A., Dodson E.J. Acta Crystallogr D Biol Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 32.Davis R.H., de Serres F.J. Methods Enzymol. 1970;17:79–143. [Google Scholar]
- 33.Colot H.V., Park G., Turner G.E., Ringelberg C., Crew C.M., Litvinkova L., Weiss R.L., Borkovich K.A., Dunlap J.C. Proc. Natl. Acad. Sci. U.S.A. 2006;103:10352–10357. doi: 10.1073/pnas.0601456103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Garceau N.Y., Liu Y., Loros J.J., Dunlap J.C. Cell. 1997;89:469–476. doi: 10.1016/s0092-8674(00)80227-5. [DOI] [PubMed] [Google Scholar]
- 35.Ahel I., Ahel D., Matsusaka T., Clark A.J., Pines J., Boulton S.J., West S.C. Nature. 2008;451:81–85. doi: 10.1038/nature06420. [DOI] [PubMed] [Google Scholar]
- 36.Timinszky G., Till S., Hassa P.O., Hothorn M., Kustatscher G., Nijmeijer B., Colombelli J., Altmeyer M., Stelzer E.H., Scheffzek K., Hottiger M.O., Ladurner A.G. Nat. Struct. Mol. Biol. 2009;16:923–929. doi: 10.1038/nsmb.1664. [DOI] [PubMed] [Google Scholar]
- 37.Kumaran D., Eswaramoorthy S., Studier F.W., Swaminathan S. Protein Sci. 2005;14:719–726. doi: 10.1110/ps.041132005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tada M., Kanai F., Tanaka Y., Sanada M., Nannya Y., Tateishi K., Ohta M., Asaoka Y., Seto M., Imazeki F., Yoshida H., Ogawa S., Yokosuka O., Omata M. Cancer Sci. 2010;101:1261–1269. doi: 10.1111/j.1349-7006.2010.01500.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Maas N.M., Van de Putte T., Melotte C., Francis A., Schrander-Stumpel C.T., Sanlaville D., Genevieve D., Lyonnet S., Dimitrov B., Devriendt K., Fryns J.P., Vermeesch J.R. J. Med. Genet. 2007;44:562–569. doi: 10.1136/jmg.2007.049510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Han W.D., Mu Y.M., Lu X.C., Xu Z.M., Li X.J., Yu L., Song H.J., Li M., Lu J.M., Zhao Y.L., Pan C.Y. Endocr. Relat. Cancer. 2003;10:217–224. doi: 10.1677/erc.0.0100217. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.