HIV-1 Tat and Host AFF4 Recruit Two Transcription Elongation Factors into a Bifunctional Complex for Coordinated Activation of HIV-1 Transcription (original) (raw)
. Author manuscript; available in PMC: 2011 May 14.
SUMMARY
Recruitment of the P-TEFb kinase by HIV-1 Tat to the viral promoter triggers the phosphorylation and escape of RNA polymerase II from promoter-proximal pausing. It is unclear, however, if Tat recruits additional host factors that further stimulate HIV-1 transcription. Using a sequential affinity-purification scheme, we have identified human transcription factors/coactivators AFF4, ENL, AF9, and elongation factor ELL2 as components of the Tat-P-TEFb complex. Through the bridging functions of Tat and AFF4, P-TEFb and ELL2 combine to form a bifunctional elongation complex that greatly activates HIV-1 transcription. Without Tat, AFF4 can mediate the ELL2-P-TEFb interaction, albeit inefficiently. Tat overcomes this limitation by bringing more ELL2 to P-TEFb and stabilizing ELL2 in a process that requires active P-TEFb. The ability of Tat to enable two different classes of elongation factors to cooperate and coordinate their actions on the same polymerase enzyme explains why Tat is such a powerful activator of HIV-1 transcription.
INTRODUCTION
Transcriptional elongation by RNA polymerase (Pol) II is recognized as a major rate-limiting step for controlling the expression of many metazoan genes (Core and Lis, 2008). In the absence of stimuli, negative elongation factors (N-TEF) pause Pol II shortly after initiation. Signal-induced reversion of this block requires P-TEFb, a heterodimer composed of CDK9 and cyclin T1 (CycT1; or the minor forms T2 or K) (Peterlin and Price, 2006). P-TEFb phosphorylates the C-terminal domain (CTD) of Pol II and N-TEF, leading to the production of full-length (FL) RNA transcripts (Peterlin and Price, 2006). Besides P-TEFb, transcriptional elongation can also be stimulated by several other factors, including ELL1/2, TFIIS, TFIIF, and the elongins, all of which enhance the processivity of Pol II through mechanisms different from that of P-TEFb (Sims et al., 2004). It is yet to be shown if any of these factors cooperate with P-TEFb to coordinate their stimulation of Pol II elongation.
Our understanding of the control of elongation, particularly by P-TEFb, has benefited greatly from the investigations of the HIV-1 virus. Pausing of Pol II near the transcription start site on the integrated proviral DNA is a major rate-limiting step for HIV-1 gene expression. To antagonize this restriction, the HIV-1 Tat protein recruits host P-TEFb to the TAR RNA element located at the 5′ end of all viral transcripts (Peterlin and Price, 2006). The localized P-TEFb phosphorylates Pol II and N-TEF to produce the FL HIV transcripts that are key for viral gene expression and replication.
In uninfected cells, P-TEFb functions as a general transcription factor required for the expression of a vast array of genes (Chao and Price, 2001). As such, P-TEFb activity is subjected to stringent cellular control through the interactions with multiple factors (Zhou and Yik, 2006). For example, more than half of nuclear P-TEFb exists in a catalytically inactive complex called 7SK snRNP that also contains the 7SK snRNA, the CDK9 kinase inhibitor HEXIM1, and the LARP7/PIP7S and MePCE proteins (He et al., 2008; Jeronimo et al., 2007; Nguyen et al., 2001; Yang et al., 2001; Yik et al., 2003). In addition, a major portion of P-TEFb also exists in a complex with the bromodomain protein Brd4, which recruits P-TEFb to cellular promoters through contacting acetylated histones (Jang et al., 2005; Yang et al., 2005). Through alternately interacting with these positive and negative regulators, P-TEFb is kept in a functional equilibrium that shifts in accordance to the cellular transcriptional demands and decisions between growth and differentiation (Zhou and Yik, 2006).
Given that P-TEFb forms distinct complexes under different conditions, we performed sequential affinity purifications to identify factors that may interact with P-TEFb in the presence of Tat. Our experiments have identified elongation factor ELL2 and transcription factors/coactivators AFF4, ENL, and AF9 as proteins that exist in a single complex with Tat and P-TEFb. Under normal conditions, ELL2 is a short-lived protein whose stability is maintained through interacting with P-TEFb in a process that is mediated by AFF4 and requires active P-TEFb. However, the AFF4 function is inadequate for activated HIV-1 transcription. Tat overcomes this limitation by recruiting more ELL2 to P-TEFb, resulting in the stabilization of ELL2 and synergistic activation of HIV-1 transcription by Tat and ELL2. Our data support the model that Tat and AFF4 recruit distinct elongation factors into a bifunctional complex, allowing ELL2 and P-TEFb to coordinate their actions and greatly stimulate the processivity of Pol II.
RESULTS
ELL2, AFF4, ENL, and AF9 Associate with the Tat-P-TEFb Complex
To identify cellular factors that control the Tat/P-TEFb-mediated activation of HIV-1 transcription, we performed affinity purifications to isolate factors associated with the Tat-P-TEFb complex. Nuclear extracts (NEs) from an engineered human HEK293-based cell line stably expressing Flag-tagged CDK9 (CDK9-F) and inducibly expressing HA-tagged Tat (Tat-HA) were subjected to sequential coimmunoprecipitations (coIPs) with anti-Flag and then anti-HA beads. CDK9-F was expressed at about the same level as the endogenous CDK9, while Tat-HA was produced under the control of a doxycycline-inducible promoter to a level similar to that of CDK9.
Inspection of the purified materials by SDS-PAGE followed by staining with silver reveals no detectable band prior to the induction of Tat-HA expression by doxycycline (Figure 1A, lane 2). However, upon the induction, several new bands besides CycT1, Cdk9-F, and Tat-HA appeared (lane 1), and the pattern was unchanged by pretreating NEs with RNase A prior to the purification (data not shown). Analyses by mass spectrometry revealed that the new bands contained proteins ELL2, AFF4, ENL, and AF9. Table S1 (available online) lists the number of recovered peptides and percent coverage for all the proteins.
Figure 1. P-TEFb Exists in Two Multisubunit Complexes Containing ELL2/AFF4/Tat/P-TEFb and ELL2/AFF4/P-TEFb, Respectively.

(A) CDK9-F, Tat-HA, and their associated factors (lane 1) were isolated through sequential immunoprecipitations (anti-Flag and then anti-HA) from NEs of TTAC-8 cells upon the induction of Tat-HA expression. The immunoprecipitates (IPs) were analyzed on a silver-stained SDS gel, with their identities indicated on the left. NE derived from TTAC-8 cells prior to the induction of Tat-HA was used in a parallel procedure for control (lane 2). MW, molecular weight.
(B) The αFlag and αHA IP analyzed in (A) were examined by western blotting for the indicated proteins.
(C) The αFlag and αHA sequential IPs derived from NE of HEK293 cells expressing the indicated proteins were analyzedby western blotting as in (B).
(D) IPs obtained with the indicated antibodies were examined as in (B).
(E) The αFlag IPs derived from NEs of ELL2-F-expressing cells were analyzed as in (B).
(F) The parental HEK293 cells and the HEK293-based cell lines expressing the indicated proteins were subjected to anti-Flag immunoprecipitation. The IPs were analyzed by western blotting for the presence of the indicated proteins.
ELL2 is related to ELL1 (49% identical and 66% similar), which was originally identified as a fusion partner of mixed lineage leukemia (MLL) in acute myeloid leukemia (Thirman et al., 1994). It was subsequently rediscovered as a transcription factor that increases the catalytic rate and suppresses transient pausing of Pol II during elongation (Shilatifard et al., 1996). ELL2 possesses similar transcriptional activity as ELL1, and both are expressed in many of the same tissues (Shilatifard et al., 1997).
AFF4 is a member of the AF4 family of transcription factors/coactivators. Like ELL1, it is also translocated to MLL in infant acute lymphoblastic leukemia (Taki et al., 1999). AFF4 was later found to associate with P-TEFb (Estable et al., 2002). However, ectopically expressed AFF4 failed to activate HIV-1 replication or transcription, for the reason that will become clear below.
ENL and AF9 are also fusion partners of MLL and are involved in MLL-associated leukemia (Harper and Aplan, 2008). Although both proteins have previously been reported to interact with P-TEFb, other MLL fusion partners (e.g., AFF4 and AFF1), and the H3K79 methyltransferase DOT1L (Bitoun et al., 2007; Mueller et al., 2007, 2009), to our knowledge this is the first time that they are specifically linked to Tat. Consistently, Sobhian et al. (2010) have also identified ENL and AF9 as Tat-associated proteins. Despite the implication of these two proteins as critical for Tat/P-TEFb to stimulate HIV-1 transcription (Sobhian et al., 2010; and our unpublished data), exactly how they accomplish this task remains mostly unknown. Given the demonstrated interactions of ENL and AF9 with DOT1L, a thorough investigation is needed in the future to study the mechanism by which ENL and AF9 contribute to Tat transactivation and the possible involvement of DOT1L and H3K79 methylation in this process. For the current study, however, we decide to concentrate on the roles of ELL2 and AFF4 in modulating the Tat/P-TEFb-dependent HIV transcription.
ELL2, AFF4, Tat, and P-TEFb Exist in a Single Complex
First, the interactions of ELL2 and AFF4 with the Tat-P-TEFb complex purified through sequential coIPs were confirmed by western blotting using protein-specific antibodies (Figure 1B). To determine whether ELL2 and AFF4 exist in the same or different Tat-P-TEFb complexes, we coexpressed ELL2-F and Tat-HA in HEK293 cells and performed sequential coIPs with anti-Flag and then anti-HA beads. Western analysis detected the associations of endogenous AFF4 and CDK9 with the affinity-purified ELL2-F-Tat-HA complex (Figure 1C). Together, these results indicate that ELL2, AFF4, Tat, and P-TEFb likely exist in a single multisubunit complex.
Tat-Independent Interactions of AFF4 and ELL2 with P-TEFb
It is important to point out that even in the absence of Tat, the anti-CDK9 immunoprecipitates (IPs) derived from HEK293 NE also contained AFF4 and ELL2 (Figure 1D). Likewise, ectopically expressed ELL2-F coprecipitated AFF4, CycT1, and CDK9 under Tat(−) conditions (Figure 1E). Thus, ELL2 and AFF4 can interact with P-TEFb independently of Tat. However, as will be shown below, Tat further enhances the ELL2-P-TEFb interaction.
Next, we performed anti-CDK9 immunodepletion and found that ~60% of AFF4 and ~40% of ELL2 were codepleted from NEs (Figure S1). Thus, a major fraction of nuclear ELL2 and AFF4 is normally sequestered in the P-TEFb-containing complex.
The ELL2/AFF4/Tat/P-TEFb-Containing Complex Does Not Contain Brd4 or HEXIM1
The identification of a P-TEFb-containing complex raises the question of whether it may be related to the two major P-TEFb complexes described in the past. Using the total CDK9-F IP harboring all the CDK9-associated factors as a reference, we performed western analysis to determine whether the affinity-purified ELL2/AFF4/Tat/P-TEFb complex might contain Brd4, the P-TEFb recruitment factor for general transcription (Jang et al., 2005; Yang et al., 2005), or HEXIM1, a signature subunit and inhibitor of CDK9 within the 7SK snRNP (Yik et al., 2003). When the levels of CycT1, a subunit shared among all the known P-TEFb-containing complexes, were normalized to about the same level, neither Brd4 nor HEXIM1 was detected in the anti-ELL2-F IP even after a prolonged exposure (Figure 1F). In contrast, abundant Brd4 and HEXIM1 were readily detected in the total CDK9-F IP. Thus, the ELL2/AFF4/Tat/P-TEFb-containing complex (and also the Tat-free ELL2/AFF4/P-TEFb complex, data not shown) is physically distinct from 7SK snRNP or the Brd4-P-TEFb complex.
ELL2 Depletion Reduces Both Basal and Tat-Activated HIV-1 Transcription
To determine how the associations of ELL2 and AFF4 with Tat-P-TEFb regulate HIV-1 transcription, we employed short hairpin (sh)RNAs to reduce ELL2 expression and examined the impact on basal and Tat-dependent HIV-1 transcription. Compared to an ineffective control shRNA (shELL2 #10), the two effective shRNAs (shELL2 #1 and #8, with the latter causing ~75% ELL2 depletion) caused a significant reduction in both basal and Tat-dependent HIV-1 LTR-driven luciferase activity from the transfected reporter construct (Figure 2A). Notably, the reduction in viral LTR activity mediated by shELL2 #1 and #8 correlated well with the reduction in ELL2 expression (compare Figures 2A and 2B). This general dependence on ELL2 for HIV-1 transcription, which was also observed with a stably integrated HIV-1 LTR-luciferase construct (data not shown), is highly reminiscent of the situation involving P-TEFb, whose loss of function has also been shown to reduce both basal and Tat-activated HIV-1 transcription (Mancebo et al., 1997; Zhou et al., 1998; Zhu et al., 1997).
Figure 2. Tat Increases the Levels of ELL2 and the ELL2/P-TEFb-Containing Complex and Synergizes with ELL2 but Not ELL1 to Activate TAR-DependentHIV-1 Transcription.

(A) Luciferase activities were measured in extracts of cells cotransfected with the indicated shELL2-expressing constructs, the HIV-1 LTR-luciferase reporter gene, and a vector expressing Tat-HA or nothing. The activity in cells expressing shELL2 #10 but not Tat was set to 1. The error bars represent mean ± SD.
(B) Western analyses of the levels of ELL2-F and α-tubulin in cells transfected with the indicated shELL2-expressing constructs.
(C) Luciferase activities were measured as in (A) in cells transfected with either WT HIV-1 LTR-luciferase reporter or a ΔTAR mutant construct, together with the indicated plasmids expressing ELL2-F (0.5 ug/well) and/or Tat-HA (0.01 ug/well).
(D) Luciferase activities were measured as in (A) in cells transfected with the HIV-1 LTR-luciferase reporter and the indicated ELL1-F-, ELL2-F-, and/or Tat-HA-expressing plasmids.
(E) Western analysis of the indicated proteins in NE of cells cotransfected with the indicated cDNA constructs or an empty vector (vec. or “−”). Tat-HA was transfected in 2-fold increments.
(F) ELL1-F, ELL2-F, and their associated CDK9 were isolated by anti-Flag IP from NE as analyzed in (E) and examined by western blotting.
Ectopically Expressed ELL2, but Not ELL1, Synergizes with Tat to Activate TAR-Dependent HIV-1 Transcription
When expressed from a transfected plasmid, ELL2-F increased basal HIV-1 LTR activity from a reporter construct that contains either wild-type (WT) or a mutant (Δ) TAR element by 1.3- and 2.0-fold, respectively (Figure 2C). Further analysis indicates a dose-dependent activation of the HIV-1 LTR by ELL2-F (Figure S2A). Tat-HA, on the other hand, increased HIV-1 transcription 64-fold in a strictly TAR-dependent manner. Remarkably, the combination of ELL2-F and Tat-HA increased HIV-1 transcription 284-fold from the WT TAR construct (Figure 2C), demonstrating a strong synergistic effect of the two on the LTR. The synergism was also observed under conditions of elevated ELL2-F expression (Figure S2B). However, when the ΔTAR reporter construct was used, the Tat/ELL2 synergism was mostly lost (Figure 2C). Finally, despite the strong homology between ELL1 and ELL2, it is interesting to note that only ELL2, but not ELL1, displayed a significant synergism with Tat in activating HIV-1 LTR (Figure 2D).
Tat Increases the Amount of ELL2, but Not ELL1, Associated with P-TEFb
To define the mechanism of this selectivity of Tat for ELL2, we examined the interactions of ELL1 and ELL2 with P-TEFb either in the absence or presence of Tat. Western analysis indicates that the expression of just a small amount of Tat significantly elevated the level of the coexpressed ELL2-F, but not ELL1-F, in NE (Figure 2E). Concurrently, there was a major increase in the amount of CDK9 bound to the immunoprecipitated ELL2-F but not ELL1-F (Figure 2F). Importantly, Tat stimulated the association with P-TEFb by both the ectopically expressed ELL2-F and endogenous ELL2 proteins (see below).
AFF4 Synergizes with ELL2 to Activate General Transcription
Because the Tat-P-TEFb complex also contains AFF4, we investigated the contribution of this factor to transcription from the HIV-1 LTR as well as several other viral and cellular promoters. Remarkably, just like the Tat/ELL2 pair, the coexpression of AFF4 and ELL2 also synergistically activated HIV-1 transcription (Figure 3A). However, the AFF4/ELL2 synergism was neither restricted to the HIV-1 LTR nor TAR-dependent. Rather, the effect was also detected using several other viral and cellular promoter constructs, although not as pronounced as that with the HIV-1 LTR (Figure 3A). Unlike Tat, which is a sequence-specific transcription factor, neither AFF4 nor ELL2 is known to display any sequence-specific DNA/RNA-binding activity. This difference explains the observations that the transcriptional synergism displayed by Tat/ELL2 was HIV-1 TAR dependent, whereas the synergism by AFF4/ELL2 was not.
Figure 3. Ectopically Expressed AFF4 Acts Like Tat to Synergize with ELL2 to Stimulate Transcriptional Elongation through Promoting ELL2 Accumulation and Association with P-TEFb.

(A) Luciferase activities were measured in extracts of cells transfected with the indicated promoter-luciferase constructs together with the ELL2-F- or/and F-AFF4-expressing plasmids as indicated. For each promoter construct, the level of activity detected in the absence of ELL2-F and F-AFF4 was set to 1. The error bars represent mean ± SD.
(B and C) Luciferase activities were measured and analyzed as in (A) in cells transfected with the indicated reporter and cDNA constructs.
(D and E) NEs of cells transfected with the indicated cDNA constructs (top panel) and anti-HA immunoprecipitates (IP) derived from NE (bottom panel) were analyzed by western blotting (WB) for the indicated proteins.
(F) Luciferase reporter assay was performed as in (A) in cells transfected with the indicated reporter and cDNA constructs.
(G) Extracts from the same cells as analyzed in (F) were examined by western blotting for the indicated proteins.
(H) Transcription reactions containing CDK9-depleted NE, template HIV-1 LTR-G400, and the indicated purified proteins were performed. The 400 nt RNA transcribed from a G-less cassette located at ~1 kb downstream of the HIV-1 promoter is indicated.
(I) mRNAs isolated from cells as analyzed in (A) were subjected to RT-PCR analysis with primers that amplify the two indicated regions. RT, reverse transcriptase.
AFF4 Synergizes with ELL2 to Activate Basal but not Tat-Dependent HIV-1 Transcription
Our subsequent analyses indicate that AFF4 and ELL2 synergistically activated only basal but not Tat-activated HIV-1 transcription (Figure 3B). Similarly, while ectopically expressed AFF4 stimulated basal HIV-1 transcription by up to 17-fold, it enhanced the Tat-dependent transcription by only 2-fold (Figure 3C).
Two questions are raised by the data in Figures 3A–3C: (1) What is the molecular basis of the AFF4/ELL2 synergism? (2) Why did ectopically expressed AFF4 have different effects on basal and Tat-activated HIV transcription? To address the first question, we examined the levels of AFF4 and ELL2 that were expressed either alone or together. Just like the Tat-induced stable accumulation of ELL2, the coexpression of F-AFF4 and ELL2-HA also significantly increased the ELL2-HA level in both NEs and whole-cell extracts (WCEs) (Figure 3D, top panel; and Figure S3). As will be demonstrated below, this was largely caused by AFF4-induced stabilization of ELL2. Correlating with more ELL2 in the cell, there was a marked increase in the amount of CDK9 bound to the immunoprecipitated ELL2-HA (Figure 3D, bottom panel). Notably, the coexpression of F-AFF4 and ELL2-HA also increased the F-AFF4 levels ~2-fold (Figure 3D, top panel; and Figure S3), probably due to the AFF4/ELL2-induced general increase in transcription (Figure 3A).
Tat and Ectopically Expressed AFF4 Increase the Levels of ELL2 and ELL2/P-TEFb-Containing Complex by a Common Mechanism
Why didn’t the ectopically expressed AFF4 further promote Tat transactivation? Since both proteins elevated the ELL2 level, we tested whether they might exert an additive effect. While the coexpression of F-AFF4 or Tat with ELL2-HA increased the latter’s level and association with P-TEFb, simultaneous expression of ELL2-HA, F-AFF4, and Tat did not further enhance these effects (Figure 3E). This absence of additive effects suggests that Tat and AFF4 used a common mechanism to promote ELL2 accumulation and interaction with P-TEFb and that their effects were likely saturated under the current conditions. The fact that overexpressed AFF4 failed to further enhance Tat trans-activation also agrees with the previous observation that AFF4 overexpression cannot activate HIV-1 replication (Estable et al., 2002), which is Tat dependent.
AFF4 Synergizes More Efficiently with ELL2 than ELL1 to Activate Transcriptional Elongation In Vitro and In Vivo
Besides promoting the ELL2-P-TEFb interaction in the cell, AFF4 and Tat also had a similar ability to distinguish between ELL1 and ELL2. Like Tat, AFF4 synergized more strongly with ELL2 than with ELL1 (Figure 3F). This is likely due to the AFF4-induced accumulation of ELL2 but not ELL1 (Figure 3G), which correlates with more ELL2 bound to P-TEFb.
So far, the stimulatory effect of AFF4/ELL2 has been demonstrated in only the luciferase reporter assay. To confirm that the stimulation indeed occurs at the transcriptional level, we first performed an in vitro transcription assay that employs the well-characterized HIV-1 LTR-G400 template for detecting elongation through a G-less cassette (G400) inserted at ~1 kb downstream of the start site (Zhou and Sharp, 1995). The reactions also contained HeLa NE, in which P-TEFb and its associated factors were immunodepleted with anti-CDK9 antibodies. While the addition of isolated P-TEFb or an equal molar mixture of AFF4/ELL2 into the depleted NE only had a minor effect, the introduction of P-TEFb plus AFF4/ELL2 greatly rescued transcription (Figure 3H), confirming that the positive effect of AFF4/ELL2 is indeed at the elongation level.
Lastly, the AFF4/ELL2 stimulation of promoter-distal (i.e., elongation) but not promoter-proximal transcription (i.e., initiation) was also confirmed by RT-PCR analysis of mRNA transcribed from the HIV LTR-luciferase reporter gene at two different locations. While the AFF4/ELL2 coexpression did not affect the abundance of transcripts mapping to the 5′ end of the mRNA (HIV sequence +1 to +80), it increased the level of transcripts corresponding to the 3′ end (+1719 to +1826) of the luciferase gene (Figure 3I).
AFF4 Knockdown Causes Stronger Inhibition of Basal than Tat-Dependent HIV-1 Transcription and Decreases the Levels of ELL2 and ELL2/P-TEFb-Containing Complex In Vivo
Further support for the idea that AFF4 and Tat act similarly to increase the level of ELL2/P-TEFb-containing complex in vivo came from an experiment involving shRNA depletion of AFF4. Although shAFF4 affected basal HIV-1 transcription quite severely (5-fold), it had only a mild effect (2-fold) on Tat-activated transcription (Figure 4A), probably because the loss of AFF4 was largely compensated by the presence of Tat to maintain the level of ELL2/P-TEFb complex.
Figure 4. AFF4 Bridges the ELL2-P-TEFb Interaction and Is Required for Stable Accumulation of ELL2.

(A) Luciferase activities were measured in cells transfected with the HIV-1 LTR-luciferase reporter gene and constructs expressing shAFF4 or/and Tat-HA. The level of activity detected in the absence of shAFF4 and Tat-HA was set to 1. The error bars represent mean ± SD.
(B) NEs (left panel) of HEK293 cells either harboring an empty vector (−) or stably expressing shAFF4 and IP (right panel) obtained from NE with the indicated antibodies were examined by western blotting for the presence of the indicated proteins.
(C) NE and anti-Flag IP derived from cells transfected with an empty vector (−) or the construct expressing either the full-length (FL) or truncated (Δ1-300) F-AFF4 were analyzed by western blotting for the indicated proteins.
(D) The indicated proteins were incubated with immobilized CycT1-HA/CDK9 in vitro, and the bound proteins were eluted and analyzed by western blotting (left and middle panels). Ten percent of the input FL and Δ1-300 F-AFF4 proteins were examined by anti-Flag western blotting (right panel).
Consistent with the finding that AFF4 promotes ELL2 accumulation in vivo, shRNA depletion of AFF4 caused the codepletion of ELL2 in HEK293 cells (Figure 4B, left panel). Concurrently, there was a decrease in the amounts of ELL2 bound to the immunoprecipitated CDK9 (right panel), indicating a reduction in the ELL2/P-TEFb complex in KD cells.
AFF4 Mediates the ELL2-P-TEFb Interaction
To determine how the AFF4 depletion could decrease the ELL2/P-TEFb-containing complex in vivo, we asked whether AFF4 might mediate the ELL2-P-TEFb interaction. To this end, the interaction of ELL2-F with the CycT1-HA/Cdk9 heterodimer was tested in the presence of either the FL or N-terminally truncated (Δ1-300) F-AFF4 in vitro. All four factors, highly purified from transfected cells under stringent conditions involving high salt (1.0 M KCl) and detergent (0.5% NP-40), were confirmed by western blotting to be free of any associated factors that are present in the ELL2/AFF4/P-TEFb complex (data not shown). Δ1-300 F-AFF4 was chosen as a control because it failed to interact with P-TEFb but had nearly wild-type association with ELL2 in vivo (Figure 4C), suggesting the existence of separable ELL2- and P-TEFb-binding domains in AFF4.
In the binding reactions (Figure 4D), purified ELL2-F bound to the immobilized CycT1-HA/CDK9 in the presence of FL but not Δ1-300 F-AFF4 (left panel). Without ELL2-F, FL but not Δ1-300 F-AFF4 interacted with CycT1-HA/CDK9 directly (middle panel). Thus, through directly contacting P-TEFb and ELL2, AFF4 mediates the ELL2-P-TEFb interaction, which explains the significantly decreased ELL2/P-TEFb complex in AFF4 KD cells (Figure 4B).
Tat Further Increases the Amount of ELL2 Bound to P-TEFb without Affecting the AFF4-P-TEFb Binding
Given that AFF4 mediated the ELL2-P-TEFb interaction without Tat, we tested whether Tat could affect the interaction. When anti-CDK9 IPs containing all endogenous proteins were isolated from cells expressing or not Tat-HA from a retroviral vector and analyzed by western blotting, the presence of Tat markedly increased the association of ELL2 but not ELL1 with P-TEFb (Figure 5A, top left panel). This again was accompanied by the accumulation of more endogenous ELL2, but not ELL1, in the cell (bottom panels).
Figure 5. Tat Increases the Amount of ELL2 Bound to P-TEFb, Leading to an Enhanced Association of the ELL2/AFF4/P-TEFb-Containing Complex with the HIV-1 Chromatin Template.

(A) NEs from HEK293 cells infected with retroviruses either expressing (+) or not (−) Tat-HA were subjected to western blotting with the indicated antibodies (top right) and immunoprecipitation with anti-CDK9 (top left), anti-ELL2 (bottom left), anti-ELL1 antibodies (bottom right), or a nonspecific rabbit IgG. IPs were analyzed by western blotting for the presence of the indicated proteins.
(B) Anti-Flag IP derived from cells stably expressing CDK9-F and harboring either an empty vector (−) or the shELL2 #8-expressing plasmid were analyzed by western blotting for the indicated proteins.
(C) Chromatin immunoprecipitation (ChIP) with anti-Flag, anti-CDK9, and anti-AFF4 antibodies was performed in cells containing the integrated HIV-1 LTR-luciferase reporter gene and stably expressing ELL2-F. Three regions corresponding to the promoter, interior, and 3′ UTR of the integrated reporter gene (bottom panel) were PCR amplified from the precipitated and purified DNA. Amplified signals from 5% and 10% of the input chromatin were also shown.
(D) ChIP assay was performed as in (C) at the GAPDH locus with the indicated antibodies. The region close to the 3′ end of the gene was PCR amplified from the precipitated DNA.
While Tat brought more ELL2 to P-TEFb, it did not significantly affect the AFF4-P-TEFb binding (Figure 5A, top left panel), which also remained unchanged upon shRNA depletion of ELL2 (Figure 5B). Thus, the autonomous AFF4-P-TEFb binding occurs independently of Tat and ELL2 within the P-TEFb-containing complexes.
The dependence on Tat to recruit more ELL2 to P-TEFb explains why the previous attempts by others and us to isolate P-TEFb-associated factors under Tat(−) conditions failed to identify ELL2. More importantly, it indicates that although AFF4 can mediate the ELL2-P-TEFb interaction, it is insufficient for activated HIV-1 transcription, probably due to AFF4’s relatively low level or/and efficiency. Consistent with this view, AFF4 overexpression has been shown to significantly increase the amount of ELL2 bound to P-TEFb (Figure 3).
To reveal the functional consequence of the Tat-induced increase in the ELL2/AFF4/P-TEFb-containing complex, the chromatin immunoprecipitation assay was conducted on a stably integrated HIV-1 LTR-luciferase reporter gene. The presence of Tat significantly increased the occupancy of not only CDK9 but also ELL2-F and AFF4 at the HIV-1 promoter, the interior of the luciferase gene, and the 3′UTR region (Figure 5C). In contrast, Tat did not promote the bindings of these factors to the control GAPDH locus (Figure 5D).
Physical Association with Active P-TEFb Is Required for Tat to Promote ELL2 Accumulation
Since P-TEFb is an integral component of the ELL2/AFF4-containing complexes, we asked whether its kinase activity is required for the complex formation and stable accumulation of ELL2. When cells were treated with the CDK9 inhibitors DRB or flavopiridol (FVP), the Tat-induced ELL2 accumulation was abolished (Figure 6A). To test whether the physical association with P-TEFb is also critical, the Tat mutant C22G, which cannot bind P-TEFb (Garber et al., 1998), was coexpressed with ELL2-F. Compared to WT Tat, C22G could neither promote the accumulation of ELL2-F nor enhance the interactions of CDK9 and CycT1 with the immunoprecipitated ELL2-F (Figure 6B). Thus, the physical association with active P-TEFb is required for Tat to promote ELL2 accumulation, suggesting that the elevated ELL2 level is a result of more ELL2 sequestered in the Tat/P-TEFb-containing complex.
Figure 6. Active P-TEFb Is Required for ELL2 Accumulation and Interaction with P-TEFb.

(A) NEs from HEK293 cells transfected with the indicated cDNA constructs and treated with the indicated drugs were analyzed by western blotting for the levels of ELL2-F and α-tubulin.
(B and C) NEs (left panel in B and top panel in C) and anti-Flag IP (right panel in B and bottom panel in C) derived from NEs of cells transfected with the indicated cDNA constructs were analyzed by western blotting for the presence of the indicated proteins.
(D) F1C2 cells stably expressing CDK9-F were either untreated or treated with the indicated drugs. FVP, flavopiridol. NEs (left panel) and anti-Flag IP (right panel) were analyzed by western blotting.
The Kinase-Inactive CDK9 Mutant Blocks the Tat-Induced ELL2 Accumulation and Fails to Interact with ELL2
Further support for the dependence on active P-TEFb for Tat-induced ELL2 accumulation and interaction with TEFb came from binding studies employing D167N, a kinase-inactive and dominant-negative CDK9 mutant (De Falco et al., 2000). D167N prevented Tat from elevating the ELL2-HA level and interacted with a significantly reduced amount of ELL2-HA under both Tat(+) and Tat(−) conditions (Figure 6C).
To test whether the Tat-free ELL2/AFF4/P-TEFb-containing complex also requires active P-TEFb for stability, cells were treated with DRB or FVP. The inhibition of CDK9 markedly reduced the association of ELL2 with P-TEFb but only slightly affected the autonomous AFF4-P-TEFb binding (Figure 6D). Together, the data indicate that ELL2 is stabilized by interacting with active P-TEFb, irrespective of whether the interaction is mediated by AFF4 alone or further enhanced by Tat.
ELL2 Is Stabilized by the Inhibition of the Proteosome or the Presence of Tat or AFF4
The above data have revealed a critical role for Tat and AFF4 in promoting ELL2 accumulation and interaction with P-TEFb, which are likely responsible for Tat and AFF4 to synergize with ELL2 to activate transcription. To define the mechanism of ELL2 accumulation, we treated cells with the proteosome inhibitor MG132 and found that both the ectopically expressed ELL2-F and its endogenous counterpart are short-lived proteins, whose half-lives were significantly prolonged by the drug (Figures 7A and 7B).
Figure 7. ELL2 Is a Short-Lived Protein Whose Stability Can Be Significantly Enhanced by Tat or AFF4.
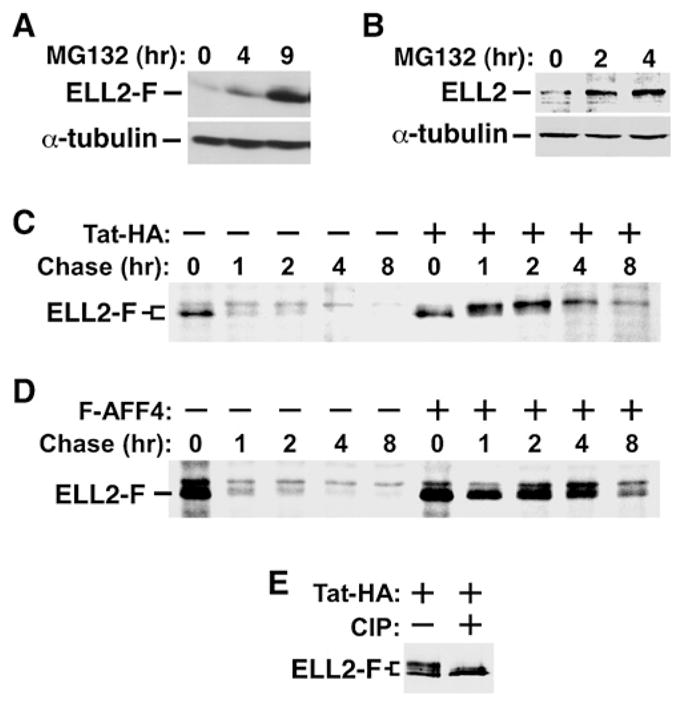
(A and B) HEK293 cells containing an ELL2-F-expressing vector (A) or nothing
(B) were treated with MG132 for the indicated periods of time. ELL2-F and its endogenous counterpart were detected by anti-Flag (A) and anti-ELL2 (B) western blotting, with α-tubulin serving as a loading control.
(C and D) The ELL2-F-producing cells transfected with either an empty vector (−) or a construct expressing Tat-HA (B) or F-AFF4 (C) were pulse labeled with 35S-labeled methionine and L-cysteine and then chased for the indicated periods of time. ELL2-F was then immunoprecipitated and analyzed by SDS-PAGE followed by autoradiography.
(E) ELL2-F affinity purified from cells coexpressing Tat-HA were incubated with calf intestine phosphatase (CIP) and analyzed by anti-Flag western blotting.
A pulse-chase experiment employing 35S-labeled methionine and cysteine was performed to determine whether Tat could affect the stability of the coexpressed ELL2-F. In the absence of Tat, most of ELL2-F had a half-life of less than 1 hr, although a minor species with slightly slower mobility was considerably more stable (Figure 7C). Interestingly, the presence of Tat significantly increased the abundance of this stable ELL2-F species and, as a result, extended the half-life of the overall ELL2-F population to more than 4 hr (Figure 7C). The Tat-induced mobility shift of ELL2 is also evident in other experiments (e.g., Figure 6C, lanes 2 and 3) and likely caused by phosphorylation, as the treatment of ELL2-F isolated from Tat-expressing cells with calf intestine phosphatase (CIP) abolished the shift (Figure 7E). Based on the demonstrations that active P-TEFb is essential for Tat-induced ELL2 accumulation and interaction with P-TEFb (Figures 6A–6C), it is possible that CDK9 plays a role in phosphorylating ELL2.
Since ectopically expressed AFF4 also promoted ELL2 accumulation, we performed another pulse-chase experiment to determine whether AFF4 could stabilize ELL2. Indeed, expression of F-AFF4 increased the half-life of the overall ELL2-F population from less than 1 hr to more than 4 hr (Figure 7D). Like Tat, F-AFF4 also reduced the mobility of ELL2-F, although only a very small fraction of ELL2-F was affected. Together, these data illustrate that Tat and AFF4 similarly regulate the stability and P-TEFb-binding activity of ELL2.
DISCUSSION
P-TEFb was identified more than a decade ago as a specific cellular cofactor for Tat activation of HIV-1 transcription (Mancebo et al., 1997; Wei et al., 1998; Zhu et al., 1997). Since then, this landmark discovery has provided the basic framework for our understanding of Tat function in the HIV-1 life cycle, and P-TEFb has gained universal acceptance as a functional Tat partner. Here, we expand this conventional view of HIV transcriptional control by showing that the Tat-P-TEFb complex contains four additional components, ELL2, AFF4, ENL, and AF9. The identification of these factors relied on a combinatorial approach of double epitope tagging and sequential immunoprecipitations. This approach allows the selective isolation of factors that are directly involved in Tat/P-TEFb-mediated HIV-1 transactivation, and can be adapted readily for future characterization of protein complexes containing at least two known subunits. While the precise roles of ENL and AF9 in Tat transactivation await further investigation, AFF4 and ELL2 are shown to cooperate with Tat/P-TEFb to stimulate HIV-1 transcription and thus join P-TEFb as Tat partners essential for Tat transactivation.
Like P-TEFb, ELL2 is also a potent elongation factor. However, its unique contribution to Tat transactivation has remained unnoticed till this day. This is likely due to the fact that both the association with the Tat-P-TEFb complex and stability of ELL2 depend on active P-TEFb (Figure 6). Therefore, the contributions to Tat function by these two elongation factors are intertwined, and any molecular or pharmacological manipulations that reduce P-TEFb expression or activity also negatively affect ELL2, thus erasing their contributions simultaneously. In light of this, our traditional view of the role of P-TEFb in Tat transactivation must be revised. Namely, the stimulatory effect on basal and Tat-activated HIV-1 transcription that has previously been assigned to P-TEFb alone should in fact be attributed to both P-TEFb and ELL2.
P-TEFb stimulates elongation by phosphorylating the Pol II CTD and negative elongation factors DSIF and NELF, which converts Pol II into an elongation-competent form and antagonizes the inhibitory effects of NELF and DSIF (Peterlin and Price, 2006). Employing a different mechanism, ELL2 promotes elongation by keeping the 3′OH of nascent mRNA in alignment with the catalytic site, thus preventing Pol II backtracking (Shilatifard et al., 1996, 1997). Although both are considered general transcription factors, it is unclear whether they work independently or cooperatively during Pol II elongation of any DNA template. The identification of a Tat/AFF4-mediated interaction between ELL2 and P-TEFb provides for the first time, to our knowledge, strong evidence in support of a coordinated promotion of Pol II elongation on HIV-1 proviral DNA by different classes of elongation factors. By forming a bifunctional elongation complex, ELL2 and P-TEFb can act cooperatively on the same polymerase enzyme. The ability of Tat to promote AFF4 recruitment of two distinct elongation factors into a single complex explains why Tat is such a powerful activator of HIV-1 transcription.
Notably, a functional connection between ELL2 and P-TEFb has been implicated in previous studies. For example, ELL2 is required for producing the secretory-specific form of IgH mRNA in plasma cells through promoting exon skipping and the use of a proximal poly(A) site (Martincic et al., 2009). Factors that enhance the Pol II elongation rate can often facilitate exon skipping, presumably by reducing the time in which splice sites are offered to the splicing apparatus. Interestingly, the ELL2-promoted processing of IgH mRNA correlates with a significant increase in Ser2 phosphorylation of the Pol II CTD (Shell et al., 2007). Prior to the current study, it would have been difficult to explain how ELL2, which acts by modulating the catalytic rate of Pol II, can induce Ser2 phosphorylation, which is thought to be performed by P-TEFb (Sims et al., 2004). The ELL2-P-TEFb interaction described here now provides an explanation of this apparent paradox. An additional functional coupling between ELL2 and P-TEFb has been detected in Drosophila melanogaster. Upon silencing of CDK9 expression in larvae, a significantly reduced amount of Drosophila ELL, which is most similar to human ELL2, was found on the chromosomes (Eissenberg et al., 2007).
The current study has revealed a strong correlation between the increased ELL2-P-TEFb interaction, irrespective of whether it is mediated by AFF4 or further promoted by Tat, and the enhanced ELL2 stability. The accumulation of stable ELL2 could be a cause or consequence of more ELL2/P-TEFb-containing complex. The observations that the enhanced ELL2 stability depended on not only the CDK9 kinase activity but also the physical association with P-TEFb indicate that the accumulation of stable ELL2 is very likely a direct consequence of more ELL2 sequestered and thus protected against proteolysis in the P-TEFb-containing complexes. Given that the stabilization correlated with a mobility shift of ELL2 (Figures 6C, 7C, and 7D), it will be interesting to determine whether the phosphorylation of ELL2 by P-TEFb may be involved.
Besides the Tat-containing and Tat-free ELL2/AFF4/P-TEFb complexes described here, nuclear P-TEFb also occurs in the 7SK snRNP and the Brd4-containing complex (Zhou and Yik, 2006). The latter two have been shown to contain P-TEFb molecules that are in storage or are being transported to a chromatin template, respectively (Zhou and Yik, 2006). The notion that Brd4 only recruits P-TEFb to a promoter region but does not participate in P-TEFb-mediated elongation is based on the different distribution patterns of Brd4 and P-TEFb throughout the transcription unit, especially outside the promoter-proximal region (Jang et al., 2005). Furthermore, the Brd4-bound P-TEFb is incompatible with Tat transactivation, since Tat directly competes with Brd4 for binding to P-TEFb and the overexpressed Brd4 interferes with Tat function (Yang et al., 2005). Like Tat, several MLL chimeras (e.g., MLL-ENL, MLL-AFF1, and MLL-AFF4) have recently been reported to recruit an ELL/P-TEFb-containing complex similar to the one described here to the MLL target loci to stimulate transcription and induce oncogenic transformation (Lin et al., 2010; Mueller et al., 2009; Yokoyama et al., 2010). These observations, coupled with our current data, make the ELL2/AFF4/P-TEFb- and ELL2/Tat/AFF4/P-TEFb-containing complexes likely candidates for the forms of P-TEFb actively engaged in general and Tat-dependent HIV-1 transcription, respectively.
EXPERIMENTAL PROCEDURES
Antibodies
The rabbit polyclonal anti-ELL2, -AFF4, and -CycT1 antibodies were purchased from Bethyl Laboratories, Abcam, and Santa Cruz Biotechnology, respectively. The antibodies against CDK9, HEXIM1, and Brd4 have been described previously (Yang et al., 2005; Yik et al., 2003).
Generation of TTAC-8 Cells that Stably Express CDK9-F and Inducibly Express Tat-HA
The procedure is essentially as described (Yang et al., 2001), with minor modifications described in the Supplemental Information.
Purification of ELL2, AFF4, ENL, and AF9 as Proteins Associated with Tat-P-TEFb
NEs prepared from TTAC-8 cells were incubated overnight with anti-Flag M2 agarose beads (Sigma). The beads were washed extensively with buffer D (20 mM HEPES-KOH [pH 7.9], 15% glycerol, 0.2 mM EDTA, 0.2% NP-40, 1 mM dithiothreitol, and 1 mM phenylmethylsulfonyl fluride) containing 0.3 M KCl (D0.3M), and the immobilized proteins were eluted with 0.5 μg/ml Flag peptide dissolved in D0.3M. The eluate was then subjected to a second precipitation with anti-HA beads (Sigma) for 2 hr. The beads were washed extensively with D0.3M and then eluted by a low pH solution (200 mM glycine, pH 2.5). The neutralized eluate (with 1/20 volume of 2M Tris-HCl, pH. 8.8) was subjected to analyses by SDS-PAGE followed by silver staining, and the bands were excised for identification by mass spectrometry.
Generation of AFF4 and ELL2 Knockdown Cells
The procedures have been described previously (He et al., 2008). The shRNA sequences used in the procedures are in the Supplemental Information.
Chromatin Immunoprecipitation Assay
The assay was performed essentially as described (Yang et al., 2005), with minor modifications described in the Supplemental Information.
Pulse-Chase Analysis
Prior to the analysis, cells expressing ELL2-F alone (~2 × 106 cells per time point) or together with Tat-HA (~1 × 106 cells per time point) were starved in the conditioned medium lacking methionine and cysteine (Invitrogen) and supplemented with 10% dialyzed fetal calf serum (FBS). After 30 min at 37°C, the pulse medium containing EXPRES35S Protein Labeling Mix (Perkin Elmer) was added to each well at the concentration of 0.5 mCi/mL, followed by incubation for 30 min at 37°C. Immediately thereafter, the plates were washed with the complete medium plus 10% regular FBS and chased for 1–8 hr. Cells were directly lysed in 1× SDS loading buffer and centrifuged. The supernatants were diluted 5-fold with buffer D0.3M, precleared, and subjected to anti-Flag immunoprecipitation. After extensive washing, the precipitates were eluted with 1× SDS loading buffer and subjected to analysis by SDS-PAGE followed by autoradiography.
Supplementary Material
2
Acknowledgments
We thank Dr. Q. Zhong of UC Berkeley for providing T-REx 293 cell line and pcDNA4/TO vector, Dr. R. Roeder of Rockefeller University for anti-AFF4 antibody, and Dr. C. Milcarek of the University of Pittsburgh for anti-ELL2 antibody. This work is supported by grants from the National Institutes of Health (NIH) (R01AI41757-11 and R01AI41757-11S1) and the UC Cancer Research Coordinating Committee (027342) to Q.Z., and a NIH grant (P50 GM82250) to N.J.K. and T.A.
Footnotes
Supplemental Information includes three figures, one table, Supplemental Experimental Procedures, and Supplemental References and can be found with this article at doi:10.1016/j.molcel.2010.04.013.
References
- Bitoun E, Oliver PL, Davies KE. The mixed-lineage leukemia fusion partner AF4 stimulates RNA polymerase II transcriptional elongation and mediates coordinated chromatin remodeling. Hum Mol Genet. 2007;16:92–106. doi: 10.1093/hmg/ddl444. [DOI] [PubMed] [Google Scholar]
- Chao S-H, Price DH. Flavopiridol inactivates P-TEFb and blocks most RNA polymerase II transcription in vivo. J Biol Chem. 2001;276:31793–31799. doi: 10.1074/jbc.M102306200. [DOI] [PubMed] [Google Scholar]
- Core LJ, Lis JT. Transcription regulation through promoter-proximal pausing of RNA polymerase II. Science. 2008;319:1791–1792. doi: 10.1126/science.1150843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Falco G, Bagella L, Claudio PP, De Luca A, Fu Y, Calabretta B, Sala A, Giordano A. Physical interaction between CDK9 and B-Myb results in suppression of B-Myb gene autoregulation. Oncogene. 2000;19:373–379. doi: 10.1038/sj.onc.1203305. [DOI] [PubMed] [Google Scholar]
- Eissenberg JC, Shilatifard A, Dorokhov N, Michener DE. Cdk9 is an essential kinase in Drosophila that is required for heat shock gene expression, histone methylation and elongation factor recruitment. Mol Genet Genomics. 2007;277:101–114. doi: 10.1007/s00438-006-0164-2. [DOI] [PubMed] [Google Scholar]
- Estable MC, Naghavi MH, Kato H, Xiao H, Qin J, Vahlne A, Roeder RG. MCEF, the newest member of the AF4 family of transcription factors involved in leukemia, is a positive transcription elongation factor-b-associated protein. J Biomed Sci. 2002;9:234–245. doi: 10.1007/BF02256070. [DOI] [PubMed] [Google Scholar]
- Garber ME, Wei P, KewalRamani VN, Mayall TP, Herrmann CH, Rice AP, Littman DR, Jones KA. The interaction between HIV-1 Tat and human cyclin T1 requires zinc and a critical cysteine residue that is not conserved in the murine CycT1 protein. Genes Dev. 1998;12:3512–3527. doi: 10.1101/gad.12.22.3512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harper DP, Aplan PD. Chromosomal rearrangements leading to MLL gene fusions: clinical and biological aspects. Cancer Res. 2008;68:10024–10027. doi: 10.1158/0008-5472.CAN-08-2208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He N, Jahchan NS, Hong E, Li Q, Bayfield MA, Maraia RJ, Luo K, Zhou Q. A La-related protein modulates 7SK snRNP integrity to suppress P-TEFb-dependent transcriptional elongation and tumorigenesis. Mol Cell. 2008;29:588–599. doi: 10.1016/j.molcel.2008.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang MK, Mochizuki K, Zhou M, Jeong HS, Brady JN, Ozato K. The bromodomain protein Brd4 is a positive regulatory component of P-TEFb and stimulates RNA polymerase II-dependent transcription. Mol Cell. 2005;19:523–534. doi: 10.1016/j.molcel.2005.06.027. [DOI] [PubMed] [Google Scholar]
- Jeronimo C, Forget D, Bouchard A, Li Q, Chua G, Poitras C, Therien C, Bergeron D, Bourassa S, Greenblatt J, et al. Systematic analysis of the protein interaction network for the human transcription machinery reveals the identity of the 7SK capping enzyme. Mol Cell. 2007;27:262–274. doi: 10.1016/j.molcel.2007.06.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin C, Smith ER, Takahashi H, Lai KC, Martin-Brown S, Florens L, Washburn MP, Conaway JW, Conaway RC, Shilatifard A. AFF4, a component of the ELL/P-TEFb elongation complex and a shared subunit of MLL chimeras, can link transcription elongation to leukemia. Mol Cell. 2010;37:429–437. doi: 10.1016/j.molcel.2010.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mancebo HS, Lee G, Flygare J, Tomassini J, Luu P, Zhu Y, Peng J, Blau C, Hazuda D, Price D, Flores O. P-TEFb kinase is required for HIV Tat transcriptional activation in vivo and in vitro. Genes Dev. 1997;11:2633–2644. doi: 10.1101/gad.11.20.2633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martincic K, Alkan SA, Cheatle A, Borghesi L, Milcarek C. Transcription elongation factor ELL2 directs immunoglobulin secretion in plasma cells by stimulating altered RNA processing. Nat Immunol. 2009;10:1102–1109. doi: 10.1038/ni.1786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller D, Bach C, Zeisig D, Garcia-Cuellar MP, Monroe S, Sreekumar A, Zhou R, Nesvizhskii A, Chinnaiyan A, Hess JL, Slany RK. A role for the MLL fusion partner ENL in transcriptional elongation and chromatin modification. Blood. 2007;110:4445–4454. doi: 10.1182/blood-2007-05-090514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller D, Garcia-Cuellar MP, Bach C, Buhl S, Maethner E, Slany RK. Misguided transcriptional elongation causes mixed lineage leukemia. PLoS Biol. 2009;7:e1000249. doi: 10.1371/journal.pbio.1000249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen VT, Kiss T, Michels AA, Bensaude O. 7SK small nuclear RNA binds to and inhibits the activity of CDK9/cyclin T complexes. Nature. 2001;414:322–325. doi: 10.1038/35104581. [DOI] [PubMed] [Google Scholar]
- Peterlin BM, Price DH. Controlling the elongation phase of transcription with P-TEFb. Mol Cell. 2006;23:297–305. doi: 10.1016/j.molcel.2006.06.014. [DOI] [PubMed] [Google Scholar]
- Shell SA, Martincic K, Tran J, Milcarek C. Increased phosphorylation of the carboxyl-terminal domain of RNA polymerase II and loading of polyadenylation and cotranscriptional factors contribute to regulation of the ig heavy chain mRNA in plasma cells. J Immunol. 2007;179:7663–7673. doi: 10.4049/jimmunol.179.11.7663. [DOI] [PubMed] [Google Scholar]
- Shilatifard A, Lane WS, Jackson KW, Conaway RC, Conaway JW. An RNA polymerase II elongation factor encoded by the human ELL gene. Science. 1996;271:1873–1876. doi: 10.1126/science.271.5257.1873. [DOI] [PubMed] [Google Scholar]
- Shilatifard A, Duan DR, Haque D, Florence C, Schubach WH, Conaway JW, Conaway RC. ELL2, a new member of an ELL family of RNA polymerase II elongation factors. Proc Natl Acad Sci USA. 1997;94:3639–3643. doi: 10.1073/pnas.94.8.3639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sims RJ, 3rd, Belotserkovskaya R, Reinberg D. Elongation by RNA polymerase II: the short and long of it. Genes Dev. 2004;18:2437–2468. doi: 10.1101/gad.1235904. [DOI] [PubMed] [Google Scholar]
- Sobhian B, Laguette N, Nakamura M, Kiernan R, Benkirane M. HIV-1 Tat assembles a multifunctional transcription elongation complex and stably associates with the 7SK snRNP. Mol Cell. 2010;38:439–451. doi: 10.1016/j.molcel.2010.04.012. this issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taki T, Kano H, Taniwaki M, Sako M, Yanagisawa M, Hayashi Y. AF5q31, a newly identified AF4-related gene, is fused to MLL in infant acute lymphoblastic leukemia with ins(5;11)(q31;q13q23) Proc Natl Acad Sci USA. 1999;96:14535–14540. doi: 10.1073/pnas.96.25.14535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thirman MJ, Levitan DA, Kobayashi H, Simon MC, Rowley JD. Cloning of ELL, a gene that fuses to MLL in a t(11;19)(q23;p13.1) in acute myeloid leukemia. Proc Natl Acad Sci USA. 1994;91:12110–12114. doi: 10.1073/pnas.91.25.12110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei P, Garber ME, Fang SM, Fischer WH, Jones KA. A novel CDK9-associated C-type cyclin interacts directly with HIV-1 Tat and mediates its high-affinity, loop-specific binding to TAR RNA. Cell. 1998;92:451–462. doi: 10.1016/s0092-8674(00)80939-3. [DOI] [PubMed] [Google Scholar]
- Yang Z, Zhu Q, Luo K, Zhou Q. The 7SK small nuclear RNA inhibits the CDK9/cyclin T1 kinase to control transcription. Nature. 2001;414:317–322. doi: 10.1038/35104575. [DOI] [PubMed] [Google Scholar]
- Yang Z, Yik JH, Chen R, He N, Jang MK, Ozato K, Zhou Q. Recruitment of P-TEFb for stimulation of transcriptional elongation by the bromodomain protein Brd4. Mol Cell. 2005;19:535–545. doi: 10.1016/j.molcel.2005.06.029. [DOI] [PubMed] [Google Scholar]
- Yik JHN, Chen R, Nishimura R, Jennings JL, Link AJ, Zhou Q. Inhibition of P-TEFb (CDK9/Cyclin T) kinase and RNA polymerase II transcription by the coordinated actions of HEXIM1 and 7SK snRNA. Mol Cell. 2003;12:971–982. doi: 10.1016/s1097-2765(03)00388-5. [DOI] [PubMed] [Google Scholar]
- Yokoyama A, Lin M, Naresh A, Kitabayashi I, Cleary ML. A Higher-Order Complex Containing AF4 and ENL Family Proteins with P-TEFb Facilitates Oncogenic and Physiologic MLL-Dependent Transcription. Cancer Cell. 2010;17:198–212. doi: 10.1016/j.ccr.2009.12.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Q, Sharp PA. Novel mechanism and factor for regulation by HIV-1 Tat. EMBO J. 1995;14:321–328. doi: 10.1002/j.1460-2075.1995.tb07006.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Q, Yik JH. The Yin and Yang of P-TEFb regulation: implications for human immunodeficiency virus gene expression and global control of cell growth and differentiation. Microbiol Mol Biol Rev. 2006;70:646–659. doi: 10.1128/MMBR.00011-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Q, Chen D, Pierstorff E, Luo K. Transcription elongation factor P-TEFb mediates Tat activation of HIV-1 transcription at multiple stages. EMBO J. 1998;17:3681–3691. doi: 10.1093/emboj/17.13.3681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Pe’ery T, Peng J, Ramanathan Y, Marshall N, Marshall T, Amendt B, Mathews MB, Price DH. Transcription elongation factor P-TEFb is required for HIV-1 tat transactivation in vitro. Genes Dev. 1997;11:2622–2632. doi: 10.1101/gad.11.20.2622. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
2