Innate or Adaptive Immunity? The Example of Natural Killer Cells (original) (raw)
. Author manuscript; available in PMC: 2011 May 9.
Published in final edited form as: Science. 2011 Jan 7;331(6013):44–49. doi: 10.1126/science.1198687
Abstract
Natural killer (NK) cells were originally defined as effector lymphocytes of innate immunity endowed with constitutive cytolytic functions. More recently, a more nuanced view of NK cells has emerged. NK cells are now recognized to express a repertoire of activating and inhibitory receptors that is calibrated to ensure self-tolerance while allowing efficacy against assaults such as viral infection and tumor development. Moreover, NK cells do not react in an invariant manner but rather adapt to their environment. Finally, recent studies have unveiled that NK cells can also mount a form of antigen-specific immunologic memory. NK cells thus exert sophisticated biological functions that are attributes of both innate and adaptive immunity, blurring the functional borders between these two arms of the immune response.
The immune system is classically divided into innate and adaptive immunity. The distinctive features of innate immunity commonly refer to a broadly distributed variety of myeloid and lymphoid cells that can exert rapid effector function through a limited repertoire of germline-encoded receptors. In contrast, adaptive immunity in mammals is characterized by two types of lymphocytes, T and B cells, clonally expressing a large repertoire of antigen receptors that are produced by site-specific somatic recombination, that is, T cell receptor (TCR) and antibody/B cell receptor (BCR). Functionally, naive T and B cells encounter antigens in specialized lymphoid organs and undergo a process of cell division and maturation before exerting their effector function. Natural killer (NK) cells represent a subgroup of white blood cells. Since their identification in 1975 (1, 2), NK cells have been classified as lymphocytes on the basis of their morphology, their expression of many lymphoid markers, and their origin from the common lymphoid progenitor cell in the bone marrow. NK cells, however, are generally considered to be components of innate immune defense because they lack antigen-specific cell surface receptors. In addition, despite the extreme rarity of convincing cases of selective NK cell deficiency in humans (Online Mendelian Inheritance in Man database 609981) (3), NK cells have been shown in humans and mice to participate in the early control against virus infection, especially herpesvirus infection (4), and in tumor immunosurveillance (5). The lack of gross abnormalities in X-linked severe combined immunodeficiency (SCID-X1) patients who have undergone hematopoietic stem cell transplantation (HSCT) or IL2RG gene therapy, but remain unexpectedly NK cell deficient, has supported the possibility that NK cells might exert redundant function (6). However, the presence of NK cells in nonhuman mammals and NK cell orthologs in other vertebrates argues for their importance (7). Notably, NK cells are peculiar in their capacity to invade the uterus, where they have been shown to contribute to the development of the embryo (8). These data prompt speculation that the role of NK cells during reproduction has contributed to their selection.
How Do NK Cells Contribute to Immunity?
NK cells were originally described as cytolytic effector lymphocytes, which, unlike cytotoxic T cells, can directly induce the death of tumor cells and virus-infected cells in the absence of specific immunization; hence their name. Subsequently, NK cells have been recognized as major producers of cytokines such as interferon-γ (IFN-γ) in many physiological and pathological conditions. NK cells also produce an array of other cytokines, both proinflammatory and immunosuppressive, such as tumor necrosis factor–α (TNF-α) and interleukin (IL)–10, respectively, and growth factors such as GM-CSF (granulocyte macrophage colony-stimulating factor), G-CSF (granulocyte colony-stimulating factor), and IL-3. NK cells also secrete many chemokines, including CCL2 (MCP-1), CCL3 (MIP1-α), CCL4 (MIP1-β), CCL5 (RANTES), XCL1 (lymphotactin), and CXCL8 (IL-8) (9). Whereas the biological function of the growth factors secreted by NK cells remains to be clarified, their secretion of chemokines is key to their colocalization with other hematopoietic cells such as dendritic cells (DC) in areas of inflammation (10). Furthermore, the production of IFN-γ by NK cells helps to shape T cell responses in lymph nodes, possibly by a direct interaction between naïve T cells and NK cells migrating to secondary lymphoid compartments from inflamed peripheral tissues and by an indirect effect on DC (11) (Fig. 1). NK cell–mediated killing of target cells also impacts T cell responses, possibly by decreasing the antigenic load (12) and/or because target cell debris might promote antigen cross-presentation to CD8+ cytotoxic T cells (13) (Fig. 1). Although NK cells can positively (12, 13) or negatively (14) influence host T and B cell immunity, depending on the nature of the antigenic challenge, the emerging notion is that NK cells are not only cytolytic effector cells against microbe-infected cells or tumor cells. Rather, NK cell–mediated cytotoxicity and cytokine production impact DC, macrophages, and neutrophils (10) and endow NK cells with regulatory function affecting subsequent antigen-specific T and B cell responses. Conversely, the “natural” effector function of NK cells has been revisited. NK cells require priming by various factors, such as IL-15 presented by DC (15) or macrophages (16), IL-12 (17) or IL-18 (18), to achieve their full effector potential, highlighting the intimate regulatory interactions between NK cells and other components of the immune response. Thus, NK cells, like T and B cells, participate in the immunity in many different ways and undergo a process of functional maturation to fulfill these functions.
Fig. 1.
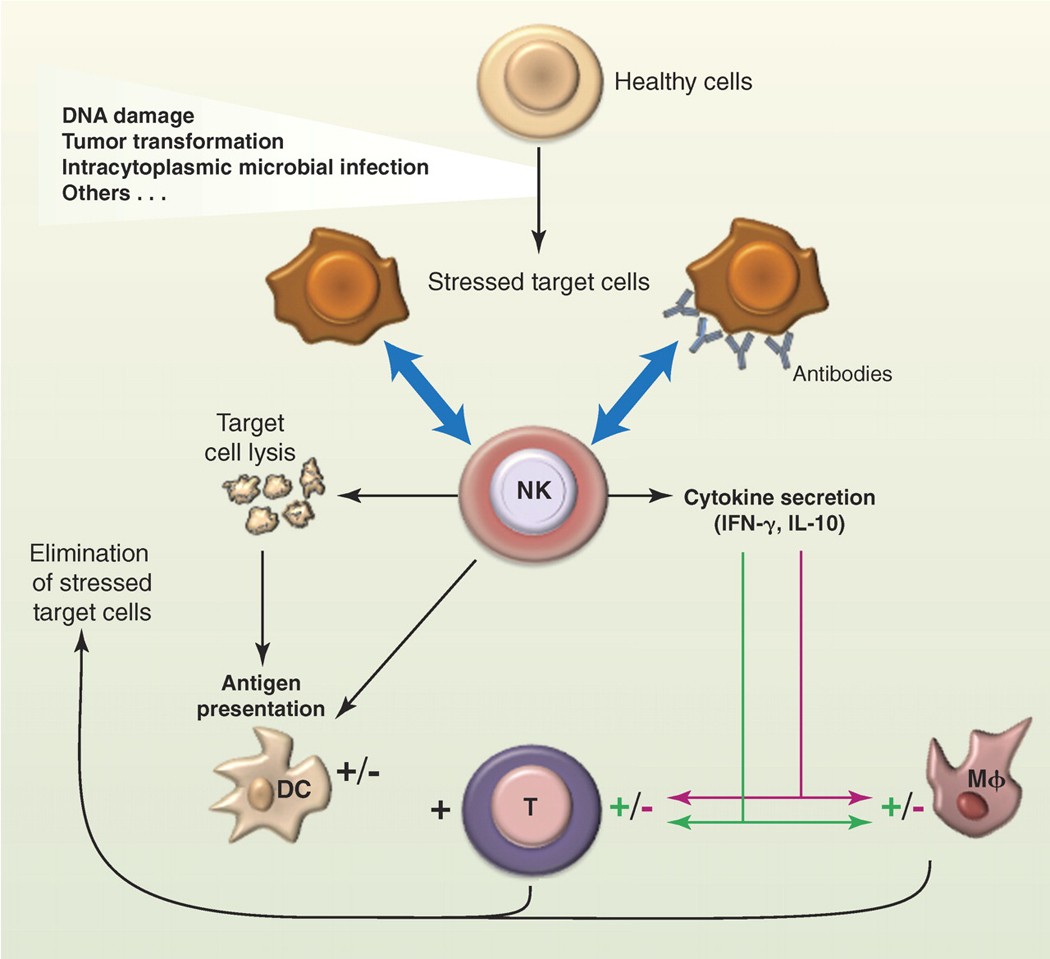
The biological functions of NK cells. NK cells can recognize a variety of stressed cells in the absence or in the presence of antibodies (blue arrows). NK cell activation triggered by this recognition can lead to the lysis of the target cell and to the production of various cytokines and chemokines depending on the nature of the stimulation. Whereas NK cells are biased to produce IFN-γ in many conditions, there are situations of chronic or systemic inflammation that promote IL-10 secretion. NK can also cross-talk with DC in many different ways, including the NK cell killing of immature DC and the promotion of DC maturation by NK cell–derived IFN-γ and TNF-α, which leads to enhanced antigen presentation to T cells. Through these biological activities, NK cells participate in the shaping of the subsequent immune response; in the depicted example, NK cells boost or dampen macrophage and T cell responses through IFN-γ (green arrows) or IL-10 secretion (red arrows), respectively.
How Are NK Cells Regulated?
NK cells are equipped with an array of receptors that can either stimulate NK cell reactivity (activating receptors) or dampen NK cell reactivity (inhibitory receptors) (19, 20). Activating receptors include receptors that interact with soluble ligands such as cytokines and receptors that interact with cell surface molecules (Fig. 2). Cytokine receptors that are coupled to the common gamma chain (γc), such as IL-15R, IL-2R, and IL-21R, are involved in NK cell development and effector function. In particular, IL-15 is required for the maturation and survival of NK cells, consistent with the absence of circulating NK cells in SCID-X1 patients and in mice lacking IL-15 or IL-15R components (21). Cytokine receptors that are linked to the adapter protein MyD88 are also important for NK cell maturation, namely IL-1R in humans (22) and IL-18R in the mouse (18).
Fig. 2.
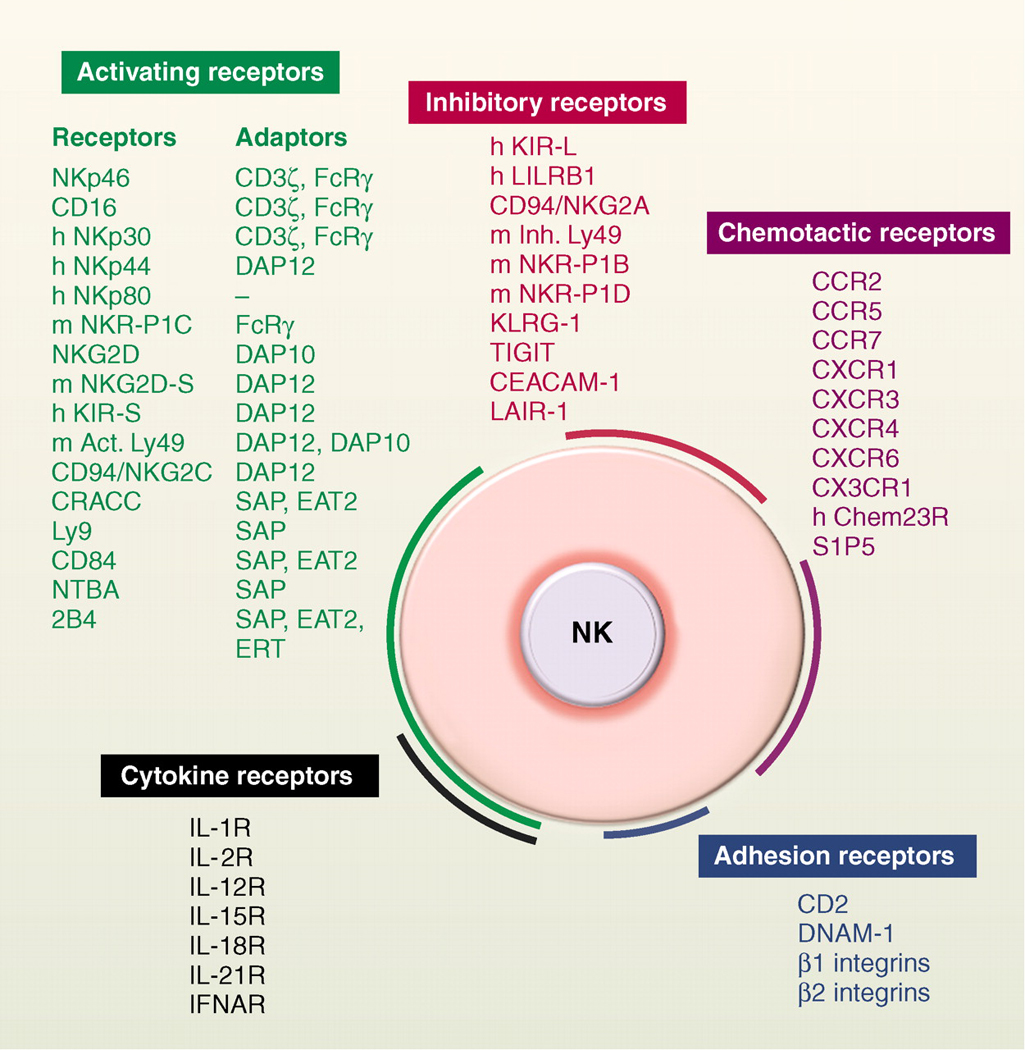
NK cell receptors. NK cells express many cell surface receptors that can be grouped into activating (green), inhibitory (red), adhesion (blue), cytokine (black) and chemotactic receptors (purple). In addition to MHC class I–specific receptors, other NK cell inhibitory receptors specific for non-MHC ligands also regulate NK cell reactivity (78). Adaptor molecules involved in the signaling cascade downstream of the engagement of activating receptors (green) are also indicated. The list of cell surface molecules involved in the regulation of mouse and human NK cell function is not exhaustive. Unless indicated (h, human; m, mouse), receptors are conserved in both species.
NK cells exert their biological functions by various means. NK cells can kill a variety of target cells, including virus-infected cells and tumors, in the absence of antibody. In the case of viruses, the mouse Ly49H activating receptor recognizes a cytomegalovirus-encoded ligand (m157) (23, 24), and NKp46 has been reported to interact with hemagglutinins derived from influenza and parainfluenza viruses (25). NK cells are also able to detect antibody-coated cells through the FcγRIIIA (CD16) cell surface receptor and to exert antibody-dependent cell cytotoxicity (ADCC) and cytokine production. CD16 is coupled to the CD3ζ and FcRγ signal transduction polypeptides bearing intracytoplasmic immunoreceptor tyrosine-based activation motifs (ITAMs). The natural cytotoxicity receptors (NKp46/NCR1, NKp44/NCR2, and NKp30/NCR3) are also potent activation receptors linked to the ITAM-bearing CD3ζ, FcRγ, or DAP12 molecules (26). In mice, the NK1.1 (Nkrp1c) molecule on CD3− cells has been a useful marker for NK cells, but its expression is confined to only certain strains of mice. NKp46 appears to be the most specific NK cell marker across mammalian species, although discrete subsets of T cells also express it (27). Accumulating data in humans and mice also indicate that NCR+ cells (NKp46+ in the mouse, NKp46+NKp44+ in humans) that produce IL-22, a cytokine noted to be important in mucosal immunity, are found in gut-associated mucosal tissue. In contrast to bona fide NK cells, these NCR+IL-22+ mucosal cells express the transcription factor RORγt, are not cytotoxic, do not secrete IFN-γ, and are not dependent on IL-15 for their development (28, 29). NCR+IL-22+ are thus clearly distinct from the conventional NK cell subsets and likely derive from a different lineage that could be related to the lymphoid tissue inducer (LTi) cells involved in the formation of lymphoid tissue (28, 29). In contrast to the ITAM-coupled antigen-specific TCR and BCR whose absence leads to a complete block in T and B cell development, respectively, NK cells still develop in the absence of ITAM-bearing molecules (30). These results highlight the redundancy of NK cell developmental pathways and may explain the robustness of this lymphoid cell compartment in most cases of immune deficiencies.
A feature of several NK cell activating receptors resides in their capacity to detect self molecules induced in conditions of cellular stress (31). This is the case for NKG2D, which interacts with various ligands that are expressed at low levels in most tissues but are overexpressed upon initiation of cellular distress, for example, after initiation of the DNA damage response (32). This is also the case for B7-H6, a ligand for NKp30 that has not been detected in healthy cells but is expressed on certain tumor cells (33).
Pioneering work showed that NK cells can detect the lack of major histocompatibility complex (MHC) class I (“missing self”), a situation that can occur when cells are perturbed by viral infection or cellular transformation (34). This “missing self” recognition is explained by the NK cell surface expression of a variety of MHC class I–specific inhibitory receptors that include killer cell immunoglobulin-like receptors (KIRs) in humans, lectin-like Ly49 molecules in mice, and CD94/NKG2A heterodimers in both species (35, 36). These MHC class I receptors belong to the large family of inhibitory receptors that mediate their function by signaling through intracytoplasmic immunoreceptor tyrosine-based inhibition motifs (ITIMs) (19). Thus, NK cells spare healthy cells that express self-MHC class I molecules and low amounts of stress-induced self molecules, whereas they selectively kill target cells “in distress” that down-regulate MHC class I molecules and/or up-regulate stress-induced self molecules such as NKG2D ligands (Fig. 3) (32).
Fig. 3.
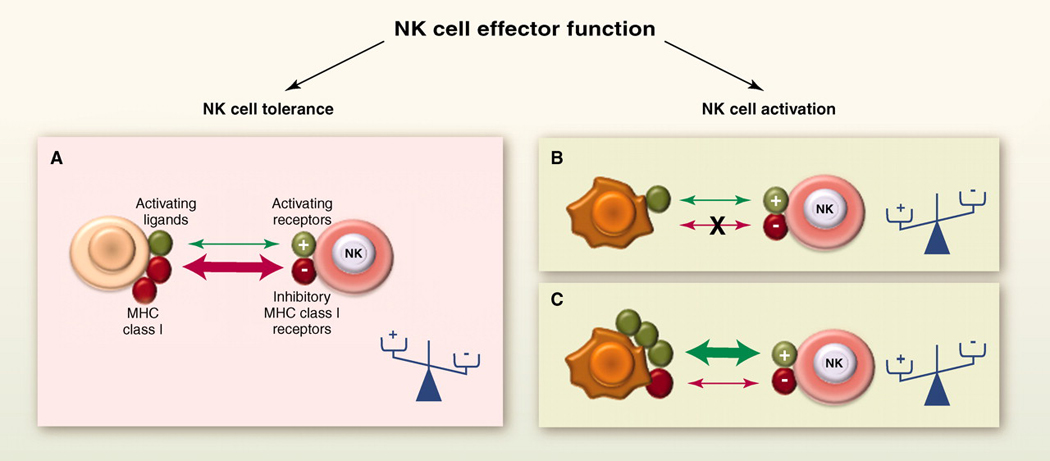
The dynamic regulation of NK cell effector function. NK cells sense the density of various cell surface molecules expressed at the surface of interacting cells. The integration of these distinct signals dictates the quality and the intensity of the NK cell response. NK cells spare healthy cells that express self-MHC class I molecules and low amounts of stress-induced self molecules (A), whereas they selectively kill target cells “in distress” that down-regulate MHC class I molecules (B) or up-regulate stress-induced self molecules (C). +, activating receptors; −, inhibitory receptors.
Why Aren’t NK Cells Self-Reactive?
Like T cells and B cells, NK cells have the potential for autoreactivity even though NK receptor genes do not undergo somatic diversification. This is because some NK cells lack inhibitory receptors that bind to the MHC class I molecules of the host (37, 38) or they express activating receptors that recognize self ligands, including MHC molecules (39–41). These patterns of expression arise because the array of receptors that individual NK cells come to express during development is largely random, and the MHC ligands recognized by these receptors are inherited independently of the receptor genes (42). Therefore, some NK cells may express activating receptors for a self ligand, yet fail to express inhibitory receptors for self-MHC molecules.
To avoid autoreactivity, an education system exists whereby such NK cells acquire self-tolerance. The potentially autoreactive NK cells are not generally clonally deleted but instead acquire a state of hyporesponsiveness to stimulation through various activating receptors. Thus, in normal mice (38) or humans (43), a fraction of NK cells lack inhibitory receptors for self-MHC, and these NK cells are unresponsive to self cells (Fig. 4A). A related situation applies in mice or humans that lack MHC class I molecules, where NK cells exist in normal numbers but fail to exert detectable autoimmunity or to kill MHC class I–deficient autologous cells in vivo or in vitro (44–46) (Fig. 4B). In both cases, the NK cells not only are unresponsive to self cells but also exhibit reduced responses to various other stimuli, including MHC class I–deficient tumor cells or cross-linking antibodies specific for activating receptors (37, 38, 43, 44, 47). By comparison, by an MHC-dependent education process described as licensing by some investigators, the NK cells that express receptors for self MHC in normal animals or humans exhibit greater responsiveness to stimulation, but their effector function against neighboring normal cells is inhibited by engagement of the MHC-specific inhibitory receptors (37, 38, 48). Whether NK responsiveness is actively induced by encounters with cells expressing MHC ligands for these NK cells (called “arming”), or hyporesponsiveness is actively induced by encounters with normal cells that lack MHC ligands and at the same time express stimulatory ligands for these NK cells (called “disarming,” or energy), or both, remain unsettled issues (48). The molecular mechanisms that govern responsiveness are also not established, except that it is clear that changes in responsiveness are not correlated with changes in the expression of the known activating receptors (37, 38, 43, 44, 47, 49).
Fig. 4.

NK cell tuning. Experimental conditions in which NK cells have been shown to adapt to their environment are schematized. In the absence of detection of MHC class I, such as when NK cells lack cognate MHC class I receptors (A) or in MHC class I–deficient hosts (B), NK cells are hyporesponsive at steady state. NK cells are rendered “anergic” by the chronic engagement of various activating receptors such as NKG2D (C), Ly49H in the mouse (D), or KIR2DS1 in humans (E). NK cells can be educated by MHC class I molecules via their cognate inhibitory receptors in trans (F) or in cis (not depicted) and primed by cytokines (G).
Experimental evidence for NK cell education in an MHC-independent scenario has been obtained using mice engineered to express ligands for activating receptors such as NKG2D (Fig. 4C) or Ly49H (50, 51) (Fig. 4D). The NK cells in these mice are tolerant to expressed ligand but retain expression of the corresponding receptor. Similarly, in humans, NK cells expressing the KIR2DS1 activating receptor specific for the human lymphocyte antigen (HLA)–C2 allotype are functional only when derived from C1/C2 or C1/C1 donors but hyporesponsive in donors homozygous for C2 (52). This suggests that in the presence of high levels of activating ligands, a negative tuning effect may occur (53) (Fig. 4E).
It is possible that some of the mechanisms that confer tolerance in mice with constitutive expression of activating ligands are the same as those that operate when NK cells lack inhibitory receptors for self-MHC. One possible mechanism for the impaired responsiveness of NK cells that are not inhibited by MHC molecules is the induction of an anergic state, as can occur in autoreactive T cells and B cells. Another is a failure of these NK cells to undergo terminal functional maturation, which may depend on interactions between MHC and inhibitory receptors on NK cells. Other possibilities include the function of inhibitory receptors for non-MHC ligands or the action of suppressor cells, but these are unlikely to fully account for these outcomes.
Whatever the mechanism (or mechanisms), it must account for the existence of intermediate states of responsiveness. NK cells vary in the number and affinity of inhibitory receptors specific for self-MHC, and the functional response of NK cells to activating stimuli was shown to increase commensurately with the number of different inhibitory receptors for self-MHC that the NK cells expressed (53, 54). Despite exhibiting greater responsiveness, NK cells with more inhibitory receptors are not autoreactive, because interactions of their inhibitory receptors with MHC class I molecules on normal cells inhibits their activity. Thus, NK cells appear to be “tuned” such that the greater effector cell inhibition that accompanies the expression of more inhibitory receptors is balanced by a greater potential responsiveness of the NK cells.
Several findings suggest that the responsiveness of mature NK cells is not fixed but may adapt to a changing environment in vivo. In the absence of infection or other disease, transfer of mature NK cells to mice with no MHC ligands led to a reduced responsiveness of the NK cells, indicating that encounters with cells lacking self-MHC, which would normally stimulate these cells, instead drive them into a hyporesponsive state (55). Conversely, transfer of NK cells from MHC-deficient mice to MHC class I+ mice resulted in increased responsiveness, specifically of those NK cells with an inhibitory receptor specific for MHC molecules in the new host, indicating that the inhibitory interaction is instrumental in increasing NK responsiveness (55, 56). Hence, persistent stimulation without inhibition results in NK cell hyporesponsiveness, whereas persistent stimulation coupled with commensurate inhibition results in NK cell responsiveness. These results suggest that NK cell tuning might occur throughout the lifetime of the NK cell under steady-state conditions. In infected animals, however, hyporesponsive NK cells are converted to a higher state of responsiveness. In fact, NK cells lacking self-MHC–specific inhibitory receptors play a more important role than other NK cells in protective responses to mouse cytomegalovirus infections (57), probably reflecting an increased responsiveness associated with infection coupled with the absence of inhibitory receptor interactions. Taken together, these findings suggest that in steady-state conditions, NK cell tuning enables those NK cells with inhibitory receptors for self-MHC to rapidly eliminate MHC class I–deficient cells that arise in the environment, whereas NK cells with fewer such receptors can be mobilized by inflammatory signals that accompany pathogen infections (38, 48).
Can NK Cell Reactivity Be Manipulated in Anticancer Treatments?
The dissection of NK cell reactivity has unveiled the basis of the recognition of tumor cells by NK cells. In mice, NK cells reject tumors that lack MHC class I expression or overexpress NKG2D ligands or costimulatory signals, a phenomenon facilitating T cell–mediated antitumor immunity. NK cells protect the host against methylcholanthrene-induced sarcomas and against B cell lymphoma arising in mice lacking perforin and β2 microglobulin (a component of MHC class I) (58). In humans, the major receptors responsible for tumor recognition by NK cells are NKp46, NKp30, NKp44, DNAM1, and NKG2D. The NK cell-mediated lysis of tumor cells involves several such receptors, depending on the malignancy. The target cell ligands recognized by some receptors have been identified, such as MICA/B and the ULBPs for NKG2D, PVR and Nectin-2 for DNAM-1, and B7-H6 for NKp30, which are primarily expressed or up-regulated on cells after activation, proliferation, or cellular transformation (31).
Several lines of evidence indicate that NK cells or their receptors have a role in immuno-surveillance of spontaneous tumors, including in humans. Indeed, tumors have evolved mechanisms to escape NK cell control such as the shedding of soluble NKG2D ligands that function as decoys for the activatingNKG2D receptor on NK cells, a phenomenon correlating with poor prognosis in human melanoma and prostate cancer (58). Mice deficient in NKG2D exhibited a higher incidence or greater severity of tumors in transgenic models of cancer (59). Furthermore, studies with mice deficient in DNAM-1, NKp46, or NKG2D demonstrate that in the presence of NK cells, tumors alter their expression of ligands (60, 61). In addition, an 11-year follow-up survey revealed that low NK lytic activity is associated with cancer risk (62).
This knowledge has prompted efforts to harness NK cell functions for an improved management of cancer patients. The seminal observation was the demonstration in humans that the success of T cell–depleted HSCT for the treatment of leukemia patients is much greater when the recipient lacks one HLA haplotype compared with the donor marrow and donor NK cells are present in the bone marrow cell infusion (63). This outcome can be attributed to “missing-self” recognition by a subset of donor alloreactive NK cells of the recipient’s tumor cells (64). These alloreactive NK cells, which express KIRs that do not recognize MHC molecules in the recipient, persist for several years and attack the recipient’s leukemic cells (graft versus leukemia reaction) but fail to cause the generalized graft-versus-host disease that alloreactive T cells can cause (65, 66). These NK cells have been shown also to promote engraftment and prevent graft-versus-host disease due to their ability to kill recipient antigen-presenting cells (63). On the basis of the education and tuning phenomena, these alloreactive NK cells would be expected to be hyporesponsive. Potentially, the infusion of large numbers of CD34+ cells provides a hematopoietic microenvironment predominantly of donor type in which the process of NK cell education and tuning would be similar to that occurring in the donor and result in generation of NK cells displaying alloreactivity against leukemic blasts. Recently, an alternative to manipulating NK cell–mediated “missing self” recognition has been set up, using the infusion of human monoclonal antibodies to KIR in cancer patients (67). These protocols are being tested in phase II clinical trials in acute myeloid leukemia and multiple myeloma. Finally, production of clinical-grade human NK cells has proven feasible, safe, and promising (65, 68), and combinations of adoptive NK cell transfer with therapeutic monoclonal antibodies are being conducted. NK cell–based therapy should benefit from a better knowledge of NK cell biodistribution and homing in vivo, identification of ligands for some activating receptors, and NK-specific immunosuppressive and immunomodulatory mechanisms. Additional studies on the role of NK cell education and KIR mismatch may also provide optimal strategies for exploiting NK cells in antitumor therapies. Moreover, genetic epidemiologic studies have shown that the expression of certain KIRs and MHC class I polymorphisms are linked to resistance to several microbes, such as human immunodeficiency virus type 1 (HIV-1) and hepatitis C, or to susceptibility to various autoimmune or inflammatory syndromes (69). As KIR can also be expressed by T cell subsets, the direct relevance of some of these data to NK cell biology remains to be firmly established. Nevertheless, these studies prompt us to extend the design of NK cell–based therapies to other disease conditions than cancer, such as infections and inflammation.
Do NK Cells Remember?
Immunological memory is a hallmark of adaptive immunity and is characterized by the long-term persistence of memory cells that rapidly undergo clonal expansion and present enhanced effector functions in response to secondary challenge. Although recent findings have shown a form of immunological memory in lower organisms that are reported to lack adaptive immunity (70), the innate immune system has been commonly considered to lack the capacity for immunological memory. Moreover, in the case of mature NK cells, their half-life has been estimated to be 17 days in steady-state conditions (71). Therefore, recent findings that at least some mature NK cells or their progeny can be long-lived and that NK cells can mount a robust recall response are quite striking.
The first evidence for NK cell memory was observed in a model of hapten-induced contact hypersensitivity in recombination activating gene–2 (Rag-2)–deficient mice, which lack T and B cells but possess NK cells (72). Hapten-induced contact hypersensitivity (CHS) was previously thought to be mediated only by CD4+ T cells after priming mice with a chemical hapten. Unexpectedly, this NK cell–mediated CHS response in Rag-2–deficient mice could be detected for at least a month after chemical priming, and the response was hapten-specific. These “memory”NK cells were unexpectedly found to reside only in the liver, but not in the spleen, and were marked by high levels of expression of cell surface Thy1 (72) and CXCR6 (73). A hapten-specific CHS response was observed in mice receiving an adoptive transfer of liver NK cells from hapten-primed mice. Although blocking the NKG2D receptor on the NK cells inhibited the CHS, the receptor responsible for hapten-specific recognition has not been identified (72, 73).
NK cell memory has also been demonstrated in viral infections. In C57BL/6 mice, the activating Ly49H receptor recognizes the mouse cytomegalovirus (MCMV) m157 glycoprotein that is displayed on the cell surface of infected cells, resulting in NK cell–mediated control of the infection (23, 24). After infection with MCMV, these Ly49H+ NK cells undergo preferential expansion (74). In experiments in which genetically marked, mature Ly49H+ NK cells were adoptively transferred into recipients infected with MCMV, the Ly49H+ NK cell population underwent contraction after control of the virus, but memory NK cells could be detected in the recipient more than a month later (75). Similar to memory T lymphocytes, upon restimulation these memory NK cells demonstrated enhanced cytolytic function and cytokine production compared with “naïve” NK cells and were more efficient at protecting MCMV-susceptible neonatal mice against infection (75). Memory NK cells isolated from the first host can be adoptively transferred to a second and even a third recipient and undergo subsequent rounds of proliferation in response to MCMV infection (75). At about 2 months after the initial infection with MCMV, memory NK cells could be detected in essentially all tissues and organs, including spleen, lymph nodes, liver, lung, and kidney (75). Although there is as yet no unique marker of memory, these long-lived MCMV-expanded NK cells stably express high levels of KLRG1, an inhibitory receptor that recognizes cadherins. Recently, memory NK cells have been described in mice after exposure to influenza, vesicular stomatitis virus (VSV), or HIV-1 (73), although a virus-specific NK receptor for these pathogens has not been identified.
In addition, recent studies have demonstrated that NK cells activated with cytokines in vitro and adoptively transferred into naïve recipients can also persist for at least a month and have an enhanced ability to produce cytokines upon restimulation (76). These findings suggest that, once activated, mature NK cells may acquire stable, heritable properties that influence their behavior during subsequent infections. Thus, NK cells appear to remember their past, a trait conventionally only considered possible for the adaptive immune system. The emerging evidence for immunological memory and the capacity for self-renewal of mature cells in the NK cell lineage raises many questions: Can NK cells expanded in response to one pathogen provide enhanced protection against other unrelated pathogens, given that NK cells possess multiple activating receptors rather than a single, dominant antigen receptor like B and T cells? Is it possible to vaccinate NK cells for enhanced host defense? What receptor systems provide for hapten-specific recognition by NK cells? What epigenetic alterations account for the longevity and enhanced effector functions demonstrated by memory NK cells? Can NK cells, like T cells, differentiate into functionally distinct subsets with regulatory roles in shaping the magnitude and nature of the immune response to different pathogens? Is memory confined to a certain subset of NK cells, as suggested by their apparent localizing in the liver as observed in some experimental systems?
Innate or Adaptive Immunity?
In addition to the above questions that they raise, recent advances in NK cell biology have thus shown that NK cells have attributes of both innate and adaptive immunity. These findings also lead to the speculation that the shared innate and adaptive features are likely not unique to NK cells. Along this line, macrophages rapidly phagocytose CD47-deficient erythrocytes, because the inhibitory macrophage receptor SIRP1α is no longer engaged by CD47, but macrophages from CD47-deficient mice do not phagocytose CD47-deficient erythrocytes (77), suggesting that macrophages have adapted to the absence of CD47 in their environment. Therefore, macrophages might undergo a process of education through the interaction of the ITIM-bearing SIRP1α with its cognate CD47 ligand, reminiscent of NK cell education through MHC class I–specific receptor engagement. Thus, notions originally restricted to T and B cells, such as diverse receptor repertoires, education, and memory, which now apply to NK cells, prompt investigation into whether other innate immune cells show similar properties. Therefore, defining “innate” as having germline-encoded receptors versus “adaptive” as having rearranged receptors appears sufficient to distinguish these two arms of immunity.
References and Notes
- 1.Herberman RB, Nunn ME, Lavrin DH. Int. J. Cancer. 1975;16:216. doi: 10.1002/ijc.2910160204. [DOI] [PubMed] [Google Scholar]
- 2.Kiessling R, Klein E, Wigzell H. Eur. J. Immunol. 1975;5:112. doi: 10.1002/eji.1830050208. [DOI] [PubMed] [Google Scholar]
- 3.Eidenschenk C, et al. Am. J. Hum. Genet. 2006;78:721. doi: 10.1086/503269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lee SH, Miyagi T, Biron CA. Trends Immunol. 2007;28:252. doi: 10.1016/j.it.2007.04.001. [DOI] [PubMed] [Google Scholar]
- 5.Smyth MJ, Hayakawa Y, Takeda K, Yagita H. Nat. Rev. Cancer. 2002;2:850. doi: 10.1038/nrc928. [DOI] [PubMed] [Google Scholar]
- 6.Fischer A. Immunity. 2007;27:835. doi: 10.1016/j.immuni.2007.11.012. [DOI] [PubMed] [Google Scholar]
- 7.Jansen CA, et al. Dev. Comp. Immunol. 2010;34:759. doi: 10.1016/j.dci.2010.02.009. [DOI] [PubMed] [Google Scholar]
- 8.Moffett-King A. Nat. Rev. Immunol. 2002;2:656. doi: 10.1038/nri886. [DOI] [PubMed] [Google Scholar]
- 9.Walzer T, Dalod M, Robbins SH, Zitvogel L, Vivier E. Blood. 2005;106:2252. doi: 10.1182/blood-2005-03-1154. [DOI] [PubMed] [Google Scholar]
- 10.Moretta A, et al. Trends Immunol. 2005;26:668. doi: 10.1016/j.it.2005.09.008. [DOI] [PubMed] [Google Scholar]
- 11.Martín-Fontecha A, et al. Nat. Immunol. 2004;5:1260. doi: 10.1038/ni1138. [DOI] [PubMed] [Google Scholar]
- 12.Robbins SH, et al. PLoS Pathog. 2007;3:e123. doi: 10.1371/journal.ppat.0030123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Krebs P, et al. Blood. 2009;113:6593. doi: 10.1182/blood-2009-01-201467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Andrews DM, et al. J. Exp. Med. 2010;207:1333. doi: 10.1084/jem.20091193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lucas M, Schachterle W, Oberle K, Aichele P, Diefenbach A. Immunity. 2007;26:503. doi: 10.1016/j.immuni.2007.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mortier E, et al. Immunity. 2009;31:811. doi: 10.1016/j.immuni.2009.09.017. [DOI] [PubMed] [Google Scholar]
- 17.Guia S, et al. Blood. 2008;111:5008. doi: 10.1182/blood-2007-11-122259. [DOI] [PubMed] [Google Scholar]
- 18.Chaix J, et al. J. Immunol. 2008;181:1627. doi: 10.4049/jimmunol.181.3.1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vivier E, Nunès JA, Vély F. Science. 2004;306:1517. doi: 10.1126/science.1103478. [DOI] [PubMed] [Google Scholar]
- 20.Bryceson YT, March ME, Ljunggren HG, Long EO. Immunol. Rev. 2006;214:73. doi: 10.1111/j.1600-065X.2006.00457.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Caligiuri MA. Blood. 2008;112:461. doi: 10.1182/blood-2007-09-077438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hughes T, et al. Immunity. 2010;32:803. doi: 10.1016/j.immuni.2010.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Arase H, Mocarski ES, Campbell AE, Hill AB, Lanier LL. Science. 2002;296:1323. doi: 10.1126/science.1070884. [DOI] [PubMed] [Google Scholar]
- 24.Smith HR, et al. Proc. Natl. Acad. Sci. U.S.A. 2002;99:8826. [Google Scholar]
- 25.Mandelboim O, et al. Nature. 2001;409:1055. doi: 10.1038/35059110. [DOI] [PubMed] [Google Scholar]
- 26.Moretta A, et al. Annu. Rev. Immunol. 2001;19:197. doi: 10.1146/annurev.immunol.19.1.197. [DOI] [PubMed] [Google Scholar]
- 27.Walzer T, et al. Proc. Natl. Acad. Sci. U.S.A. 2007;104:3384. [Google Scholar]
- 28.Vivier E, Spits H, Cupedo T. Nat. Rev. Immunol. 2009;9:229. doi: 10.1038/nri2522. [DOI] [PubMed] [Google Scholar]
- 29.Veiga-Fernandes HH, Kioussis D, Coles M. J. Exp. Med. 2010;207:269. doi: 10.1084/jem.20100105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chiesa S, et al. Blood. 2006;107:2364. doi: 10.1182/blood-2005-08-3504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bottino C, Castriconi R, Moretta L, Moretta A. Trends Immunol. 2005;26:221. doi: 10.1016/j.it.2005.02.007. [DOI] [PubMed] [Google Scholar]
- 32.Raulet DH, Guerra N. Nat. Rev. Immunol. 2009;9:568. doi: 10.1038/nri2604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Brandt CS, et al. J. Exp. Med. 2009;206:1495. doi: 10.1084/jem.20090681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kärre K, Ljunggren HG, Piontek G, Kiessling R. Nature. 1986;319:675. doi: 10.1038/319675a0. [DOI] [PubMed] [Google Scholar]
- 35.Karlhofer FM, Ribaudo RK, Yokoyama WM. Nature. 1992;358:66. doi: 10.1038/358066a0. [DOI] [PubMed] [Google Scholar]
- 36.Moretta A, et al. Annu. Rev. Immunol. 1996;14:619. doi: 10.1146/annurev.immunol.14.1.619. [DOI] [PubMed] [Google Scholar]
- 37.Fernandez NC, et al. Blood. 2005;105:4416. doi: 10.1182/blood-2004-08-3156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kim S, et al. Nature. 2005;436:709. doi: 10.1038/nature03847. [DOI] [PubMed] [Google Scholar]
- 39.Lanier LL. Annu. Rev. Immunol. 2005;23:225. doi: 10.1146/annurev.immunol.23.021704.115526. [DOI] [PubMed] [Google Scholar]
- 40.Moretta A, et al. J. Exp. Med. 1995;182:875. doi: 10.1084/jem.182.3.875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Stewart CA, et al. Proc. Natl. Acad. Sci. U.S.A. 2005;102:13224. [Google Scholar]
- 42.Parham P. Nat. Rev. Immunol. 2005;5:201. doi: 10.1038/nri1570. [DOI] [PubMed] [Google Scholar]
- 43.Anfossi N, et al. Immunity. 2006;25:331. doi: 10.1016/j.immuni.2006.06.013. [DOI] [PubMed] [Google Scholar]
- 44.Liao N-S, Bix M, Zijlstra M, Jaenisch R, Raulet D. Science. 1991;253:199. doi: 10.1126/science.1853205. [DOI] [PubMed] [Google Scholar]
- 45.Höglund P, et al. Eur. J. Immunol. 1998;28:370. doi: 10.1002/(SICI)1521-4141(199801)28:01<370::AID-IMMU370>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 46.Zimmer J, et al. J. Exp. Med. 1998;187:117. doi: 10.1084/jem.187.1.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cooley S, et al. Blood. 2007;110:578. doi: 10.1182/blood-2006-07-036228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Raulet DH, Vance RE. Nat. Rev. Immunol. 2006;6:520. doi: 10.1038/nri1863. [DOI] [PubMed] [Google Scholar]
- 49.Höglund P, Brodin P. Nat. Rev. Immunol. 2010;10:724. doi: 10.1038/nri2835. [DOI] [PubMed] [Google Scholar]
- 50.Tripathy SK, et al. J. Exp. Med. 2008;205:1829. doi: 10.1084/jem.20072446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sun JC, Lanier LL. J. Exp. Med. 2008;205:1819. doi: 10.1084/jem.20072448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fauriat C, Ivarsson MA, Ljunggren HG, Malmberg KJ, Michaëlsson J. Blood. 2010;115:1166. doi: 10.1182/blood-2009-09-245746. [DOI] [PubMed] [Google Scholar]
- 53.Brodin P, Kärre K, Höglund P. Trends Immunol. 2009;30:143. doi: 10.1016/j.it.2009.01.006. [DOI] [PubMed] [Google Scholar]
- 54.Joncker NT, Fernandez NC, Treiner E, Vivier E, Raulet DH. J. Immunol. 2009;182:4572. doi: 10.4049/jimmunol.0803900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Joncker NT, Shifrin N, Delebecque F, Raulet DH. J. Exp. Med. 2010;207:2065. doi: 10.1084/jem.20100570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Elliott JM, Wahle JA, Yokoyama WM. J. Exp. Med. 2010;207:2073. doi: 10.1084/jem.20100986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Orr MT, Murphy WJ, Lanier LL. Nat. Immunol. 2010;11:321. doi: 10.1038/ni.1849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Terme M, Ullrich E, Delahaye NF, Chaput N, Zitvogel L. Nat. Immunol. 2008;9:486. doi: 10.1038/ni1580. [DOI] [PubMed] [Google Scholar]
- 59.Guerra N, et al. Immunity. 2008;28:571. doi: 10.1016/j.immuni.2008.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Iguchi-Manaka A, et al. J. Exp. Med. 2008;205:2959. doi: 10.1084/jem.20081611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Elboim M, et al. J. Immunol. 2010;184:5637. doi: 10.4049/jimmunol.0901644. [DOI] [PubMed] [Google Scholar]
- 62.Imai K, Matsuyama S, Miyake S, Suga K, Nakachi K. Lancet. 2000;356:1795. doi: 10.1016/S0140-6736(00)03231-1. [DOI] [PubMed] [Google Scholar]
- 63.Ruggeri L, et al. Science. 2002;295:2097. doi: 10.1126/science.1068440. [DOI] [PubMed] [Google Scholar]
- 64.Parham P, McQueen KL. Nat. Rev. Immunol. 2003;3:108. doi: 10.1038/nri999. [DOI] [PubMed] [Google Scholar]
- 65.Ljunggren HG, Malmberg KJ. Nat. Rev. Immunol. 2007;7:329. doi: 10.1038/nri2073. [DOI] [PubMed] [Google Scholar]
- 66.Pende D, et al. Blood. 2009;113:3119. doi: 10.1182/blood-2008-06-164103. [DOI] [PubMed] [Google Scholar]
- 67.Romagné F, et al. Blood. 2009;114:2667. doi: 10.1182/blood-2009-02-206532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Miller JS, et al. Blood. 2005;105:3051. doi: 10.1182/blood-2004-07-2974. [DOI] [PubMed] [Google Scholar]
- 69.Khakoo SI, Carrington M. Immunol. Rev. 2006;214:186. doi: 10.1111/j.1600-065X.2006.00459.x. [DOI] [PubMed] [Google Scholar]
- 70.Kurtz J. Trends Immunol. 2005;26:186. doi: 10.1016/j.it.2005.02.001. [DOI] [PubMed] [Google Scholar]
- 71.Jamieson AM, Isnard P, Dorfman JR, Coles MC, Raulet DH. J. Immunol. 2004;172:864. doi: 10.4049/jimmunol.172.2.864. [DOI] [PubMed] [Google Scholar]
- 72.O’Leary JG, Goodarzi M, Drayton DL, von Andrian UH. Nat. Immunol. 2006;7:507. doi: 10.1038/ni1332. [DOI] [PubMed] [Google Scholar]
- 73.Paust S, et al. Nat. Immunol. 2010;11:1127. doi: 10.1038/ni.1953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Dokun AO, et al. Nat. Immunol. 2001;2:951. doi: 10.1038/ni714. [DOI] [PubMed] [Google Scholar]
- 75.Sun JC, Beilke JN, Lanier LL. Nature. 2009;457:557. doi: 10.1038/nature07665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Cooper MA, et al. Proc. Natl. Acad. Sci. U.S.A. 2009;106:1915. [Google Scholar]
- 77.Wang H, et al. Proc. Natl. Acad. Sci. U.S.A. 2007;104:13744. [Google Scholar]
- 78.Kumar V, McNerney ME. Nat. Rev. Immunol. 2005;5:363. doi: 10.1038/nri1603. [DOI] [PubMed] [Google Scholar]
- 79.The authors apologize for not having been able to quote all relevant papers due to space limitations and thank C. Beziers-Lafosse (CIML) for excellent graphic assistance as well as members of their laboratories for help and discussions. E.V. and S.U. are supported by grants from the Agence Nationale de la Recherche (ANR), Ligue Nationale Contre le Cancer (Equipe Labellisée “La Ligue”), Fondation Del Duca, and institutional grants from INSERM, CNRS, and Université de la Méditerranée to the CIML. E.V. is a scholar from the Institut Universitaire de France and is a cofounder and shareholder of Innate-Pharma. D.H.R. is supported by NIH grants AI035021, AI039642, and CA093678 and is a scientific board member and shareholder of Innate-Pharma. A.M. is supported by grants awarded by Associazione Italiana per la Ricerca sul Cancro (AIRC), Ministero dell’Istruzione, dell’Università e della Ricerca, and a Special Project from the AIRC and is a cofounder and shareholder of Innate-Pharma. M.A.C. is supported by U.S. National Cancer Institute grants CA16058, CA95426, and CA68458. L.Z. is supported by Institut National du Cancer, ANR, Ligue Nationale Contre le Cancer (Equipe Labellisée “La Ligue”), and INFLARE EU grants. L.L.L. is an American Cancer Society professor and is supported by NIH grants AI068129, CA095137, and AI066897 and is a consultant for Novo Nordisk and SBI Biotech Co., Ltd. W.M.Y. is supported by the Howard Hughes Medical Institute and NIH grants AI33903, AI34385, AI51345, and AI5716.