Cdc53 is a scaffold protein for multiple Cdc34/Skp1/F-box protein complexes that regulate cell division and methionine biosynthesis in yeast (original) (raw)
Abstract
In budding yeast, ubiquitination of the cyclin-dependent kinase (Cdk) inhibitor Sic1 is catalyzed by the E2 ubiquitin conjugating enzyme Cdc34 in conjunction with an E3 ubiquitin ligase complex composed of Skp1, Cdc53 and the F-box protein, Cdc4 (the SCFCdc4 complex). Skp1 binds a motif called the F-box and in turn F-box proteins appear to recruit specific substrates for ubiquitination. We find that Skp1 interacts with Cdc53 in vivo, and that Skp1 bridges Cdc53 to three different F-box proteins, Cdc4, Met30, and Grr1. Cdc53 contains independent binding sites for Cdc34 and Skp1 suggesting it functions as a scaffold protein within an E2/E3 core complex. F-box proteins show remarkable functional specificity in vivo: Cdc4 is specific for degradation of Sic1, Grr1 is specific for degradation of the G1 cyclin Cln2, and Met30 is specific for repression of methionine biosynthesis genes. In contrast, the Cdc34–Cdc53–Skp1 E2/E3 core complex is required for all three functions. Combinatorial control of SCF complexes may provide a basis for the regulation of diverse cellular processes.
Keywords: SCF ubiquitin ligase, glucose, methionine, cyclin, Cdk, protein–protein interaction, Met30, Grrl, Cdc4
Ubiquitin-dependent proteolysis is an important regulatory mechanism that controls many cellular processes (for review, see Hochstrasser 1996). In this pathway, ubiquitin is transferred from a ubiquitin activating enzyme (E1) to a ubiquitin-conjugating enzyme (E2) and, often in conjunction with a ubiquitin ligase (E3), ultimately conjugated in an isopeptide linkage to a lysine residue of a substrate protein. Reiteration of the ubiquitin transferase reaction results in formation of a polyubiquitin chain on the substrate, which is then recognized by the 26S proteasome, and rapidly degraded. Specificity in protein ubiquitination often derives from E3 ubiquitin ligases (Hershko et al. 1983). In some cases, an E3 facilitates recognition of the target protein by an E2, whereas in others an E3 accepts a ubiquitin thioester from an E2 and directly transfers ubiquitin to the substrate (Scheffner et al. 1995). Although substrate recognition is a key aspect of ubiquitin mediated proteolysis, the identification of E3 enzymes has been problematic because the few known E3 families bear no sequence relationship to each other.
Ubiquitin-dependent proteolysis is essential for two major cell cycle transitions, the G1- to S-phase transition and the metaphase to anaphase transition (for review, see Hershko 1997). Key targets of the ubiquitin proteolytic pathway at these transitions include cyclins, which are positive regulators of cyclin-dependent kinases (Cdks), and Cdk inhibitors, which are negative regulators of Cdks. In budding yeast, a single Cdk, Cdc28 (a.k.a. Cdk1) is activated in G1 phase by the G1 cyclins Cln1–Cln3, and in S through M phase by the mitotic cyclins, Clb1–Clb6 (for review, see Nasmyth 1996). Mitotic cyclins and other proteins that regulate mitosis are targeted for degradation by a cell cycle regulated E3 ubiquitin ligase called the anaphase promoting complex (APC) or cyclosome (for review, see Hershko 1997). In contrast, G1 cyclins and Cdk inhibitors are degraded via a constitutive ubiquitination pathway in which substrates are targeted by phosphorylation.
Genetic analysis in budding yeast has revealed several components required for degradation of the Cdk inhibitor Sic1: Cdc4, a WD40 repeat protein (Yochem and Byers 1987); Cdc34, an E2 ubiquitin-conjugating enzyme (Goebl et al. 1988); Cdc53, a protein that forms a complex with Cdc4 and Cdc34 (Mathias et al. 1996; Willems et al. 1996); and Skp1, a protein that binds to a motif in Cdc4 called the F-box (Bai et al. 1996). The F-box motif was originally identified in Cdc4 and two mammalian proteins, Cyclin F and Skp2, and is also found in many other eukaryotic proteins (Bai et al. 1996). Cells lacking functional Cdc4, Cdc34, Cdc53, or Skp1 arrest in G1 phase because the Clb–Cdc28 inhibitor Sic1 is not degraded, and so the onset of Clb–Cdc28 activity and initiation of DNA replication cannot occur (Nugroho and Mendenhall 1994; Schwob et al. 1994; Bai et al. 1996). In late G1 phase, phosphorylation of Sic1 by the Cln–Cdc28 kinases targets the inhibitor for ubiquitination and degradation (Schwob et al. 1994; Schneider et al. 1996; Tyers 1996; Verma et al. 1997a). An E3 complex has been assembled from recombinant Skp1, Cdc53, and Cdc4 (the SCFCdc4 complex), and in association with Cdc34 this complex is sufficient for ubiquitination of phosphorylated Sic1 in vitro (Feldman et al. 1997; Skowrya et al. 1997). Within the SCFCdc4 complex, Skp1 facilitates the interaction of Cdc4 with Cdc53 and, in turn, the WD40 repeats of Cdc4 confer specific recognition of phosphorylated Sic1 (Skowrya et al. 1997). Many other important regulatory proteins are degraded via the SCFCdc4 pathway, including the Cln–Cdc28 inhibitor Far1 (Henchoz et al. 1997), the replication protein Cdc6 (Drury et al. 1997), and the transcription factor Gcn4 (Kornitzer et al. 1994).
In addition to their role in Sic1 proteolysis, Cdc34, Cdc53, and Skp1 mediate Cln degradation in vivo (Deshaies et al. 1995; Yaglom et al. 1995; Bai et al. 1996; Willems et al. 1996). Cln degradation is probably triggered by autocatalytic Cdc28 dependent phosphorylation of the Cln subunit in Cln–Cdc28 complexes (Lanker et al. 1996). Consistent with this requirement, Cdc53 forms a tight complex with phosphorylated Cln2 in yeast lysates (Willems et al. 1996). Cln1 and Cln2 degradation also requires Grr1, another F-box protein that interacts with Skp1 in vivo and in vitro (Barral et al. 1995; Li and Johnston 1997; Skowrya et al. 1997). The ability of two distinct F-box proteins, Cdc4 and Grr1, to confer substrate specificity on a common ubiquitination pathway lead to the hypothesis that F-box proteins are substrate-specific recruitment factors (Bai et al. 1996). In support of this idea, Grr1 is able to recruit phosphorylated Cln1 and Cln2, but not phosphorylated Sic1, into Cdc34–Cdc53–Skp1 complexes in vitro (Skowrya et al. 1997).
Finally, Skp1 has other functions that have not yet been linked directly to proteolysis. Skp1 acts with Grr1 and Cdc53 to mediate the induction of HXT glucose transporter genes by glucose, although Cdc34 is apparently dispensible in this pathway (Li and Johnston 1997). Furthermore, Skp1 has a G2/M function because certain conditional alleles cause arrest in G2, and because it interacts genetically and physically with the kinetochore component Ctf13 (Connelly and Hieter 1996; Stemmann and Lechner 1996; Kaplan et al. 1997).
Through analysis of Cdc53-interacting proteins, we have determined that Cdc53 forms distinct complexes with Skp1, Cdc34, and the F-box proteins Cdc4, Grr1, and Met30 in vivo. We find that Cdc53 serves as a scaffold protein that links Skp1/F-box proteins and Cdc34, whereas the F-box proteins serve to confer functional specificity on the core Cdc34—Cdc53—Skp1 complex.
Results
Interactions of Cdc53, Skp1, Cdc4, and Met30 in the two-hybrid system
To identify proteins that interact with Cdc53, twohybrid screens were carried out with full length Cdc53 and two Cdc53 deletion mutants (Fig. 1A). Two of the Cdc53 fusion proteins, Gal4DBD–Cdc53 and Gal4DBD–Cdc53Δ581–664, recovered multiple independent isolates of Skp1, Cdc4, and Met30 from Gal4AD genomic and cDNA libraries (Fig. 1B,C). None of the positive clones recovered interacted with Gal4DBD–Cdc53Δ1–280, suggesting that the amino-terminal region of Cdc53 was important for these interactions (see below). Met30 was originally isolated as a methionine-dependent repressor of methionine biosynthesis gene expression, and has a similar overall structure as Cdc4, with an amino-terminal F-box and carboxy-terminal WD40 repeats (Thomas et al. 1995; Bai et al. 1996). All of the Met30 and Cdc4 isolates that interacted with Cdc53 contained the F-box motif, suggesting the F-box may mediate interactions with Cdc53. In fact, two of three independent Met30 isolates contained just the F-box and a small amount of flanking region (Fig. 1C). Similarly, three independent Cdc4 isolates encompassed the F-box but lacked more amino-terminal sequences. Cdc4 and Met30 isolates, missing some or all of the WD40 repeats, did, however, interact more weakly with Cdc53 than the full-length proteins (Fig. 1B,C), which may reflect an auxiliary role for the WD40 repeats. Because Cdc4 binds Skp1 via the F-box motif (Bai et al. 1996), we directly tested for a Met30–Skp1 interaction in the two-hybrid system. The F-box of Met30 was both necessary and sufficient for interaction of Met30 with Skp1 (Fig. 1D). As for the Cdc53–Met30 interaction, the WD40 repeats of Met30 were required for maximal interaction with Skp1. Despite the known physical interaction of Cdc34 and Cdc53 (Mathias et al. 1996; Willems et al. 1996), Cdc34 was not isolated in the Cdc53 two-hybrid screens, nor did it interact in direct two-hybrid tests with Cdc53 (data not shown). In summary, two-hybrid analysis revealed a Cdc53–Skp1 interaction and suggested that Cdc53–F-box protein interactions may be bridged by Skp1.
Figure 1.


Cdc53 two-hybrid interactions. (A) Cdc53 two-hybrid screens were carried out with three Cdc53 fusion proteins: Gal4DBD–Cdc53, Gal4DBD–Cdc53Δ1–280, and Gal4DBD–Cdc53Δ581–664. (B) Interaction of two-hybrid Gal4AD isolates with Gal4DBD fusions in a β-galactosidase filter assay. (C) Schematic of Cdc53 interacting proteins and two-hybrid isolates. (D) Two-hybrid interactions of LexADBD–Met30 derivatives with VP16AD–Skp1. Interactions of the indicated constructs were quantitated by liquid β-galactosidase assays in Miller units.
Genetic interactions between CDC53 and SKP1
To assess the in vivo relevance of the Cdc53–Skp1 two-hybrid interaction, we tested for genetic interactions between the cdc53-1 mutation and the skp1-11 and skp1-12 mutations. At a semi-permissive temperature of 30°C both the cdc53-1 skp1-11 and cdc53-1 skp1-12 double mutants were inviable, whereas either single mutant grew as well as the wild-type strain (Fig. 2A). At a permissive temperature of 25°C, cdc53-1 skp1-12 double mutants had a severe growth defect, and accumulated multiple hyperpolarized buds (Fig. 2B), akin to the arrest phenotype of single mutants in the Cdc34 pathway (Mathias et al. 1996). Other pairwise synthetic lethal interactions between cdc4-1, cdc34-2, and cdc53-1 have been demonstrated previously (Mathias et al. 1996). Finally, we found that overproduction of CDC53 rescued skp1 temperature-sensitive strains (data not shown). This genetic evidence suggested that the Cdc53–Skp1 two-hybrid interaction reflects a common function of Cdc53 and Skp1 in vivo.
Figure 2.


Genetic interaction between CDC53 and SKP1. (A) cdc53 skp1 double mutants are inviable at the semipermissive temperature. The indicated spore clones of a representative tetratype tetrad were grown at 30°C for 2 days. (B) Photomicrographs of cells from a representative tetratype tetrad grown at 25°C.
Association of Cdc53 with Cdc34, Skp1, and Cdc4 in yeast lysates
Next, we determined whether endogenous levels of Cdc53 and Skp1 form a complex in yeast lysates. To minimize possible disruption of complexes by antibodies, we used epitope-tagged versions of Cdc53 and Skp1. Immunoprecipitation of MYC-tagged Cdc53, followed by immunoblotting with polyclonal antibodies directed against Skp1, revealed a specific association between Cdc53 and Skp1 (Fig. 3A, lane 2). Cdc4 and Cdc34 were also present in the Cdc53 complexes, consistent with the observation that Cdc4 and Cdc53 cofractionate with polyhistidine-tagged Cdc34 (Mathias et al. 1996). In the reciprocal coimmunoprecipitation experiment, Cdc53 specifically associated with HA-tagged Skp1, as did Cdc4 and Cdc34 (Fig. 3B, lane 2). Taken together, these results indicated that Cdc53 forms a multiprotein complex in vivo with Skp1, Cdc4, and Cdc34.
Figure 3.
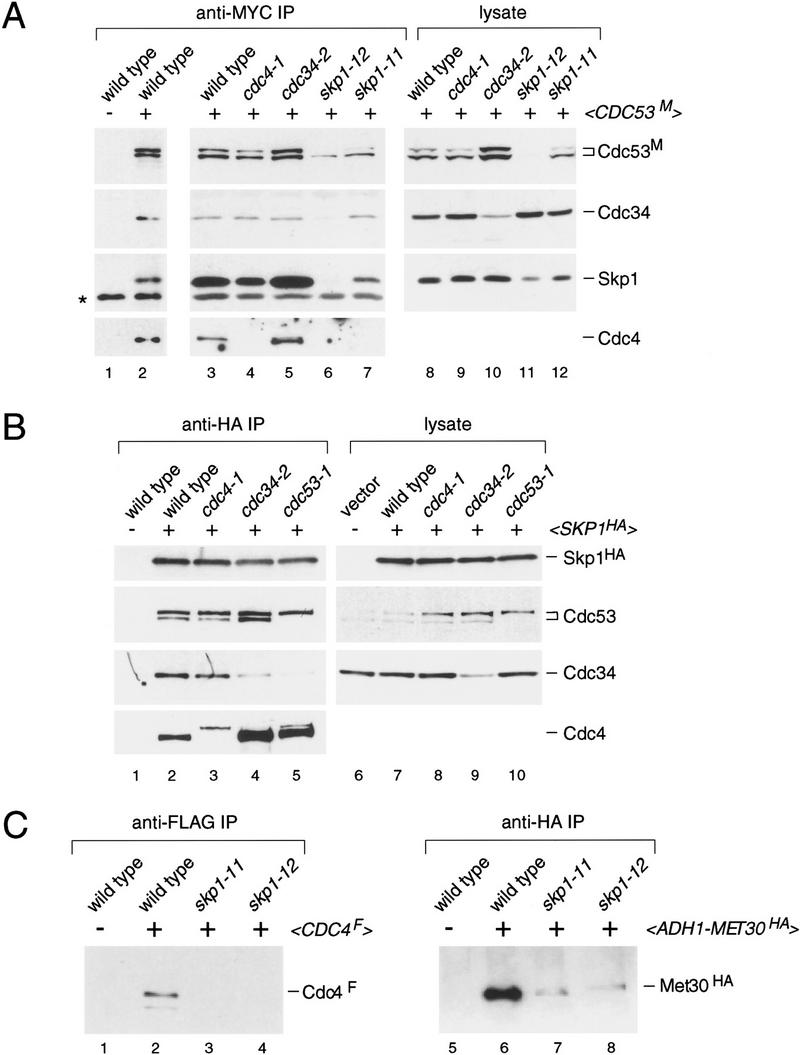
Characterization of Cdc53 complexes in yeast lysates. (A) Effects of temperature-sensitive mutations on the composition of Cdc53 immune complexes. The indicated strains containing <_CDC53 TRP1 CEN_> or <_CDC53M TRP1 CEN_> plasmids were arrested at 37°C for 2 hr. Anti-MYC immunoprecipitates from each strain were immunoblotted and sequentially probed with antibodies against Cdc4, Cdc34, Skp1, and the MYC epitope. The antibody against Cdc4 did not reliably detect Cdc4 in lysates and, therefore, these panels were omitted (see part C, below). (B) Effects of temperature-sensitive mutations on the composition of Skp1 immune complexes. Analysis was as above except that strains contained either empty vector or <_SKP1HA LEU2 CEN_> plasmids. Anti-HA immunoprecipitates were probed as in A. The cause of the lower mobility form of Cdc4 in lanes 3 and 5 has not been determined. (C) Abundance of Cdc4 and Met30 in skp1 mutants. Wild-type, skp1-11, skp1-12 strains containing either empty vector, <_CDC4F TRP1 CEN_> or <_pADH1–MET30HA TRP1_ 2μ> plasmids, were analyzed as above. Anti-FLAG and anti-HA immunoprecipitates were probed with antibodies against Cdc4 and the HA epitope, respectively.
To determine if any of these protein–protein interactions correlated with function in vivo, we examined the composition of the complex in strains bearing various temperature-sensitive alleles. In one set of experiments, Cdc53 immune complexes were immunoblotted with antibodies against Cdc4, Cdc34, and Skp1 (Fig. 3A). In cdc4-1 and skp1-11 mutants, Cdc4 was not detected in Cdc53 immune complexes; however, the absence of Cdc4 from the complexes was attributable, at least in part, to decreased Cdc4 abundance in the mutants (see Fig. 3C). The skp1-12 mutation severely decreased the abundance of Cdc4, Cdc53, and Skp1 itself, and so the absence of associated proteins in Cdc53 complexes from skp1-12 cells was not informative.
In another set of experiments, Skp1 immune complexes from temperature-sensitive strains were immunoblotted with antibodies against Cdc4, Cdc34, and Cdc53 (Fig. 3B). In this configuration, the amount of Cdc4 in the complex was also reduced by the cdc4-1 mutation. In contrast, the amount of Cdc4 in the complex was increased by both the cdc34-2 and cdc53-1 mutations. Interestingly, Cdc4 mobility was altered in cdc4-1 and cdc53-1 strains, but the cause of this mobility shift has not been determined. Relative to the abundance of Cdc34 in lysates, the amount of Cdc34 in Skp1 complexes was severely compromised by the cdc53-1 mutation, suggesting that Cdc53 may bridge the Cdc34–Skp1 interaction (see below).
As the anti-Cdc4 antibodies we used could not reliably detect Cdc4 in yeast lysates, we were unable to determine directly whether the skp1 mutations reduced the abundance of Cdc4. Immunoprecipitation of a FLAG-tagged version of Cdc4, followed by immunoblotting with anti-Cdc4 polyclonal antibody, however, revealed that Cdc4 abundance is greatly diminished in skp1-11 and skp1-12 strains (Fig. 3C). The abundance of another F-box protein, Met30, was similarly reduced by the skp1-11 and skp1-12 mutations (Fig. 3C). As noted above, the abundance of Cdc53 is also decreased by the skp1-12 mutation. Thus, Skp1 may function, at least in part, to stabilize both Cdc53 and F-box proteins. Overall, each of the temperature-sensitive mutations perturbed the mutual interactions by altering the abundance of a given component in lysates and/or the immune complexes.
Interaction of Cdc53 with multiple F-box proteins
To corroborate the Cdc53–Met30 and Skp1–Met30 two-hybrid interactions, we determined whether Met30 formed complexes with Cdc53 and Skp1 in yeast lysates. For this purpose, we used an HA-tagged version of Met30 expressed from the constitutive ADH1 promoter. Immunoprecipitation of Met30, followed by immunoblotting against Cdc53 and Skp1, revealed the presence of both Cdc53 and Skp1 in Met30 immune complexes (Fig, 4A).
Because Grr1 functions with Skp1 and Cdc53 to mediate Cln1/2 degradation and glucose regulation (Barral et al. 1995; Bai et al. 1996; Willems et al. 1996; Li and Johnston 1997), we asked if Grr1 interacted with Cdc53 in yeast lysates. We found that Cdc53 specifically coimmunoprecipitated with an HA-tagged version of Grr1 (Fig. 4A). Skp1 was also present in the Grr1 immune complexes, as shown previously (Li and Johnston 1997). In a control experiment, FLAG-tagged Cdc4 immune complexes also contained Cdc53 and Skp1, thereby completing the set of all possible pairwise coimmunoprecipitations between Cdc4, Cdc34, Cdc53, and Skp1 (Fig. 3A,B; Mathias et al. 1996). We were unable to reproducibly detect Cdc34 in the various F-box protein immune complexes, perhaps because each of these complexes necessarily contains only a fraction of the total Cdc34, Cdc53, and Skp1.
Figure 4.



Interactions of different F-box proteins. (A) Interaction of Met30, Grr1, and Cdc4 with Cdc53 and Skp1. The indicated immunoprecipitates from wild-type cells containing either vector, <_pADH1–MET30HA TRP1_ 2μ>, <_pADH1–GRR1HA TRP1_ 2μ> and <_CDC4F TRP1 CEN_ > plasmids were probed as indicated with antibodies against Cdc53, Skp1, Cdc4, and the HA epitope. IgG light chain is indicated by an asterisk. (B) Different F-box proteins do not interact with each other. Gal4AD–Met30 (A10), Gal4AD–Grr1 and Gal4AD–Cdc4 were tested against Gal4DBD–Grr1 and Gal4DBD–Cdc53 in the two-hybrid system by a β-galactosidase filter assay. (C) Effects of MET30 or GRR1 overexpression. Strains of the indicated genotype containing an empty vector plasmid <_pADH1–MET30HA TRP1_ 2μ> (left) or <_pADH1–GRR1HA TRP1_ 2μ> (right) plasmids were grown at 30°C for 3 days and photographed.
To determine whether Cdc53 forms independent complexes with the various F-box proteins, we tested for interactions between Cdc4, Met30, and Grr1 in the two-hybrid system. A Gal4DBD–Grr1 fusion known to interact with Gal4AD–Skp1 (Li and Johnston 1997) failed to interact with either Gal4AD–Cdc4 or VP16AD–Met30 fusions, or with a Gal4AD–Grr1 fusion (Fig. 4B). Each of the F-box protein fusions interacted with a Gal4DBD–Cdc53 fusion as expected. We have also been unable to detect Cdc4 in either Met30 or Grr1 immune complexes in direct coprecipitation experiments (data not shown). Thus, the available evidence suggests that Skp1 and Cdc53 form mutually exclusive complexes with at least three different F-box proteins in vivo.
The ability of Cdc53 to interact with multiple F-box proteins in independent complexes raised the possibility that different F-box proteins may compete for binding to a Cdc34–Cdc53–Skp1 core complex. We tested this possibility by overexpressing MET30 or GRR1 in cdc4-1, cdc34-2, and cdc53-1 temperature-sensitive strains. Overexpression of MET30 dramatically impaired growth of a cdc4-1 strain at 30°C, and caused a mild growth defect in cdc34-2 and cdc53-1 strains (Fig. 4C), but had no effect on either skp1-11 or skp1-12 strains (data not shown). Although overexpression of GRR1 did not affect growth of a cdc4-1 strain, the growth of cdc34-2 and cdc53-1 strains was retarded at 30°C (Fig. 4C). It has been noted previously that high-level expression of GRR1 is lethal in skp1-12 strains at 30°C (Li and Johnston 1997), and high-level expression of Cdc4 causes inviability of cdc34 and cdc53 strains at 23°C (Mathias et al. 1996). Taken together, the above results suggest various F-box proteins may compete for binding to a Cdc34–Cdc53–Skp1 core complex in vivo, and that the relative stoichiometry of the various complexes may be critical for viability.
Cdc53 is scaffold for Cdc34 and Skp1/F-box proteins
To identify potential protein–protein interaction domains of Cdc53, we constructed a series of Cdc53 deletion mutants using natural and engineered restriction sites (see Materials and Methods). Each of the mutant proteins was expressed in yeast to similar levels as wild-type Cdc53 (Fig. 5A). The ability of each Cdc53 mutant protein to interact with Cdc34, Skp1, and the three F-box proteins Cdc4, Grr1, and Met30 was assessed by immunoblot analysis of MYC-tagged Cdc53 immune complexes with specific polyclonal antibodies (Fig. 5A). In this experiment, each of the interactions detected involved approximately wild-type levels of Cdc53 (which was expressed from a low copy plasmid) and endogenous levels of each of the associated proteins. Deletion of an amino-terminal region of Cdc53 (residues 9–280) completely disrupted Skp1 binding. In parallel, the binding of all three F-box proteins was specifically disrupted. Importantly, Cdc34 still interacted with Cdc53Δ9–280, eliminating the possibility that the truncated protein was simply misfolded and entirely nonfunctional. Conversely, deletion of an internal region of Cdc53 (residues 448–748) abrogated Cdc34 binding but did not affect binding of Skp1 or any of the F-box proteins. The correlation between the Cdc53–Skp1 interaction and Cdc53–F-box protein interactions is most easily explained by a bridging role for Skp1. Furthermore, the independent nonoverlapping binding regions in Cdc53 indicate that the protein–protein interactions within Cdc53 complexes occur in a modular fashion.
Figure 5.
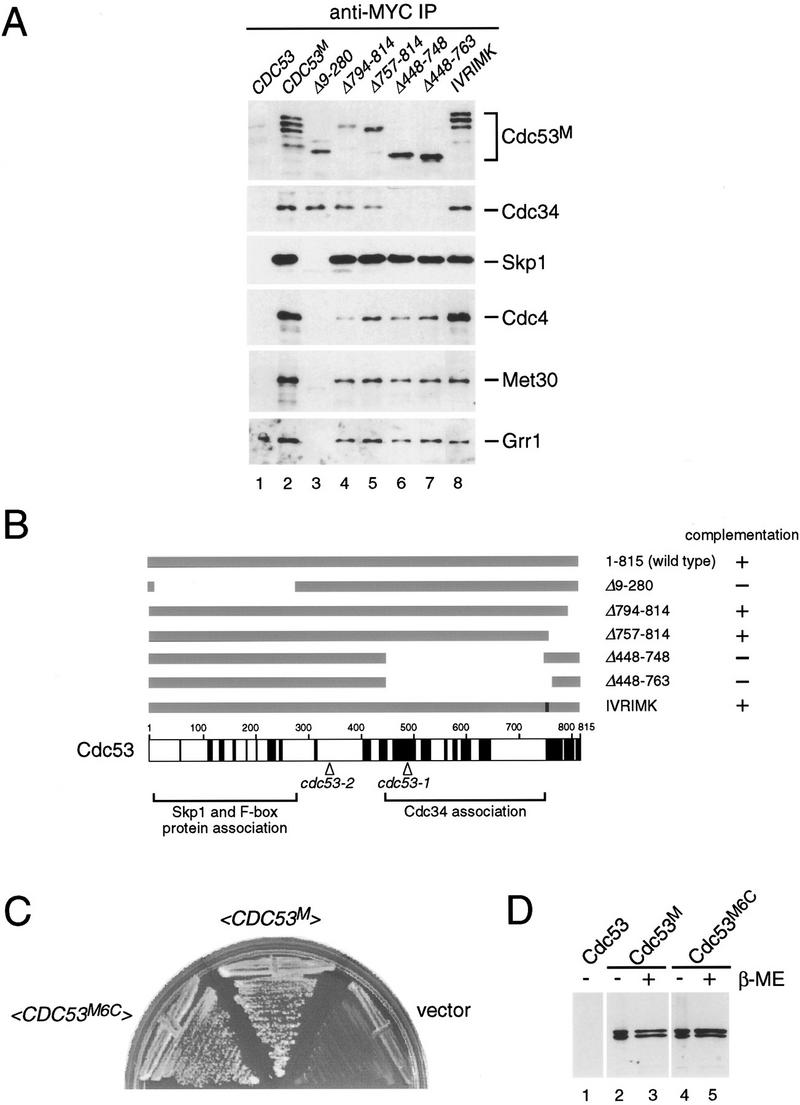
Mutational analysis of Cdc53. (A) Deletion analysis of Cdc53 protein–protein interaction domains. Cells were transformed with untagged Cdc53 (lane 1), MYC-tagged Cdc53 (lane 2) or MYC-tagged versions of the indicated Cdc53 mutants (lanes 3–8). All of the proteins were expressed from the wild-type CDC53 promoter on a CEN plasmid. Lysates from each strain were immunoprecipitated with anti-MYC antibody, immunoblotted, and probed with antibodies specific to each of the indicated proteins. (B) Schematic representation of Cdc53 mutant proteins and their ability to complement a cdc53 deletion strain. Regions of amino acid sequence conservation in the Cdc53 family are indicated in black (see Materials and Methods). The positions of the cdc53-1 (R488C) and cdc53-2 (G340D) point mutations, and the regions required for binding to Skp1/F-box proteins and Cdc34 are also indicated. (C) Cdc53 does not contain essential cysteine residues. A cdc53::ADE2 deletion strain containing a <_CDC53HA URA3 CEN_> plasmid was transformed with <_CDC53M6C TRP1 CEN_>, <_CDC53M TRP1 CEN_>, or an empty vector plasmid, plated on 5-FOA medium to select for Ura− cells, and photographed after 2 days. (D) Cysteine residues in Cdc53 are not required for posttranslational modification of Cdc53. Lysates from strains containing a <_CDC53M TRP1 CEN_> or <_CDC53M6C TRP1 CEN_> plasmid were separated by SDS-PAGE in the absence or presence of reducing agent and immunoblotted with anti-MYC antibody.
Importantly, Cdc53 mutants that were unable to bind either Skp1/F-box proteins or Cdc34 could not complement a cdc53 deletion strain, whereas mutants unaffected in protein–protein interactions could complement (Fig. 5B). To determine if the Skp1/F-box protein and Cdc34 binding domains of Cdc53 corresponded to conserved regions of Cdc53, we aligned 15 different members of the Cdc53 family (Fig. 5B; see Materials and Methods for details of the sequence alignment). Sequence similarity within the Cdc53 family is highest over a broad internal region and a narrow region at the extreme carboxyl terminus. Surprisingly, the latter region is not required for binding to Skp1/F-box proteins or Cdc34, nor for viability (Fig. 5A,B). The internal conserved region, however, overlaps with the Cdc34 binding site. There is relatively poor conservation in the amino terminus of Cdc53, despite the fact that this region contains the Skp1 binding site, suggesting that only a subset of the Cdc53 family may interact with Skp1.
On the basis of the sequence alignment, we mutated many conserved charged residues in Cdc53 to alanines, but none of the mutants had any overt phenotype (data not shown). For instance, mutation of the most conserved stretch in the entire protein, IVRIMK (residues 755–760), to polyalanine did not cause an obvious defect in Cdc53 function or in binding to Skp1/F-box proteins or Cdc34 (Fig. 5A,B). To further explore the structure/function relationship of Cdc53, we determined the sequence of two temperature-sensitive alleles of CDC53 (Mathias et al. 1996). The cdc53-1 mutation causes an R488C substitution whereas the cdc53-2 mutation causes a G340D substitution. Both mutations alter highly conserved residues, even though G340 does not lie within a window of conserved residues. Interestingly, the cdc53-1 mutation occurs within the Cdc34 binding region. In conjunction with the defective Skp1–Cdc34 interaction in cdc53-1 strains (Fig. 3B), this result strongly suggests that the cdc53-1 mutation specifically perturbs the Cdc34 binding site.
Cdc53 does not form essential ubiquitin thioesters in vivo
In addition to target protein recognition, some E3 ubiquitin ligases participate in the catalytic transfer of ubiquitin onto substrates through formation of ubiquitin thioesters on cysteine residues (Scheffner et al. 1995). Therefore, we determined whether any of the cysteine residues in Cdc53 were required for function in vivo. Simultaneous mutation of all six cysteine residues in Cdc53 to alanines (Cdc53M6C) did not impair complementation of a cdc53 deletion strain (Fig. 5C). Cdc53 is covalently conjugated to a ubiquitin-cross reactive species in vivo (Willems et al. 1996), which is not ubiquitin itself, but is Rub1, a ubiquitin-related protein (M. Estelle, pers. comm.). Therefore, we compared the extent of modification of the cysteineless Cdc53 to wild-type Cdc53. The mutant protein was modified by Rub1 to the same extent as wild type, and the modification was not sensitive to reducing agents, demonstrating that this modification does not occur on cysteine residues (Fig. 5D). Although this mutational analysis does not rule out thioester formation on Cdc53 in vivo, such intermediates clearly cannot be essential for viability. Cdc53 thus appears to act as a noncatalytic scaffold protein for Cdc34 and Skp1/F-box proteins.
Cdc34, Cdc53, and Skp1 function in methionine repression
To assess the biological significance of the Cdc53–Met30 and Skp1–Met30 interactions, we determined whether Cdc34, Cdc53, or Skp1 were required for proper regulation of methionine biosynthesis genes. We examined the regulation of MET25, which encodes homocysteine synthase and is one of several methionine regulated genes. MET25 is activated by the Cbf1–Met4–Met28 transcriptional complex and repressed by Met30 (Thomas et al. 1995; Kuras et al. 1996). As expected, methionine repressed MET25 expression in wild-type cells (Fig. 6A). As MET30 is an essential gene, we used an antimorphic allele called MET30-1 as a positive control for derepression in methionine medium (Thomas et al. 1995). As shown previously, MET25 was incompletely repressed by methionine in MET30-1 cells. Strikingly, repression of MET25 by methionine was severely compromised in cdc53-1 cells and completely defective in cdc34-2, skp1-11, and skp1-12 cells (Fig. 6A). The derepression of MET25 in methionine medium did not depend on G1 phase cell cycle arrest because derepression did not occur in cdc4-1 cells that arrest at the identical point in G1, and yet did occur in skp1-12 mutants that arrest in G2 phase. Because MET25 was effectively repressed with wild-type kinetics in cdc4-1 cells, methionine biosynthesis gene regulation specifically requires the F-box protein Met30. We were unable to assess the role of Grr1 in methionine repression because grr1Δ strains were inviable when grown on standard B medium used for analysis of methionine repression (data not shown).
Figure 6.

Specificity of F-box protein function. (A) Methionine repression is mediated by Met30, Cdc34, Cdc53, and Skp1 but not Cdc4. The indicated strains were grown in methionine-free medium and then repressed with 1.0 mm methionine for the indicated times. MET25 expression was determined by Northern analysis and normalized to ACT1 expression. Values are expressed as percent of the signal at t = 0. (B) Grr1 specifically mediates Cln2 degradation. Cln2 stability was examined in the indicated strains carrying a <_pGAL1–CLN2HA URA3 LEU2 CEN_> plasmid by repression of the GAL1 promoter. Cln2HA was detected by immunoblotting with anti-HA antibody. Exposures were adjusted to give approximately equal Cln2HA signals at t = 0. Cln2HA was quantitated by densitometry and normalized to Cdc28 signals from the same blot probed with anti-Cdc28 antibody. Values are expressed as percent of the signal at t = 0. (C) Cdc4 does not overlap with Grr1 in Cln2 degradation. Cln2 stability was determined by [35S]methionine/cysteine pulse-chase analysis in the indicated strains. The 35S-labeled Cln2HA signal was quantitated by PhosphorImager and normalized to the 35S-labeled total lysate signal for each sample. Values are expressed as percent of the signal at t = 0.
F-box protein function in Cln2 degradation
It has been shown previously that Cln2 is stabilized in grr1Δ, cdc34-2, cdc53-1, and skp1-12 strains (Barral et al. 1995; Deshaies et al. 1995; Bai et al. 1996; Willems et al. 1996). To directly assess the specificity of F-box protein function in Cln2 degradation, we compared the half-life of Cln2 in cdc4-1, MET30-1, and grr1Δ strains. Because Grr1 and Met30 are required for proper responses to glucose and methionine, respectively, we carried out half-life experiments in each F-box mutant strain by both glucose repression of a pGAL1–CLN2HA construct and [35S]methionine/cysteine pulse-chase analysis of endogenous Cln2. In promoter shut-off experiments, Cln2 half-life was greatly increased in a grr1Δ strain as expected (Barral et al. 1995), but was not altered in either cdc4-1 or MET30-1 strains (Fig. 6B). Virtually identical results were obtained in pulse-chase experiments, as Cln2 was stabilized only in the grr1Δ strain (Fig. 6C). To determine whether Cdc4 might have any ancilliary role in Cln2 degradation, we also examined the half-life of Cln2 in a grr1Δ cdc4-1 double mutant strain by pulse-chase analysis. Cln2 was no more stable in the cdc4-1 grr1Δ double mutant than in the grr1Δ single mutant (Fig. 6C). Thus, Grr1 is the primary mediator of Cln2 degradation in vivo. Finally, consistent with previous results (Schwob et al. 1994; Bai et al. 1996), we found that Sic1 degradation in vivo required Cdc4, but not Grr1 or Met30 (data not shown).
Discussion
Architecture of Skp1–Cdc53–F-box protein (SCF) complexes
Cdc53 forms multiprotein complexes in vivo with Cdc34, Skp1, and three independent F-box proteins, Cdc4, Met30, and Grr1. To simplify description of the various F-box containing complexes, we have adopted the term SCF, for Skp1–Cdc53–F-box protein complex (Feldman et al. 1997; Skowrya et al. 1997). The specific F-box complexes described here are thus designated SCFCdc4, SCFMet30, and SCFGrr1. Formally, SCF complexes are E3 ubiquitin ligases, as they interact with both substrates and an E2 enzyme, Cdc34 (Hershko 1997). The following observations bear on the general architecture of SCF complexes in vivo: (1) the Cdc34–Cdc53 interaction is independent of Skp1 binding; (2) the Skp1/F-box protein–Cdc53 interaction is independent of Cdc34 binding; (3) the Skp1–Cdc34 interaction is abrogated by the cdc53-1 mutation, which lies in the Cdc34 binding site; (4) the F-box of Met30 is sufficient for interaction with Skp1 and Cdc53 in the two-hybrid system; (5) the binding region for Skp1 on Cdc53 precisely overlaps that of three different F-box proteins, Cdc4, Grr1, and Met30. These in vivo observations are consistent with the assembly of recombinant SCF complexes in vitro (Bai et al. 1996; Feldman et al. 1997; Skowrya et al. 1997). We envisage that Cdc53 is a scaffold protein that bridges Cdc34 to Skp1, which in turn binds to various F-box proteins, which in turn target various substrates (Fig. 7). Although the various protein–protein interactions within SCF complexes are largely modular in nature, the interaction of Skp1 with Grr1 in the two-hybrid system requires both the F-box and the leucine rich repeats of Grr1 (Li and Johnston 1997). We also observe that the WD40 repeats of Cdc4 and Met30 are required for maximal interaction with Skp1. Thus, Skp1 may recruit F-box proteins to Cdc53 complexes in conjunction with other domains within F-box proteins.
Figure 7.

Multiple F-box proteins recruit different substrates to an E2/E3 core ubiquitination complex. The hatched box in Cdc53 represents a conserved region found in all Cdc53 homologs that overlaps with the Cdc34 binding site in Cdc53. The F-box protein-substrate interactions are most firmly established for Cln1, Cln2, and Sic1. P indicates a phosphorylation dependent interaction, WD40 indicates WD40 repeats, and LRR indicates leucine rich repeats. See text for details.
Unlike some other E3 ligases, Cdc53 does not form essential ubiquitin thioester intermediates. We have also mutated several conserved cysteine residues in Cdc4 (residues 454, 455, 468, and 562) and found that such mutants provide full Cdc4 function in vivo, even when combined with a cysteineless Cdc53 mutant (data not shown). In addition, the only conserved cysteine residue in Skp1 is nonessential (S. Elledge, pers. comm.). Thus, SCF-mediated ubiquitin transfer may rely solely on Cdc34 for catalytic activity. Genetic evidence suggests that Cdc34 is the only E2 able to provide function in Sic1 degradation, Cln degradation, and methionine repression, indicating that Cdc34 may be specific for SCF complexes. Glucose induction of HXT1 expression, however, occurs normally in cdc34-2 strains, suggesting that either another E2 is required or that the cdc34-2 allele is able to function in HXT1 induction (Li and Johnston 1997). It will be of interest to determine whether the conserved central region in other Cdc53 homologs mediates interactions with E2 enzymes.
Finally, although three distinct SCF complexes conform to the generic SCF architecture, the Skp1–Ctf13 interaction occurs in the context of the CBF3 kinetochore complex, which is composed of p23Skp1, p58Ctf13, p64Cep3, and p110Ndc10 (Connelly and Hieter 1996; Stemmann and Lechner 1996; Kaplan et al. 1997). Surprising, Skp1 is not an obligate structural component of this complex, but rather appears to faciliate phosphorylation dependent conversion of Ctf13 into an active form (Kaplan et al. 1997). It is possible that the Skp1–F-box interaction may be a widely utilized protein motif that functions in contexts other than ubiquitin dependent proteolysis.
F-box proteins confer specificity on SCF function
The dramatic cell cycle arrest phenotype caused by the inability to degrade Sic1 has, to some extent, obscured the pleiotropic functions of the Cdc34–Cdc53–Skp1 E2/E3 core complex. Specific functions are confered on the E2/E3 core complex by its association with different F-box proteins. SCFCdc4 is required for specific degradation of Sic1 and other cell-cycle regulators. SCFGrr1 mediates specific degradation of Cln1 and Cln2, and is also required for transcriptional induction of glucose transporter genes (Barral et al. 1995; Li and Johnston 1997). As grr1 mutants have many other defects, it seems likely that SCFGrr1 will regulate several other pathways as well (Li and Johnston 1997). Here, we have described a third SCF complex, SCFMet30, and demonstrated that in addition to Met30, Cdc34, Cdc53, and Skp1 are required for appropriate repression of methionine biosynthesis genes.
The specificity of each SCF complex for different cellular processes is demonstrated by a remarkable absence of cross talk. The cdc4-1 mutation does not affect MET25 repression and conversely, the MET30-1 mutation does not affect either Sic1 or Cln2 degradation. Similarly, Sic1 degradation requires only Cdc4, whereas Cln2 degradation requires only Grr1. It should be pointed out that this latter result is at odds with a model in which Clb–Cdc28 activity instigates Cln degradation, an issue that has yet to be resolved (Blondel and Mann 1996).
The growth defects caused by high level expression of CDC4, GRR1, or MET30 in various SCF mutants suggests that different F-box proteins may be in equilibrium with a limiting amount of the E2/E3 core complex. There are other indications that F-box proteins may be subject to stringent regulation. The Skp1–Grr1 interaction is stimulated by glucose (Li and Johnston 1997), and Ctf13 levels appear to be precisely controlled by the SCFCdc4 pathway (Kaplan et al. 1997). Furthermore, Ctf13 is activated by phosphorylation in a Skp1 dependent manner (Kaplan et al. 1997). It will be of interest to determine whether the Skp1–Met30 interaction is modulated by methionine, and to determine whether other F-box proteins are regulated by phosphorylation.
It seems likely that other SCF complexes will regulate many other processes because yeast contain at least nine additional F-box proteins of unknown function, at least two of which associate with Cdc53 (A. Willems and A. Shevchenko, unpubl.). Yeast also contain three identifiable Cdc53 homologs and a Skp1 homolog, all of unknown function. The use of specific adaptor proteins to recruit multiple substrates to core ubiquitination complexes may be a common theme, as two putative adaptor proteins for the APC, Cdc20, and Hct1/Cdh1, have recently been identified (Schwab et al. 1997; Visintin et al. 1997) and shown to confer substrate specificity on APC function (Visintin et al. 1997).
Substrates of SCF complexes
To date, only SCFCdc4 has been unequivocally shown to directly mediate ubiquitination of target proteins. Ubiquitination of phosphorylated Sic1 has been reconstituted in vitro, in both a yeast extract system and in a purified system with recombinant proteins (Feldman et al. 1997; Skowrya et al. 1997; Verma et al. 1997b). Recently, phosphorylation-dependent ubiquitination of Far1 by SCFCdc4 has been achieved in yeast extracts (Henchoz et al. 1997). Substantial evidence also suggests that SCFCdc4 also targets Cdc6, Ctf13 and, Gcn4 for degradation in a phosphorylation-dependent manner (Kornitzer et al. 1994; Drury et al. 1997; Kaplan et al. 1997).
Although ubiquitination of Cln1/2 has not yet been reconstituted in vitro, SCFGrr1 specifically binds to phosphorylated Cln1/2, consistent with Grr1-dependent degradation of Cln1/2 in vivo (Barral et al. 1995; Skowyra et al. 1997). Somewhat surprisingly, Cln3 is not stabilized in grr1 strains (Barral et al. 1995), but is stabilized in cdc34-2 strains (Yaglom et al. 1995), perhaps indicating that another F-box protein may target Cln3 for degradation. A negative regulator of HXT expression called Rgt1 is a possible target of the SCFGrr1 complex; however, it is not yet known if Rgt1 is regulated by ubiquitin-dependent proteolysis (Li and Johnston 1997). The requirement for SCFMet30 function in methionine repression also implicates ubiquitin-dependent proteolysis, but, again, this remains to be proven. Because Met30 forms a complex with the transactivator Met4, it is possible that Met30 targets Met4 for degradation, although other components of the Met4 transcriptional complex are also candidate targets (Kuras et al. 1996).
Diverse functions of SCF complexes
In instances where function is known, F-box protein complexes have emerged as key regulators of cell division in other organisms. In Schizosaccharomyces pombe, a Cdc4 homolog called Pop1 regulates DNA replication by targeting a Cdk inhibitor, Rum1, and a Cdc6 homolog, Cdc18, for ubiquitin dependent proteolysis (Kominami and Toda 1997). In Caenorhabditis elegans, a Cdc53 homolog called CUL-1 appears to negatively regulate activators of division as cul-1 mutants exhibit hyperplasia in all tissues (Kipreos et al. 1996). As in yeast, degradation of mammalian G1 cyclins and Cdk inhibitors is phosphorylation dependent (Clurman et al. 1996; Won and Reed 1996; Diehl et al. 1997; Sheaff et al. 1997) and, therefore, it will be of prime importance to determine the role of SCF complexes in these pathways. It is likely that SCF architecture will be conserved, as human Skp1 forms a specific complex with human Cul-1 and binds to cyclin A-Cdk2 through its associated F-box protein, Skp2 (Zhang et al. 1995; Lisztwan et al. 1998; Y. Xiong, pers. comm.).
The regulation of various transcription factors by SCFCdc4, SCFGrr1, and SCFMet30 suggests that SCF pathways may be a common means of transcriptional control, particularly as gene expression is often regulated by proteolysis (Pahl and Baeuerle 1996). Transcriptional regulation of methionine biosynthesis by SCF complexes is conserved in fungi as the Met30 homologs Scon2 and SconB, and the Skp1 homolog SconC regulate sulfur metabolism in Neurospora and Aspergillus, respectively (Natorff et al. 1993; Kumar and Paietta 1995). Another human Cdc53 homolog, Cul-2, may also regulate transcription as it physically associates with the VHL tumour suppressor protein in a complex with elongin B and elongin C, two transcriptional elongation factors that have similarity to ubiquitin and Skp1, respectively (Pause et al. 1997). Intriguingly, another F-box protein, elongin A, participates in a complex with elongin B and elongin C (Aso et al. 1996).
Finally, F-box proteins are implicated in developmental decisions, often in the context of transcriptional control. A Cdc4 homolog in C. elegans, SEL-10, influences a lateral cell fate decision through negative regulation of the notch homolog LIN-12, possibly via ubiquitin-dependent degradation (Hubbard et al. 1997). In Arabidopsis, the F-box protein UFO controls floral development through regulation of a floral organ identity gene APETALA3 (Lee et al. 1997). The analogous F-box protein in Antirrhinum, FIMBRIATA, functions in a similar manner, and moreover, physically interacts with three Skp1 homologs (Ingram et al. 1997).
SCF complexes represent a versatile regulatory system that has been exploited in many cellular processes. At least seven metazoan homologs of Cdc53 have been identified and are generically refered to as cullins, after the C. elegans Cdc53 homolog, CUL-1 (Kipreos et al. 1996). Similarly, database searches reveal several Skp1 homologs and a large number of metazoan proteins containing putative F-box motifs. If the combinatorial control of SCF complexes defined to date applies to these homologs, then many other SCF pathways remain to be elucidated.
Materials and methods
Plasmids
Plasmids were constructed by use of standard molecular cloning techniques (Table 1). For two-hybrid screens, the CDC53 open reading frame was cloned into the _Bam_HI site of pAS2 (provided by S. Elledge, Baylor College of Medicine, Houston, TX) to create a Gal4DBD–Cdc53 fusion. Versions that lacked either the amino-terminal 280 residues (Gal4DBD–Cdc53Δ1–280) or internal residues 581–664 (Gal4DBD–Cdc53Δ581–664) were created by digestion with _Nco_I or _Kpn_I, respectively and religating. Gal4AD–Cdc4Δ566–779 is a truncated CDC4 PCR product cloned into the _Bam_HI site of pGAD424 (Skowyra et al. 1997). To test the Skp1–Met30 interaction in the two-hybrid system, a SKP1 fragment was cloned into the _Bam_HI site of pVAD1, to create a VP16–Skp1 fusion. LexA–Met30 derivatives were based on pLEXM30-4 (Thomas et al. 1995). Met30 was tagged at the amino terminus with an HA epitope by insertion of a MET30 fragment encoding amino acids 7–640 from pLEXM30-4 into a pADH1-HA expression plasmid (Li and Johnston 1997). Cdc4 was tagged at the amino terminus with a FLAG epitope by site directed mutagenesis. A CDC53 deletion construct was made by replacing an internal _Bgl_II fragment of pGEM53-8 (Mathias et al. 1996) with an ADE2 fragment. To allow for negative selection of wild-type CDC53 in the cdc53Δ shuffle strain a 3.6-kb _Eco_RI fragment of CDC53 was cloned into a <_URA3 CEN_> plasmid. Mutagenesis of Cdc53 was carried out in pMT843, as described previously (Willems et al. 1996) and restriction sites incorporated during mutagenesis were used to construct the deletions shown in Figure 5. The version of Cdc53 in which all six cysteine residues are replaced by alanines (C59A, C157A, C412A, C606A, C754A, and C774A) was created in a single site-directed mutagenesis reaction.
Table 1.
Plasmids used in this study
| Plasmid | Relevant characteristics | Source |
|---|---|---|
| pBF339 | pADH1HA TRP1 2μ | Li and Johnston (1997) |
| pBF494 | pADH1HA–GRR1 _Δ_N TRP1 2μ | Li and Johnston (1997) |
| pBM2576 | pADH1–GAL4DBD–GRR1 _Δ_N TRP1 | Li and Johnston (1997) |
| pBM2868 | pADH1–GAL4AD–GRR1 _Δ_N TRP1 | Li and Johnston (1997) |
| pMT634 | pGAL1–CLN2–HA LEU2 URA3 CEN | Willems et al. (1996) |
| pMT817 | _CDC53–C–Not_I TRP1 CEN | Willems et al. (1996) |
| pMT843 | CDC53M TRP1 CEN | Willems et al. (1996) |
| pMT915 | pADH1–GAL4AD–CDC4 _Δ_3WD LEU2 2μ | this study |
| pMT918 | pADH1–GAL4DBD–CDC53 TRP1 2μ | this study |
| pMT951 | CDC53HA URA3 CEN | this study |
| pMT954 | pADH1–GAL4DBD–CDC53 _Δ_1–280 TRP1 2μ | this study |
| pMT955 | pADH1–GAL4DBD–CDC53 _Δ_581–664 TRP1 2μ | this study |
| pMT1511 | SKP1HA LEU2 CEN | P. Heiter (University of British ColumbiaVancouver, Canada) |
| pMT1514 | cdc53::ADE2 in pGEM3 | this study |
| pMT1567 | CDC4F TRP1 CEN | this study |
| pMT1707 | pADH1HA–MET30 TRP1 2μ | this study |
| pMT1850 | CDC53 _MΔ_9–280 TRP1 CEN | this study |
| pMT1854 | CDC53 _MΔ_448–763,H767A TRP1 CEN | this study |
| pMT1856 | CDC53 _MΔ_448–748 TRP1 CEN | this study |
| pMT1857 | CDC53 _MΔ_757–815 TRP1 CEN | this study |
| pMT1858 | CDC53 _MΔ_794–815 TRP1 CEN | this study |
| pMT1859 | CDC53MIVRIMK TRP1 CEN | this study |
| pMT1861 | CDC53M6C TRP1 CEN | this study |
| pMT1589 | LEXADBD–MET30 TRP1 2μ | this study |
| pLexM30-4(297-540) | LEXADBD–MET30 _Δ_297–540 TRP1 2μ | this study |
| pLexM30-4(158-297) | LEXADBD–MET30 _Δ_158–297 TRP1 2μ | this study |
| pLexM30-4(158-540) | LEXADBD–MET30 _Δ_158–540 TRP1 2μ | this study |
| pVAD1-SKP1 | VAD–SKP1 LEU2 2μ | this study |
| pSE1111 | pADH1–GAL4AD–SNF1 LEU2 2μ | S. Elledge |
| pSE1112 | pADH1–GAL4DBD–SNF4 TRP1 2μ | S. Elledge |
Yeast strains and culture
Yeast strains are listed in Table 2. All strains were isogenic with the W303 background except for Y187, Y190, Y533, Y555, and WX131-2C. Standard methods were used for yeast culture and transformation (Kaiser et al. 1994). A cdc53Δ shuffle strain was constructed by deleting one copy of CDC53 with pMT1514 in a K699 a/α diploid, transforming with pMT951, sporulating and isolating a Ura+ Ade+ segregant. Complementation of the shuffle strain by various <_CDC53M TRP1 CEN_ > plasmids was tested by plating on 0.1% FOA medium. cdc53 skp1 double mutants were generated in crosses of MTY871 with Y553 and Y555 (Bai et al. 1996). The MET30-1 strain (CC786-1A) was created by crossing W303-1B with CM100-1A (Thomas et al. 1995).
Table 2.
Yeast strains used in this study
| Strain | Relevant genotype | Source |
|---|---|---|
| K699 | MATa ade2-1 can1-100 his3-1,15 leu2-3,112 trp1-1 ura3 | K. Nasmyth (Research Institute of Molecular Pathology, Vienna, Austria) |
| MTY668 | MATa cdc4-1 | this study |
| MTY670 | MATa cdc34-2 | Willems et al. (1996) |
| MTY871 | MATa cdc53-1 | Willems et al. (1996) |
| MTY1243 | cdc53::ADE2 | this study |
| MTY1293 | cdc53-1 skp1-11 | this study |
| MTY1294 | cdc53-1 skp1-12 | this study |
| MTY1280 | grr1::LEU2 | |
| MTY1310 | cdc4-1 grr1::LEU2 | |
| Y187 | MATa ade2-101 his3-Δ200 leu2-3,112 trp1-901 ura3-52 gal4Δ gal80Δ URA3::GAL–lacZ LYS2::GAL–HIS3 | S. Elledge |
| Y190 | as for Y187 but _MAT_α | S. Elledge |
| Y553 | MATα skp1-11 | Bai et al. (1996) |
| Y555 | MATα skp1-12 | Bai et al. 1996) |
| WX131-2c | MATα cdc53-2 trp1-7 ura3-52 ade2 | M. Goebl (Indiana University School of Medicine, Indianapolis) |
| CC786-1A | ade2 his3 leu2 ura3 trp1 MET30-1 | this study |
| C170 | ade2 his3 leu2 trp1 met4::TRP1 ura3::lexA op–lacZ::URA3 | Kuras et al. (1996) |
Two-hybrid analysis
Strain Y187 expressing Gal4DBD fusions was transformed with a yeast Gal4AD–cDNA library (provided by S. Elledge) or a Gal4AD genomic DNA library (James et al. 1996) and screened as described (Durfee et al. 1993). With the cDNA library, Gal4DBD–Cdc53 recovered one positive clone (Met30–A10) from 140,000 transformants, and Gal4DBD–Cdc53Δ581–664 recovered two positive clones (Skp1–C23, Skp1–C24) from 225,000 transformants. Gal4DBD–Cdc53Δ1–280 recovered no positive clones from 427,000 transformants. With the genomic DNA library, Gal4DBD–Cdc53 recovered five positive clones (Cdc4–F20, Cdc4–F23, Cdc4–F24; Met30–F15, Met30–F19) from 1,000,000 transformants and Gal4DBD–Cdc53Δ581–664 recovered seven positive clones (Cdc4–H1, Cdc4–H8; Met30–H6, Met30–H9, Met30–H17; Skp1–H11, Skp1–H13) from 500,000 transformants. Some clones were isolated several independent times, but all unique clones are represented in Figure 1.
RNA analysis
Preparation of yeast lysates and analysis of total RNA were carried out as described previously (Willems et al. 1996). MET25 mRNA expression was assayed in cultures grown in B media supplemented with 0.1 mm methionine (Thomas et al. 1995). At a density of 0.5 × 107 cells/ml cultures were shifted to 37°C for 2 hr, repressed with 1.0 mm methionine, and time points taken for RNA extraction. Northern blots were probed with a 1.3-kb MET25 fragment and a 0.6-kb ACT1 fragment. mRNA abundance was quantitated on a Molecular Dynamics Storm PhosphorImager.
Protein analysis
Immunoblots were processed for ECL detection as described (Willems et al. 1996) and where indicated, immunoreactivity was quantitated by densitometry and NIH Image software. Antibodies against Cdc4, Cdc34, and Cdc53 (Mathias et al. 1996), Cdc28 (Tyers et al. 1992) and Skp1 (Bai et al. 1996) were used as described. Anti-Grr1 antiserum was adsorbed against polyacrylamide to eliminate background binding and used at 1:100 (Flick and Johnston 1991). Anti-Met30 antiserum was raised against recombinant Gst–Met30 (residues 297–640, which encompass the WD40 repeats), affinity purified and used at a dilution of 1:100. 9E10 anti-MYC and 12CA5 anti-HA monoclonal antibodies were produced as ascites fluid and used at a dilution of 1:10,000. Anti-FLAG M2 antibody conjugated to Sepharose beads (Kodak) and HRP-conjugated secondary antibodies (Amersham) were used according to manufacturer’s instructions.
For promoter shut-off of pGAL1–CLN2HA constructs, cultures grown in galactose for 2 hr at 37°C were rapidly pelleted, rinsed, and resuspended in glucose medium at 37°C for the indicated timecourse (Willems et al. 1996). Northern analysis of the CLN2 mRNA confirmed that the GAL1 promoter was rapidly repressed in all strains. Determination of Cln2 stability by pulse-chase analysis was carried out as described (Amon et al. 1994). Briefly, 40 ml of culture was grown to 1 × 107 cells/ml in selective medium that contained 0.1 mm methionine. Cells were washed by filtration, resuspended in 1 ml of medium lacking methionine, incubated for 5 min, pulse-labeled with [35S]-methionine/cysteine (ICN) for 5 min, and chased for various times with cold 1 mm methionine/cysteine and 1 μg/ml of cycloheximide. Cells were killed by addition of 10% TCA, and disrupted by glass bead lysis followed by boiling in 1% SDS. Extracts were diluted to RIPA conditions, incubated with 1 μl of 12CA5 anti-HA antibody for 2 hr on ice, and immune complexes collected on protein A–Sepharose for 2 hr. For each pulse-chase series, equivalent amounts of radioactivity were immunoprecipitated for each sample. Immunoprecipitated proteins were separated by 10% SDS-PAGE and the dried gel exposed to a PhosphorImager screen. 35S-labeled Cln2HA signal was quantitated with a Molecular Dynamics Storm PhosphorImager and normalized to total radioactivity in the lysate.
Sequence analysis
Regions of sequence conservation between Cdc53 homologs identified in database searches were determined by amino acid alignment with ClustalW (Thompson 1994). Conserved residues with a weight of 10 or higher were identified by analysis of 15 full-length homologs with the Wisconsin Package program Pretty. Black lines in Figure 5B indicate the central residue of an 11 residue window containing four or more such conserved residues.
Acknowledgments
S. Elledge, W. Harper, M. Goebl, M. Estelle, J. Rosamond, P. James, R. Deshaies, P. Hieter, A. Hershko, W. Krek, Y. Xiong, M. Johnston, F. Li, D. Kornitzer, Y. Surdin-Kerjan, B. Andrews, X. Tang, and L. Harrington provided reagents, communication of unpublished results, and/or critical comments on the manuscript. We are particularly grateful to A. Amon for invaluable advice on pulse-chase experiments, J. Lamb for sharing his unpublished data, and H. Cheng for excellent technical assistance. E.P. is a recipient of a National Sciences and Engineering Research Council of Canada (NSERC) studentship and A.W. is a recipient of an Ontario Graduate Scholarship studentship. L.K. and D.T. are supported by grants from the Centre National de la Recherche Scientifique and the Association pour la Recherche sur le Cancer. M.T. is supported by the National Cancer Institute of Canada (NCI) and the Medical Research Council of Canada and is a Research Scientist of the NCI of Canada.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL tyers@mshri.on.ca; FAX (416) 586-8857.
References
- Amon A, Irniger S, Nasmyth K. Closing the cell cycle circle in yeast: G2 cyclin proteolysis initiated at mitosis persists until the activation of G1 cyclins in the next cycle. Cell. 1994;77:1037–1050. doi: 10.1016/0092-8674(94)90443-x. [DOI] [PubMed] [Google Scholar]
- Aso T, Haque D, Barstead RJ, Conaway RC, Conaway JW. The inducible elongin A elongation activation domain: Structure, function and interaction with the elongin BC complex. EMBO J. 1996;15:5557–5566. [PMC free article] [PubMed] [Google Scholar]
- Bai C, Sen P, Hofmann K, Ma L, Goebl M, Harper JW, Elledge SJ. SKP1 connects cell cycle regulators to the ubiquitin proteolysis machinery through a novel motif, the F-box. Cell. 1996;86:263–274. doi: 10.1016/s0092-8674(00)80098-7. [DOI] [PubMed] [Google Scholar]
- Barral Y, Jentsch S, Mann C. G1 cyclin turnover and nutrient uptake are controlled by a common pathway in yeast. Genes & Dev. 1995;9:399–409. doi: 10.1101/gad.9.4.399. [DOI] [PubMed] [Google Scholar]
- Blondel M, Mann C. G2 cyclins are required for the degradation of G1 cyclins in yeast. Nature. 1996;384:279–282. doi: 10.1038/384279a0. [DOI] [PubMed] [Google Scholar]
- Clurman BE, Sheaff RJ, Thress K, Groudine M, Roberts JM. Turnover of cyclin E by the ubiquitin-proteasome pathway is regulated by cdk2 binding and cyclin phosphorylation. Genes & Dev. 1996;10:1979–1990. doi: 10.1101/gad.10.16.1979. [DOI] [PubMed] [Google Scholar]
- Connelly C, Hieter P. Budding yeast SKP1 encodes an evolutionarily conserved kinetochore protein required for cell cycle progression. Cell. 1996;86:275–285. doi: 10.1016/S0092-8674(00)80099-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshaies RJ, Chau V, Kirschner M. Ubiquitination of the G1 cyclin Cln2p by a Cdc34p-dependent pathway. EMBO J. 1995;14:303–312. doi: 10.1002/j.1460-2075.1995.tb07004.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diehl JA, Zindy F, Sherr CJ. Inhibition of cyclin D1 phosphorylation on threonine-286 prevents its rapid degradation via the ubiquitin-proteasome pathway. Genes & Dev. 1997;11:957–972. doi: 10.1101/gad.11.8.957. [DOI] [PubMed] [Google Scholar]
- Drury LS, Perkins G, Diffley JFX. The Cdc4/34/53 pathway targets Cdc6p for proteolysis in budding yeast. EMBO J. 1997;16:5966–5976. doi: 10.1093/emboj/16.19.5966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durfee T, Becherer K, Chen P-L, Yeh S-H, Yang Y, Kilburn AE, Lee WH, Elledge SJ. The retinoblastoma protein associates with the protein phosphatase type 1 catalytic subunit. Genes & Dev. 1993;7:555–569. doi: 10.1101/gad.7.4.555. [DOI] [PubMed] [Google Scholar]
- Feldman RMR, Correll CC, Kaplan KB, Deshaies RJ. A complex of Cdc4, Skp1, and Cdc53/Cullin catalyzes ubiquitination of the phosphorylated CDK inhibitor Sic1. Cell. 1997;91:221–230. doi: 10.1016/s0092-8674(00)80404-3. [DOI] [PubMed] [Google Scholar]
- Flick JS, Johnston M. Grr1 of Saccharomyces cerevisiae is required for glucose repression and encodes a protein with leucine-rich repeats. Mol Cell Biol. 1991;11:5101–5112. doi: 10.1128/mcb.11.10.5101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goebl MG, Yochem J, Jentsch S, McGrath JP, Varshavsky A, Byers B. The yeast cell cycle gene CDC34 encodes a ubiquitin-conjugating enzyme. Science. 1988;241:1331–1335. doi: 10.1126/science.2842867. [DOI] [PubMed] [Google Scholar]
- Henchoz S, Chi Y, Catarin B, Herskowitz I, Deshaies RJ, Peter M. Phosphorylation and ubiquitin-dependent degradation of the cyclin-dependent kinase inhibitor Far1p in budding yeast. Genes & Dev. 1997;11:3046–3060. doi: 10.1101/gad.11.22.3046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hersko A. Roles of ubiquitin-mediated proteolysis in cell cycle control. Curr Opin Cell Biol. 1997;9:788–799. doi: 10.1016/s0955-0674(97)80079-8. [DOI] [PubMed] [Google Scholar]
- Hershko A, Heller H, Elias S, Ciechanover A. Components of ubiquitin-protein ligase system. Resolution, affinity purification, and role in protein breakdown. J Biol Chem. 1983;258:8206–8214. [PubMed] [Google Scholar]
- Hochstrasser M. Ubiquitin-dependent protein degradation. Annu Rev Genet. 1996;30:405–439. doi: 10.1146/annurev.genet.30.1.405. [DOI] [PubMed] [Google Scholar]
- Hubbard E, Wu G, Kitajewski J, Greenwald I. sel-10, a negative regulator of lin-12 activity in Caenorhabditis elegans, encodes a member of the CDC4 family of proteins. Genes & Dev. 1997;11:3182–3193. doi: 10.1101/gad.11.23.3182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingram GC, Doyle S, Carpenter R, Schultz EA, Simon R, Coen ES. Dual role for fimbriata in regulating floral homeotic genes and cell division in Antirrhinum. EMBO J. 1997;16:6521–6534. doi: 10.1093/emboj/16.21.6521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- James P, Halladay J, Craig EA. Genomic libraries and a host strain designed for highly efficient two-hybrid selection in yeast. Genetics. 1996;144:1425–1436. doi: 10.1093/genetics/144.4.1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaiser C, Michaelis S, Mitchell A. Methods in yeast genetics. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1994. [Google Scholar]
- Kaplan KB, Hyman AA, Sorger PK. Regulating the yeast kinetochore by ubiquitin-dependent degradation and Skp1p-mediated phosphorylation. Cell. 1997;91:491–500. doi: 10.1016/s0092-8674(00)80435-3. [DOI] [PubMed] [Google Scholar]
- Kipreos ET, Lander LE, Wing JP, He WW, Hedgecock EM. cul-1 is required for cell cycle exit in C. elegans and identifies a novel gene family. Cell. 1996;85:829–839. doi: 10.1016/s0092-8674(00)81267-2. [DOI] [PubMed] [Google Scholar]
- Kominami K, Toda T. Fission yeast WD-repeat protein Pop1 regulates genome ploidy through ubiquitin-proteasome-mediated degradation of the CDK inhibitor Rum1 and the S-phase initiator Cdc18. Genes & Dev. 1997;11:1548–1560. doi: 10.1101/gad.11.12.1548. [DOI] [PubMed] [Google Scholar]
- Kornitzer D, Raboy B, Kulka RG, Fink GR. Regulated degradation of the transcription factor Gcn4. EMBO J. 1994;13:6021–6030. doi: 10.1002/j.1460-2075.1994.tb06948.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, Paietta JV. The sulfur controller-2 negative regulatory gene of Neurospora crassa encodes a protein with beta-transducin repeats. Proc Natl Acad Sci. 1995;92:3343–3347. doi: 10.1073/pnas.92.8.3343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuras L, Cherest H, Surdin KY, Thomas D. A heteromeric complex containing the centromere binding factor 1 and two basic leucine zipper factors, Met4 and Met28, mediates the transcription activation of yeast sulfur metabolism. EMBO J. 1996;15:2519–2529. [PMC free article] [PubMed] [Google Scholar]
- Lanker S, Valdivieso MH, Wittenberg C. Rapid degradation of the G1 cyclin Cln2 induced by CDK-dependent phosphorylation. Science. 1996;271:1597–1601. doi: 10.1126/science.271.5255.1597. [DOI] [PubMed] [Google Scholar]
- Lee I, Wolfe DS, Nilsson O, Weigel D. A LEAFY co-regulator encoded by UNUSUAL FLORAL ORGANS. Curr Biol. 1997;7:95–104. doi: 10.1016/s0960-9822(06)00053-4. [DOI] [PubMed] [Google Scholar]
- Li FN, Johnston M. Grr1 of S. cerevisiae is connected to the ubiquitination machinery through Skp1: Coupling glucose sensing to gene expression and the cell cycle. EMBO J. 1997;16:5629–5638. doi: 10.1093/emboj/16.18.5629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisztwan J, Marti A, Suterluty H, Gstaiger M, Wirbelauer C, Krek W. Association of human Cul-1 and ubiquitin-conjugating enzyme Cdc34 with the F-box protein p45SKP2: Evidence for evolutionary conservation in the subunit composition of the Cdc34-SCF pathway. EMBO J. 1998;17:368–383. doi: 10.1093/emboj/17.2.368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathias N, Johnson SL, Winey M, Adams AE, Goetsch L, Pringle JR, Byers B, Goebl M. Cdc53p acts in concert with Cdc4p and Cdc34p to control the G1-to-S- phase transition and identifies a conserved family of proteins. Mol Cell Biol. 1996;16:6634–6643. doi: 10.1128/mcb.16.12.6634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nasmyth K. At the heart of the budding yeast cell cycle. Trends Genet. 1996;12:405–412. doi: 10.1016/0168-9525(96)10041-x. [DOI] [PubMed] [Google Scholar]
- Natorff R, Balinska M, Paszewski A. At least four regulatory genes control sulphur metabolite repression in Aspergillus nidulans. Mol & Gen Genet. 1993;238:185–192. doi: 10.1007/BF00279546. [DOI] [PubMed] [Google Scholar]
- Nugroho TT, Mendenhall MD. An inhibitor of yeast cyclin-dependent protein kinase plays an important role in ensuring the genomic integrity of daughter cells. Mol Cell Biol. 1994;14:3320–3328. doi: 10.1128/mcb.14.5.3320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pahl HL, Baeuerle PA. Control of gene expression by proteolysis. Curr Opin Cell Biol. 1996;8:340–347. doi: 10.1016/s0955-0674(96)80007-x. [DOI] [PubMed] [Google Scholar]
- Pause A, Lee S, Worrell RA, Chen DY, Burgess WH, Linehan WM, Klausner RD. The von Hippel–Lindau tumor-suppressor gene product forms a stable complex with human CUL-2, a member of the Cdc53 family of proteins. Proc Natl Acad Sci. 1997;94:2156–2161. doi: 10.1073/pnas.94.6.2156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheffner M, Nuber U, Huibregtse JM. Protein ubiquitination involving an E1-E2-E3 enzyme ubiquitin thioester cascade. Nature. 1995;373:81–83. doi: 10.1038/373081a0. [DOI] [PubMed] [Google Scholar]
- Schneider BL, Yang Q-H, Futcher AB. Linkage of replication to Start by the Cdk inhibitor Sic1. Science. 1996;272:560–562. doi: 10.1126/science.272.5261.560. [DOI] [PubMed] [Google Scholar]
- Schwab M, Lutum AS, Seufert W. Yeast Hct1 is a regulator of Clb2 cyclin proteolysis. Cell. 1997;90:683–693. doi: 10.1016/s0092-8674(00)80529-2. [DOI] [PubMed] [Google Scholar]
- Schwob E, Bohm T, Mendenhall MD, Nasmyth K. The B-type cyclin kinase inhibitor p40SIC1 controls the G1 to S transition in S. cerevisiae. Cell. 1994;79:233–244. doi: 10.1016/0092-8674(94)90193-7. [DOI] [PubMed] [Google Scholar]
- Sheaff RJ, Groudine M, Gordon M, Roberts JM, Clurman BE. Cyclin E-CDK2 is a regulator of p27Kip1. Genes & Dev. 1997;11:1464–1478. doi: 10.1101/gad.11.11.1464. [DOI] [PubMed] [Google Scholar]
- Skowrya D, Craig KL, Tyers M, Elledge SJ, Harper JW. F-box proteins are receptors that recruit phosphorylated substrates to the SCF ubiquitin-ligase complex. Cell. 1997;91:221–230. doi: 10.1016/s0092-8674(00)80403-1. [DOI] [PubMed] [Google Scholar]
- Stemmann O, Lechner J. The Saccharomyces cerevisiae kinetochore contains a cyclin-CDK complexing homologue, as identified by in vitro reconstitution. EMBO J. 1996;15:3611–3620. [PMC free article] [PubMed] [Google Scholar]
- Thomas D, Kuras L, Barbey R, Cherest H, Blaiseau PL, Surdin-Kerjan Y. Met30p, a yeast transcriptional inhibitor that responds to S- adenosylmethionine, is an essential protein with WD40 repeats. Mol Cell Biol. 1995;15:6526–6534. doi: 10.1128/mcb.15.12.6526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyers M. The cyclin-dependent kinase inhibitor p40SIC1 imposes the requirement for Cln G1 cyclin function at Start. Proc Natl Acad Sci. 1996;93:7772–7776. doi: 10.1073/pnas.93.15.7772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyers M, Tokiwa G, Nash R, Futcher B. The Cln3-Cdc28 kinase complex of S. cerevisiae is regulated by proteolysis and phosphorylation. EMBO J. 1992;11:1773–1784. doi: 10.1002/j.1460-2075.1992.tb05229.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verma R, Annan RS, Huddleston MJ, Carr SA, Reynard G, Deshaies RJ. Phosphorylation of Sic1p by G1 Cdk required for its degradation and entry into S phase. Science. 1997a;278:455–460. doi: 10.1126/science.278.5337.455. [DOI] [PubMed] [Google Scholar]
- Verma R, Feldman RM, Deshaies RJ. SIC1 is ubiquitinated in vitro by a pathway that requires CDC4, CDC34, and cyclin/CDK activities. Mol Biol Cell. 1997b;8:1427–1437. doi: 10.1091/mbc.8.8.1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visintin R, Prinz S, Amon A. Cdc20 and Cdh1, a familiy of substrate-specific activators of APC-dependent proteolysis. Science. 1997;278:460–463. doi: 10.1126/science.278.5337.460. [DOI] [PubMed] [Google Scholar]
- Willems AR, Lanker S, Patton EE, Craig KL, Nason TF, Mathias N, Kobayashi R, Wittenburg C, Tyers M. Cdc53 targets phosphorylated G1 cyclins for degradation by the ubiquitin proteolytic pathway. Cell. 1996;86:453–463. doi: 10.1016/s0092-8674(00)80118-x. [DOI] [PubMed] [Google Scholar]
- Won KA, Reed SI. Activation of cyclin E/CDK2 is coupled to site-specific autophosphorylation and ubiquitin-dependent degradation of cyclin E. EMBO J. 1996;15:4182–4193. [PMC free article] [PubMed] [Google Scholar]
- Yaglom J, Linskens MH, Sadis S, Rubin DM, Futcher B, Finley D. p34CDC28-mediated control of Cln3 cyclin degradation. Mol Cell Biol. 1995;15:731–741. doi: 10.1128/mcb.15.2.731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yochem J, Byers B. Structural comparison of the yeast cell division cycle gene CDC4 and a related pseudogene. J Mol Biol. 1987;195:233–245. doi: 10.1016/0022-2836(87)90646-2. [DOI] [PubMed] [Google Scholar]
- Zhang H, Kobayashi R, Galaktionov K, Beach D. p19Skp1 and p45Skp2 are essential elements of the cyclin A-CDK2 S phase kinase. Cell. 1995;82:915–925. doi: 10.1016/0092-8674(95)90271-6. [DOI] [PubMed] [Google Scholar]