Toward defining the preclinical stages of Alzheimer's disease: Recommendations from the National Institute on Aging-Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease (original) (raw)
. Author manuscript; available in PMC: 2012 May 1.
Published in final edited form as: Alzheimers Dement. 2011 Apr 21;7(3):280–292. doi: 10.1016/j.jalz.2011.03.003
Abstract
The pathophysiological process of Alzheimer's disease (AD) is thought to begin many years before the diagnosis of AD dementia. This long “preclinical” phase of AD would provide a critical opportunity for therapeutic intervention; however, we need to further elucidate the link between the pathological cascade of AD and the emergence of clinical symptoms. The National Institute on Aging and the Alzheimer's Association convened an international workgroup to review the biomarker, epidemiological, and neuropsychological evidence, and to develop recommendations to determine the factors which best predict the risk of progression from “normal” cognition to mild cognitive impairment and AD dementia. We propose a conceptual framework and operational research criteria, based on the prevailing scientific evidence to date, to test and refine these models with longitudinal clinical research studies. These recommendations are solely intended for research purposes and do not have any clinical implications at this time. It is hoped that these recommendations will provide a common rubric to advance the study of preclinical AD, and ultimately, aid the field in moving toward earlier intervention at a stage of AD when some disease-modifying therapies may be most efficacious.
Keywords: Preclinical Alzheimer's disease, Biomarker, Amyloid, Neurodegeneration, Prevention
1. Introduction
Converging evidence from both genetic at-risk cohorts and clinically normal older individuals suggests that the pathophysiological process of Alzheimer's disease (AD) begins years, if not decades, before the diagnosis of clinical dementia [1]. Recent advances in neuroimaging, cerebrospinal fluid (CSF) assays, and other biomarkers now provide the ability to detect evidence of the AD pathophysiological process in vivo. Emerging data in clinically normal older individuals suggest that biomarker evidence of amyloid beta (Aβ) accumulation is associated with functional and structural brain alterations, consistent with the patterns of abnormality seen in patients with mild cognitive impairment (MCI) and AD dementia. Furthermore, clinical cohort studies suggest that there may be very subtle cognitive alterations that are detectable years before meeting criteria for MCI, and that predict progression to AD dementia. It is also clear, however, that some older individuals with the pathophysiological process of AD may not become symptomatic during their lifetime. Thus, it is critical to better define the biomarker and/or cognitive profile that best predicts progression from the preclinical to the clinical stages of MCI and AD dementia. The long preclinical phase of AD provides a critical opportunity for potential intervention with disease-modifying therapy, if we are able to elucidate the link between the pathophysiological process of AD and the emergence of the clinical syndrome.
A recent report on the economic implications of the impending epidemic of AD, as the “baby boomer” generation ages, suggests that more than 13.5 million individuals just in the United States will manifest AD dementia by the year 2050 (http://www.alz.org/alzheimers_disease_trajectory.asp). A hypothetical intervention that delayed the onset of AD dementia by 5 years would result in a 57% reduction in the number of patients with AD dementia, and reduce the projected Medicare costs of AD from 627to627 to 627to344 billion dollars. Screening and treatment programs instituted for other diseases, such as cholesterol screening for cardiovascular and cerebrovascular disease and colonoscopy for colorectal cancer, have already been associated with a decrease in mortality because of these conditions. The current lifetime risk of AD dementia for a 65-year-old is estimated to be at 10.5%. Recent statistical models suggest that a screening instrument for markers of the pathophysiological process of AD (with 90% sensitivity and specificity) and a treatment that slows down progression by 50% would reduce that risk to 5.7%.
Both laboratory work and recent disappointing clinical trial results raise the possibility that therapeutic interventions applied earlier in the course of AD would be more likely to achieve disease modification. Studies with trans-genic mouse models suggest that Aβ-modifying therapies may have limited effect after neuronal degeneration has begun. Several recent clinical trials involving the stages of mild to moderate dementia have failed to demonstrate clinical benefit, even in the setting of biomarker or autopsy evidence of decreased Aβ burden. Although the field is already moving to earlier clinical trials at the stage of MCI, it is possible that similar to cardiac disease and cancer treatment, AD would be optimally treated before significant cognitive impairment, in the “presymptomatic” or “preclinical” stages of AD. Secondary prevention studies, which would treat “normal” or asymptomatic individuals or those with subtle evidence of impairment due to AD so as to delay the onset of full-blown clinical symptoms, are already in the planning stages. The overarching therapeutic objective of these preclinical studies would be to treat early pathological processes (e.g., lower Aβ burden or decrease neurofibrillary tangle pathology) to prevent subsequent neurodegeneration and eventual cognitive decline.
For these reasons, our working group sought to examine the evidence for a definable preclinical stage of AD, and to review the biomarker, epidemiological, and neuropsychological factors that best predict the risk of progression from asymptomatic to MCI and AD dementia. To narrow the scope of our task, we chose to specifically focus on predictors of cognitive decline thought to be due to the pathophysiological process of AD. We did not address cognitive aging in the absence of recognized pathological changes in the brain,or cognitive decline because of other common age-related brain diseases; however, we readily acknowledge that these brain diseases, in particular, cerebrovascular disease, Lewy body disease, and other neurodegenerative processes, may significantly influence clinical manifestations of AD and possibly its pathophysiology. Although there are likely lifelong characteristics and midlife risk factors that influence the likelihood of developing cognitive impairment late in life, for feasibility in current studies, we chose to focus on the 10-year period before the emergence of cognitive impairment.
Furthermore, we propose a research framework to provide a common language to advance the scientific understanding of the preclinical stages of AD and a foundation for the evaluation of preclinical AD treatments. These criteria are intended purely for research purposes, and have no clinical or diagnostic utility at the present time. We hope these criteria will enable researchers to characterize further the sequence of biological events over the course of preclinical AD, refine biomarker criteria that will best predict clinical outcome, and ultimately aid in selecting appropriate populations for preclinical therapeutic intervention.
2. Redefining the earliest stages of AD
The term “Alzheimer's disease” has referred in some contexts to the neuropathological criteria for AD and in other contexts to the clinical syndrome of progressive cognitive and behavioral impairment, typically at the stage of AD dementia. As we move toward defining the earliest stages of AD, the dissociation between these two connotations of the term “Alzheimer's disease” becomes particularly salient. It has become increasingly clear that both the underlying pathophysiological process of AD and its clinical symptomatology are best conceptualized as a continuum or a trajectory, and that these processes may evolve in parallel but temporally offset trajectories.
To facilitate the possibility of future presymptomatic/preclinical treatment of AD, our working group, as well as the other two groups, felt it was important to define AD as encompassing the underlying pathophysiological disease process, as opposed to having “AD” connote only the clinical stages of the disease [2]. To disambiguate the term “AD,” it may be useful to refer to evidence of the underlying brain disease process as AD-pathophysiological process (abbreviated as AD-P) and the clinical phases of the illness as “AD-Clinical” (abbreviated as AD-C), which would include not only AD dementia but also individuals with MCI due to AD-P. AD-P is thought to begin years before the emergence of AD-C. In particular, emerging evidence from both genetic at-risk and aging cohorts suggests that there may be a time lag of a decade or more between the beginning of the pathological cascade of AD and the onset of clinically evident impairment. We postulate that AD begins with a long asymptomatic period during which the pathophysiological process is progressing, and that individuals with biomarker evidence of early AD-P are at increased risk for developing cognitive and behavioral impairment and progression to AD dementia (AD-C). The extent to which biomarkers of AD-P predict a cognitively normal individual's subsequent clinical course remains to be clarified, and we acknowledge that some of these individuals will never manifest clinical symptoms in their lifetime. Thus, it is critical to better define the preclinical stage of AD, to determine the factors that best predict the emergence of clinical impairment and progression to eventual AD dementia, and to reveal the biomarker profile that will identify individuals most likely to benefit from early intervention.
The concept of a preclinical phase of disease should not be too foreign because medical professionals readily acknowledge that cancer can be detected at the stage of “carcinoma _in situ_” and that hypercholesterolemia and atherosclerosis can result in narrowing of coronary arteries that is detectable before myocardial infarction. It is widely acknowledged that symptoms are not necessary to diagnose human disease. Type II diabetes, hypertension, renal insufficiency, and osteoporosis are frequently detected through laboratory tests (i.e., biomarkers), and effective treatment can prevent the emergence of symptoms. Thus, we should be open to the idea that AD could one day be diagnosed preclinically by the presence of biomarker evidence of AD-P, which may eventually guide therapy before the onset of symptoms.
The difficulty in the field of AD is that we have not yet established a firm link between the appearance of any specific biomarker in asymptomatic individuals and the subsequent emergence of clinical symptomatology. If we can, however, definitively determine the risk of developing AD dementia and the temporal course of clinical progression associated with AD-P in individuals without dementia or MCI, we will open a crucial window of opportunity to intervene with disease-modifying therapy. Although we hypothesize that the current earliest detectable pathological change will be in the form of Aβ accumulation, it is possible that Aβ accumulation is necessary but not sufficient to produce the clinical manifestations of AD. It is likely that cognitive decline would occur only in the setting of Aβ accumulation plus synaptic dysfunction and/or neurodegeneration, including paired helical filament tau formation and neuronal loss. It also remains unknown whether there is a specific threshold or regional distribution of AD pathology, and/or a specific combination of biomarker abnormalities that will best predict the emergence of clinical symptoms. Evidence also suggests that additional factors, such as brain and cognitive reserve, and conversely, the presence of other age-related brain diseases, may modulate the relationship between AD-P and AD-C. We also recognize that some individuals can evidence all of the diagnostic neuropathological features of AD at autopsy but never express dementia during their life; it remains unknown whether these individuals would have manifested clinical symptoms should they have lived longer. It is also possible that some individuals are relatively resistant to AD-P because of cognitive or brain reserve, protective genetic factors, or environmental influences. Recent advances in antemortem biomarkers now allow us to test the hypothesis that many individuals with laboratory evidence of AD-P are indeed in the preclinical stages of AD, and determine which biomarker and cognitive profiles are most predictive of subsequent clinical decline and emergence of AD-C.
3. The continuum of AD
The other two working groups established by the National Institute on Aging/Alzheimer's Association are focused on developing diagnostic criteria for the clinical stages of MCI and dementia due to underlying AD-P [3–5]. Our group focused on developing research recommendations for the study of individuals who have evidence of early AD pathological changes but do not meet clinical criteria for MCI or dementia. It is likely that even this preclinical stage of the disease represents a continuum from completely asymptomatic individuals with biomarker evidence suggestive of AD-P at risk for progression to AD dementia to biomarker-positive individuals who are already demonstrating very subtle decline but not yet meeting standardized criteria for MCI (refer to accompanying MCI workgroup recommendations by Albert et al). This latter group of individuals might be classified as “Not normal, not MCI” but would be included under the rubric of preclinical AD (Fig. 1). Importantly, this continuum of preclinical AD would also encompass (1) individuals who carry one or more apolipoprotein E (APOE) _ε_4 alleles who are known to have an increased risk of developing AD dementia, at the point they are AD-P biomarker-positive, and (2) carriers of autosomal dominant mutations, who are in the presymptomatic biomarker-positive stage of their illness, and who will almost certainly manifest clinical symptoms and progress to dementia.
Fig. 1.
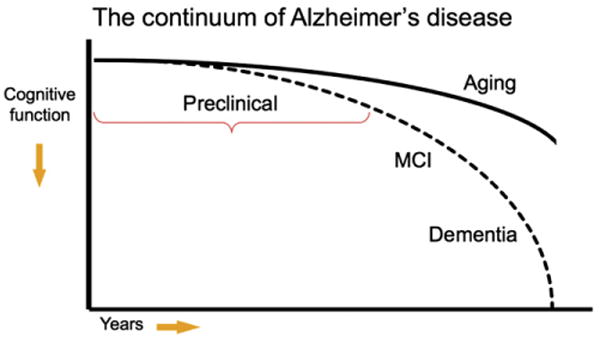
Model of the clinical trajectory of Alzheimer's disease (AD). The stage of preclinical AD precedes mild cognitive impairment (MCI) and encompasses the spectrum of presymptomatic autosomal dominant mutation carriers, asymptomatic biomarker-positive older individuals at risk for progression to MCI due to AD and AD dementia, as well as biomarker-positive individuals who have demonstrated subtle decline from their own baseline that exceeds that expected in typical aging, but would not yet meet criteria for MCI. Note that this diagram represents a hypothetical model for the pathological-clinical continuum of AD but does not imply that all individuals with biomarker evidence of AD-pathophysiological process will progress to the clinical phases of the illness.
Our group carefully considered several monikers to best capture this stage of the disease, including “asymptomatic,” “presymptomatic,” “latent,” “premanifest,” and “preclinical.” The term “preclinical” was felt to best encompass this conceptual phase of the disease process but is not meant to imply that all individuals who have evidence of early AD pathology will necessarily progress to clinical AD dementia. Individuals who are biomarker positive but cognitively normal might currently be defined as “asymptomatic at risk for AD dementia.” Indeed, our goal is to better define the factors which best predict cognitive decline in biomarker-positive individuals, so as to move toward an accurate profile of preclinical AD.
4. Models of the pathophysiological sequence of AD
To facilitate the discussion of the concept of a preclinical stage of AD, we propose a theoretical model of the pathophysiological cascade of AD (Fig. 2). It is important to acknowledge that this model, although based on the prevailing evidence, may be incorrect, is certainly incomplete, and will evolve as additional laboratory and clinical studies are completed. Indeed, this model should be viewed as an initial attempt to bring together multiple areas of research into our best estimate of a more coherent whole.
Fig. 2.
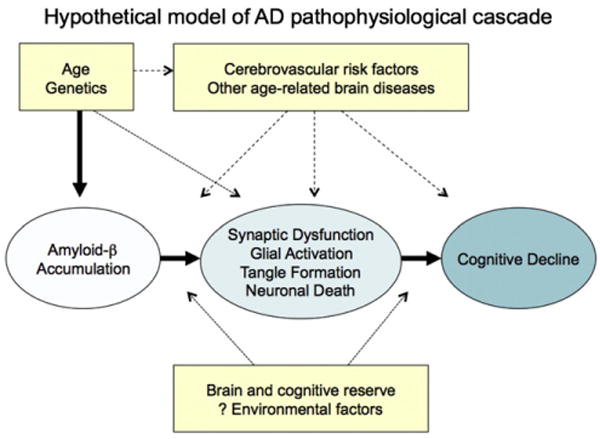
Hypothetical model of the Alzheimer's disease (AD) pathophysiological sequence leading to cognitive impairment. This model postulates that amyloid beta (Aβ) accumulation is an “upstream” event in the cascade that is associated with “downstream” synaptic dysfunction, neurodegeneration, and eventual neuronal loss. Note that although recent work from animal models suggests that specific forms of Aβ may cause both functional and morphological synaptic changes, it remains unknown whether Aβ is sufficient to incite the neurodegenerative process in sporadic late-onset AD. Age and genetics, as well as other specific host factors, such as brain and cognitive reserve, or other brain diseases may influence the response to Aβ and/or the pace of progression toward the clinical manifestations of AD.
The proposed model of AD views Aβ peptide accumulation as a key early event in the pathophysiological process of AD. However, we acknowledge that the etiology of AD remains uncertain, and some investigators have proposed that synaptic, mitochondrial, metabolic, inflammatory, neuronal, cytoskeletal, and other age-related alterations may play an even earlier, or more central, role than Aβ peptides in the pathogenesis of AD [6,7]. There also remains significant debate in the field as to whether abnormal processing versus clearance of Aβ42 is the etiologic event in sporadic, late-onset AD [8]. Some investigators have suggested that sequestration of Aβ into fibrillar forms may even serve as a protective mechanism against oligomeric species, which may be the more synaptotoxic forms of Aβ [9–11]. However, of all the known autosomal dominant, early onset forms of AD are thought to be, at least in part, due to alterations in amyloid precursor protein (APP) production or cleavage. Similarly, trisomy 21 invariably results in AD-P in individuals who have three intact copies of the APP coding region located on chromosome 21. Finally, APOE, the major genetic risk factor for late-onset AD, has been implicated in amyloid trafficking and plaque clearance. Both autopsy and biomarker studies (see later in the text) similarly suggest that Aβ42 accumulation increases with advanced aging, the greatest risk factor for developing AD. At this point, it remains unclear whether it is meaningful or feasible to make the distinction between Aβ as a risk factor for developing the clinical syndrome of AD versus Aβ accumulation as an early detectable stage of AD because current evidence suggests that both concepts are plausible.
Also, it is clear that synaptic depletion, intracellular hyperphosphorylated forms of tau, and neuronal loss invariably occur in AD, and at autopsy, these markers seem to correlate better than plaque counts or total Aβ load with clinical impairment. Although we present evidence later that the presence of markers of “upstream” Aβ accumulation is associated with markers of “downstream” pathological change, including abnormal tau, neural dysfunction, glial activation, and neuronal loss and atrophy, it remains to be proven that Aβ accumulation is sufficient to incite the downstream pathological cascade of AD. It remains unknown whether this neurodegenerative process could be related to direct synaptic toxicity due to oligomeric forms of Aβ, disruption of axonal trajectories from fibrillar forms of Aβ, or a “second hit” that results in synaptic dysfunction, neurodegeneration, neurofibrillary tangle formation, and eventually neuronal loss.
Epidemiological data suggest there are significant modulating factors that may alter the pace of the clinical expression of AD-P, although evidence that these factors alter the underlying pathophysiological process itself is less secure. Large cohort studies have implicated multiple health factors that may increase the risk for developing cognitive decline and dementia thought to be caused by AD [12]. In particular, vascular risk factors such as hypertension, hypercholesterolemia, and diabetes have been associated with an increased risk of dementia, and may contribute directly to the effect of AD pathology on the aging brain [13,14]. Depressive symptomatology, apathy, and chronic psychological distress have also been linked to increased risk of manifesting MCI and dementia [15–17]. It also remains unclear whether there are specific environmental exposures, such as head trauma, that may influence the progression of the pathophysiological sequence or the clinical expression of the pathology. On the positive side, there is some evidence that engagement in specific activities, including cognitive, physical, leisure, and social activity, may be associated with decreased risk of MCI and AD dementia [18].
The temporal lag between the appearance of AD-P and the emergence of AD-C also may be altered by factors such as brain or cognitive reserve [19]. The concept of reserve was originally invoked to provide an explanation for the observation that the extent of AD histopathological changes at autopsy did not always align with the degree of clinical impairment, and can be thought of as the ability to tolerate higher levels of brain injury without exhibiting clinical symptoms. “Brain reserve” refers to the capacity of the brain to withstand pathological insult, perhaps because of greater synaptic density or larger number of healthy neurons, such that sufficient neural substrate remains to support normal function. In contrast, “cognitive reserve” is thought to represent the ability to engage alternate brain networks or cognitive strategies to cope with the effects of encroaching pathology. It is not clear, however, that the data support a sharp demarcation between these two constructs because many factors, such as higher socioeconomic status or engagement in cognitively stimulating activities, may contribute to both forms of reserve. Higher education and socioeconomic status have been associated with lower age-adjusted incidence of AD diagnosis. Recent studies suggest that high reserve may primarily influence the capability of individuals to tolerate their AD-P for longer periods, but may also be associated with rapid decline after a “tipping point” is reached and compensatory mechanisms begin to fail [20,21].
5. Biomarker model of the preclinical stage of AD
A biomarker model has been recently proposed in which the most widely validated biomarkers of AD-P become abnormal and likewise reach a ceiling in an ordered manner [22]. This biomarker model parallels the hypothetical pathophysiological sequence of AD discussed previously, and is particularly relevant to tracking the preclinical stages of AD (Fig. 3). Biomarkers of brain Aβ amyloidosis include reductions in CSF Aβ42 and increased amyloid tracer retention on positron emission tomography (PET) imaging. Elevated CSF tau is not specific to AD and is thought to be a biomarker of neuronal injury. Decreased fluorodeoxyglucose 18F (FDG) uptake on PET with a temporoparietal pattern of hypometabolism is a biomarker of AD-related synaptic dysfunction. Brain atrophy on structural magnetic resonance imaging (MRI) in a characteristic pattern involving the medial temporal lobes, paralimbic and temporoparietal cortices is a biomarker of AD-related neurodegeneration.
Fig. 3.
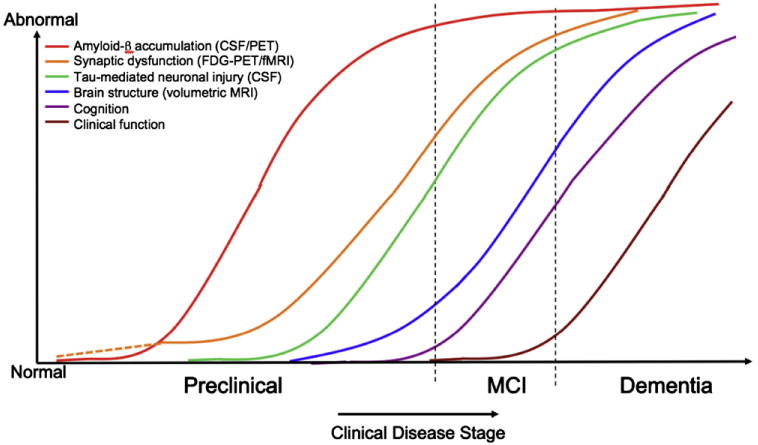
Hypothetical model of dynamic biomarkers of the AD expanded to explicate the preclinical phase: Aβ as identified by cerebrospinal fluid Aβ42 assay or PET amyloid imaging. Synaptic dysfunction evidenced by fluorodeoxyglucose (F18) positron emission tomography (FDG-PET) or functional magnetic resonance imaging (fMRI), with a dashed line to indicate that synaptic dysfunction may be detectable in carriers of the _ε_4 allele of the apolipoprotein E gene before detectable Aβ deposition. Neuronal injury is evidenced by cerebrospinal fluid tau or phospho-tau, brain structure is evidenced by structural magnetic resonance imaging. Biomarkers change from normal to maximally abnormal (y-axis) as a function of disease stage (x-axis). The temporal trajectory of two key indicators used to stage the disease clinically, cognitive and behavioral measures, and clinical function are also illustrated. Figure adapted with permission from Jack et al [22].
This biomarker model was adapted from the original graph proposed by Jack et al [22] to expand the preclinical phase, and has the following features: (1) Aβ accumulation biomarkers become abnormal first and a substantial Aβ load accumulates before the appearance of clinical symptoms. The lag phase between Aβ accumulation and clinical symptoms remains to be quantified, but current theories suggest that the lag may be for more than a decade. Similar to the hypothetical pathophysiological model described previously, interindividual differences in this time lag are likely caused by differences in brain reserve, cognitive reserve, and the added contributions of coexisting pathologies. Note that in this biomarker model, brain Aβ accumulation is necessary but not sufficient to produce the clinical symptoms of MCI and dementia, (2) biomarkers of synaptic dysfunction, including FDG and functional MRI (fMRI), may demonstrate abnormalities very early, particularly in APOE gene _ε_4 allele carriers, who may manifest functional abnormalities before detectable Aβ deposition [23–25]. The severity and change over time in these synaptic markers correlate with clinical symptoms during MCI and AD dementia, (3) structural MRI is thought to become abnormal a bit later, as a marker of neuronal loss, and MRI retains a close relationship with cognitive performance through the clinical phases of MCI and dementia [26], (4) none of the biomarkers is static; rates of change in each biomarker change over time and follow a nonlinear time course, which is hypothesized to be sigmoid shaped, and (5) anatomic information from imaging biomarkers provides useful disease staging information in that the topography of disease-related imaging abnormalities changes in a characteristic manner with disease progression.
6. Biomarker and autopsy evidence linking AD pathology to early symptomatology
Several multicenter biomarker initiatives, including the Alzheimer's Disease Neuroimaging Initiative; the Australian Imaging, Biomarkers and Lifestyle Flagship Study of Aging; as well as major biomarker studies in preclinical populations at several academic centers, are ongoing. These studies have already provided preliminary evidence that biomarker abnormalities consistent with AD pathophysiological process are detectable before the emergence of overt clinical symptomatology and are predictive of subsequent cognitive decline. Many of the recent studies have focused on markers of Aβ using either CSF assays of Aβ42 or PET amyloid imaging with radioactive tracers that bind to fibrillar forms of Aβ. Both CSF and PET amyloid imaging studies suggest that a substantial proportion of clinically normal older individuals demonstrate evidence of Aβ accumulation [27–32]. The exact proportion of “amyloid-positive” normal individuals is dependent on the age and genetic background of the cohort, but ranges from approximately 20% to 40% and is very consonant with large postmortem series [33,34]. Furthermore, there is evidence that the AD-P detected at autopsy is related to episodic memory performance even within the “normal” range [35]. Interestingly, the percentage of “amyloid-positive”normal individuals at autopsy detected at a given age closely parallels the percentage of individuals diagnosed with AD dementia a decade later [36,37] (Fig. 4). Similarly, genetic at-risk cohorts demonstrate evidence of Aβ accumulation many years before detectable cognitive impairment [38–41]. These data support the hypothesis that there is a lengthy temporal lag between the appearance of detectable AD-P and the emergence of AD-C.
Fig. 4.
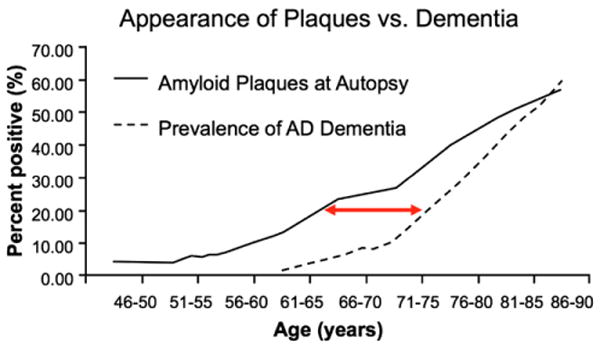
Postulated temporal lag of approximately a decade between the deposition of Aβ (% of individuals with amyloid plaques in a large autopsy series [68]) and the clinical syndrome of AD dementia (estimated prevalence from three epidemiological studies [69–71]). Figure courtesy of Mark Mintun and John Morris, Washington University.
Multiple groups have now reported that cognitively normal older individuals with low CSF Aβ1–42 or high PET amyloid binding demonstrate disruption of functional networks [42–44] and decreased brain volume [45–49], consistent with the patterns seen in AD. There have been variable reports in the previously published data thus far, regarding whether Aβ-positive individuals demonstrate lower neuropsychological test scores at the time of biomarker study [50–54], which may represent heterogeneity in where these individuals fall on the preclinical continuum, the cognitive measures evaluated, and the degree of cognitive reserve in the cohorts. A few early studies have reported that Aβ positivity in clinically normal older individuals is associated with an increased rate of atrophy [55] and an increased risk of cognitive decline and progression to dementia [56–62]. Multiple studies focused on other biomarkers, including volumetric MRI, FDG-PET, or plasma biomarkers, in cohorts of clinically normal older individuals have also reported evidence that these markers are predictive of cognitive decline (refer [63,64] for recent examples). Additional longitudinal studies are clearly needed to confirm these findings and to elucidate the combination of factors that best predict likelihood and rate of decline, and to better understand individual diff-erences in risk for decline.
As a complement to longitudinal studies in the population at risk by virtue of age, researchers continue to detect and track the biological and cognitive changes associated with the predisposition to AD in cognitively normal people at differential genetic risk for AD alone or in conjunction with other risk factors (such as a person's reported family history of the disease). To date, the best established genetic risk factors for AD include common allelic variants of APOE; the major late-onset AD susceptibility gene; uncommon early-onset AD-causing mutations in the presenilin 1, presenilin 2, and APP genes; and trisomy 21 (Down syndrome). Biomarker studies in presymptomatic carriers of these genetic risk factors have revealed evidence of Aβ accumulation on CSF and PET amyloid imaging, as well as FDG-PET hypometabolism, fMRI abnormalities, and brain atrophy that may precede symptoms by more than a decade.
7. Cognitive studies
Despite the clear potential of biomarkers for detecting evidence of the AD pathophysiological process, it is important not to lose sight of the potential that behavioral markers hold for early identification. Tests developed by both neuropsychological and cognitive aging researchers have provided evidence that normal aging is accompanied by declines in speed of information processing, executive function (working memory, task switching, inhibitory function), and reasoning. Studies that have conducted assessments of cognitive function at multiple time points before dementia have also shown consistently a long period of gradual cognitive decline in episodic memory as well as nonmemory domains progressing up to a decade before onset of dementia. Importantly, in studies that have modeled the curve of cognitive change versus time, the preclinical trajectory suggests not only a long- and slow rate of presymptomatic change but also a period of acceleration of performance decrement that may begin several years before MCI onset [65]. Recent studies also suggest that self-report of subtle cognitive decline, even in the absence of significant objective impairment on testing, may portend future decline in older individuals. Despite the existence of multiple studies spanning thousands of participants, the promise of both subjective and objective cognitive measures for assessing risk of progression to AD in individual elders has not yet been fully realized. It is likely that measured change in cognition over time will be more sensitive than any one-time measure. Additional longitudinal studies of older individuals, perhaps combining biomarkers with measures sensitive to detecting very subtle cognitive decline, are clearly needed.
8. Caveats
Although the aforementioned studies provide compelling evidence that markers of Aβ in “normal” older individuals are associated with other brain alterations consistent those seen in AD dementia, and that specific factors may accurately predict those individuals who are at a higher risk of progression to AD-C, it is important to note several potential confounding issues in the majority of these studies. It is likely that many of these studies suffer from cohort biases. In particular, the biomarker and cognitive studies likely are not representative of the general older population because they are typically “samples of convenience,” that is, volunteer cohorts who tend to come from highly educated and socioeconomic status backgrounds. These individuals may also be less likely to harbor typical age-related comorbidities that may influence the rate of cognitive decline. Older individuals who are willing to participate in such intensive studies may also represent the “volunteer gene,” and may be more actively engaged than the typical aging population. Conversely, these cohorts may include individuals who self-select for this research because of subjective concerns about their own memory function or positive family history, as reflected by the high rate of _APOE ε_4 carriers in some of these cohorts.
It is also important to note that although these biomarkers have revolutionized the field of early AD, these markers are merely “proxies” for the underlying disease and may not fully reflect the biological processes in the living brain. For example, both CSF and PET amyloid imaging markers seem to be estimates of the deposition of fibrillar forms of Aβ, and may not provide information about oligomeric forms, which may be the relevant species for synaptic toxicity. Similarly, our proxy measurements for synaptic dysfunction, such as fMRI or FDG-PET, are indirect measurements of neural function. Other markers of neurodegeneration such as CSF tau and volumetric MRI are not specific to the AD process. Finally, it is important to acknowledge that the relationship between biomarkers and cognition may vary significantly across age and genetic cohorts. In particular, the dissociation between the presence or absence of AD-P and clinical symptomatology in the oldest-old needs to be better understood.
Finally, it is important to re-emphasize that although Aβ deposition and neuritic plaque formation are required for the diagnosis of definite AD, and that current evidence suggests that Aβ accumulation is an early detectable stage of the pathological-clinical continuum of AD, the role of Aβ as the etiologic agent in sporadic late-onset AD remains to be proven. There may be pathophysiological events that are “upstream” of Aβ accumulation yet to be discovered, and the relationship between Aβ and neurodegeneration is not yet clear. In particular, the failure of biologically active Aβ-lowering therapies to demonstrate clinical benefit thus far is of concern. Thus, it is important to continue research in alternative pathophysiological pathways and therapeutic avenues.
9. Draft operational research framework for staging preclinical AD
To facilitate future studies, we propose draft operational research criteria to define study cohorts at risk for developing AD dementia for use in (1) longitudinal natural history studies to determine whether the presence of Aβ markers, either in isolation or in combination with additional markers of neurodegeneration, is predictive of cognitive decline in clinically normal older individuals, and (2) clinical trials of potential disease-modifying agents to investigate effects on biomarker progression and/or the emergence of clinical symptoms.
We emphasize again that this framework is not intended to serve as diagnostic criteria for clinical purposes. Use of these biomarkers in the clinical setting is currently unwarranted because many individuals who satisfy the proposed research criteria may not develop the clinical features of AD in their lifetime. Inappropriate use of this information in this context could be associated with unwarranted concern because there is currently insufficient information to relate preclinical biomarker evidence of AD to subsequent rates of clinical progression with any certainty.
These research criteria are based on the postulate that AD is characterized by a sequence of biological events that begins far in advance of clinical dementia. On the basis of current evidence from both genetic at-risk and older cohort studies, we put forth the hypothesis that Aβ accumulation, or the stage of cerebral amyloidosis, is currently one of the earliest measurable stages of AD, and occurs before any other evidence of cognitive symptomatology. We postulate that the presence of biomarker “positivity” for Aβ in clinically normal older individuals, particularly in combination with evidence of abnormality on other biomarkers of AD-P, may have implications for the subsequent course of AD-C and the responsiveness to treatments targeting AD-P.
Recognizing that the preclinical stages of AD represent a continuum, including individuals who may never progress beyond the stage of Aβ accumulation, we further suggest the following staging schema (see Table 1), which may prove useful in defining research cohorts to test specific hypotheses. Research cohorts could be selected on the basis of these staging criteria, to optimize the ability to ascertain the specific outcomes important for a given type (e.g., natural history or treatment trial) and duration of the study. Evidence of “downstream” biomarkers or subtle cognitive symptoms in addition to evidence of Aβ accumulation may increase the likelihood of rapid emergence of cognitive symptomatology and clinical decline to MCI within several years. The presence of one or more of these additional biomarkers would indicate that individuals are already experiencing early neurodegeneration, and as such, it is possible that amyloid-modifying therapies may be less efficacious after the downstream pathological process is set in motion. There are specific circumstances, however, such as pharmaceutical industry trials that may require a cognitive or clinical endpoint, rather than relying solely on biomarker outcomes. In these cases, it may be advantageous to enrich the study population with individuals in late preclinical stages of AD with evidence of very subtle cognitive change, who would be most likely to rapidly decline and manifest MCI within a short period (see Fig. 5). We recognize that these stages will likely require further modification as new findings emerge, and that the feasibility of delineating these stages in recruiting clinical research cohorts remains unclear. It may be easiest to recruit individuals on the basis of Aβ positivity and perform post hoc analyses to determine the predictive value of specific combinations of biomarker abnormalities. These proposed research criteria are intended to facilitate the standardized collection of new data to better define the spectrum of preclinical AD, and to elucidate the endophenotype of individuals who are most likely to progress toward AD-C.
Table 1. Staging categories for preclinical AD research.
| Stage | Description | Aβ (PET or CSF) | Markers of neuronal injury (tau, FDG, sMRI) | Evidence of subtle cognitive change |
|---|---|---|---|---|
| Stage 1 | Asymptomatic cerebral amyloidosis | Positive | Negative | Negative |
| Stage 2 | Asymptomatic amyloidosis + “downstream” neurodegeneration | Positive | Positive | Negative |
| Stage 3 | Amyloidosis + neuronal injury + subtle cognitive/behavioral decline | Positive | Positive | Positive |
Fig. 5.
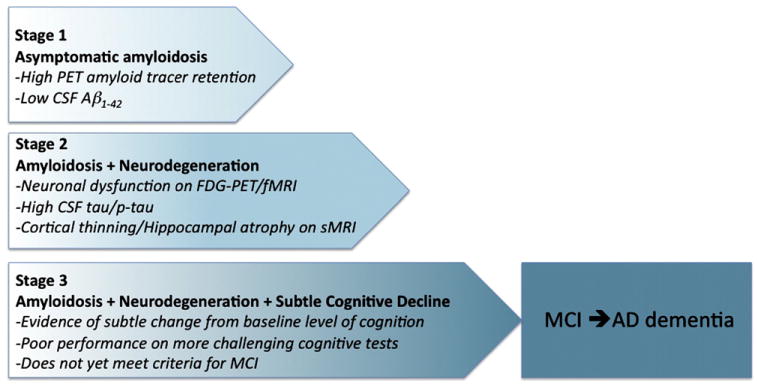
Graphic representation of the proposed staging framework for preclinical AD. Note that some individuals will not progress beyond Stage 1 or Stage 2. Individuals in Stage 3 are postulated to be more likely to progress to MCI and AD dementia. Abbreviations: AD, Alzheimer's disease; Ab, amyloid beta; PET, position emission tomography; CSF, cerebrospinal fluid; FDG, fluorodeoxyglucose, fMRI, functional magnetic resonance imaging, sMRI, structural magnetic resonance imaging.
9.1. Stage 1: The stage of asymptomatic cerebral amyloidosis
These individuals have biomarker evidence of Aβ accumulation with elevated tracer retention on PET amyloid imaging and/or low Aβ42 in CSF assay, but no detectable evidence of additional brain alterations suggestive of neurodegeneration or subtle cognitive and/or behavioral symptomatology. The standards for determining “amyloid-positivity” are still evolving (refer to the next section). Although recent work suggests there may be a CSF Aβ42 cutoff value that is predictive of progression from MCI to AD dementia [66], it is unknown whether a similar threshold will be optimal in prediction of decline in individuals with normal or near normal cognition. Similarly, using PET imaging techniques, it remains unknown whether a summary numeric threshold within an aggregate cortical region or within specific anatomic region will provide the most useful predictive value. Recent data suggest that although CSF Aβ42 is strongly inversely correlated with quantitative PET amyloid imaging measures (distribution value ratio or standardized uptake value), there are some individuals who demonstrate decreased CSF Aβ42 and who would not be considered amyloid positive on PET scans [67]. It remains unclear whether this finding reflects different thresholds used across these techniques or if decreased CSF Aβ42 is an earlier marker of accumulation. In addition, there may be genetic effects that are specific to CSF or PET markers of Aβ.
As mentioned previously, we note that the currently available CSF and PET imaging biomarkers of Aβ primarily provide evidence of amyloid accumulation and deposition of fibrillar forms of amyloid. Although limited, current data suggest that soluble or oligomeric forms of Aβ are likely in equilibrium with plaques, which may serve as reservoirs, but it remains unknown whether there is an identifiable preplaque stage in which only soluble forms of Aβ are present. Because laboratory data increasingly suggest that oligomeric forms of amyloid may be critical in the pathological cascade, there is ongoing work to develop CSF and plasma assays for oligomeric forms of Aβ. There are also emerging data from genetic-risk cohorts that suggest early synaptic changes may be present before evidence of amyloid accumulation using currently available amyloid markers. Thus, it may be possible in the future to detect a stage of disease that precedes stage 1.
9.2. Stage 2: Amyloid positivity + evidence of synaptic dysfunction and/or early neurodegeneration
These individuals have evidence of amyloid positivity and presence of one or more markers of “downstream” AD-P-related neuronal injury. The current markers of neuronal injury with the greatest validation are: (1) elevated CSF tau or phospho-tau, (2) hypometabolism in an AD-like pattern (i.e., posterior cingulate, precuneus, and/or temporoparietal cortices) on FDG-PET, and (3) cortical thinning/gray matter loss in a specific anatomic distribution (i.e., lateral and medial parietal, posterior cingulate, and lateral temporal cortices) and/or hippocampal atrophy on volumetric MRI. Future markers may also include fMRI measures of default network connectivity. Although previous studies have demonstrated that, on average, amyloid-positive individuals demonstrate significantly greater abnormalities on these markers as compared with amyloid-negative individuals, there is significant interindividual variability. We hypothesize that amyloid-positive individuals with evidence of early neurodegeneration may be farther down the trajectory (i.e., in later stages of preclinical AD). It remains unclear whether it will be feasible to detect differences among these other biomarkers of AD-P, but there is some evidence that early synaptic dysfunction, as assessed by functional imaging techniques such as FDG-PET and fMRI, may be detectable before volumetric loss.
9.3. Stage 3: Amyloid positivity + evidence of neurodegeneration + subtle cognitive decline
We postulate that individuals with biomarker evidence of amyloid accumulation, early neurodegeneration, and evidence of subtle cognitive decline are in the last stage of preclinical AD, and are approaching the border zone with the proposed clinical criteria for MCI. These individuals may demonstrate evidence of decline from their own baseline (particularly if proxies of cognitive reserve are taken into consideration), even if they still perform within the “normal” range on standard cognitive measures. There is emerging evidence that more sensitive cognitive measures, particularly with challenging episodic memory measures, may detect very subtle cognitive impairment in amyloid-positive individuals. It remains unclear whether self-complaint of memory decline or other subtle neurobehavioral changes will be a useful predictor of progression, but it is possible that the combination of biomarkers and subjective assessment of subtle change will prove to be useful.
10. Need for additional study
We propose a general framework with biomarker criteria for the study of the preclinical phase of AD; however, more work is needed to clarify the optimal CSF assays, PET or MRI analytic techniques, and in particular, the specific thresholds needed to meet these criteria. There are significant challenges in implementing standardized biomarker “cut-off” values across centers, studies, and countries. Work to standardize and validate both fluid-based and imaging biomarker thresholds is ongoing in multiple academic and pharmaceutical industry laboratories, as well as in several multicenter initiatives. These criteria will need to be validated in large multicenter natural history studies, or as provisional criteria for the planning of preventative clinical trials. For instance, it will be important to establish the test–retest and cross-center reliability of biomarker measurements, further characterize the sequence of biomarker changes, and the extent to which these biomarkers predict subsequent clinical decline or clinical benefit. In particular, there is an important need to evaluate methods for determining “amyloid-positivity” because it remains unclear whether there is a biologically relevant continuum of Aβ accumulation, or whether there is a clear threshold or “cut-off” value that could be defined on the basis of predictive value for subsequent clinical decline, as has been suggested in several CSF studies [28,66]. It also remains unknown whether these thresholds should be adjusted for age or genotype. After these thresholds are established, it may be most feasible to select research cohorts for large studies solely on the basis of “amyloid-positivity” on CSF or PET amyloid imaging, and to use additional biomarker and cognitive measures for post hoc analyses to determine additional predictive value.
Although recent advances in biomarkers have revolutionized our ability to detect evidence of early AD-P there is still a need for novel biomarker development. In particular, although the current biomarkers provide evidence of Aβ deposition, an in vivo marker of oligomeric forms of Aβ would be of great value. Imaging markers of intraneuronal pathology, including specific markers of specific forms of tau/tangles and alpha-synuclein, are also needed. In addition, more sensitive imaging biomarkers that can detect early synaptic dysfunction and functional and structural disconnection, such as fMRI and diffusion tensor imaging, may one day prove to be useful to track early response to amyloid-lowering therapies. Finally, we may be able to use the currently available biomarkers as a new “gold standard” to re-evaluate simple blood and urine markers that were discarded on the basis of excessive overlap between clinically normal and AD patients. The significant proportion of clinically normal individuals who are “amyloid-positive” on both CSF and PET imaging may have confounded previous studies attempting to differentiate “normal” controls from patients with AD.
Similarly, additional work is required to identify and validate neuropsychological and neurobehavioral measures to detect the earliest clinical manifestations of AD. We need to develop sensitive measures in multiple cognitive and behavioral domains that will reveal evidence of early synaptic dysfunction in neural networks vulnerable to AD pathology. We also need to develop measures of very early functional changes in other domains, including social interaction, mood, psychomotor aspects of function, and decision making. These measures would allow us to link better the pathological processes to the emergence of clinical symptoms, and may be particularly useful to monitor response to potential disease-modifying therapies in these very early stages.
The proposed criteria apply primarily to individuals at risk by virtue of advanced age because inclusion criteria for trials in autosomal dominant mutation carriers and homozygous _APOE ε_4 carriers will be likely defined primarily on genetic status. Trials in genetic-risk populations might use these criteria to stage individuals within the preclinical phase of AD. In genetic-risk cohorts, it may even be possible to detect an even earlier stage of presymptomatic AD, before the point when there is already detectable cerebral amyloidosis. Several FDG-PET and fMRI studies have suggested that evidence of synaptic dysfunction may be present in young and middle-aged _APOE ε_4 carriers (see Fig. 3), and there may be other biological alterations that are present before significant deposition of fibrillar forms of amyloid that would be preferentially responsive to presymptomatic intervention.
The emerging concept of preclinical AD and the role of biomarkers in the detection and tracking in this stage of the disease have important implications for the development of effective treatments. Therapies for preclinical AD would be intended to postpone, reduce the risk of, or completely prevent the clinical stages of the disorder. As recently noted, the use of clinical endpoints in clinical trials of such treatments would require large numbers of healthy volunteers, large amounts of money, and many years of study. Researchers have raised the possibility of evaluating biomarker endpoints for these treatments in cognitively normal people at increased risk for AD because these studies might be performed more rapidly than otherwise possible. Subjects enrolled in these studies could include individuals with autosomal dominant mutation carriers (with essentially a 100% chance of developing clinical AD) or those at increased risk of developing sporadic AD (e.g., _APOE ε_4 carriers or subjects with biomarker evidence of preclinical AD pathology). The use of biomarkers rather than clinical outcomes could accelerate progress in these trials; however, regulatory agencies must be assured that a given biomarker is “reasonably likely” to predict a clinically meaningful outcome before they would grant approval for treatments tested in trials using biomarkers as surrogate endpoints. Research strategies have been proposed to provide this evidence by embedding the most promising biomarkers in preclinical AD trials of people at the highest imminent risk of clinical onset to establish a link between a biomarker effect and the onset of clinical symptoms of AD. We envision the time when the scientific means and accelerated regulatory approval pathway support multiple preclinical AD trials using biomarkers to identify subjects and provide shorter term outcomes, such that demonstrably effective treatments to ward off the clinical stages of AD are found as quickly as possible. There are several burgeoning efforts to design and conduct clinical trials in both genetic at-risk and amyloid-positive older individuals, including the Dominantly Inherited Alzheimer Network (study of familial AD), the Alzheimer Prevention Initiative, and Anti-Amyloid Treatment in Asymptomatic AD (A4) trial being considered by the Alzheimer's Disease Cooperative Study.
Finally, the ethical and practical implications surrounding the issues of future implementation of making a “diagnosis” of AD at a preclinical stage need to be studied, should the postulates put forth previously prove to be correct. Although at this point our recommendations are strictly for research purposes only, the public controversy surrounding the identification of asymptomatic individuals with evidence of AD-P raised several important points that the field must consider. In particular, the poignant question of “why would anyone want to know they have AD a decade before they might develop symptoms, if there is nothing they can do about it?” should be carefully considered well before any results from research is translated into clinical practice. First, there may be important reasons, including social and financial planning, why some individuals would want to know their likelihood of developing AD dementia within the next decade, even in the absence of an available disease-modifying therapy. It is our hope, however, that the advances in preclinical detection of AD-P will enable earlier, more effective treatment, just as nearly all of therapeutic gains in cancer, cardiovascular disease, osteoporosis, and diabetes involve treatment before significant clinical symptoms are present. It is entirely possible that promising drugs, particularly amyloid-modifying agents, will fail to affect the clinical course of AD at the stage of dementia or even MCI, when the neurodegenerative process is well entrenched, but may be efficacious at the earliest stages of the AD-P, before the onset of symptoms.
The definitive studies to determine whether the majority of asymptomatic individuals with evidence of AD-P are indeed destined to develop AD dementia, to elucidate the biomarker and/or cognitive endophenotype that is most predictive of cognitive decline, and to determine whether intervention with potential disease-modifying therapies in the preclinical stages of AD will prevent dementia are likely to take more than a decade to fully accomplish. Thus, we must move quickly to test the postulates put forth previously, and adjust our models and study designs as new data become available. Because potential biologically active treatments may be associated with small but significant risk of adverse side effects, we will need to determine whether we can predict the emergence of cognitive symptoms with sufficient certainty to appropriately weigh the risk/benefit ratios to begin treatment in asymptomatic individuals. It is clear that many questions remain to be answered, and that there may be additional factors which will influence the probability of developing clinical AD. However, the considerable progress made over the past two decades now enables a strategic path forward to test these hypotheses, move the field toward earlier intervention, and ultimately, toward the prevention of AD dementia.
Acknowledgments
The chair (Reisa Sperling) acknowledges the invaluable assistance of Dr. Cerise Elliott at National Institute on Aging, as well as thoughtful input solicited from several individuals, in particular, Drs. Keith Johnson, Dorene Rentz, Peter Davies, Deborah Blacker, Steve Salloway, Sanjay Asthana, and Dennis Selkoe, as well as the helpful public commentary provided by our colleagues in the field.
Reisa Sperling has served as a site investigator and/or consultant to several companies developing imaging biomarkers and pharmacological treatments for early AD, including Avid, Bayer, Bristol-Myers-Squibb, Elan, Eisai, Janssen, Pfizer, and Wyeth. Paul Aisen serves on a scientific advisory board for NeuroPhage; serves as a consultant to Elan Corporation, Wyeth, Eisai Inc., Bristol-Myers Squibb, Eli Lilly and Company, NeuroPhage, Merck & Co., Roche, Amgen, Abbott, Pfizer Inc., Novartis, Bayer, Astellas, Dainippon, Biomarin, Solvay, Otsuka, Daiichi, AstraZeneca, Janssen, and Medivation Inc.; receives research support from Pfizer Inc. and Baxter International Inc.; and has received stock options from Medivation Inc. and NeuroPhage. Clifford Jack serves as a consultant for Eli Lilly, Eisai, and Élan; is an investigator in clinical trials sponsored by Baxter and Pfizer Inc.; and owns stock in Johnson and Johnson. Denise Park has received research support from Avid Pharmaceuticals. Eric Siemers is an employee of Eli Lilly and Company, which acquired Avid Pharmaceuticals. Yaakov Stern has consulted to Bayer Pharmaceuticals and has received research support from Bayer, Janssen, Eli Lilly, and Elan. Maria Carrillo is an employee of the Alzheimer's Association and reports no conflicts. Bill Thies is an employee of the Alzheimer's Association and reports no conflicts. Creighton Phelps is an employee of the U.S. Government and reports no conflicts.
References
- 1.Morris JC. Early-stage and preclinical Alzheimer disease. Alzheimer Dis Assoc Disord. 2005;19:163–5. doi: 10.1097/01.wad.0000184005.22611.cc. [DOI] [PubMed] [Google Scholar]
- 2.Dubois B, Feldman HH, Jacova C, Cummings JL, Dekosky ST, Barberger-Gateau P, et al. Revising the definition of Alzheimer's disease: a new lexicon. Lancet Neurol. 2010;9:1118–27. doi: 10.1016/S1474-4422(10)70223-4. [DOI] [PubMed] [Google Scholar]
- 3.Jack CR, Jr, Albert MS, Knopman DS, McKhann GM, Sperling RA, Carrillo MC, et al. Introduction to the recommendations from the National Institute on Aging-Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement. 2011;7:257–62. doi: 10.1016/j.jalz.2011.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Albert MS, DeKosky ST, Dickson D, Dubois B, Feldman HH, Fox NC, et al. The diagnosis of mild cognitive impairment due to Alzheimer's disease: recommendations from the National Institute on Aging–Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement. 2011;7:270–9. doi: 10.1016/j.jalz.2011.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McKhann GM, Knopman DS, Chertkow H, Hyman BT, Jack CR, Jr, Kawas CH, et al. The diagnosis of dementia due to Alzheimer's disease: recommendations from the National Institute on Aging– Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement. 2011;7:263–9. doi: 10.1016/j.jalz.2011.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pimplikar SW, Nixon RA, Robakis NK, Shen J, Tsai LH. Amyloid-independent mechanisms in Alzheimer's disease pathogenesis. J Neurosci. 2010;30:14946–54. doi: 10.1523/JNEUROSCI.4305-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Herrup K. Reimagining Alzheimer's disease-an age-based hypothesis. J Neurosci. 2010;30:16755–62. doi: 10.1523/JNEUROSCI.4521-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mawuenyega KG, Sigurdson W, Ovod V, Munsell L, Kasten T, Morris JC, et al. Decreased Clearance of CNS {beta}-Amyloid in Alzheimer's Disease. Science. 2010;330:1774. doi: 10.1126/science.1197623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Selkoe DJ. Alzheimer's disease is a synaptic failure. Science. 2002;298:789–91. doi: 10.1126/science.1074069. [DOI] [PubMed] [Google Scholar]
- 10.Lee HG, Casadesus G, Zhu X, Takeda A, Perry G, Smith MA. Challenging the amyloid cascade hypothesis: senile plaques and amyloid-beta as protective adaptations to Alzheimer disease. Ann N Y Acad Sci. 2004;1019:1–4. doi: 10.1196/annals.1297.001. [DOI] [PubMed] [Google Scholar]
- 11.Shankar GM, Li S, Mehta TH, Garcia-Munoz A, Shepardson NE, Smith I, et al. Amyloid-beta protein dimers isolated directly from Alzheimer's brains impair synaptic plasticity and memory. Nat Med. 2008;14:837–42. doi: 10.1038/nm1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yaffe K, Fiocco AJ, Lindquist K, Vittinghoff E, Simonsick EM, Newman AB, et al. Predictors of maintaining cognitive function in older adults: the Health ABC study. Neurology. 2009;72:2029–35. doi: 10.1212/WNL.0b013e3181a92c36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Arvanitakis Z, Wilson RS, Bienias JL, Evans DA, Bennett DA. Diabetes mellitus and risk of Alzheimer disease and decline in cognitive function. Arch Neurol. 2004;61:661–6. doi: 10.1001/archneur.61.5.661. [DOI] [PubMed] [Google Scholar]
- 14.Craft S. The role of metabolic disorders in Alzheimer disease and vascular dementia: two roads converged. Arch Neurol. 2009;66:300–5. doi: 10.1001/archneurol.2009.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ganguli M, Du Y, Dodge HH, Ratcliff GG, Chang CC. Depressive symptoms and cognitive decline in late life: a prospective epidemio-logical study. Arch Gen Psychiatry. 2006;63:153–60. doi: 10.1001/archpsyc.63.2.153. [DOI] [PubMed] [Google Scholar]
- 16.Wilson RS, Arnold SE, Schneider JA, Kelly JF, Tang Y, Bennett DA. Chronic psychological distress and risk of Alzheimer's disease in old age. Neuroepidemiology. 2006;27:143–53. doi: 10.1159/000095761. [DOI] [PubMed] [Google Scholar]
- 17.Onyike CU, Sheppard JM, Tschanz JT, Norton MC, Green RC, Steinberg M, et al. Epidemiology of apathy in older adults: the Cache County Study. Am J Geriatr Psychiatry. 2007;15:365–75. doi: 10.1097/01.JGP.0000235689.42910.0d. [DOI] [PubMed] [Google Scholar]
- 18.Wilson RS, Scherr PA, Schneider JA, Tang Y, Bennett DA. Relation of cognitive activity to risk of developing Alzheimer disease. Neurology. 2007;69:1911–20. doi: 10.1212/01.wnl.0000271087.67782.cb. [DOI] [PubMed] [Google Scholar]
- 19.Stern Y. Cognitive reserve. Neuropsychologia. 2009;47:2015–28. doi: 10.1016/j.neuropsychologia.2009.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fotenos AF, Mintun MA, Snyder AZ, Morris JC, Buckner RL. Brain volume decline in aging: evidence for a relation between socioeconomic status, preclinical Alzheimer disease, and reserve. Arch Neurol. 2008;65:113–20. doi: 10.1001/archneurol.2007.27. [DOI] [PubMed] [Google Scholar]
- 21.Wilson RS, Barnes LL, Aggarwal NT, Boyle PA, Hebert LE, Mendes de Leon CF, et al. Cognitive activity and the cognitive morbidity of Alzheimer disease. Neurology. 2010;75:990–6. doi: 10.1212/WNL.0b013e3181f25b5e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jack CR, Jr, Knopman DS, Jagust WJ, Shaw LM, Aisen PS, Weiner MW, et al. Hypothetical model of dynamic biomarkers of the Alzheimer's pathological cascade. Lancet Neurol. 2010;9:119–28. doi: 10.1016/S1474-4422(09)70299-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Reiman EM, Chen K, Alexander GE, Caselli RJ, Bandy D, Osborne D, et al. Functional brain abnormalities in young adults at genetic risk for late-onset Alzheimer's dementia. Proc Natl Acad Sci U S A. 2004;101:284–9. doi: 10.1073/pnas.2635903100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Filippini N, MacIntosh BJ, Hough MG, Goodwin GM, Frisoni GB, Smith SM, et al. Distinct patterns of brain activity in young carriers of the APOE-epsilon4 allele. Proc Natl Acad Sci U S A. 2003;106:7209–14. doi: 10.1073/pnas.0811879106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sheline YI, Morris JC, Snyder AZ, Price JL, Yan Z, D'Angelo G, et al. APOE4 allele disrupts resting state fMRI connectivity in the absence of amyloid plaques or decreased CSF Abeta42. J Neurosci. 2010;30:17035–40. doi: 10.1523/JNEUROSCI.3987-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vemuri P, Wiste HJ, Weigand SD, Knopman DS, Trojanowski JQ, Shaw LM, et al. Serial MRI and CSF biomarkers in normal aging, MCI, and AD. Neurology. 2010;75:143–51. doi: 10.1212/WNL.0b013e3181e7ca82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rowe CC, Ellis KA, Rimajova M, Bourgeat P, Pike KE, Jones G, et al. Amyloid imaging results from the Australian Imaging, Biomarkers and Lifestyle (AIBL) study of aging. Neurobiol Aging. 2010;31:1275–83. doi: 10.1016/j.neurobiolaging.2010.04.007. [DOI] [PubMed] [Google Scholar]
- 28.Mintun MA, Larossa GN, Sheline YI, Dence CS, Lee SY, Mach RH, et al. [11C]PIB in a nondemented population: potential antecedent marker of Alzheimer disease. Neurology. 2006;67:446–52. doi: 10.1212/01.wnl.0000228230.26044.a4. [DOI] [PubMed] [Google Scholar]
- 29.Jack CR, Jr, Lowe VJ, Senjem ML, Weigand SD, Kemp BJ, Shiung MM, et al. 11C PiB and structural MRI provide complementary information in imaging of Alzheimer's disease and amnestic mild cognitive impairment. Brain. 2008;131(Pt 3):665–80. doi: 10.1093/brain/awm336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gomperts SN, Rentz DM, Moran E, Becker JA, Locascio JJ, Klunk WE, et al. Imaging amyloid deposition in Lewy body diseases. Neurology. 2008;71:903–10. doi: 10.1212/01.wnl.0000326146.60732.d6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.De Meyer G, Shapiro F, Vanderstichele H, Vanmechelen E, Engelborghs S, De Deyn PP, et al. Diagnosis-independent Alzheimer disease biomarker signature in cognitively normal elderly people. Arch Neurol. 2010;67:949–56. doi: 10.1001/archneurol.2010.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Montine TJ, Peskind ER, Quinn JF, Wilson AM, Montine KS, Galasko D. Increased cerebrospinal fluid F(2)-isoprostanes are associated with aging and latent Alzheimer's disease as identified by biomarkers. Neuromolecular Med. 2011;13:37–43. doi: 10.1007/s12017-010-8126-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Arriagada PV, Marzloff K, Hyman BT. Distribution of Alzheimer-type pathologic changes in nondemented elderly individuals matches the pattern in Alzheimer's disease. Neurology. 1992;42:1681–8. doi: 10.1212/wnl.42.9.1681. [DOI] [PubMed] [Google Scholar]
- 34.Morris JC, Storandt M, McKeel DW, Jr, Rubin EH, Price JL, Grant EA, et al. Cerebral amyloid deposition and diffuse plaques in “normal” aging: evidence for presymptomatic and very mild Alzheimer's disease. Neurology. 1996;46:707–19. doi: 10.1212/wnl.46.3.707. [DOI] [PubMed] [Google Scholar]
- 35.Bennett D, Schneider J, Arvanitakis Z, Kelly J, Aggarwal N, Shah R, et al. Neuropathology of older persons without cognitive impairment from two community-based studies. Neurology. 2006;66:1837–44. doi: 10.1212/01.wnl.0000219668.47116.e6. [DOI] [PubMed] [Google Scholar]
- 36.Brookmeyer R, Gray S, Kawas C. Projections of Alzheimer's disease in the United States and the public health impact of delaying disease onset. Am J Public Health. 1998;88:1337–42. doi: 10.2105/ajph.88.9.1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Alzheimer's Association. 2009 Alzheimer's disease facts and figures. Alzheimers Dement. 2009;5:234–70. doi: 10.1016/j.jalz.2009.03.001. [DOI] [PubMed] [Google Scholar]
- 38.Moonis M, Swearer JM, Dayaw MP, St George-Hyslop P, Rogaeva E, Kawarai T, et al. Familial Alzheimer disease: decreases in CSF Abeta42 levels precede cognitive decline. Neurology. 2005;65:323–5. doi: 10.1212/01.wnl.0000171397.32851.bc. [DOI] [PubMed] [Google Scholar]
- 39.Klunk WE, Price JC, Mathis CA, Tsopelas ND, Lopresti BJ, Ziolko SK, et al. Amyloid deposition begins in the striatum of presenilin-1 mutation carriers from two unrelated pedigrees. J Neurosci. 2007;27:6174–84. doi: 10.1523/JNEUROSCI.0730-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ringman JM, Younkin SG, Pratico D, Seltzer W, Cole GM, Geschwind DH, et al. Biochemical markers in persons with preclinical familial Alzheimer disease. Neurology. 2008;71:85–92. doi: 10.1212/01.wnl.0000303973.71803.81. [DOI] [PubMed] [Google Scholar]
- 41.Reiman EM, Chen K, Liu X, Bandy D, Yu M, Lee W, et al. Fibrillar amyloid-beta burden in cognitively normal people at 3 levels of genetic risk for Alzheimer's disease. Proc Natl Acad Sci U S A. 2009;106:6820–5. doi: 10.1073/pnas.0900345106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sperling RA, Laviolette PS, O'Keefe K, O'Brien J, Rentz DM, Pihlajamaki M, et al. Amyloid deposition is associated with impaired default network function in older persons without dementia. Neuron. 2009;63:178–88. doi: 10.1016/j.neuron.2009.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hedden T, Van Dijk KR, Becker JA, Mehta A, Sperling RA, Johnson KA, et al. Disruption of functional connectivity in clinically normal older adults harboring amyloid burden. J Neurosci. 2009;29:12686–94. doi: 10.1523/JNEUROSCI.3189-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sheline YI, Raichle ME, Snyder AZ, Morris JC, Head D, Wang S, et al. Amyloid plaques disrupt resting state default mode network connectivity in cognitively normal elderly. Biol Psychiatry. 2010;67:584–7. doi: 10.1016/j.biopsych.2009.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fjell AM, Walhovd KB, Fennema-Notestine C, McEvoy LK, Hagler DJ, Holland D, et al. Brain atrophy in healthy aging is related to CSF levels of Abeta1-42. Cereb Cortex. 2010;20:2069–79. doi: 10.1093/cercor/bhp279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dickerson BC, Bakkour A, Salat DH, Feczko E, Pacheco J, Greve DN, et al. The cortical signature of Alzheimer's disease: regionally specific cortical thinning relates to symptom severity in very mild to mild AD dementia and is detectable in asymptomatic amyloid-positive individuals. Cereb Cortex. 2009;19:497–510. doi: 10.1093/cercor/bhn113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Desikan RS, Sabuncu MR, Schmansky NJ, Reuter M, Cabral HJ, Hess CP, et al. Selective disruption of the cerebral neocortex in Alzheimer's disease. PLoS One. 2010;5:e12853. doi: 10.1371/journal.pone.0012853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Becker JA, Rentz D, Carmasin JS, Hedden T, Hamdi I, Buckner RL, et al. Amyloid deposition and brain volume across the continuum of aging and AD. Ann Neurol. 2011 in press. [Google Scholar]
- 49.Oh H, Mormino EC, Madison C, Hayenga A, Smiljic A, Jagust WJ. beta-Amyloid affects frontal and posterior brain networks in normal aging. Neuroimage. 2011;54:1887–95. doi: 10.1016/j.neuroimage.2010.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pike KE, Savage G, Villemagne VL, Ng S, Moss SA, Maruff P, et al. Beta-amyloid imaging and memory in non-demented individuals: evidence for preclinical Alzheimer's disease. Brain. 2007;130(Pt 11):2837–44. doi: 10.1093/brain/awm238. [DOI] [PubMed] [Google Scholar]
- 51.Mormino EC, Kluth JT, Madison CM, Rabinovici GD, Baker SL, Miller BL, et al. Episodic memory loss is related to hippocampal-mediated {beta}-amyloid deposition in elderly subjects. Brain. 2009;132:1310–23. doi: 10.1093/brain/awn320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Aizenstein HJ, Nebes RD, Saxton JA, Price JC, Mathis CA, Tsopelas ND, et al. Frequent amyloid deposition without significant cognitive impairment among the elderly. Arch Neurol. 2008;65:1509–17. doi: 10.1001/archneur.65.11.1509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rentz DM, Locascio JJ, Becker JA, Moran EK, Eng E, Buckner RL, et al. Cognition, reserve, and amyloid deposition in normal aging. Ann Neurol. 2010;67:353–64. doi: 10.1002/ana.21904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Villemagne VL, Pike KE, Chetelat G, Ellis KA, Mulligan RS, Bourgeat P, et al. Longitudinal assessment of Abeta and cognition in aging and Alzheimer disease. Ann Neurol. 2011;69:181–92. doi: 10.1002/ana.22248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Schott JM, Bartlett JW, Fox NC, Barnes J Investigators ftAsDNI. Increased brain atrophy rates in cognitively normal older adults with low cerebrospinal fluid Aβ-42. Ann Neurol. 2011 doi: 10.1002/ana.22315. in press. [DOI] [PubMed] [Google Scholar]
- 56.Fagan AM, Roe CM, Xiong C, Mintun MA, Morris JC, Holtzman DM. Cerebrospinal fluid tau/beta-amyloid(42) ratio as a prediction of cognitive decline in nondemented older adults. Arch Neurol. 2007;64:343–9. doi: 10.1001/archneur.64.3.noc60123. [DOI] [PubMed] [Google Scholar]
- 57.Li G, Sokal I, Quinn JF, Leverenz JB, Brodey M, Schellenberg GD, et al. CSF tau/Abeta42 ratio for increased risk of mild cognitive impairment: a follow-up study. Neurology. 2007;69:631–9. doi: 10.1212/01.wnl.0000267428.62582.aa. [DOI] [PubMed] [Google Scholar]
- 58.Storandt M, Mintun MA, Head D, Morris JC. Cognitive decline and brain volume loss as signatures of cerebral amyloid-beta peptide deposition identified with Pittsburgh compound B: cognitive decline associated with Abeta deposition. Arch Neurol. 2009;66:1476–81. doi: 10.1001/archneurol.2009.272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Resnick SM, Sojkova J, Zhou Y, An Y, Ye W, Holt DP, et al. Longitudinal cognitive decline is associated with fibrillar amyloid-beta measured by [11C]PiB. Neurology. 2010;74:807–15. doi: 10.1212/WNL.0b013e3181d3e3e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Morris JC, Roe CM, Grant EA, Head D, Storandt M, Goate AM, et al. Pittsburgh Compound B imaging and prediction of progression from cognitive normality to symptomatic Alzheimer disease. Arch Neurol. 2009;66:1469–75. doi: 10.1001/archneurol.2009.269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Villemagne VL, Pike KE, Darby D, Maruff P, Savage G, Ng S, et al. Abeta deposits in older non-demented individuals with cognitive decline are indicative of preclinical Alzheimer's disease. Neuropsychologia. 2008;46:1688–97. doi: 10.1016/j.neuropsychologia.2008.02.008. [DOI] [PubMed] [Google Scholar]
- 62.Chételat G, Villemagne VL, Pike KE, Ellis KA, Bourgeat P, Jones G, et al. Independent contribution of temporal b-amyloid deposition to memory decline in non-dementedelderly. Brain. 2011;134:798–807. doi: 10.1093/brain/awq383. [DOI] [PubMed] [Google Scholar]
- 63.Vemuri P, Wiste HJ, Weigand SD, Shaw LM, Trojanowski JQ, Weiner MW, et al. MRI and CSF biomarkers in normal, MCI, and AD subjects: predicting future clinical change. Neurology. 2009;73:294–301. doi: 10.1212/WNL.0b013e3181af79fb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yaffe K, Weston A, Graff-Radford NR, Satterfield S, Simonsick EM, Younkin SG, et al. Association of plasma beta-amyloid level and cognitive reserve with subsequent cognitive decline. JAMA. 2011;305:261–6. doi: 10.1001/jama.2010.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Howieson DB, Carlson NE, Moore MM, Wasserman D, Abendroth CD, Payne-Murphy J, et al. Trajectory of mild cognitive impairment onset. J Int Neuropsychol Soc. 2008;14:192–8. doi: 10.1017/S1355617708080375. [DOI] [PubMed] [Google Scholar]
- 66.Shaw LM, Vanderstichele H, Knapik-Czajka M, Clark CM, Aisen PS, Petersen RC, et al. Cerebrospinal fluid biomarker signature in Alzheimer's disease neuroimaging initiative subjects. Ann Neurol. 2009;65:403–13. doi: 10.1002/ana.21610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Fagan AM, Mintun MA, Mach RH, Lee SY, Dence CS, Shah AR, et al. Inverse relation between in vivo amyloid imaging load and cerebrospinal fluid Abeta42 in humans. Ann Neurol. 2006;59:512–9. doi: 10.1002/ana.20730. [DOI] [PubMed] [Google Scholar]
- 68.Braak H, Braak E. Neuropathological staging of Alzheimer-related changes. Acta Neuropathol. 1991;82:239–59. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- 69.Hebert LE, Scherr PA, Beckett LA, Albert MS, Pilgrim DM, Chown MJ, et al. Age-specific incidence of Alzheimer's disease in a community population. JAMA. 1995;273:1354–9. [PubMed] [Google Scholar]
- 70.Ganguli M, Dodge HH, Chen P, Belle S, DeKosky ST. Ten-year incidence of dementia in a rural elderly US community population: the MoVIES Project. Neurology. 2000;54:1109–16. doi: 10.1212/wnl.54.5.1109. [DOI] [PubMed] [Google Scholar]
- 71.Kukull WA, Higdon R, Bowen JD, McCormick WC, Teri L, Schellenberg GD, et al. Dementia and Alzheimer disease incidence: a prospective cohort study. Arch Neurol. 2002;59:1737–46. doi: 10.1001/archneur.59.11.1737. [DOI] [PubMed] [Google Scholar]