Mechanical stress-strain sensors embedded in cardiac cytoskeleton: Z disk, titin, and associated structures (original) (raw)
. Author manuscript; available in PMC: 2011 Dec 18.
Published in final edited form as: Am J Physiol Heart Circ Physiol. 2006 Apr;290(4):H1313–H1325. doi: 10.1152/ajpheart.00816.2005
Abstract
Cardiac muscle is equipped with intricate intrinsic mechanisms to regulate adaptive remodeling. Recent and extensive experimental findings powered by novel strategies for screening protein-protein interactions, improved imaging technologies, and versatile transgenic mouse methodologies reveal that Z disks and titin filaments possess unexpectedly complicated sensory and modulatory mechanisms for signal reception and transduction. These mechanisms employ molecules such as muscle-enriched LIM domain proteins, PDZ-LIM domain proteins, myozenin gene family members, titin-associated ankyrin repeat family proteins, and muscle-specific ring finger proteins, which have been identified as potential molecular sensor components. Moreover, classic transmembrane signaling processes, including mitogen-activated kinase, protein kinase C, and calcium signaling, also involve novel interactions with the Z disk/titin network. This compartmentalization of signaling complexes permits alteration of receptor-dependent transcriptional regulation by direct sensing of intrinsic stress. Newly identified mechanical stress sensors are not limited to Z-disk region and to I-band and M-band regions of titin but are also embedded in muscle-specific membrane systems such as the costamere, intercalated disks, and caveolae-like microdomains. This review summarizes current knowledge of this rapidly developing area with focus on how the heart adjusts physiological remodeling process to meet with mechanical demands and how this process fails in cardiac pathologies.
Keywords: heart, actin cytoskeleton, transmembrane signaling, proteosome
MECHANICAL SENSORS AND COMPLEXITY OF CONTROL OF CARDIAC FUNCTION
Control mechanisms regulating cardiac function reveal unexpected complexity and ingenuity in design. An excellent analogy is the dazzling complexity found under the hood of modern automobiles. Sensors wired to microchips regulating power output and economical fuel flow are widely distributed and surround an engine and power transmission system. Similarly, a cardiac myocyte is not a simple carbohydrate-consuming power generator. Apart from the classic intrinsic control mechanisms such as the autoregulation in the Frank-Starling relation and extrinsic control by baroreflex regulation, the heart possesses extensive flexibility to adjust to short- and long-term hemodynamic demands while maintaining optimal and effective energy consumption. The loss of these adaptive mechanisms promotes cardiac failure. At a cellular level, the plasticity of postnatal cardiomyocytes has been well known as cellular hypertrophy and molecular remodeling. In this way, molecular interpretation of mechanical stress-sensing machinery has attracted the attention of many cardiovascular scientists. Early research focus was to find how second messengers and signaling cascades work in cardiomyocytes and how these pathways regulate transcriptional mechanisms of a series of genes that are in some cases protooncogenes and in other cases cardiac-specific genes, such as myosin heavy and light chains, cardiac and skeletal actins, troponin subunits, and ion channels. The working hypothesis was relatively simple, which was illustrated by arrows connecting signal molecules between sarcolemma, cytoplasm, and the nucleus (Fig. 1_A_). However, scientists have been naturally realizing that passive diffusion of signal molecules cannot explain cardiomyocyte biology, and the subcelluar compartmentalization of signal machineries has become a popular research focus (Fig. 1_B_).
Fig. 1.

Mechanical stress sensing and subcellular domains in cardiomyocytes. The simple classical view of cardiac stress signaling (A) has been evolving. Mechanical sensor functions have been linked to distinctive subcellular domains (B).
Z DISKS AS THE “CENTRAL STRUCTURE” OF SARCOMERE
The Z disk is commonly illustrated as a simple structure at the lateral border of the sarcomere unit. Actin-containing thin filaments from neighboring sarcomeres overlap at the Z disk and are cross-linked by tight interactions with α-actinin. However, an alternative view places Z disks at the center of sarcomeric structure. Indeed, in the early steps of myofibrillogenesis, Z-disk precursors (Z bodies) form as a patchy α-actinin-rich electrically dense body distributed just beneath the plasma membrane (102, 110). These Z bodies are initially connected with stress fiber-like structures that contain actin and nonmuscle myosin similarly to adhesion junctions found in nonmyocytes. This myofilament antecedent-bridging Z body matures to a striated structure consisting of three filamentous components: thin filaments with actin and actin-binding proteins, thick filaments with oligomerized muscle-type myosin, and the giant titin molecule (22, 51). In addition, intermediate filaments (predominantly desmin) circumscribe the myofilaments densely at the Z disk in cardiomyocytes (22). Intermediate filaments intermingle between the laterally aligned myofilaments so that Z disks line up and form a support for synchronized and cost-effective contraction and relaxation between muscle fibers. Intermediate filaments also link Z disks to desmosomes, focal adhesion junctions, and the nucleus.
Classically, α-actinin was considered an exclusive component of Z disk embedded tightly inside of its lattice structure; however, scores of recent studies have provided evidence of an unpredicted complexity and wealth of Z disk-associated molecules in cardiomyocytes (Fig. 2). It is now clear that the Z disk is not a simple mechanical joint but an as yet incompletely understood protein-rich functional structure. An important issue is that it is apparent in some cases there is poorly documented use of the term “Z-disk localization” to characterize protein distribution. The widely employed strategy of double immunostaining with α-actinin or vinculin as a reference protein under epifluorescent and confocal microscopy does not provide sufficient spatial resolution to determine real molecular compartmentalization. Moreover, computer-aided imaging and postprocessing of microscopic images may be misleading.
Fig. 2.

Cardiac Z disk/titin cytoskeletal structure-associated proteins. An increasing number of molecules have been identified on or in the vicinity of cardiac Z disk/titin cytoskeleton. Many of these proteins have been linked to intrinsic mechanical sensor sensor/signal modulator functions. See text for details. MYOZ2, myozenin 2 (carsarin 1); Cn, calcineurin; PDZ-3LIM, one-PDZ and three-LIM domain protein; PDZ-1LIM, one-PDZ and one-LIM domain protein; MLP/CRP3, muscle-specific LIM protein/cysteine-rich protein 3; FHL2, four-and-a-half LIM protein 2; MAPRs, muscle ankyrin repeat proteins; MURFs, muscle-specific ring-finger proteins.
The accumulation of signaling-related proteins at Z disks and their physiological significance were extensively and thoroughly reviewed in a recent article by Pyle and Solaro (101). The following section extends this information with focus on LIM proteins enriched in this subcellular compartment.
LIM PROTEINS SEGREGATING AT Z DISKS: POTENTIAL STRESS SENSOR MEDIATORS
Cytoplasmic LIM-Only Proteins
Proteins interacting with or localizing in close proximity to cardiac Z disk include a subset of adaptor molecules that have putative protein binding motifs. One such protein is muscle-specific LIM protein (MLP, i.e., CSPR3), which was originally identified by Caroni’s laboratory as a skeletal muscle gene expressing coincidently with the appearance of differentiated skeletal muscle, downregulated in the adult skeletal muscle, and reinduced after surgical denervation and was a candidate molecule as a regulator of myogenic differentiation (5). Subsequent studies identified that MLP is highly expressed in the developing and adult myocardium. An MLP-null mouse demonstrated severe cardiac dysfunction and histological changes closely resembling those in human dilated cardiomyopathy (6). MLP is also known as the cysteine-rich protein 3 (CRP3) because it was independently found as a member of a closely related CRP protein family that had been studied by Beckerle’s laboratory (104). The prototype CRP1 was found localized at adhesion plaques of chick embryo fibroblasts as a protein associated with F-actin and zyxin, another LIM protein. Members of the CRP gene family [CRP1, CRP2, MLP/CRP3, and HLP (128)] have common domain structures with two LIM domains arrayed in tandem. The LIM domain, which derived its name from three homeo-domain proteins (Caenorhabditis elegans Lin-11, rat Isl-1, and C. elegans Mec-3) and has been recognized in a variety of cytoplasmic and nuclear functional molecules, defines a double zinc finger structure containing a characteristic cysteine-rich seqence, (CysX2CysX16–23HisX2CysX2CysX2CysX16–23CysX2–3) with about 50 amino acids; the LIM domain has been known as a potent protein-protein interaction motif (10, 26).
Human MLP/CRP3 (CSRP3) mutations have been linked to the pathogenesis of cardiomyopathy. We first identified the Trp4Arg mutation in a subset of European dilated cardiomyopathy patient population (69). Subsequently, Leu44Pro, Ser54Arg/Glu55Gly, and Cys58Gly amino acid changes were found in unrelated patients with familial hypertrophic cardiomyopathy (49). Furthermore, a Lys69Arg mutation was identified in an infant with an early-onset dilated cardiomyopathy (90). Among these amino acid changes, protein interaction analyses indicated that Trp4Arg, Lys69Arg, and Cys58Gly decrease or abolish MLP/CRP3 interactions with T-cap/telethonin (69), α-actinin-2 (49), and nebulin-related anchoring protein (N-RAP) (48), respectively (see below for more details about MLP/CRP3-interacting proteins). Because these genetic changes are heterologous, they may cause haplo-insufficiencies or dominant negative effects in cardiomyopathy patients. Along this line, a decrease in MLP/CRP3 protein level was found in end-stage failing hearts collected generally from patients with dilated and ischemic cardiomyopathy (135). The low MLP/CRP3 protein expression level in human failing hearts was found without significant changes in mRNA levels.
Several cytoskeletal proteins and signal regulators have been identified as partners associating with MLP/CRP3. Beckerle’s group originally demonstrated the direct interaction of MLP/CRP3 with zyxin and α-actinin using a protein overlay on blotted membranes (81). Experiments performed in Perriard’s laboratory, which employed a solid-phase protein-binding assay, together with dot-blot protein overlay and coimmunoprecipitation analyses, indicated that MLP/CRP3 interacts with N-RAP (38): N-RAP is a nebulin-related NH2-terminal LIM protein that also binds to actin, vinculin, and tailin (82). On the other hand, with the use of yeast two-hybrid protein interaction screening confirmed by coimmunoprecipitation and an in vitro GST-fusion protein pull-down assay, our group (69) found that MLP/CRP3 interacts with T-cap/telethonin, which caps titin filaments at the Z disk. MLP/CRP3 binds to α1-spectrin as well, as identified by a yeast two-hybrid screen and confirmed by coimmunoprecipitation and GST pull-down analyses (42). Furthermore, MLP/CRP3 interaction with several muscle basic helix-loop-helix transcriptional regulators such as myogenic differentiation antigen (MyoD), muscle regulatory factor 4 (MRF4), and myogenin was indicated by using in vitro protein-interaction assays, coimmunoprecipitaion, and a mammalian two-hybrid system (70), which is consistent with early studies showing that overexpresssion of MLP/CRP3 distributes inhomogeneously both in the cytosol and in the nucleus (4). More recently, MLP/CRP3 interaction with a calcium/calmodulin-dependent phosphatase calcineurin (Cn) was detected by MLP/CRP3 immunoblotting after immunoprecipitation of mouse ventricular lysates with an anti-Cn antibody (55). The potential significance of MLP/CRP3 binding with these molecules is discussed below.
Because MLP-null mice develop dilated cardiomyopathy with progressive heart failure (6) and, as indicated above, mutations of MLP/CRP3 (CSRP3) and downregulation of MLP/CRP3 have been found in human patients, it has been questioned how MLP/CRP3 defects induce (or are associated with) the initiation and progress of cardiomyopathy and heart failure. Intrigued by the interaction of MLP/CRP3 with various Z disk proteins and MLP/CRP3 localization in the close vicinity of Z disk (MLP/CRP3 immunostaining flanks Z disk at its edge) (6, 56), we investigated the ultrastructure and the mechanical properties of MLP-null mouse myocardium. Electric microscopy revealed strikingly disorganized and widened Z disks in the MLP-null myocardium. Physiological characterization of cardiac papillary muscle obtained from 2-wk-old MLP-null mice identified a selective defect in passive elastic properties (69), which was most obvious in the low range of strain where titin is the major source of passive stress in the myocardium (51). Furthermore, MLP-null neonatal cardiomyocytes cultured on elastic silicon membranes showed a complete loss of induction by stretch of brain natriuretic peptide (BNP) mRNA, a heart failure marker (69), whereas BNP induction by Gq-protein coupling receptor (GqPCR) agonist stimulation remained intact (69). Notably, a recent study from Wollert and Drexler’s laboratory (55) showed BNP induction was nearly completely blunted in the noninfarct zone myocardium obtained from the 6-wk post-myocardial infarct (post-MI) heterozygous MLP-null mice, suggesting defects in selective signalings to regulate some genes in the hearts with a reduced amount or null of MLP protein.
Physiological characteristics of MLP-null myocardium indicate that the absence of MLP/CRP3 induces abnormal intrinsic elastic properties of titin, perhaps via dislocating T-cap/telethonin, a common MLP/CRP3 and titin interacting protein from Z disks. Importantly, T-cap/telethonin dropout from Z disks was confirmed in a cardiac biopsy specimen obtained from a dilated cardiomyopathy patient with the Trp4Arg MLP/CRP3 mutation (69). We hypothesized that either the conformational changes of titin itself or the reduction of passive strain generated by titin and projected to hypothetical Z-disk mechanical stress sensors (presumably the MLP/CRP3-T-cap/telethonin-Z-disk domain titin macromolecular complex) results in an inability of cardiomyocytes with MLP/CRP3 defects to properly sense passive mechanical stresses. We viewed the passive mechanical stress as corresponding to the end-diastolic ventricular wall stress, as titin elastic elements are stretched at the highest level at end diastole during a cardiac cycle. Reduction of diastolic wall stress by enhancing sarcoplasmic reticulum calcium uptake indeed prevented the development of dilated cardiomyopathy phenotypes in MLP-null mice (87); this therapeutic strategy was further tested in BIO14.6 cardiomyopathic hamsters (58) and in post-MI chronic heart failure rats (62) by using a somatic gene transfer strategy. There is an associated yet distinctive possibility that MLP/CRP3 itself is a unique signal molecule and/or a site of docking of other signal molecules. This possibility is discussed below.
A major question remains: what is the molecular identity of the derailed signal pathways linked to titin-Z disk mechanical stress sensors in null MLP/CRP3 or reduced MLP/CRP3 myocardium? MLP/CRP3 itself is an obvious candidate as a signal mediator, in light of the previous finding that MLP/CRP3 interacts with MyoD, MRF4, and myogenin and induces C2C12 myoblast differentiation (70). CRP family members have a conserved potential nuclear localization sequence, and retention of CRPs in the cytoplasm by interacting with α-actinin and/or zyxin has been hypothesized (14). Of interest is that zyxin is another Z-disk vicinity-localized LIM domain protein with a nuclear export signal sequence. It is tempting to speculate that zyxin retention of MLP/CRP3 at or adjacent to the Z disk is regulated by mechanical stress. In this regard, enhanced nuclear staining of MLP was reported in a rat model of right ventricular hypertrophy exposed to surgical pulmonary hypertension (37); however, our preliminary study did not detect obvious nuclear translocation of MLP/CRP3 in the left ventricles collected from hearts chronically stressed by aortic coarctation (unpublished observations). Intriguingly, recent studies by Sussman’s laboratory demonstrated that zyxin transduces anti-apoptotic signal to the nucleus, which was activated by cGMP elevation, cooperatively with Akt in cardiomyocytes (65). Whether such zyxin-dependent signaling is involved in the pathogenesis of MLP/CRP3-defective myocardium remains to be determined. On the other hand, Wollert and Drexler’s laboratory (55) reported Cn as another direct MLP/CRP3-binding protein in the myocardium. In this study, MI surgery in heterozygous MLP-null mice induced dissociation of Cn from Z disks and blunted activation of Cn-dependent signaling. Further studies are required to more fully understand the physiological significance of this signaling pathway; e.g., the binding affinity and stoichiometric ratio of MLP-Cn interaction should be determined together with the identification of interaction domains. Cn-dependent signal can be analyzed in MLP/CRP3 complete null cardiomyocytes by applying other forms of in vivo and in vitro mechanical stresses.
Muscle PDZ-LIM Proteins
PDZ-LIM proteins have emerged as a family of candidate molecules functioning as adaptors in translating mechanical stress signals from the Z disk to the nucleus. The PDZ domain is recognized as repeated sequences of ~90 amino acids and is the most widely distributed protein-binding motif (93). Proteins with the PDZ domain accumulate largely at or near plasma membranes, yet some are distributed in the nucleus. Whereas there are PDZ domain-only proteins, a subset of PDZ domain proteins have additional protein interacting motifs. Among them, PDZ-LIM proteins constitute a gene family with two protein adaptor motifs: PDZ and LIM. Two major types of PDZ-LIM proteins have been recognized in cardiomyocytes. One type are PDZ-1LIM proteins represented by α-actinin-associated LIM domain protein (ALP, i.e., PDLIM3), which has an NH2-terminal PDZ domain and a single LIM domain at the COOH terminus. The other type constitutes the enigma gene family with one NH2-terminal PDZ domain and three tandem LIM domains on the COOH-terminal side (PDZ-3LIM proteins). The 36-kDa PDZ-1LIM protein ALP was found at Z disks in neonatal rodent cardiomyocytes and in embryonic chick cardiomyocytes and is less expressed in adult rodent hearts (56, 94). In vitro experiments demonstrated that ALP enhanced the ability of α-actinin to cross-link F-actin (94). Thus cardiac ALP protein may facilitate actin-filament anchoring to Z disks and promote their mechanical stability. Indeed, right ventricular dominant cardiomyopathy was recognized in ALP-null mice, whereas embryonic ALP expression is prominent in the trabeculae of right ventricles (94). Interestingly, although ALP binds to α-actinin (56, 125), ALP localization at Z disks is independent of its association with α-actinin, suggesting there exist other interacting molecules of ALP at Z disks (56). In fact, ALP is more readily detectable at intercalated disks in adult mouse hearts (94) in a distribution that does not overlap with the density distribution of α-actinin in cardiomyocytes. The physiological role of ALP at intercalated disks has not been fully understood yet.
Enigma (PDLIM7), which is the prototype of enigma gene family, was originally identified as a binding protein of human insulin receptor (124) and the Ret/ptc2 oncogene derived from c-Ret receptor tyrosine kinase (36), and later the association of enigma with actin filaments and its interaction with β-tropomyosin (TPM2) were identified (52). Enigma localizes at the boundary of Z disks and I bands (52). Enigma homolog (ENH, i.e., PDLIM5) is the closest gene family member of enigma and was originally identified as a protein kinase C (PKC) binding protein (72); ENH is highly expressed in cardiomyocytes and localizes at Z disks in neonatal and adult rodent cardiomyocytes (92). PKC binding to the prototype gene enigma has also been confirmed (72). Another enigma family protein, LIM domain-binding factor 3 (LDB3), was found in cardiomyocytes independently by three groups. The human and mouse sequences of LDB3 were found by Faulkner’s laboratory and named as Z band alternatively spliced PDZ-motif protein (ZASP) due to its highly variable alternative splicing (41); Chen’s laboratory (133) independently identified splicing variants of mouse homologs of LDB3 by in silico screening of LIM proteins enriched in the heart and named this gene as Cypher; Olson’s laboratory also isolated mouse sequence of LDB3 (named Oracle) during their process of differential screening of genes expressed specifically in the heart (95). LDB3 localizes at Z disks (41, 133), and LDB3 binding to α-actinin through its PDZ domain was identified (133). As is the case with other enigma gene family members, LDB3 has PKC-interacting capacity, but its isotype specificity for PKC binding is lower than that of ENH and enigma (133).
A mouse with a genetic deletion of LDB3 developed embryonic-neonatal myopathy and died prematurely, most likely due to respiratory distress (132). Notably, frequent mutations of LDB3 have been found in patients with dilated cardiomyopathy: Towbin’s laboratory screened 100 probands and found 5 distinctive amino acid changes (118), whereas Kimura’s laboratory identified an Asp626Asn mutation in 1 of 96 unrelated cardiomyopathy patients (7). Although in none of the cases was familial size sufficient to establish phenotype-genotype relationship, the Asp626Asn mutation was demonstrated to increase the affinity of LDB3 to PKC (7), suggesting a disturbance of the adaptor function of LDB3 for PKC may play a role in the pathogenesis of a subset of dilated cardiomyopathy.
TRANSMEMBRANE SIGNALING INTERMINGLED WITH Z DISK-TITIN STRESS SENSORS
MAP Kinase Signaling and Four-And-A-Half LIM Proteins
It is widely accepted that the cytoplasmic mytogen-activated protein kinase (MAP) kinase/MAP kinase kinase (MKK)/MKK kinase cascades constitute major stress (including mechanical stress)-activated signal pathways in cardiomyocytes (11, 113). This cascade demonstrates several modes of activation, including transmembrane receptor stimulation by soluble or diffusible ligands, cell engagement to the extracellular matrix, and cell-cell communication through the outer membrane protein recognition or gap junctions. Our previous study documented phase-dependent coordinated activations of MAP kinase cascades in left ventricles stressed by pressure overload (127). However, how cytosolic signal molecules move to the nucleus upon activation and whether there are mechanisms to modulate this signaling transition by mechanical stresses remains largely elusive.
Recent studies by Molkentin’s laboratory provided the first evidence that a titin/Z disk-associated scaffolding protein modulates the cardiac MAP kinase cascade (99) (Fig. 3). Four-and-a-half LIM (FHL) proteins constitute a gene family with five identified members: FHL1, FHL2, FHL3, FHL4, and ACT. Among these, FHL2 [also named as downregulated in rhabdomyosarcoma LIM protein (DRAL) or skeletal muscle LIM protein 3 (SLIM3)] is most prominently expressed in cardiomyocytes, whereas FHL1 and FHL3 are more abundant in skeletal muscles (21, 71). Immunological signals of FHL2 in adult and neonatal cardiomyocytes demonstrated their localization at Z disks (99) and more precisely at the I-band region of sarcomeres in the proximity of Z disks (the titin N2B region of the I band) (75). FHL2 was identified as an ERK2-binding molecule in cardiomyocytes (99); ERK2 binding to FHL1 and FHL3 was also confirmed (99). The coimmunoprecipitation analysis by Molkentin’s laboratory showed FHL2 had a higher affinity to the phosphorylated and active form of ERK2 compared with the nonphosphorylated form of this molecule. Adenovirus-mediated overexpression of FHL2 in cardiomyocytes suppressed nuclear translocation of the phosphorylated ERK2 and inhibited ERK-mediated transcriptional activations, suggesting that FHL2 may work as a cytoplasmic tether of activated ERK2. Alternatively, several studies in other cell types report that FHL2 shuttles between the cytoplasm and the nucleus and regulates transcription [e.g., FHL2 may antagonize part of rhomegakaryotic acute leukemia (MAL)/myocardin-related transcriptional factor (MRTF) signal transduction (98); FHL2 on the other cooperates with CBP/p300 and β-catenin (74)], which hints at the possibility that FHL2 and activated ERK2 cotranslocate to the nucleus and regulate gene expression. In addition, FHL2 has been reported to interact with α7β1-integrin (109), an abundant integrin isotype both in skeletal and cardiac muscles (103). FHL2 is also found at the M band (75) so that there may be multiple cytoplamsmic FHL2 pools, including sarcomere I and M bands and sarcolemmal adhesion plaques. FHL2-null mice were independently generated and characterized by two laboratories (20, 71). Consistent with the hypothesis that FHL2 negatively regulates ERK-mediated signaling, Williams’ laboratory found 7-day chronic infusion of isoproterenol augmented cardiac hypertrophy in FHL2-null mice (71), whereas left ventricular hypertrophy induced by surgical aortic coarctation in FHL2-null mice generated by Chen’s laboratory was indistinguishable from wild-type control animals (20). This phenotypic difference may be due to the complexity and redundancy of signal cascades activated by pressure overload (57, 59).
Fig. 3.
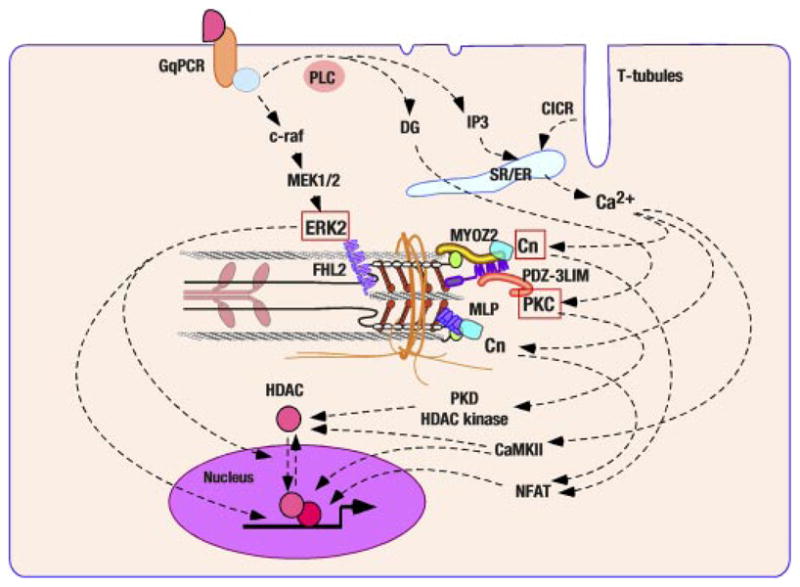
Z disk/I band titin associated signal cascades. Several nodal molecules of transmembrane signaling have been mapped on Z disks and I band titin. Mitogen-activated protein (MAP) kinase cascade interact with I-band titin through ERK2-FHL2 association. Calcium signal activates calcineurin (Cn) and subsequently nuclear factor of activated T cells (NFAT). The interaction of Cn with two distinctive Z disk proteins has been revealed (MYZO2 and MLP/CRP3). A subset of PKC isotypes localize at Z disk or its vicinity and PDZ-3LIM proteins (enigma, enigma homolog, and LIM domain-binding factor 3) may be critical for their sequestration.
PKC and Emerging Z Disk PKC Adaptors
Two “novel”PKC (nPKC) isotypes, nPKCδ and nPKCε, both of which are highly expressed in cardiomyocytes, translocate from the cytoplasm to the cardiac particulate fraction by GqPCR agonist (such as phenylephrine or endothelin-1) stimulations (23). Both acute (96) and chronic pressure overload (28) have also shown to induce PKCε translocation to the particulate fraction in rodent myocardium. Furthermore, earlier studies by Yazaki’s laboratory demonstrated that PKC inhibition suppressed stretch-induced hypertrophy in cultured neonatal cardiomyocytes and that PKC-dependent c-Raf activation was at least partially responsible for this direct mechanical stress-induced in vitro hypertrophic responses (126). Collectively, PKCs (nPKC isotypes, in particular) have been suggested as stress signal transducers in cardiomyocytes. A recent study from Solaro’s laboratory also pointed out that PKCs critically regulate actin thin filaments through its functional interaction with an actin-capping protein (CapZ), which localizes at Z disks (100).
Multiple immunohistological studies using isotype-specific antibodies have convincingly documented that PKCε rapidly moves from the cytosol to Z disks accompanied by its activation (28–30, 61). Hereby, PKC isotype-selective adaptor proteins, which are collectively named as receptors for activated C-kinase (RACKs), obviously play significant roles to regulate the activations and subcellular compartmentalization of distinctive PKC isotypes (31). A Golgi membrane complex-associated protein β′-COP has been proposed as a PKCε-specific RACK, and its cross-striated pattern of localization was reported in cardiomyocytes (25); however, Z-disk localization of β′-COP has not been established nor has the direct interaction of β′-COP with Z disk proteins been known. In this regard, as mentioned above, enigma family PDZ-3LIM proteins, which directly bind to PKCs with some isotype selectivities, localize apparently at Z disks (Fig. 3). Further studies are required to determine whether enigma family PDZ-3LIM proteins are essential components of PKC localization at Z disks and to test whether this molecular complex plays a critical role in mechanical stress sensing.
Calcium-Activated Signaling Associating With Z Disks
An unequivocally significant role of calcium-Cn signaling in cardiac (perhaps largely pathological) hypertrophy has been established (119, 120). The recent identification of calsarcins (also known as myozenins) as a novel Cn-binding protein family has introduced a novel molecular mechanism in which calcium-dependent signaling occurs or is modulated through Z-disk interaction (Fig. 3). Calsarcin1 [myozenin 2 (MYOZ2)] was found as a gene expressed uniquely in the adult heart and slow skeletal muscles among the members of calsarcin (myozenin) gene family, which were identified as Cn-binding proteins using a yeast two-hybrid screening by Olson’s laboratory (47). Z-disk localization of MYOZ2 was further demonstrated (47). Studies by Olson’s laboratory and others (40, 46, 47, 114) identified calsarcins (myozenins) bind to α-actinin (ACTN2), γ-filamin (FLNC), telethonin/T-cap (TCAP), and LDB-3 (ZASP/Cypher/Oracle); the skeletal muscle type of calsarcin calsarcin 2 [myozenin 1 (MYOZ1)] was identified by Faulkner et al. (40) as a filamin-, actinin-, and telethonin-binding protein at the Z disk (FATZ) and also identified by Takada et al. (114) as a filamin- and actinin-binding Z-disk protein myozenin. Accordingly, MYOZ2 was proposed as a cardiac Cn-adaptor protein localized at Z disks. Indeed, high-level accumulation of cytoplasmic Cn at Z disks is documented (47). In this connection, Z-disk association of an immediate downstream transcriptional factor of Cn, nuclear factor of activated T cells (NFAT), was also suggested, although this study utilized a strategy of in vitro enhanced green fluorescence protein-tagged NFAT expression in dissociated skeletal muscle fibers (80).
MYOZ2-null mouse was generated by Olson’s laboratory, and exaggerated left ventricular hypertrophy was found in this mutant animal exposed to chronic aortic coarctation (45). Diminished Cn-dependent transcriptional regulation was shown in stressed MYOZ2-null mouse hearts. These data suggested that the loss of MYOZ2 per se or Z-disk abnormalities induced by MYOZ2 ablation enhanced calcium-Cn-dependent signaling. Olson’s group also indicated that Z disks may serve as a subcellular compartment for cytoplasmic Cn sequestration. Intriguingly, the Z-disk localization of Cn was retained in the MYOZ2-null myocardium (45), suggesting other Z disk proteins also interact with Cn. As described above, MLP/CRP3 may be one such Cn-binding molecule at or in the vicinity of Z disks (55). The essential requirement of MLP/CRP3 for Cn sequestration at Z disks has to be established utilizing MLP/CRP3-null cardiomyocytes.
TITIN ELASTIC DOMAINS AND INTERACTING POTENTIAL SIGNAL MEDIATORS
The I-band region of titin is highly flexible and has unique elemental structures (such as N2B, N2A, and PEVK domains). As mentioned previously, there is a possibility that mechanical stretch defects found in MLP-null myocardium are exclusively due to the alternation of the intrinsic elastic properties of titin, which may directly be sensed by its interacting signal molecules. In this regard, a series of molecules, such as muscle ankyrin repeat family proteins (MARPs) (85), myopalladin (13), and p94 calpain (112), have been identified as unique I-band titin-associating proteins. More experiments are required to test the attractive hypothesis that titin itself functions as a mechanical sensor together with its interacting molecules at coiled domains. Recent papers (51, 86) have reviewed this topic.
“THE OTHER END OF TITIN” STORY: M LINE AND STRESS SENSORS FOR NUCLEAR REGULATION AND PROTEIN TURNOVER
The M-band, which is the central structure of thick filament and where the COOH end of titin anchors, is recognized as another potential molecular center of mechanical stress sensing. It appears likely that both transcriptional regulation and ubiquitination-dependent protein degradation are involved in this stress sensory machinery. M-band titin (the COOH terminus of titin) has a putative serine-threonine kinase domain that overlaps with the COOH terminus of another titin molecule that symmetrically extends its NH2 end to the other side of a sarcomere (51). T-cap, which localizes at Z disk, was proposed as a substrate of titin kinase during myofibrillar formation (83); however, because of the physical distance between the M band and Z disk (~0.9 μm at the slack length of the cardiac sarcomere), the physiological significance of titin kinase activity in mature myofilaments had been unclear. Yet in search of binding molecules to the COOH terminus of titin, Gautel’s group discovered a novel molecular complex at the M band (76). This complex consists of a zinc-finger protein (nbr1), which binds directly to M-band titin, and p62 (another zinc-finger protein). These proteins were identified as novel substrates of titin kinase (a recombinant kinase fragment with a mutation to constitutively activate kinase domain was used in this study, the _K_m of p62 was 10 times less than those of nbr1 and T-cap in vitro, so that p62 is a less likely substrate of titin kinase) (76). It is intriguing that one of the titin mutations, Arg279Trp, found in patients with hereditary myopathy, reduced titin interaction with nbr1 significantly (76). The nbr1 and p62 interact with each other and both have the ubiquitin-associated (UBA) domain. Accordingly, Gautel and collaborators (76) further identified the muscle-specific ring finger-2 (MURF-2) as another component of the titin COOH-terminal multimolecular complex (titin COOH-terminal kinase domain-nbr1-p62-MURF-2) (Fig. 4). On the other hand, an independent yeast two-hybrid screening by Labeit’s group (19) has determined MURF-1, another member of highly homologous MURF family genes, directly binds to the titin COOH-terminal kinase domain and found that MURF-2 and MURF-3 interact indirectly with this domain (through hetero-oligomerization of MURF family proteins).
Fig. 4.
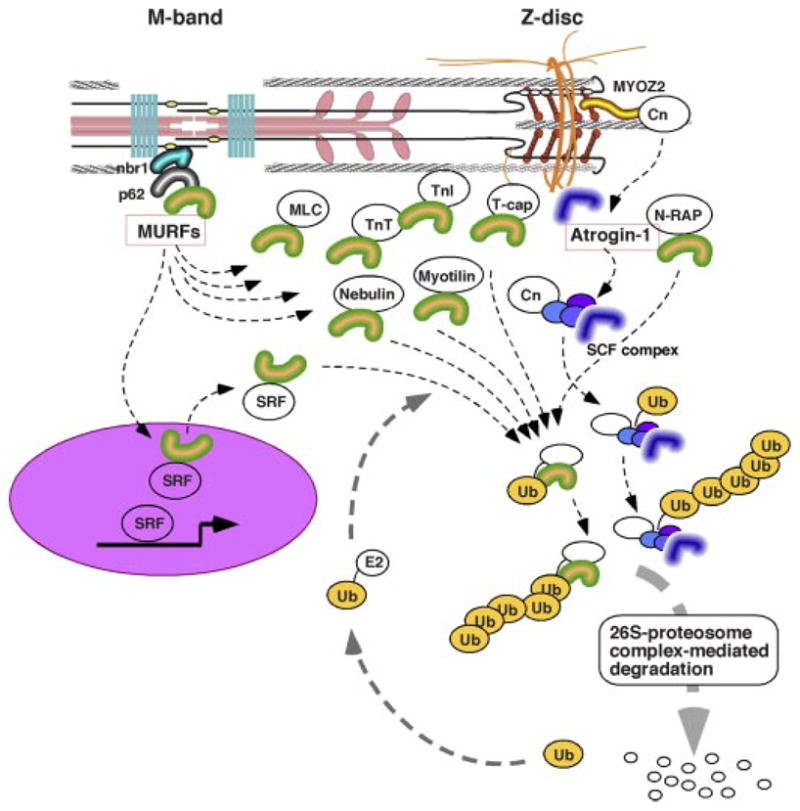
Sarcomere protein turnover and Z disk/M-band-associated ubiquitin (Ub) ligase complexes. Ubiqutination-dependent protein turnover may critically regulate turnover of cardiac proteins. Selective localizations of E3 ubiquitin ligase complex components have been identified: MURFs at M-band titin and atrogin-1 at Z disks. In addition to their direct interaction with sarocomere proteins, M-band MURF may regulate the SRF transcriptional factor, and atrogin-1 binds and induces degradation of Cn at Z disk. Collectively, sarcomere-associated ubiquitine ligase components may serve as critical regulators of both cardiac protein synthesis and degradation.
MURF-1 null mice generated by Glass’ laboratory showed significant resistance to denervation-induced skeletal muscle atrophy (16), which strongly indicates that cardiac MURFs may also control cardiac mass through a ubiquitination-dependent protein degradation pathway. In cardiomyocytes, both MURF1 and MURF2 distribute in the cytoplasm and the nucleus (76, 84), and indeed yeast two-hybrid screening of muscle cDNA libraries performed by several laboratories have identified multiple MURF-interacting proteins in both cytoplasmic and nuclear locations, which include the serum response factor (SRF) (76), glucocorticoid modulatory element binding protein-1 (84), PKC-adaptor RACK1 (8), cardiac and skeletal troponin I (67, 121), troponin T, nebulin, N-RAP, myosin light chain, myotilin, T-cap, and a series of enzymes related to energy metabolism (121) (Fig. 4).
With the use of beating-arrested cultured neonatal cardiomyocytes treated with either a cardioplegic medium containing 2,3-butanedione monoxime or an L-type calcium channel blocker verapamil, Gautel’s laboratory (76) provided experimental data supporting their hypothesis that constant mechanical stress on myofilaments given by spontaneous rhythmic beating maintains the stability of M-band titin-nbr1-p62-MURF2 protein complex, holding MURF2 in the cytoplasm. Gautel and his colleagues further propose that cytoplasmic retention of MURF2 is important to maintain the nuclear level of SRF, which binds to MURF2 and may be targeted to 26S proteosomes by ubiquitination. Yet more studies are needed to establish the in vivo significance of this emerging and attractive hypothesis related to the M-band titin-associated mechanical stress-sensory system. Moreover, a recent study by Patterson’s laboratory identified that Atrogin-1 [muscle atrophy F-box protein (FBXO32)] localizes at cardiac Z disks and regulates the protein level of Cn (78), providing a new mechanistic view that multiple active protein degradation systems are associated with the cardiac cytoskeleton and involved in on-demand regulation of assembled sarcomere numbers that determines the size of cardiomyocytes.
COSTAMERE: “FOOT STRUCTURE” LINKING Z DISK CYTOSKELETON TO SARCOLEMMA AND EXTRACELLULAR MATRIX
Costameres are riblike perisarcolemmal subcellular structures aligned predominantly with Z disks. They function as a striated muscle adhesion plaque to extracellular matrix (ECM), similarly to the focal adhesions in nonmuscle cell types. Vinculin is historically a signature marker protein of this structure. Costameres have been thought as the lateral force transmission foot structure connected to ECM proteins and also to the neighboring cardiomyocytes. The general recognition is that this lateral force transmission is an important element of the coordinated contraction within cardiac muscle layers. However, as opposed to this outward function, there has emerged a new concept that the costamere also plays an important role as an inward lateral mechanical sensor of cardiomyocytes (39, 108). Two distinct macromolecular protein complexes, the integrin complex and the dystrophin-glycoprotein complex (DGC), may play major regulatory roles at costameres.
Signal transduciton through an integrin complex has been extensively studied in nonmuscle cells at focal adhesions (89), as well as in cardiomyocytes (103). The cytoplasmic domain of integrin provides critical anchors for the actin cytoskeleton and for a multi-molecular molecular complex formed at the costamere, which includes nonreceptor type tyrosine kinases such as focal adhesion kinase (FAK), src-family tyrosin kinases, and integrin-linked protein kinase (ILK), cytoskeletal proteins such as talin and vinculin, and signal mediators such as small Mr GTP-binding proteins (rac, rho, Cdc42) (39, 89, 103, 108). It is well recognized that the integrin complex is important for heart formation and for maintaining its physiological functions; e.g., cardiac-restricted deletion of β1-integrin resulted in dilated cardiomyopathy (111), and mice with heterologous vinculin deletion did not tolerate an increase in left ventricular afterload (129). Although FAK has been thought of as a key signaling molecule associated with integrins in cardiomyocytes (103), ILK also constitutes a potentially significant molecular module associated with the cytoplasmic domain of integrin (122): ILK is known to phosphorylate the Ser473 residue of Akt and to regulate its enzymatic activity. ILK also forms a ternary complex with PINCH1, a five LIM domain protein, and CH-ILKBP, a parvin family gene, at the costamere. The genetic ablation of either ILK, PINCH1, or parvin in Drosophila and C. elegans resulted in the disruption of integrin-actin linkage and causes defective attachment of cells to ECM (122), suggesting a critical role of ILK at adhesion plaques in functional linkage of cardiac myocytes to the ECM. Although ventricular specific ablation of PINCH1 did not induce obvious heart defects (79), it would be interesting to challenge PINCH1-null or ILK-ablated cardiomyocytes with mechanical stresses.
The potential importance of ILK was further emphasized by a recent study conducted by Srivastava’s group (15). In this study, thymosin β4 (TMSB4X), a 43-amino acid ubiquitous actin-binding protein that buffers ATP-actin monomers in the process of actin filament polymerizing (32), was identified as another independent binding partner of ILK and PINCH. Externally applied thymosin β4 was taken up via an unknown molecular mechanism, enhanced migration capability and augmented the survival of cultured cardiomyocytes (15), perhaps through the activation of the Akt signal cascade (113). Moreover, systemic infusion of thymosin β4 immediately after coronary artery ligation reduced the size of myocardial infarction in mice (15). Thus it is tempting to speculate that ILK-Akt signal constitutes a part of integrin-dependent mechanical stress sensory function in cardiomyocytes and promotes cell survival. Nevertheless, the activation of ILK-Akt signaling by mechanical stress remains to be characterized, whereas rapid activation of FAK has been extensively analyzed and reported in vivo with acute pressure overload in rat hearts (43) and with cyclical stretch of isolated rat neonatal cardiomyocytes in vitro (115).
Another notable molecule interacting with integrin at constameres is a striated muscle-specific protein melusin [integrin-β1-binding protein 2 (ITGB1BP2)], which was isolated by yeast two-hybrid screening of a neonatal rat heart library with the cytoplasmic domain of β1-integrin as a bait (18). Melusin has two cysteine-rich zinc-binding domains, potential SH2, SH3-binding domains, and a calsequestrin-like COOH-terminal acidic amino acid cluster. Genetic ablation analysis of melusin in mice resulted in an attenuated hypertrophic response in hearts exposed to 7-day pressure overload with subsequent chronic development of chamber dilation (17). In contrast, cardiac melusin transgenic mice showed a moderate level of cardiac hypertrophy and a resistance to the induction of ventricular dilation after long-term pressure overload (27). In melusin-null mice, surgical coarctation of the aorta failed an acute (within 10 min) induction of glycogen synthase kinase 3β (GSK3β) Ser9 phosphorylation, and Akt Ser473 phosphorylation, whereas ERK1/2 and p38 activations remained sensitive (17). This result indicates that melusin may selectively link the integrin molecular complex to the Akt-GSKβ signal cascade, which not only is one of the major regulatory pathways of cardiac hypertrophy (53) but also plays a significant role in cardiomyocyte survival (63). Further clarification is needed to determine whether melusin constitutes an integrin-mediated pure mechanical stress sensor of alterations in ECM engagement.
An alternative explanation of the data from the melusin knockout and transgenic mouse studies could be that melusin plays a role in the intergrin-dependent enhancement of trans-membrane receptor signaling (such as β-adrenergic receptor signaling). Surgical coarctation of the aorta stimulates cardiomyocytes in multiple ways. For example, within a few minutes of surgical aortic coarctation in rodents, myocardial cAMP content increases and cAMP-dependent kinase (PKA) activity increases (68), which is partly due to the reflex of sympathetic efferent limb and the acute release of locally stored catecholamines (33). It has been shown that β-adrenergic agonists inhibit GSK3β activities in adipocytes (91) and this effect has been confirmed in cardiomyocytes (54). Furthermore, transgenic activation of GSK3β suppressed in vivo β-adrenergic cardiac hypertrophy (3). The molecular link between β-adrenergic receptor and Akt-GSK3β signal has not been fully established; however, β2-adrenergic receptor and both G protein-dependent and G protein-independent signal cascades may be involved (131). As the proximal interaction between integrins and adrenergic receptors has been revealed (103), it is tempting to speculate that integrin-melusin complex stabilizes and augments β-adrenergic receptor-activated Akt-GSK3β signaling in mechanically stressed hearts.
In addition to the integrin-complex, DGC constitutes a mechanistic element of the cardiac costamere (39, 77). A physiological role of DGC has been emphasized by the development of marked cardiomyopathy in Duchenne and Baker muscular dystrophy patients as well as in animal models of muscular dystrophies (the mdx mouse, which is a genetic model of Duchenne and Baker muscular dystrophy; δ-sarcoglycan-deficient BIO14.6/TO2 hamster strains, and mouse strains with genetic ablations of sarcoglycan genes, which are genetic models of Limb-Girdle Muscular Dystrophies) (2, 35). It is well accepted that DGC complex is an essential component to stabilize the sacrolemma against physical stress (24); however, the inward mechanical stress sensor function of DGC has not been characterized sufficiently (77). In this regard, mislocalization and dysregulation of NO synthase (NOS) has been linked to dystrophin defects in skeletal muscle cells, and the association of endothelial type NOS (eNOS) with δ-sarcoglycan and γ-sarcoglycan has been identified in cardiomyocytes (77). An eNOS abnormality in response to mechanical stress may be a part of pathogenesis of dystrophic cardiomyocytes, as indicated by a study in which adult cardiomyocytes that were stretched in vitro demonstrated a nitric oxide-dependent calcium mobilization (97). Interestingly, there is a lower amount of DGC accumulation in a less well-studied foot structure along the sarcolemma that overlies M bands (M-band sarcolemma) (39). From the potential existence of an M-band stress sensor described above, the M-band sarcolemma and a role of the M-band DSG need further investigation.
INTERCALATED DISK AND STRETCH SENSING
Intercardiomyocyte adhesion structures formed at intercalated disk may also be functional as mechanical stress sensors. “Fascia adherens junctions” are where _N_-cadherin forms cell-cell contacts and scaffolds multi-molecular complexes anchoring actin cytoskeleton, whereas “desmosomes” are constituted with desmosomal cadherins, including desmogleins and desmocollins, as intercellular adaptor molecules (22). Desmosomes are internally linked to desmin, an intermediate filament. Both fascia adherens and desmosomes enriched at intercalated disks could serve as ideal structures to sense and process mechanical stress projected to the longitudinal direction of individual cardiomyocytes. Intriguingly some of the molecules constituting these intercellular adhesion structures have been linked to cardiac diseases such as Naxos disease and Carvajal syndrome, as discussed in detail in recent reviews (22, 106).
So what are the identities of molecules forming mechanical stress sensors potentially at intercalated disks? One such candidate is a prominent MLP-binding protein N-RAP with an NH2-terminal LIM domain and a COOH-terminal actin-binding domain, which is enriched at the intercalated disks (38) and perhaps associated with actin-binding molecular components at the fascia adherens (130). N-RAP expression is highly upregulated and found abnormally distributed in MLP-null myocardium (38). As stated previously, we proposed MLP/CRP3 constitutes a mechanical stress sensor mechanism in cardiomyocytes together with T-cap/telethonin at Z disks; however, N-RAP-MLP or N-RAP-MLP-telethonin/T-captitin macromolecular complexes formed at intercalated disks may also serve as a stress sensor.
In addition, sequestration of multiple transcriptional factors at the intercalated disk is of interest from a point of view of mechanical stress sensing. Clearly cadherins serve as a prominent cytoplasmic docking site of the β-catenin transcriptional regulator (22). The fascia adherens sink may cooperate with canonical Wnt signaling and adjust the stabilization of cytoplasmic β-catenin pool. Zonula occludens-1 (ZO-1, i.e., TJP1), one of the family members of PDZ proteins, which was originally identified as a tight-junction component, is also enriched at intercalated disks and may sequester several transcriptional regulators (50). ZO-1 interacts with α-catenin and β-catenin, as well as the COOH tail of a gap junction protein connexin 43 (Cx43: GJA1) (50). The formation of cardiac ZO-1 complex is affected by ionic conditions (calcium and pH) (34, 123) and regulated by other signaling molecules (50) including c-src (116). A recent study showed that ZO-1 binds to ZONAB (a ZO-1 associated Y-box transcriptional factor) (12), and ZONAB is a part of a density-dependent regulatory system of cell proliferation (12, 44). Cardiomyocytes may utilize such an adhesion molecule-dependent intercellular communication system to sense mechanical stress.
“Gap junctions” constitute the third intercellular adhesion structure at the intercalated disks, along with “fascia adherens junctions” and “desmosomes” (22). This structure demonstrates remarkable high plasticity (106). Extremely rapid protein turnover rate of connexins, a family of gap junction channel proteins, represented by Cx43 has been revealed, and studies from Saffitz and colleagues (134) showed that pulsative mechanical stretch induced marked upregulation of Cx43 in cultured rat neonatal cardiomyocytes. An obvious outcome of density changes in gap junction is the modulation of intercellular electrical conduction velocity, which is directly linked to arrhythmogenesis (64). However, as connexins form diffusion pores that pass ~1,000-Da molecules relatively nonselectively, which may include ions and second messengers, the mechanical stress-induced quantitative changes (e.g., connexin induction) and qualitative changes (e.g., phosphorylation of Cx43 by interacting kinases) in gap junction proteins may work as a not yet well-explored stress sensory system for caridomyocytes.
SARCOLEMMAL RECEPTORS AND F-ACTIN ASSEMBLY AS DIRECT MECHANICAL SENSOR MACHINERIES
Recent studies have revealed a startling molecular mechanism of mechanical stress sensing in membrane receptors. Komuro’s group (136) reported an agonist-independent direct activation of angiotensin II type 1 receptor (AT1R) in statically 20% stretched HEK293 cells that overexpressed AT1R. The stretch mobilized receptor-coupling Gαq11 to the cytoplasm, enhanced the production of inositol phosphates, and activated ERK signaling. Compelling evidence using AT1R mutant without angiotensin II binding activity and murine cardiomyocytes in which the angiotensingen gene was ablated supported the notion that stretch-induced AT1R activation is ligand independent (136). This study may motivate us to revisit a previously proposed working hypothesis of the mechanical stress-induced activation of cardiac AT1R (mechanical stretch induces the release of angiotensin II from cardiomyocytes and activates cellular hypertrophy) (105).
Although Komuro’s study (136) did not identify the molecular mechanism for AT1R to sense mechanical stretch, selective association of AT1R with caveolae or lipid rafts may constitute a unique sarcolemmal mechanical stress sensor mechanism. Transactivation of tyrosine kinase receptors by AT1R at caveolae-like microdomains has been shown in vascular smooth muscle cells (117). Activated AT1R is transported to the caveolae-like microdomain and transactivates epidermal growth factor receptors (EGFRs), which is mediated by the assembly of microtubles accompanied by the accumulation of rac1 small _M_r GTPase at the caveolae-like microdomain in vascular smooth muscle cells (137). In cardiomyocytes, ligand-dependent trasactivation of EGFR by AT1R has been partly explained by the activation of metalloprotease12 (ADAM12) and the subsequent cleavage of membrane-anchored heparin-binding EGF (HB-EGF) (9). However, whether the AT1R-EGFRs transactivation can also be explained by the direct interaction of these receptor molecules at caveolae-like microdomain, particularly in the setting of mechanical stress exposure, has not been tested.
A related, but distinct, mechanical stress sensory system has been indicated in the caveolae-like microdomain of cardiomyocytes by Brown’s laboratory (66). In this study, static stretch of cultured neonatal cardiomyocytes segregated rac1 and rhoA small _M_r GTPases to the caveolae-like microdomain, and subsequent actin cytoskeletal rearrangement was essential for the nuclear translocation of ERK1/2 and the induction of cardiomyocyte hypertrophy. These findings are consistent with earlier observations by Komuro and Yazaki’s laboratory in which two rho inactivators, a dominant negative mutant of rhoA and a rho GDP dissociation inhibitor, suppressed mechanical stretch-induced hypertrophy in cultured neonatal rat cardiomyocytes (1). In this regard, the rhoA-rho kinase signal cascade is a potent regulator of cardiomyocyte hypertrophy (60, 107). Mechanical stretch-induced rac1 or rhoA-regulated actin cytoskeletal rearrangement may activate another transcriptional regulatory system. Previously, Treisman’s group discovered that cytoplasmic actin assembly/disassembly that is regulated by rho GTPase controls one of MADS-box transcriptional factor, SRF, and that a myocardin-related SRF coactivator MAL is involved in this process in NIH3T3 cells (88). Olson’s laboratory has extended their analysis of this signal pathway and found that a muscle-specific actin-binding protein named striated muscle activator of rho signaling (STARS) cooperates with rho and regulates MAL/MRTFs in fibroblasts as well as in cultured rat neonatal cardiomyocytes (73). Notably, although STARS significantly affects MAL/MRTFs activities, STARS is not an upstream regulator of rho (73). Taken together, the assembly/disassembly of the sarcolemmal actin cytoskeleton regulated by small _M_r GTP-binding protein (rho, rac, cdc42), and STARS may serve as another not yet well-analyzed molecular mechanism of mechanical stress sensing in cardiomyocytes (Fig. 5).
Fig. 5.
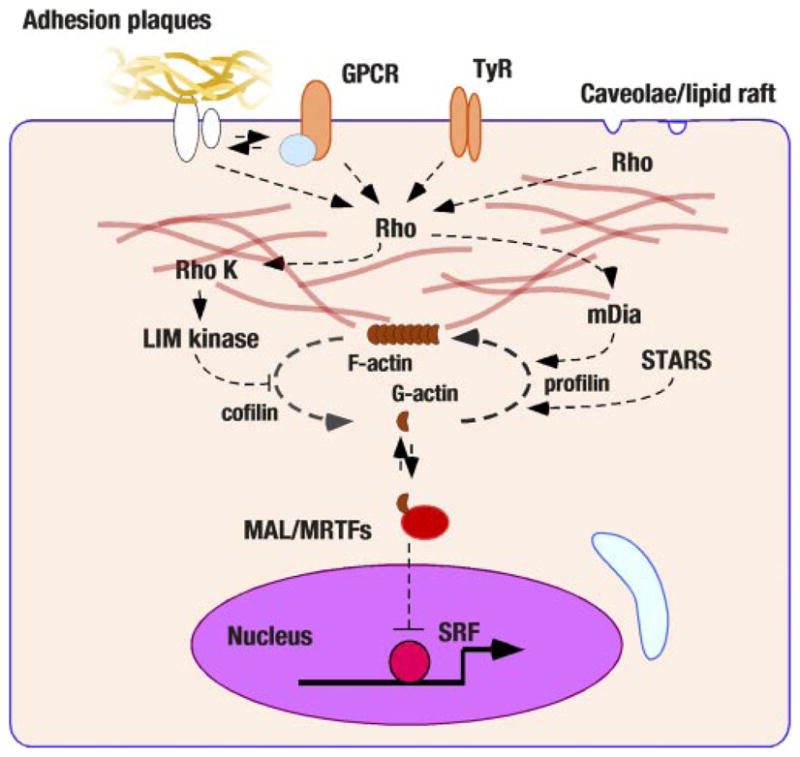
Sarcolemmal actin assembly and nuclear signaling. Recent studies have revealed that cytoplasmic actin assembly/disassembly may regulate transcriptional factors. The rho GTPase is a nodal molecule of this pathway and has been linked to various sarcolemmal activators (G protein coupling receptors, GPCR; tyrosine-kinase receptors, TyR; adhesion plaque complexes; and the caveolae-like structure). Cytoplasmic actin assembly has been found to regulate SRF transcriptional activities through G-actin association with megakaryotic acute leukemia (MAL)/myocardin-related transcriptional factors (MRTF) proteins. STARS (striated muscle activator of rho signaling) also regulates actin disassembly.
CONCLUSION
Cardiomyocytes are specially differentiated cells for rhythmic power generation with a wide ranging capacity for physiological adaptation. The sarcomere structure is made of three filaments (thin, thick, and titin elastic filaments) marked with Z disk, I band, A band, and M band. It is increasingly clear that the unique striated appearance of cardiac muscle cells is not a simple lattice structure but is equipped with sensors and well-organized signal regulatory systems. The molecular machineries discussed in this article are perhaps the tip of the iceberg. There are substantial limitations in analytical resolution using current technologies. For example, we have not been able to measure intracellular molecule distribution accurately enough (particularly in living cells); there are no versatile microscales to measure molecular distances between native proteins in situ. Although new technologies such as fluorescent resonant energy transfer or bioluminescence resonant energy transfer have become available, their use has been limited to recombinant molecules. No microstrain gauges exist to measure mechanical stresses developed inside of cytoskeletal compartments. Development of such advanced technologies will open a new era of true subcellular molecular biology. Until then, current approaches permit continued investigation of how the pieces of this fascinating puzzle fit together.
Acknowledgments
Colleagues of Drs. Kenneth R. Chien and John Ross Jr.’s laboratories at UCSD and Dr. Shun’ichi Kuroda’s laboratory (Osaka University, Japan) contributed to studies discussed in this article. The author is grateful to Dr. John Solaro for thorough editorial assistance.
GRANTS
This work is support by American Heart Association National Center Scientist Development Grant 0335079N and National Heart, Lung, and Blood Institute Grant R01 HL-081401.
References
- 1.Aikawa R, Komuro I, Yamazaki T, Zou Y, Kudoh S, Zhu W, Kadowaki T, Yazaki Y. Rho family small G proteins play critical roles in mechanical stress-induced hypertrophic responses in cardiac myocytes. Circ Res. 1999;84:458–466. doi: 10.1161/01.res.84.4.458. [DOI] [PubMed] [Google Scholar]
- 2.Allamand V, Campbell KP. Animal models for muscular dystrophy: valuable tools for the development of therapies. Hum Mol Genet. 2000;9:2459–2467. doi: 10.1093/hmg/9.16.2459. [DOI] [PubMed] [Google Scholar]
- 3.Antos CL, McKinsey TA, Frey N, Kutschke W, McAnally J, Shelton JM, Richardson JA, Hill JA, Olson EN. Activated glycogen synthase-3 beta suppresses cardiac hypertrophy in vivo. Proc Natl Acad Sci USA. 2002;99:907–912. doi: 10.1073/pnas.231619298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Arber S, Caroni P. Specificity of single LIM motifs in targeting and LIM/LIM interactions in situ. Genes Dev. 1996;10:289–300. doi: 10.1101/gad.10.3.289. [DOI] [PubMed] [Google Scholar]
- 5.Arber S, Halder G, Caroni P. Muscle LIM protein, a novel essential regulator of myogenesis, promotes myogenic differentiation. Cell. 1994;79:221–231. doi: 10.1016/0092-8674(94)90192-9. [DOI] [PubMed] [Google Scholar]
- 6.Arber S, Hunter JJ, Ross J, Jr, Hongo M, Sansig G, Borg J, Perriard JC, Chien KR, Caroni P. MLP-deficient mice exhibit a disruption of cardiac cytoarchitectural organization, dilated cardiomyopathy, and heart failure. Cell. 1997;88:393–403. doi: 10.1016/s0092-8674(00)81878-4. [DOI] [PubMed] [Google Scholar]
- 7.Arimura T, Hayashi T, Terada H, Lee SY, Zhou Q, Takahashi M, Ueda K, Nouchi T, Hohda S, Shibutani M, Hirose M, Chen J, Park JE, Yasunami M, Hayashi H, Kimura A. A Cypher/ZASP mutation associated with dilated cardiomyopathy alters the binding affinity to protein kinase C. J Biol Chem. 2004;279:6746–6752. doi: 10.1074/jbc.M311849200. [DOI] [PubMed] [Google Scholar]
- 8.Arya R, Kedar V, Hwang JR, McDonough H, Li HH, Taylor J, Patterson C. Muscle ring finger protein-1 inhibits PKCε activation and prevents cardiomyocyte hypertrophy. J Cell Biol. 2004;167:1147–1159. doi: 10.1083/jcb.200402033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Asakura M, Kitakaze M, Takashima S, Liao Y, Ishikura F, Yoshinaka T, Ohmoto H, Node K, Yoshino K, Ishiguro H, Asanuma H, Sanada S, Matsumura Y, Takeda H, Beppu S, Tada M, Hori M, Higashiyama S. Cardiac hypertrophy is inhibited by antagonism of ADAM12 processing of HB-EGF: metalloproteinase inhibitors as a new therapy. Nat Med. 2002;8:35–40. doi: 10.1038/nm0102-35. [DOI] [PubMed] [Google Scholar]
- 10.Bach I. The LIM domain: regulation by association. Mech Dev. 2000;91:5–17. doi: 10.1016/s0925-4773(99)00314-7. [DOI] [PubMed] [Google Scholar]
- 11.Baines CP, Molkentin JD. STRESS signaling pathways that modulate cardiac myocyte apoptosis. J Mol Cell Cardiol. 2005;38:47–62. doi: 10.1016/j.yjmcc.2004.11.004. [DOI] [PubMed] [Google Scholar]
- 12.Balda MS, Garrett MD, Matter K. The ZO-1-associated Y-box factor ZONAB regulates epithelial cell proliferation and cell density. J Cell Biol. 2003;160:423–432. doi: 10.1083/jcb.200210020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bang ML, Mudry RE, McElhinny AS, Trombitas K, Geach AJ, Yamasaki R, Sorimachi H, Granzier H, Gregorio CC, Labeit S. Myopalladin, a novel 145-kilodalton sarcomeric protein with multiple roles in Z-disc and I-band protein assemblies. J Cell Biol. 2001;153:413–427. doi: 10.1083/jcb.153.2.413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Beckerle MC. Zyxin: zinc fingers at sites of cell adhesion. Bioessays. 1997;19:949–957. doi: 10.1002/bies.950191104. [DOI] [PubMed] [Google Scholar]
- 15.Bock-Marquette I, Saxena A, White MD, Dimaio JM, Srivastava D. Thymosin beta4 activates integrin-linked kinase and promotes cardiac cell migration, survival and cardiac repair. Nature. 2004;432:466–472. doi: 10.1038/nature03000. [DOI] [PubMed] [Google Scholar]
- 16.Bodine SC, Latres E, Baumhueter S, Lai VK, Nunez L, Clarke BA, Poueymirou WT, Panaro FJ, Na E, Dharmarajan K, Pan ZQ, Valenzuela DM, DeChiara TM, Stitt TN, Yancopoulos GD, Glass DJ. Identification of ubiquitin ligases required for skeletal muscle atrophy. Science. 2001;294:1704–1708. doi: 10.1126/science.1065874. [DOI] [PubMed] [Google Scholar]
- 17.Brancaccio M, Fratta L, Notte A, Hirsch E, Poulet R, Guazzone S, De Acetis M, Vecchione C, Marino G, Altruda F, Silengo L, Tarone G, Lembo G. Melusin, a muscle-specific integrin beta1-interacting protein, is required to prevent cardiac failure in response to chronic pressure overload. Nat Med. 2003;9:68–75. doi: 10.1038/nm805. [DOI] [PubMed] [Google Scholar]
- 18.Brancaccio M, Guazzone S, Menini N, Sibona E, Hirsch E, De Andrea M, Rocchi M, Altruda F, Tarone G, Silengo L. Melusin is a new muscle-specific interactor for beta(1) integrin cytoplasmic domain. J Biol Chem. 1999;274:29282–29288. doi: 10.1074/jbc.274.41.29282. [DOI] [PubMed] [Google Scholar]
- 19.Centner T, Yano J, Kimura E, McElhinny AS, Pelin K, Witt CC, Bang ML, Trombitas K, Granzier H, Gregorio CC, Sorimachi H, Labeit S. Identification of muscle specific ring finger proteins as potential regulators of the titin kinase domain. J Mol Biol. 2001;306:717–726. doi: 10.1006/jmbi.2001.4448. [DOI] [PubMed] [Google Scholar]
- 20.Chu PH, Bardwell WM, Gu Y, Ross J, Jr, Chen J. FHL2 (SLIM3) is not essential for cardiac development and function. Mol Cell Biol. 2000;20:7460–7462. doi: 10.1128/mcb.20.20.7460-7462.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chu PH, Ruiz-Lozano P, Zhou Q, Cai C, Chen J. Expression patterns of FHL/SLIM family members suggest important functional roles in skeletal muscle and cardiovascular system. Mech Dev. 2000;95:259–265. doi: 10.1016/s0925-4773(00)00341-5. [DOI] [PubMed] [Google Scholar]
- 22.Clark KA, McElhinny AS, Beckerle MC, Gregorio CC. Striated muscle cytoarchitecture: an intricate web of form and function. Annu Rev Cell Dev Biol. 2002;18:637–706. doi: 10.1146/annurev.cellbio.18.012502.105840. [DOI] [PubMed] [Google Scholar]
- 23.Clerk A, Sugden PH. Untangling the Web: specific signaling from PKC isoforms to MAPK cascades. Circ Res. 2001;89:847–849. [PubMed] [Google Scholar]
- 24.Cohn RD, Campbell KP. Molecular basis of muscular dystrophies. Muscle Nerve. 2000;23:1456–1471. doi: 10.1002/1097-4598(200010)23:10<1456::aid-mus2>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- 25.Csukai M, Chen CH, De Matteis MA, Mochly-Rosen D. The coatomer protein beta′;-COP, a selective binding protein (RACK) for protein kinase Cepsilon. J Biol Chem. 1997;272:29200–29206. doi: 10.1074/jbc.272.46.29200. [DOI] [PubMed] [Google Scholar]
- 26.Dawid IB, Breen JJ, Toyama R. LIM domains: multiple roles as adapters and functional modifiers in protein interactions. Trends Genet. 1998;14:156–162. doi: 10.1016/s0168-9525(98)01424-3. [DOI] [PubMed] [Google Scholar]
- 27.De Acetis M, Notte A, Accornero F, Selvetella G, Brancaccio M, Vecchione C, Sbroggio M, Collino F, Pacchioni B, Lanfranchi G, Aretini A, Ferretti R, Maffei A, Altruda F, Silengo L, Tarone G, Lembo G. Cardiac overexpression of melusin protects from dilated cardiomyopathy due to long-standing pressure overload. Circ Res. 2005;96:1087–1094. doi: 10.1161/01.RES.0000168028.36081.e0. [DOI] [PubMed] [Google Scholar]
- 28.De Windt LJ, Lim HW, Haq S, Force T, Molkentin JD. Calcineurin promotes protein kinase C and c-Jun NH2-terminal kinase activation in the heart. Cross-talk between cardiac hypertrophic signaling pathways. J Biol Chem. 2000;275:13571–13579. doi: 10.1074/jbc.275.18.13571. [DOI] [PubMed] [Google Scholar]
- 29.Disatnik MH, Buraggi G, Mochly-Rosen D. Localization of protein kinase C isozymes in cardiac myocytes. Exp Cell Res. 1994;210:287–297. doi: 10.1006/excr.1994.1041. [DOI] [PubMed] [Google Scholar]
- 30.Dorn GW, 2nd, Souroujon MC, Liron T, Chen CH, Gray MO, Zhou HZ, Csukai M, Wu G, Lorenz JN, Mochly-Rosen D. Sustained in vivo cardiac protection by a rationally designed peptide that causes epsilon protein kinase C translocation. Proc Natl Acad Sci USA. 1999;96:12798–12803. doi: 10.1073/pnas.96.22.12798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dorn GW, II, Mochly-Rosen D. Intracellular transport mechanisms of signal transducers. Annu Rev Physiol. 2002;64:407–429. doi: 10.1146/annurev.physiol.64.081501.155903. [DOI] [PubMed] [Google Scholar]
- 32.Dos Remedios CG, Chhabra D, Kekic M, Dedova IV, Tsubakihara M, Berry DA, Nosworthy NJ. Actin binding proteins: regulation of cytoskeletal microfilaments. Physiol Rev. 2003;83:433–473. doi: 10.1152/physrev.00026.2002. [DOI] [PubMed] [Google Scholar]
- 33.Drake-Holland AJ, Noble MI, Lab MJ. Acute pressure overload cardiac arrhythmias are dependent on the presence of myocardial tissue catecholamines. Heart. 2001;85:576. doi: 10.1136/heart.85.5.576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Duffy HS, Ashton AW, O’Donnell P, Coombs W, Taffet SM, Delmar M, Spray DC. Regulation of connexin43 protein complexes by intracellular acidification. Circ Res. 2004;94:215–222. doi: 10.1161/01.RES.0000113924.06926.11. [DOI] [PubMed] [Google Scholar]
- 35.Durbeej M, Campbell KP. Muscular dystrophies involving the dystrophin-glycoprotein complex: an overview of current mouse models. Curr Opin Genet Dev. 2002;12:349–361. doi: 10.1016/s0959-437x(02)00309-x. [DOI] [PubMed] [Google Scholar]
- 36.Durick K, Wu RY, Gill GN, Taylor SS. Mitogenic signaling by Ret/ptc2 requires association with enigma via a LIM domain. J Biol Chem. 1996;271:12691–12694. doi: 10.1074/jbc.271.22.12691. [DOI] [PubMed] [Google Scholar]
- 37.Ecarnot-Laubriet A, De Luca K, Vandroux D, Moisant M, Bernard C, Assem M, Rochette L, Teyssier JR. Downregulation and nuclear relocation of MLP during the progression of right ventricular hypertrophy induced by chronic pressure overload. J Mol Cell Cardiol. 2000;32:2385–2395. doi: 10.1006/jmcc.2000.1269. [DOI] [PubMed] [Google Scholar]
- 38.Ehler E, Horowits R, Zuppinger C, Price RL, Perriard E, Leu M, Caroni P, Sussman M, Eppenberger HM, Perriard JC. Alterations at the intercalated disk associated with the absence of muscle LIM protein. J Cell Biol. 2001;153:763–772. doi: 10.1083/jcb.153.4.763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ervasti JM. Costameres: the Achilles’ heel of Herculean muscle. J Biol Chem. 2003;278:13591–13594. doi: 10.1074/jbc.R200021200. [DOI] [PubMed] [Google Scholar]
- 40.Faulkner G, Pallavicini A, Comelli A, Salamon M, Bortoletto G, Ievolella C, Trevisan S, Kojic S, Dalla Vecchia F, Laveder P, Valle G, Lanfranchi G. FATZ, a filamin-, actinin-, and telethonin-binding protein of the Z-disc of skeletal muscle. J Biol Chem. 2000;275:41234–41242. doi: 10.1074/jbc.M007493200. [DOI] [PubMed] [Google Scholar]
- 41.Faulkner G, Pallavicini A, Formentin E, Comelli A, Ievolella C, Trevisan S, Bortoletto G, Scannapieco P, Salamon M, Mouly V, Valle G, Lanfranchi G. ZASP: a new Z-band alternatively spliced PDZ-motif protein. J Cell Biol. 1999;146:465–475. doi: 10.1083/jcb.146.2.465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Flick MJ, Konieczny SF. The muscle regulatory and structural protein MLP is a cytoskeletal binding partner of betaI-spectrin. J Cell Sci. 2000;113:1553–1564. doi: 10.1242/jcs.113.9.1553. [DOI] [PubMed] [Google Scholar]
- 43.Franchini KG, Torsoni AS, Soares PH, Saad MJ. Early activation of the multicomponent signaling complex associated with focal adhesion kinase induced by pressure overload in the rat heart. Circ Res. 2000;87:558–565. doi: 10.1161/01.res.87.7.558. [DOI] [PubMed] [Google Scholar]
- 44.Frankel P, Aronheim A, Kavanagh E, Balda MS, Matter K, Bunney TD, Marshall CJ. RalA interacts with ZONAB in a cell density-dependent manner and regulates its transcriptional activity. EMBO J. 2005;24:54–62. doi: 10.1038/sj.emboj.7600497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Frey N, Barrientos T, Shelton JM, Frank D, Rutten H, Gehring D, Kuhn C, Lutz M, Rothermel B, Bassel-Duby R, Richardson JA, Katus HA, Hill JA, Olson EN. Mice lacking calsarcin-1 are sensitized to calcineurin signaling and show accelerated cardiomyopathy in response to pathological biomechanical stress. Nat Med. 2004;10:1336–1343. doi: 10.1038/nm1132. [DOI] [PubMed] [Google Scholar]
- 46.Frey N, Olson EN. Calsarcin-3, a novel skeletal muscle-specific member of the calsarcin family, interacts with multiple Z-disc proteins. J Biol Chem. 2002;277:13998–14004. doi: 10.1074/jbc.M200712200. [DOI] [PubMed] [Google Scholar]
- 47.Frey N, Richardson JA, Olson EN. Calsarcins, a novel family of sarcomeric calcineurin-binding proteins. Proc Natl Acad Sci USA. 2000;97:14632–14637. doi: 10.1073/pnas.260501097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gehmlich K, Geier C, Osterziel KJ, Van der Ven PF, Furst DO. Decreased interactions of mutant muscle LIM protein (MLP) with N-RAP and alpha-actinin and their implication for hypertrophic cardiomyopathy. Cell Tissue Res. 2004;317:129–136. doi: 10.1007/s00441-004-0873-y. [DOI] [PubMed] [Google Scholar]
- 49.Geier C, Perrot A, Ozcelik C, Binner P, Counsell D, Hoffmann K, Pilz B, Martiniak Y, Gehmlich K, van der Ven PF, Furst DO, Vornwald A, von Hodenberg E, Nurnberg P, Scheffold T, Dietz R, Osterziel KJ. Mutations in the human muscle LIM protein gene in families with hypertrophic cardiomyopathy. Circulation. 2003;107:1390–1395. doi: 10.1161/01.cir.0000056522.82563.5f. [DOI] [PubMed] [Google Scholar]
- 50.Giepmans BN. Gap junctions and connexin-interacting proteins. Cardiovasc Res. 2004;62:233–245. doi: 10.1016/j.cardiores.2003.12.009. [DOI] [PubMed] [Google Scholar]
- 51.Granzier HL, Labeit S. The giant protein titin: a major player in myocardial mechanics, signaling, and disease. Circ Res. 2004;94:284–295. doi: 10.1161/01.RES.0000117769.88862.F8. [DOI] [PubMed] [Google Scholar]
- 52.Guy PM, Kenny DA, Gill GN. The PDZ domain of the LIM protein enigma binds to beta-tropomyosin. Mol Biol Cell. 1999;10:1973–1984. doi: 10.1091/mbc.10.6.1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hardt SE, Sadoshima J. Glycogen synthase kinase-3beta: a novel regulator of cardiac hypertrophy and development. Circ Res. 2002;90:1055–1063. doi: 10.1161/01.res.0000018952.70505.f1. [DOI] [PubMed] [Google Scholar]
- 54.Hardt SE, Tomita H, Katus HA, Sadoshima J. Phosphorylation of eukaryotic translation initiation factor 2Bepsilon by glycogen synthase kinase-3beta regulates beta-adrenergic cardiac myocyte hypertrophy. Circ Res. 2004;94:926–935. doi: 10.1161/01.RES.0000124977.59827.80. [DOI] [PubMed] [Google Scholar]
- 55.Heineke J, Ruetten H, Willenbockel C, Gross SC, Naguib M, Schaefer A, Kempf T, Hilfiker-Kleiner D, Caroni P, Kraft T, Kaiser RA, Molkentin JD, Drexler H, Wollert KC. Attenuation of cardiac remodeling after myocardial infarction by muscle LIM protein-calcineurin signaling at the sarcomeric Z-disc. Proc Natl Acad Sci USA. 2005;102:1655–1660. doi: 10.1073/pnas.0405488102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Henderson JR, Pomies P, Auffray C, Beckerle MC. ALP and MLP distribution during myofibrillogenesis in cultured cardiomyocytes. Cell Motil Cytoskeleton. 2003;54:254–265. doi: 10.1002/cm.10102. [DOI] [PubMed] [Google Scholar]
- 57.Hoshijima M, Chien KR. Mixed signals in heart failure: cancer rules. J Clin Invest. 2002;109:849–855. doi: 10.1172/JCI15380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hoshijima M, Ikeda Y, Iwanaga Y, Minamisawa S, Date MO, Gu Y, Iwatate M, Li M, Wang L, Wilson JM, Wang Y, Ross J, Jr, Chien KR. Chronic suppression of heart-failure progression by a pseudophosphorylated mutant of phospholamban via in vivo cardiac rAAV gene delivery. Nat Med. 2002;8:864–871. doi: 10.1038/nm739. [DOI] [PubMed] [Google Scholar]
- 59.Hoshijima M, Minamisawa S, Yasukawa H, Chien KR. Cardiovascular signaling pathways. In: Chein KR, editor. Molecular Basis of Cardiovascular Diseases: A Companion to Braunwald’s Heart Disease. 2. Philadelphia, PA: Sauders; 2004. pp. 273–292. [Google Scholar]
- 60.Hoshijima M, Sah VP, Wang Y, Chien KR, Brown JH. The low molecular weight GTPase Rho regulates myofibril formation and organization in neonatal rat ventricular myocytes. Involvement of Rho kinase. J Biol Chem. 1998;273:7725–7730. doi: 10.1074/jbc.273.13.7725. [DOI] [PubMed] [Google Scholar]
- 61.Huang XP, Pi Y, Lokuta AJ, Greaser ML, Walker JW. Arachidonic acid stimulates protein kinase C-epsilon redistribution in heart cells. J Cell Sci. 1997;110:1625–1634. doi: 10.1242/jcs.110.14.1625. [DOI] [PubMed] [Google Scholar]
- 62.Iwanaga Y, Hoshijima M, Gu Y, Iwatate M, Dieterle T, Ikeda Y, Date MO, Chrast J, Matsuzaki M, Peterson KL, Chien KR, Ross J., Jr Chronic phospholamban inhibition prevents progressive cardiac dysfunction and pathological remodeling after infarction in rats. J Clin Invest. 2004;113:727–736. doi: 10.1172/JCI18716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Juhaszova M, Zorov DB, Kim SH, Pepe S, Fu Q, Fishbein KW, Ziman BD, Wang S, Ytrehus K, Antos CL, Olson EN, Sollott SJ. Glycogen synthase kinase-3beta mediates convergence of protection signaling to inhibit the mitochondrial permeability transition pore. J Clin Invest. 2004;113:1535–1549. doi: 10.1172/JCI19906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kanno S, Saffitz JE. The role of myocardial gap junctions in electrical conduction and arrhythmogenesis. Cardiovasc Pathol. 2001;10:169–177. doi: 10.1016/s1054-8807(01)00078-3. [DOI] [PubMed] [Google Scholar]
- 65.Kato T, Muraski J, Chen Y, Tsujita Y, Wall J, Glembotski CC, Schaefer E, Beckerle M, Sussman MA. Atrial natriuretic peptide promotes cardiomyocyte survival by cGMP-dependent nuclear accumulation of zyxin and Akt. J Clin Invest. 2005;115:2716–2730. doi: 10.1172/JCI24280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kawamura S, Miyamoto S, Brown JH. Initiation and transduction of stretch-induced RhoA and Rac1 activation through caveolae: cytoskeletal regulation of ERK translocation. J Biol Chem. 2003;278:31111–31117. doi: 10.1074/jbc.M300725200. [DOI] [PubMed] [Google Scholar]
- 67.Kedar V, McDonough H, Arya R, Li HH, Rockman HA, Patterson C. Muscle-specific RING finger 1 is a bona fide ubiquitin ligase that degrades cardiac troponin I. Proc Natl Acad Sci USA. 2004;101:18135–18140. doi: 10.1073/pnas.0404341102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Klug GA, Knudson MB, Cartier LJ, Gollnick PD. Cardiac contractility, cAMP concentration, cAMP-dependent protein kinase, and phosphorylase activation during acute pressure overload. Pflügers Arch. 1984;402:216–221. doi: 10.1007/BF00583338. [DOI] [PubMed] [Google Scholar]
- 69.Knoll R, Hoshijima M, Hoffman HM, Person V, Lorenzen-Schmidt I, Bang ML, Hayashi T, Shiga N, Yasukawa H, Schaper W, Mc-Kenna W, Yokoyama M, Schork NJ, Omens JH, McCulloch AD, Kimura A, Gregorio CC, Poller W, Schaper J, Schultheiss HP, Chien KR. The cardiac mechanical stretch sensor machinery involves a Z disc complex that is defective in a subset of human dilated cardiomyopathy. Cell. 2002;111:943–955. doi: 10.1016/s0092-8674(02)01226-6. [DOI] [PubMed] [Google Scholar]
- 70.Kong Y, Flick MJ, Kudla AJ, Konieczny SF. Muscle LIM protein promotes myogenesis by enhancing the activity of MyoD. Mol Cell Biol. 1997;17:4750–4760. doi: 10.1128/mcb.17.8.4750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kong Y, Shelton JM, Rothermel B, Li X, Richardson JA, Bassel-Duby R, Williams RS. Cardiac-specific LIM protein FHL2 modifies the hypertrophic response to beta-adrenergic stimulation. Circulation. 2001;103:2731–2738. doi: 10.1161/01.cir.103.22.2731. [DOI] [PubMed] [Google Scholar]
- 72.Kuroda S, Tokunaga C, Kiyohara Y, Higuchi O, Konishi H, Mizuno K, Gill GN, Kikkawa U. Protein-protein interaction of zinc finger LIM domains with protein kinase C. J Biol Chem. 1996;271:31029–31032. doi: 10.1074/jbc.271.49.31029. [DOI] [PubMed] [Google Scholar]
- 73.Kuwahara K, Barrientos T, Pipes GC, Li S, Olson EN. Muscle-specific signaling mechanism that links actin dynamics to serum response factor. Mol Cell Biol. 2005;25:3173–3181. doi: 10.1128/MCB.25.8.3173-3181.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Labalette C, Renard CA, Neuveut C, Buendia MA, Wei Y. Interaction and functional cooperation between the LIM protein FHL2, CBP/p300, and beta-catenin. Mol Cell Biol. 2004;24:10689–10702. doi: 10.1128/MCB.24.24.10689-10702.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lange S, Auerbach D, McLoughlin P, Perriard E, Schafer BW, Perriard JC, Ehler E. Subcellular targeting of metabolic enzymes to titin in heart muscle may be mediated by DRAL/FHL-2. J Cell Sci. 2002;115:4925–4936. doi: 10.1242/jcs.00181. [DOI] [PubMed] [Google Scholar]
- 76.Lange S, Xiang F, Yakovenko A, Vihola A, Hackman P, Rostkova E, Kristensen J, Brandmeier B, Franzen G, Hedberg B, Gunnarsson LG, Hughes SM, Marchand S, Sejersen T, Richard I, Edstrom L, Ehler E, Udd B, Gautel M. The kinase domain of titin controls muscle gene expression and protein turnover. Science. 2005;308:1599–1603. doi: 10.1126/science.1110463. [DOI] [PubMed] [Google Scholar]
- 77.Lapidos KA, Kakkar R, McNally EM. The dystrophin glycoprotein complex: signaling strength and integrity for the sarcolemma. Circ Res. 2004;94:1023–1031. doi: 10.1161/01.RES.0000126574.61061.25. [DOI] [PubMed] [Google Scholar]
- 78.Li HH, Kedar V, Zhang C, McDonough H, Arya R, Wang DZ, Patterson C. Atrogin-1/muscle atrophy F-box inhibits calcineurin-dependent cardiac hypertrophy by participating in an SCF ubiquitin ligase complex. J Clin Invest. 2004;114:1058–1071. doi: 10.1172/JCI22220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Liang X, Zhou Q, Li X, Sun Y, Lu M, Dalton N, Ross J, Jr, Chen J. PINCH1 plays an essential role in early murine embryonic development but is dispensable in ventricular cardiomyocytes. Mol Cell Biol. 2005;25:3056–3062. doi: 10.1128/MCB.25.8.3056-3062.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Liu Y, Cseresnyes Z, Randall WR, Schneider MF. Activity-dependent nuclear translocation and intranuclear distribution of NFATc in adult skeletal muscle fibers. J Cell Biol. 2001;155:27–39. doi: 10.1083/jcb.200103020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Louis HA, Pino JD, Schmeichel KL, Pomies P, Beckerle MC. Comparison of three members of the cysteine-rich protein family reveals functional conservation and divergent patterns of gene expression. J Biol Chem. 1997;272:27484–27491. doi: 10.1074/jbc.272.43.27484. [DOI] [PubMed] [Google Scholar]
- 82.Luo G, Herrera AH, Horowits R. Molecular interactions of N-RAP, a nebulin-related protein of striated muscle myotendon junctions and intercalated disks. Biochemistry. 1999;38:6135–6143. doi: 10.1021/bi982395t. [DOI] [PubMed] [Google Scholar]
- 83.Mayans O, van der Ven PF, Wilm M, Mues A, Young P, Furst DO, Wilmanns M, Gautel M. Structural basis for activation of the titin kinase domain during myofibrillogenesis. Nature. 1998;395:863–869. doi: 10.1038/27603. [DOI] [PubMed] [Google Scholar]
- 84.McElhinny AS, Kakinuma K, Sorimachi H, Labeit S, Gregorio CC. Muscle-specific RING finger-1 interacts with titin to regulate sarcomeric M-line and thick filament structure and may have nuclear functions via its interaction with glucocorticoid modulatory element binding protein-1. J Cell Biol. 2002;157:125–136. doi: 10.1083/jcb.200108089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Miller MK, Bang ML, Witt CC, Labeit D, Trombitas C, Watanabe K, Granzier H, McElhinny AS, Gregorio CC, Labeit S. The muscle ankyrin repeat proteins: CARP, ankrd2/Arpp and DARP as a family of titin filament-based stress response molecules. J Mol Biol. 2003;333:951–964. doi: 10.1016/j.jmb.2003.09.012. [DOI] [PubMed] [Google Scholar]
- 86.Miller MK, Granzier H, Ehler E, Gregorio CC. The sensitive giant: the role of titin-based stretch sensing complexes in the heart. Trends Cell Biol. 2004;14:119–126. doi: 10.1016/j.tcb.2004.01.003. [DOI] [PubMed] [Google Scholar]
- 87.Minamisawa S, Hoshijima M, Chu G, Ward CA, Frank K, Gu Y, Martone ME, Wang Y, Ross J, Jr, Kranias EG, Giles WR, Chien KR. Chronic phospholamban-sarcoplasmic reticulum calcium ATPase interaction is the critical calcium cycling defect in dilated cardiomyopathy. Cell. 1999;99:313–322. doi: 10.1016/s0092-8674(00)81662-1. [DOI] [PubMed] [Google Scholar]
- 88.Miralles F, Posern G, Zaromytidou AI, Treisman R. Actin dynamics control SRF activity by regulation of its coactivator MAL. Cell. 2003;113:329–342. doi: 10.1016/s0092-8674(03)00278-2. [DOI] [PubMed] [Google Scholar]
- 89.Miranti CK, Brugge JS. Sensing the environment: a historical perspective on integrin signal transduction. Nat Cell Biol. 2002;4:E83–E90. doi: 10.1038/ncb0402-e83. [DOI] [PubMed] [Google Scholar]
- 90.Mohapatra B, Jimenez S, Lin JH, Bowles KR, Coveler KJ, Marx JG, Chrisco MA, Murphy RT, Lurie PR, Schwartz RJ, Elliott PM, Vatta M, McKenna W, Towbin JA, Bowles NE. Mutations in the muscle LIM protein and alpha-actinin-2 genes in dilated cardiomyopathy and endocardial fibroelastosis. Mol Genet Metab. 2003;80:207–215. doi: 10.1016/s1096-7192(03)00142-2. [DOI] [PubMed] [Google Scholar]
- 91.Moule SK, Welsh GI, Edgell NJ, Foulstone EJ, Proud CG, Denton RM. Regulation of protein kinase B and glycogen synthase kinase-3 by insulin and beta-adrenergic agonists in rat epididymal fat cells. Activation of protein kinase B by wortmannin-sensitive and -insensitive mechanisms. J Biol Chem. 1997;272:7713–7719. doi: 10.1074/jbc.272.12.7713. [DOI] [PubMed] [Google Scholar]
- 92.Nakagawa N, Hoshijima M, Oyasu M, Saito N, Tanizawa K, Kuroda S. ENH, containing PDZ and LIM domains, heart/skeletal muscle-specific protein, associates with cytoskeletal proteins through the PDZ domain. Biochem Biophys Res Commun. 2000;272:505–512. doi: 10.1006/bbrc.2000.2787. [DOI] [PubMed] [Google Scholar]
- 93.Nourry C, Grant SG, Borg JP. PDZ domain proteins: plug and play! Sci STKE. 2003;179:RE7. doi: 10.1126/stke.2003.179.re7. [DOI] [PubMed] [Google Scholar]
- 94.Pashmforoush M, Pomies P, Peterson KL, Kubalak S, Ross J, Jr, Hefti A, Aebi U, Beckerle MC, Chien KR. Adult mice deficient in actinin-associated LIM-domain protein reveal a developmental pathway for right ventricular cardiomyopathy. Nat Med. 2001;7:591–597. doi: 10.1038/87920. [DOI] [PubMed] [Google Scholar]
- 95.Passier R, Richardson JA, Olson EN. Oracle, a novel PDZ-LIM domain protein expressed in heart and skeletal muscle. Mech Dev. 2000;92:277–284. doi: 10.1016/s0925-4773(99)00330-5. [DOI] [PubMed] [Google Scholar]
- 96.Paul K, Ball NA, Dorn GW, III, Walsh RA. Left ventricular stretch stimulates angiotensin II–mediated phosphatidylinositol hydrolysis and protein kinase C epsilon isoform translocation in adult guinea pig hearts. Circ Res. 1997;81:643–650. doi: 10.1161/01.res.81.5.643. [DOI] [PubMed] [Google Scholar]
- 97.Petroff MG, Kim SH, Pepe S, Dessy C, Marban E, Balligand JL, Sollott SJ. Endogenous nitric oxide mechanisms mediate the stretch dependence of Ca2+ release in cardiomyocytes. Nat Cell Biol. 2001;3:867–873. doi: 10.1038/ncb1001-867. [DOI] [PubMed] [Google Scholar]
- 98.Philippar U, Schratt G, Dieterich C, Muller JM, Galgoczy P, Engel FB, Keating MT, Gertler F, Schule R, Vingron M, Nordheim A. The SRF target gene Fhl2 antagonizes RhoA/MAL-dependent activation of SRF. Mol Cell. 2004;16:867–880. doi: 10.1016/j.molcel.2004.11.039. [DOI] [PubMed] [Google Scholar]
- 99.Purcell NH, Darwis D, Bueno OF, Muller JM, Schule R, Molkentin JD. Extracellular signal-regulated kinase 2 interacts with and is negatively regulated by the LIM-only protein FHL2 in cardiomyocytes. Mol Cell Biol. 2004;24:1081–1095. doi: 10.1128/MCB.24.3.1081-1095.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Pyle WG, Hart MC, Cooper JA, Sumandea MP, de Tombe PP, Solaro RJ. Actin capping protein: an essential element in protein kinase signaling to the myofilaments. Circ Res. 2002;90:1299–1306. doi: 10.1161/01.res.0000024389.03152.22. [DOI] [PubMed] [Google Scholar]
- 101.Pyle WG, Solaro RJ. At the crossroads of myocardial signaling: the role of Z-discs in intracellular signaling and cardiac function. Circ Res. 2004;94:296–305. doi: 10.1161/01.RES.0000116143.74830.A9. [DOI] [PubMed] [Google Scholar]
- 102.Rhee D, Sanger JM, Sanger JW. The premyofibril: evidence for its role in myofibrillogenesis. Cell Motil Cytoskeleton. 1994;28:1–24. doi: 10.1002/cm.970280102. [DOI] [PubMed] [Google Scholar]
- 103.Ross RS. Molecular and mechanical synergy: cross-talk between integrins and growth factor receptors. Cardiovasc Res. 2004;63:381–390. doi: 10.1016/j.cardiores.2004.04.027. [DOI] [PubMed] [Google Scholar]
- 104.Sadler I, Crawford AW, Michelsen JW, Beckerle MC. Zyxin and cCRP: two interactive LIM domain proteins associated with the cytoskeleton. J Cell Biol. 1992;119:1573–1587. doi: 10.1083/jcb.119.6.1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Sadoshima J, Xu Y, Slayter HS, Izumo S. Autocrine release of angiotensin II mediates stretch-induced hypertrophy of cardiac myocytes in vitro. Cell. 1993;75:977–984. doi: 10.1016/0092-8674(93)90541-w. [DOI] [PubMed] [Google Scholar]
- 106.Saffitz JE, Kleber AG. Effects of mechanical forces and mediators of hypertrophy on remodeling of gap junctions in the heart. Circ Res. 2004;94:585–591. doi: 10.1161/01.RES.0000121575.34653.50. [DOI] [PubMed] [Google Scholar]
- 107.Sah VP, Hoshijima M, Chien KR, Brown JH. Rho is required for Galphaq and alpha1-adrenergic receptor signaling in cardiomyocytes. Dissociation of Ras and Rho pathways. J Biol Chem. 1996;271:31185–31190. doi: 10.1074/jbc.271.49.31185. [DOI] [PubMed] [Google Scholar]
- 108.Samarel AM. Costameres, focal adhesions, and cardiomyocyte mechanotransduction. Am J Physiol Heart Circ Physiol. 2005;289:H2291–H2301. doi: 10.1152/ajpheart.00749.2005. [DOI] [PubMed] [Google Scholar]
- 109.Samson T, Smyth N, Janetzky S, Wendler O, Muller JM, Schule R, von der Mark H, von der Mark K, Wixler V. The LIM-only proteins FHL2 and FHL3 interact with alpha- and beta-subunits of the muscle alpha7beta1 integrin receptor. J Biol Chem. 2004;279:28641–28652. doi: 10.1074/jbc.M312894200. [DOI] [PubMed] [Google Scholar]
- 110.Sanger JW, Ayoob JC, Chowrashi P, Zurawski D, Sanger JM. Assembly of myofibrils in cardiac muscle cells. Adv Exp Med Biol. 2000;481:89–102. doi: 10.1007/978-1-4615-4267-4_6. [DOI] [PubMed] [Google Scholar]
- 111.Shai SY, Harpf AE, Babbitt CJ, Jordan MC, Fishbein MC, Chen J, Omura M, Leil TA, Becker KD, Jiang M, Smith DJ, Cherry SR, Loftus JC, Ross RS. Cardiac myocyte-specific excision of the beta1 integrin gene results in myocardial fibrosis and cardiac failure. Circ Res. 2002;90:458–464. doi: 10.1161/hh0402.105790. [DOI] [PubMed] [Google Scholar]
- 112.Sorimachi H, Kimura S, Kinbara K, Kazama J, Takahashi M, Yajima H, Ishiura S, Sasagawa N, Nonaka I, Sugita H, Maruyama K, Suzuki K. Structure and physiological functions of ubiquitous and tissue-specific calpain species. Muscle-specific calpain, p94, interacts with connectin/titin. Adv Biophys. 1996;33:101–122. doi: 10.1016/s0065-227x(96)90026-x. [DOI] [PubMed] [Google Scholar]
- 113.Sugden PH. Ras, Akt, and mechanotransduction in the cardiac myocyte. Circ Res. 2003;93:1179–1192. doi: 10.1161/01.RES.0000106132.04301.F5. [DOI] [PubMed] [Google Scholar]
- 114.Takada F, Vander Woude DL, Tong HQ, Thompson TG, Watkins SC, Kunkel LM, Beggs AH. Myozenin: an alpha-actinin- and gamma-filamin-binding protein of skeletal muscle Z lines. Proc Natl Acad Sci USA. 2001;98:1595–1600. doi: 10.1073/pnas.041609698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Torsoni AS, Constancio SS, Nadruz W, Jr, Hanks SK, Franchini KG. Focal adhesion kinase is activated and mediates the early hypertrophic response to stretch in cardiac myocytes. Circ Res. 2003;93:140–147. doi: 10.1161/01.RES.0000081595.25297.1B. [DOI] [PubMed] [Google Scholar]
- 116.Toyofuku T, Akamatsu Y, Zhang H, Kuzuya T, Tada M, Hori M. c-Src regulates the interaction between connexin-43 and ZO-1 in cardiac myocytes. J Biol Chem. 2001;276:1780–1788. doi: 10.1074/jbc.M005826200. [DOI] [PubMed] [Google Scholar]
- 117.Ushio-Fukai M, Hilenski L, Santanam N, Becker PL, Ma Y, Griendling KK, Alexander RW. Cholesterol depletion inhibits epidermal growth factor receptor transactivation by angiotensin II in vascular smooth muscle cells: role of cholesterol-rich microdomains and focal adhesions in angiotensin II signaling. J Biol Chem. 2001;276:48269–48275. doi: 10.1074/jbc.M105901200. [DOI] [PubMed] [Google Scholar]
- 118.Vatta M, Mohapatra B, Jimenez S, Sanchez X, Faulkner G, Perles Z, Sinagra G, Lin JH, Vu TM, Zhou Q, Bowles KR, Di Lenarda A, Schimmenti L, Fox M, Chrisco MA, Murphy RT, McKenna W, Elliott P, Bowles NE, Chen J, Valle G, Towbin JA. Mutations in Cypher/ZASP in patients with dilated cardiomyopathy and left ventricular non-compaction. J Am Coll Cardiol. 2003;42:2014–2027. doi: 10.1016/j.jacc.2003.10.021. [DOI] [PubMed] [Google Scholar]
- 119.Vega RB, Bassel-Duby R, Olson EN. Control of cardiac growth and function by calcineurin signaling. J Biol Chem. 2003;278:36981–36984. doi: 10.1074/jbc.R300023200. [DOI] [PubMed] [Google Scholar]
- 120.Wilkins BJ, Molkentin JD. Calcium-calcineurin signaling in the regulation of cardiac hypertrophy. Biochem Biophys Res Commun. 2004;322:1178–1191. doi: 10.1016/j.bbrc.2004.07.121. [DOI] [PubMed] [Google Scholar]
- 121.Witt SH, Granzier H, Witt CC, Labeit S. MURF-1 and MURF-2 target a specific subset of myofibrillar proteins redundantly: towards understanding MURF-dependent muscle ubiquitination. J Mol Biol. 2005;350:713–722. doi: 10.1016/j.jmb.2005.05.021. [DOI] [PubMed] [Google Scholar]
- 122.Wu C. The PINCH-ILK-parvin complexes: assembly, functions and regulation. Biochim Biophys Acta. 2004;1692:55–62. doi: 10.1016/j.bbamcr.2004.01.006. [DOI] [PubMed] [Google Scholar]
- 123.Wu JC, Tsai RY, Chung TH. Role of catenins in the development of gap junctions in rat cardiomyocytes. J Cell Biochem. 2003;88:823–835. doi: 10.1002/jcb.10390. [DOI] [PubMed] [Google Scholar]
- 124.Wu RY, Gill GN. LIM domain recognition of a tyrosine-containing tight turn. J Biol Chem. 1994;269:25085–25090. [PubMed] [Google Scholar]
- 125.Xia H, Winokur ST, Kuo WL, Altherr MR, Bredt DS. Actinin-associated LIM protein: identification of a domain interaction between PDZ and spectrin-like repeat motifs. J Cell Biol. 1997;139:507–515. doi: 10.1083/jcb.139.2.507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Yamazaki T, Komuro I, Kudoh S, Zou Y, Shiojima I, Mizuno T, Takano H, Hiroi Y, Ueki K, Tobe K, Kadowaki T, Nagai R, Yazaki Y. Mechanical stress activates protein kinase cascade of phosphorylation in neonatal rat cardiac myocytes. J Clin Invest. 1995;96:438–446. doi: 10.1172/JCI118054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Yasukawa H, Hoshijima M, Gu Y, Nakamura T, Pradervand S, Hanada T, Hanakawa Y, Yoshimura A, Ross J, Jr, Chien KR. Suppressor of cytokine signaling-3 is a biomechanical stress-inducible gene that suppresses gp130-mediated cardiac myocyte hypertrophy and survival pathways. J Clin Invest. 2001;108:1459–1467. doi: 10.1172/JCI13939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Yu TS, Moctezuma-Anaya M, Kubo A, Keller G, Robertson S. The heart LIM protein gene (Hlp), expressed in the developing and adult heart, defines a new tissue-specific LIM-only protein family. Mech Dev. 2002;116:187–192. doi: 10.1016/s0925-4773(02)00139-9. [DOI] [PubMed] [Google Scholar]
- 129.Zemljic-Harpf AE, Ponrartana S, Avalos RT, Jordan MC, Roos KP, Dalton ND, Phan VQ, Adamson ED, Ross RS. Heterozygous inactivation of the vinculin gene predisposes to stress-induced cardiomyopathy. Am J Pathol. 2004;165:1033–1044. doi: 10.1016/S0002-9440(10)63364-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Zhang JQ, Elzey B, Williams G, Lu S, Law DJ, Horowits R. Ultrastructural and biochemical localization of N-RAP at the interface between myofibrils and intercalated disks in the mouse heart. Biochemistry. 2001;40:14898–14906. doi: 10.1021/bi0107445. [DOI] [PubMed] [Google Scholar]
- 131.Zheng M, Zhu W, Han Q, Xiao RP. Emerging concepts and therapeutic implications of beta-adrenergic receptor subtype signaling. Pharmacol Ther. 2005;108:257–268. doi: 10.1016/j.pharmthera.2005.04.006. [DOI] [PubMed] [Google Scholar]
- 132.Zhou Q, Chu PH, Huang C, Cheng CF, Martone ME, Knoll G, Shelton GD, Evans S, Chen J. Ablation of Cypher, a PDZ-LIM domain Z-line protein, causes a severe form of congenital myopathy. J Cell Biol. 2001;155:605–612. doi: 10.1083/jcb.200107092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Zhou Q, Ruiz-Lozano P, Martone ME, Chen J. Cypher, a striated muscle-restricted PDZ and LIM domain-containing protein, binds to alpha-actinin-2 and protein kinase C. J Biol Chem. 1999;274:19807–19813. doi: 10.1074/jbc.274.28.19807. [DOI] [PubMed] [Google Scholar]
- 134.Zhuang J, Yamada KA, Saffitz JE, Kleber AG. Pulsatile stretch remodels cell-to-cell communication in cultured myocytes. Circ Res. 2000;87:316–322. doi: 10.1161/01.res.87.4.316. [DOI] [PubMed] [Google Scholar]
- 135.Zolk O, Caroni P, Bohm M. Decreased expression of the cardiac LIM domain protein MLP in chronic human heart failure. Circulation. 2000;101:2674–2677. doi: 10.1161/01.cir.101.23.2674. [DOI] [PubMed] [Google Scholar]
- 136.Zou Y, Akazawa H, Qin Y, Sano M, Takano H, Minamino T, Makita N, Iwanaga K, Zhu W, Kudoh S, Toko H, Tamura K, Kihara M, Nagai T, Fukamizu A, Umemura S, Iiri T, Fujita T, Komuro I. Mechanical stress activates angiotensin II type 1 receptor without the involvement of angiotensin II. Nat Cell Biol. 2004;6:499–506. doi: 10.1038/ncb1137. [DOI] [PubMed] [Google Scholar]
- 137.Zuo L, Ushio-Fukai M, Hilenski LL, Alexander RW. Microtubules regulate angiotensin II type 1 receptor and Rac1 localization in caveolae/lipid rafts: role in redox signaling. Arterioscler Thromb Vasc Biol. 2004;24:1223–1228. doi: 10.1161/01.ATV.0000132400.25045.2a. [DOI] [PubMed] [Google Scholar]