Mutant huntingtin's interaction with mitochondrial protein Drp1 impairs mitochondrial biogenesis and causes defective axonal transport and synaptic degeneration in Huntington's disease (original) (raw)
Abstract
The purpose of this study was to investigate the link between mutant huntingtin (Htt) and neuronal damage in relation to mitochondria in Huntington's disease (HD). In an earlier study, we determined the relationship between mutant Htt and mitochondrial dynamics/synaptic viability in HD patients. We found mitochondrial loss, abnormal mitochondrial dynamics and mutant Htt association with mitochondria in HD patients. In the current study, we sought to expand on our previous findings and further elucidate the relationship between mutant Htt and mitochondrial and synaptic deficiencies. We hypothesized that mutant Htt, in association with mitochondria, alters mitochondrial dynamics, leading to mitochondrial fragmentation and defective axonal transport of mitochondria in HD neurons. In this study, using postmortem HD brains and primary neurons from transgenic BACHD mice, we identified mutant Htt interaction with the mitochondrial protein Drp1 and factors that cause abnormal mitochondrial dynamics, including GTPase Drp1 enzymatic activity. Further, using primary neurons from BACHD mice, for the first time, we studied axonal transport of mitochondria and synaptic degeneration. We also investigated the effect of mutant Htt aggregates and oligomers in synaptic and mitochondrial deficiencies in postmortem HD brains and primary neurons from BACHD mice. We found that mutant Htt interacts with Drp1, elevates GTPase Drp1 enzymatic activity, increases abnormal mitochondrial dynamics and results in defective anterograde mitochondrial movement and synaptic deficiencies. These observations support our hypothesis and provide data that can be utilized to develop therapeutic targets that are capable of inhibiting mutant Htt interaction with Drp1, decreasing mitochondrial fragmentation, enhancing axonal transport of mitochondria and protecting synapses from toxic insults caused by mutant Htt.
INTRODUCTION
Huntington's disease (HD) is a monogenic, fully penetrant, fatal, progressive neurodegenerative disease characterized by motor dysfunction, involuntary movements, chorea, cognitive decline and psychiatric disturbances (1–3). The loss of body weight is a typical characteristic feature of disease progression found in HD patients (4–8). In postmortem brains from HD patients, medium spiny neuronal loss occurs in the caudate and putamen, along with pyramidal neuronal loss in the cortex and hippocampus (9–13). More recently, hypothalamic neuronal loss has also been reported in HD brains (14,15). Currently, there are no drugs or agents available that prevent, slow or cure HD pathogenesis and progression.
HD is caused by an expanded polyglutamine (polyQ) repeat within exon 1 of an HD gene that encodes for an expanded polyQ stretch in the huntingtin (Htt) protein (16). HD is inherited in an autosomal dominant manner, with age-dependence penetrance: 40 or more repeats linked with full penetrance by 65 years. Prevalence of HD is 4–10 in 100 000 individuals in the Western world (3,17). PolyQ repeats are highly polymorphic, with 6–36 in healthy persons. Individuals with 36 or more polyQ repeats are likely to develop HD (16). Genetic and epidemiological data suggest that expanded polyQ repeats are inversely correlated with disease onset.
Htt, a 350 kDa protein product of HD gene, is ubiquitously expressed in peripheral cells and both neurons and glia in the brain, but is mainly localized in the cytoplasm (16). Mutant Htt protein aggregates have been found in pathological sites in HD postmortem brains and brain specimens from HD mouse models (18–27). Mutant Htt soluble oligomers, fibrils and fibrillogenesis have also been reported in HD brains and brain tissues from mouse models and HD cells (28–36). Extensive research using cell cultures, animal models and postmortem brains from HD patients suggests that multiple cellular changes are involved in neuronal damage that characterizes HD pathogenesis (reviewed in 17), including transcriptional deregulation, altered calcium homeostatis, aberrant protein–protein interaction, abnormal mitochondrial dynamics and impaired axonal transport (17). Among these, abnormal mitochondrial dynamics and impaired axonal transport are strongly associated with HD pathogenesis and progression.
Recent studies suggest that abnormal mitochondrial dynamics are involved in HD pathogenesis (36–39) and that these abnormal mitochondrial dynamics are caused by an imbalance in highly conserved GTPase genes that are essential for mitochondrial fission (division) and fusion. In normal neurons, mitochondrial fission and fusion balance equally, maintaining mitochondrial dynamics and distribution in the neuron (40–43). However, in neurons that express mutant Htt, an imbalance between mitochondrial fission and fusion leads to abnormalities in mitochondrial structure and function (36,39). Recently, we studied abnormal mitochondrial dynamics in tissues from postmortem brains of patients at grade 3 (HD3) and grade 4 (HD4) stages of HD progression and from control subjects (36). In the striatum and cortex (HD-affected brain regions) but not in the cerebellum (non-HD-affected brain regions), we found increased expression of the fission genes, Drp1 and Fis1, and decreased expression of the fusion genes, Mfn1, Mfn2 and Opa1 (36). These results indicate that mitochondrial fragmentation and abnormal mitochondrial dynamics may be related to HD since these altered mitochondrial dynamics were not found in the cerebellum (36). Further, we also found significantly increased levels of the mitochondrial matrix gene, CypD, only in the striatum and cortex of HD3 and HD4 patients relative to control subjects, again indicating the presence of structurally damaged mitochondria in the HD-affected brain regions. However, the mechanistic link between mutant Htt and abnormal mitochondrial dynamics and axonal transport of mitochondria in neurons that express full-length human Htt with expanded polyQ repeats is not fully understood.
Dynamin-related protein 1 is evolutionarily highly conserved, a large GTPase protein reported to be involved in several important structural features of mitochondria, including shape, size, distribution, remodeling and the maintenance of mitochondria in mammalian cells (41). Further, recent research revealed that Drp1 is also associated with several mitochondrial structural and functional alterations, including fragmentation, phosphorylation, SUMOylation, ubiquitination and cell death. In addition, we recently found that Drp1 is interacted with amyloid beta (a toxic protein found in the brains of Alzheimer's disease patients), causing abnormal mitochondrial dynamics and neuronal damage in AD patients (44).
Recently, Gray et al. (18) developed BACHD transgenic mice that express the full-length (170 kb DNA) human Htt gene with 97 CAA and CAG (mixed repeats). The BACHD mice exhibit motor deficits by 2 months of age and exhibit selective, late-onset neuropathology in similar regions of the brain affected in humans with HD. Recently, several studies reported synaptic deficiencies and metabolic abnormalities in BACHD mice (45–53). Although BACHD mice have been used extensively to analyze mutant Htt-associated neurophysiological alterations and associated behavioral deficits, the model has yet to be characterized in terms of abnormal mitochondrial dynamics, defective axonal transport of mitochondria and abnormal synaptic changes.
In the current study, we used postmortem HD brains and tissues from BACHD mice to investigate mutant Htt interaction with Drp1, mitochondrial dynamics as well as Drp1 enzymatic activity. Further, using primary neurons from BACHD mice, we studied axonal transport of mitochondria and synaptic proteins. Finally, we investigated the effects of mutant Htt aggregates and oligomers in synaptic and mitochondrial deficiencies in HD postmortem brains and in BACHD mice.
RESULTS
Mutant Htt expression in 2-month-old BACHD mice
To establish the mutant Htt expression in a recently generated BACHD mouse line, we studied 2-month-old BACHD mice and age-matched, non-transgenic wild-type (WT) mice (control mice) using MAB 2166 (that recognizes both WT and mutant Htt) and 1C2 (that recognizes only expanded polyQ protein) antibodies. As shown in Figure 1 (upper panel), both WT and full-length mutant Htt were found in the 2-month-old BACHD mice (lanes 2 and 3), and only the full-length Htt was observed in WT mice (lane 1). Western blot analysis using the 1C2 antibody found four distinct bands expressed in the BACHD mice cortex: <350 kDa full-length mutant Htt and 230, 115 and 82 kDa cleaved products of mutant Htt. We also found 115 kDa in the WT mice. These results suggest that both mutant Htt were present early in the disease process in BACHD and that the two cleaved 230 and 82 kDa proteins may be specific to BACHD mice.
Figure 1.
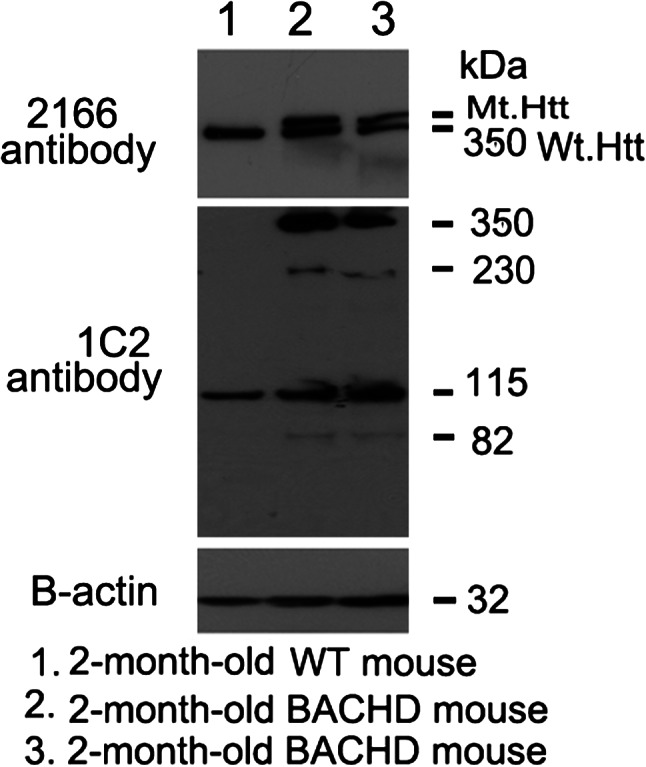
Identification of Htt in BACHD mice. Mutant Htt proteins were identified in cortical tissues from 2-month-old BACHD mice and control mice using two different Htt antibodies: MAB2166 (which recognizes both WT and mutant Htt) and 1C2 (which identifies expanded polyQ proteins). As shown, both full-length WT and mutant Htt proteins were found in the 2-month-old BACHD mice (lanes 2 and 3), and only full-length WT Htt was observed in the WT mice (lane 1). Western blot analysis using the 1C2 antibody (in the middle panel) showed that the BACHD mice cortex expressed four distinct bands: <350 kDa full-length mutant Htt, and 230, 115 and 82 kDa cleaved products of mutant Htt. A 115 kDa protein was identified in the WT mice. Bottom panel shows β-actin for equal loading.
Interaction of mutant Htt with Drp1 in HD patients
To determine whether mutant Htt interacts with Drp1, we conducted co-immunoprecipitation (IP) analysis, using cortical protein lysates from postmortem brain tissues of HD patients [Vonsattel Stage 3 (HD3) and Stage 4 (HD4)] and of age-matched control subjects, and antibodies to mutant Htt (1C2) and Drp1.
Our IP and immunoblotting analyses revealed an 82 kDa band of Drp1 expression in the IP elutes of cortical tissues from controls and HD3 and HD4 patients, with much higher expression levels in HD patient brains (Fig. 2A). Drp1 expression was much higher in lysates from HD3 and HD4 cortical samples compared with healthy controls (Fig. 2B). These findings suggest that the Drp1 antibody used for IP and immunoblotting is specific for Drp1.
Figure 2.

IP analysis in HD patients using the Drp1 antibody. (A) IP and immunoblotting analyses of Drp1 in cortical tissues from control subjects (lanes 1 and 2) and HD patients (lanes 3 and 4). (B) Immunoblotting analysis of Drp1 in control subjects (lanes 5 and 6) and HD patients (7 and 8). The Drp1 antibody recognizes the 82 kDa protein.
To determine whether the interaction of mutant Htt with Drp1 increases while HD progresses, we performed co-IP analysis with the Drp1 antibody, and ran immunoblotting analysis with the 1C2 antibody and also cortical protein lysates from control subjects and HD patients. As shown in Figure 3A, we found an 82 and a 40 kDa mutant Htt protein in IP elutes from HD3 and HD4 patients. We also found Drp1 interaction with WT Htt, but to a lesser extent, in the control subjects. As shown in Figure 3B, our immunoblotting analysis of protein lysates using the 1C2 antibody revealed an 82 and a 40 kDa mutant Htt in HD3 and HD4 patients but not in the control subjects, indicating that 1C2 is specific for expanded polyQ repeat protein in HD patients.
Figure 3.
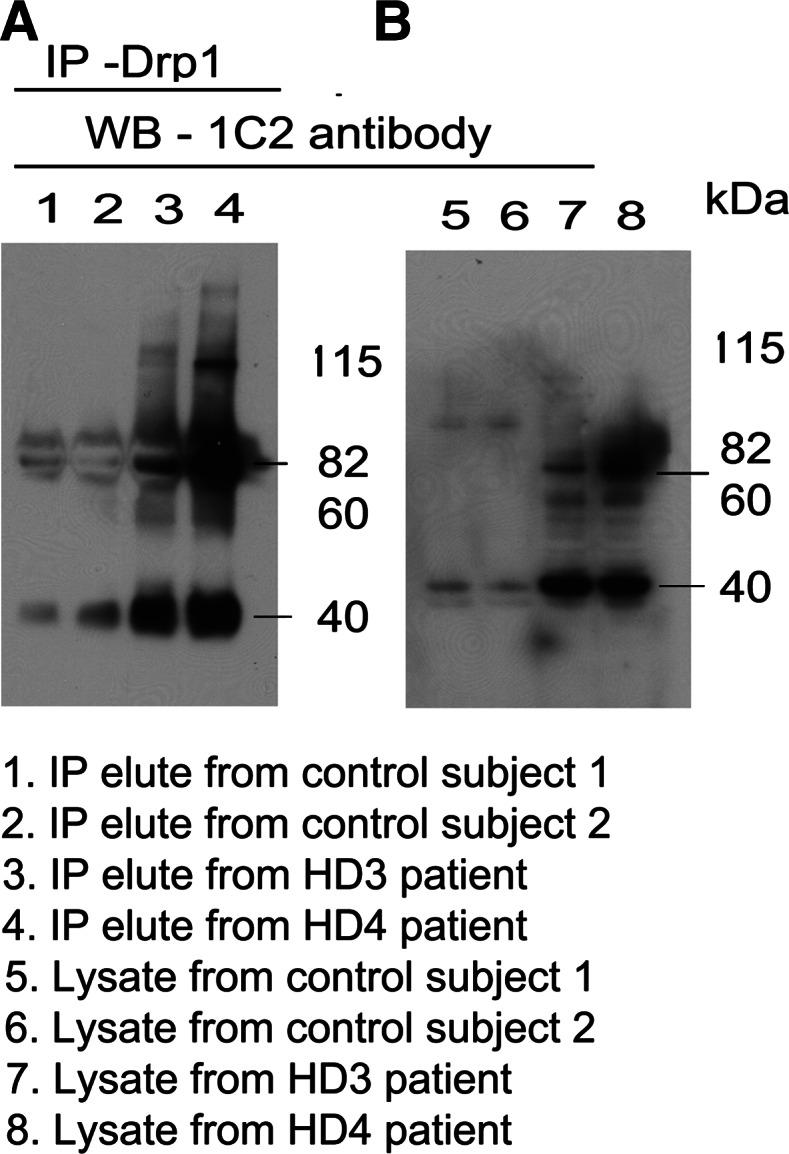
Co-IP analysis of Drp1 and mutant Htt antibodies in HD patients and control subjects. (A) IP with the Drp1antibody and immunoblotting with the mutant Htt-specific 1C2 antibody in control subjects (lanes 1 and 2) and HD patients (3 and 4). (B) Immunoblotting with the 1C2 antibody using protein lysates from control subjects (lanes 5 and 6) and HD patients (lanes 7 and 8). Mutant Htt-specific 1C2 antibody immunoreacted with two proteins: one with 82 kDa and the other with 40 kDa in IP elutes from HD patients.
Interaction of mutant Htt with Drp1 in BACHD mice
As shown in Figure 4A, we found an 82 kDa Drp1 protein in IP elutes of the cerebral cortex of WT and BACHD mice, and protein lysates from cortical WT and BACHD mice (Fig. 4B), indicating that the antibody used for IP and immunoblotting is specific for Drp1. Figure 5 shows co-IP analysis of Drp1 and immunoblotting with the 1C2 antibody. Two bands of proteins, one with an 82 kDa and the other with a 40 kDa mutant Htt protein were found in the IP elutes of the BACHD mice (lane 2), but not in those of the WT mice (lane 1) (Fig. 5A). Similar to protein in the IP elutes, we also found two bands of proteins, 82 and 40 kDa in the protein lysates of BACHD mice (lane 4; Fig. 5B).
Figure 4.
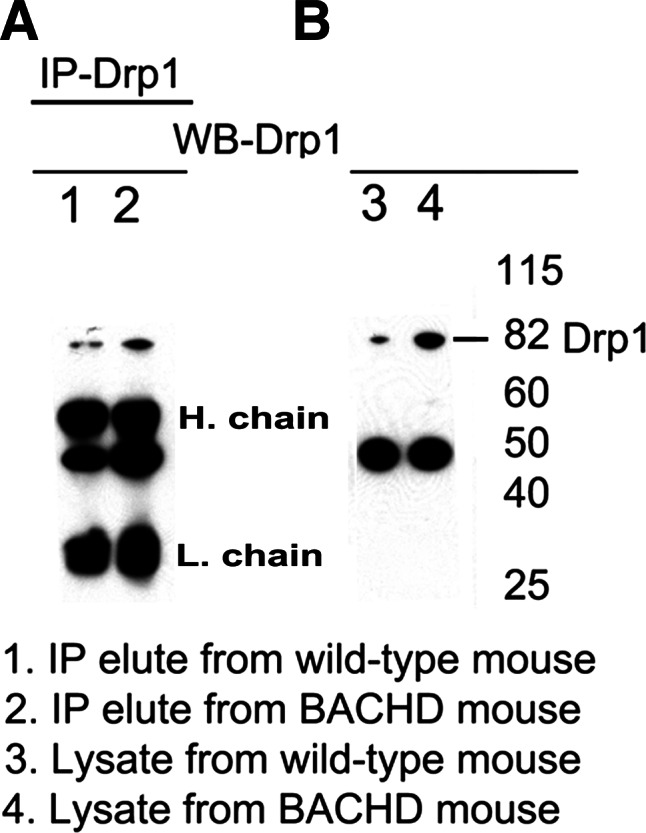
IP analysis in BACHD mice using the Drp1 antibody. (A) IP and immunoblotting analyses of Drp1 in cortical tissues from WT mice (lane 1) and BACHD mice (lane 2). (B) Immunoblotting analysis of Drp1 in WT mice (lane 3) and BACHD mice (lane 4).
Figure 5.

Co-IP analysis of Drp1 and mutant Htt antibodies in WT mice and BACHD mice. (A) IP with the Drp1 antibody and immunoblotting with the mutant Htt-specific 1C2 antibody in WT mice (lane 1) and BACHD mice (lane 2). (B) Immunoblotting with the 1C2 antibody using protein lysates from WT mice (lane 1) and BACHD mice (lane 2). The mutant Htt-specific 1C2 antibody immunoreacted with two proteins: one with 82 kDa and the other with 40 kDa in IP elutes from the BACHD mice.
Drp1 and mutant Htt in primary cortical neurons of BACHD mice
To determine whether the increased Drp1 expression in BACHD brains altered neuronal morphology or mitochondrial localization in the cytoplasm, we double-labeled primary cortical neurons taken at 4, 7 and 14 days in vitro (DIV) with Drp1 and 1C2. Increased Drp1 immunoreactivity was found in the neuronal branches and processes in the 4 DIV neurons and even higher in 7 and 14 DIV neurons. Interestingly, in the 7 and 14 DIV BACHD neurons, we found co-localization of Drp1 and 1C2 in the mitochondria of neurons, particularly at synapses, indicating mitochondrial fragmentation and degenerating synapses (Fig. 6). However in the WT neurons, 1C2 was not stained in all DIV cultures, whereas immunoreactivity of Drp1 was found in healthy branching dendrites and in the growth cones of neurons (data not shown).
Figure 6.

Time-course, double-labeling analysis of Drp1 and mutant Htt in cortical primary neurons from BACHD mice. Drp1 (green) and mutant Htt (1C2 in red) accumulate in the cell neurites and soma over time. Drp1 co-localization increased with mutant Htt in DIV 14.
Drp1 enzymatic activity in HD brains and BACHD mice
We determined whether Drp1 interaction with mutant Htt enhances GTPase Drp1 enzymatic activity in HD patients and BACHD mice. We studied Drp1 enzymatic activity in the cortex of HD3 patients (_n_= 3), HD4 patients (_n_= 3) and control subjects (_n_= 3), and in the striatum and cortex tissues of 2-month-old BACHD mice (_n_= 5) and age-matched WT control mice (_n_= 5). As shown in Figure 7A, we found significantly increased Drp1 enzymatic activity in the cortex tissues from the HD patients relative to control subjects (P < 0.005). We also found significantly increased levels of Drp1 enzymatic activity in the striatum and cerebral cortex tissues of the 2-month-old BACHD mice relative to the age-matched WT mice (striatum, P< 0.005; cortex, P< 0.05) (Fig. 7B), indicating that increased interaction of mutant Htt with Drp1 is associated with enhanced Drp1 enzymatic activity, leading to excessive fragmentation of mitochondria in HD neurons.
Figure 7.

Increased GTPase Drp1 enzymatic activity in HD patients and BACHD mice. (A) Significantly increased GTPase Drp1 enzymatic activity in the cortex of HD patients relative to control subjects. (B) Significantly increased Drp1 activity in the striatum and cortex of 2-month-old BACHD mice relative to non-transgenic littermates.
Decreased mitochondrial motility in BACHD hippocampal neurons
To determine the effect of mutant Htt on mitochondrial transport, we incubated hippocampal neurons from BACHD mice with pDsRed2-mito, at 2 DIV, and then we quantified mitochondrial motility at 12 DIV (Fig. 8). In BACHD transgenic neurons, we observed significantly decreased mitochondrial motility (20.88 ± 4.86%, mean ± SEM) relative to the WT mice neurons (36.76 ± 3.36%, mean ± SEM, P< 0.015). The number of mitochondria moving anterograde was significantly decreased in the BACHD neurons (10.04 ± 1.78%, mean ± SEM) relative to the WT mice neurons (21.58 ± 2.42%, mean ± SEM, P< 0.0009). A decrease in retrograde-moving mitochondria was also observed (10.84 ± 3.59%, mean ± SEM) in the BACHD neurons relative to the WT neurons (15.18 ± 2.38%, mean ± SE). However, this decrease did not reach statistical significance (_P_= 0.32). The average speed of mitochondrial movement was slightly elevated in the BACHD neurons (13.83 ± 2.22 µm/min, mean ± SEM, _P_= 0.66) relative to the WT neurons (12.7 ± 1.51, mean ± SEM), suggesting that mutant Htt impairs axonal transport of mitochondria in BACHD neurons.
Figure 8.

Primary neurons from BACHD mice show reduced motile mitochondria and also anterograde axonal transport of mitochondria. (A) Percent of motile mitochondria in neuronal cultures of BACHD and WT mice. (B) Mitochondrial speed in BACHD and WT neurons. (C) Kymographs show anterograde movement of mitochondria. *P < 0.02; **P < 0.0005.
Mitochondrial distribution in BACHD cortical neurons
To determine whether mutant Htt is involved in mitochondrial fragmentation and abnormal mitochondrial distribution in HD neurons, we transfected primary cortical neurons from WT and BACHD mice with MitoDsRed and GFP constructs at 2 DIV, and studied mitochondrial distribution at DIV 12. As shown in Figure 9, we found healthy, evenly distributed mitochondria along dendritic branches in the WT neurons. In contrast, the mitochondria in the BACHD mice were fragmented, and dendrites were deformed and had fewer branches.
Figure 9.

Mitochondria are more fragmented in primary cortical neurons from BACHD mice. Upper panel shows DsRed-mito (A) and GFP (B) transfected (C, merged) cortical neurons from WT mice, and lower panel shows DsRed-mito (D) and GFP (E) transfected (F, merged) cortical neurons from BACHD mice.
Drp1 dominant negative mutation and increased mitochondrial fusion in primary neurons
To determine whether Drp1 dominant negative mutation (K38A) decreases mitochondrial fragmentation, we transfected WT neurons with cDNAs of Drp1 K38A and WT-Drp1, and studied mitochondrial fragmentation. As shown in Figure 10B, we found increased mitochondrial fragmentation, concentrated mainly in the soma of neurons. In the neurons transfected with Drp1 K38A, we found elongated mitochondria distributed throughout neuron (Fig. 1C), including the soma, the neuronal process and terminals. These observations suggest that Drp1 dominant negative mutation reduces mitochondrial fragmentation and promotes mitochondrial fusion in neurons.
Figure 10.

Mitochondrial fragmentation is prevented in neurons transfected with the Drp1 dominant negative mutation K38A. (A) Hippocampal primary neuron showing mitochondrial fragmentation. (B) Primary neurons transfected with WT Drp1 showing increased fragmentation. (C) Elongated mitochondria in WT neurons transfected with the Drp1.K38A mutation.
Mutant Htt aggregates and oligomers in BACHD cortical neurons
To determine whether mutant Htt aggregates and oligomers are present in BACHD neurons, we performed immunostaining analyses using 1C2 (that recognizes expanded polyQ repeat protein) and A11 (oligomer-specific) antibodies in 10 DIV primary cortical neurons from BACHD mice. As shown in Figure 11, we found mutant Htt aggregates and oligomer immunoreactivity throughout the BACHD neurons, including in the cell body and neuronal processes. We also noticed that mutant Htt aggregates co-localize with oligomers in BACHD neurons. These findings suggest that mutant Htt aggregates and oligomers may promote neuronal damage and cell death in HD neurons.
Figure 11.

Mutant Htt aggregates co-localize with oligomers in BACHD primary neurons. (A) Immunoreactivity mutant Htt oligomers (immunolabeled with the A11 antibody). (B) Mutant Htt. (C) Co-localization of mutant aggregates and oligomers.
Decreased synaptic gene expression and increased mitochondrial fission genes in BACHD mice
To further investigate potential alterations in mitochondrial function in BACHD mice, we used quantitative real-time RT–PCR to measure mRNA expression levels of genes involved in mitochondrial dynamics and synapse formation and neuroprotective genes (Table 1), in cortical tissues from 2-month-old WT and BACHD mice. In six mitochondrial genes that we examined, four showed a statistically significant increase in mRNA expression levels, compared with WT mice. Drp1 and Fis1, which mediate mitochondrial fission, were significantly increased (7.1- and 1.8-fold, respectively), whereas Mfn1 and Mfn2 (fusion genes) were significantly decreased (−1.1- and −1.3-fold, respectively) in the BACHD mice relative to the WT mice. We also found Tomm40 (outer mitochondrial membrane protein) significantly increased (1.6-fold) in BACHD mice relative to WT mice. Additionally, we found a significant reduction in fold changes for the synaptic genes, synaptophysin (−1.2-fold) and PSD 95 (−1.3-fold) relative to WT mice. As shown in Table 1, PGC1α (−1.4-fold) and Sirt1 (NAD-dependent protein deacetylase, which regulates apoptosis) (−1.3-fold) were significantly reduced in the BACHD mice relative to WT mice.
Table 1.
mRNA fold change of synaptic, mitochondrial fission and fusion and neuroprotective genes in the cortex of BACHD mice relative to WT mice
| Mitochondrial dynamics | Synaptic and neuroprotective genes | ||
|---|---|---|---|
| Fission genes | |||
| Drp1 | 7.1** | PSD-95 | −1.3* |
| Fis1 | 1.8* | Synaptophysin | −1.2* |
| Fusion genes | |||
| Mfn1 | −1.1* | PCG1α | −1.1* |
| Mfn2 | −1.3* | Sirt1 | −1.3* |
| Tomm40 | 1.6* | ||
| Matrix gene | |||
| CypD | 1.6** |
Overall, our results indicate the presence of abnormal mitochondrial dynamics in HD progression. Up-regulation of the mitochondrial matrix gene, CypD, may be associated with mitochondrial structural damage in BACHD mice. These observations from BACHD mice concur with our earlier observations of HD postmortem brains (36).
Synaptic loss in BACHD neurons
To determine the effects of mutant Htt on synaptophysin and MAP2 levels, we performed immunostaining analysis of BACHD neurons using 10 DIV neurons from BACHD and WT mice. As shown in Figure 12A and B, immunoreactivity of synaptophysin was significantly decreased in the BACHD neurons relative to WT neurons (P< 0.001). Similar to synaptophysin, immunoreactivity of MAP2 was significantly decreased in the BACHD neurons relative to WT neurons (P< 0.01) (Fig. 12A and C), indicating that mutant Htt may be involved in synaptic degeneration.
Figure 12.

Double-labeling analysis of synaptophysin and MAP2 in BACHD and WT primary neurons. Upper panel shows immunoreactivity of synaptophysin (A), MAP2 (B), DAPI (C) and merged (D) in WT neurons. Lower panel shows immunoreactivity of synaptophysin (E), MAP2 (F), DAPI (G) and merged (H) in WT neurons. Images are taken with a confocal microscope using a 60× objective. (B) Quantification of synaptophysin in BACHD and WT neurons. Significantly decreased synaptophysin was found in BACHD neurons.
DISCUSSION
In a previous study, we examined mitochondrial electron transport chain genes, mitochondrial structural genes (fission and fusion), mitochondrial dynamics, oxidative damage, mutant Htt aggregates and oligomers in brain specimens from HD3 and HD4 patients and control subjects (36). We found abnormal mitochondrial dynamics (increased fission and decreased fusion) and altered electron transport chain genes in HD brains, both of which increased with HD progression. Mutant Htt aggregates and oligomers were significantly increased in HD3 and HD4 patients, and abnormal dynamics positively correlated with HD progression (HD3 to HD4) relative to the controls (36). Based on the postmortem brains from HD patients, we proposed that mutant Htt is associated with mitochondrial protein(s), alters dynamics of mitochondria and may cause mitochondrial fragmentation and impair axonal transport of mitochondria, ultimately leading to synaptic deficiencies and damaging HD neurons.
Using postmortem HD brains, brain tissues and primary neurons from BACHD mice, in the present study, we extended our previous investigations (i) to determine whether mutant Htt interacts with mitochondrial protein Drp1, (ii) to determine the extent that mutant Htt and Drp1 interaction influences GTPase Drp1 enzymatic activity (which is essential for mitochondrial fragmentation), (iii) to study mitochondrial axonal transport, and (iv) to study mitochondrial and synaptic deficiencies. We also investigated the relationship between mutant Htt aggregates/oligomers to abnormal mitochondrial dynamics and synaptic deficiencies in HD neurons.
We found that mutant Htt interacts with the mitochondrial protein Drp1, enhances GTPase Drp1 enzymatic activity and causes excessive mitochondrial fragmentation, and abnormal distribution, leading to defective anterograde transport of mitochondria and selective synaptic degeneration.
BACHD mice and mutant Htt
Using two different antibodies that recognize mutant Htt proteins, we found both full-length and cleaved products with expanded polyQ stretch of proteins in cortical lysates from BACHD mice. The 2166 MAB antibody recognizes full-length WT and mutant proteins. The 1C2 antibody recognized only proteins with expanded polyQ (53), and our immunoblotting results showed four different bands of 350, 230, 115 and 82 kDa sizes in BACHD mice, indicating the presence of several cleavage sites that cleave Htt proteins that contain polyQ repeat stretches in BACHD mice. However, a protein of 115 kDa was found in both WT and BACHD mice, indicating that the 1C2 antibody recognizes a protein in WT Htt. These observations agree with the earlier reports by Gray et al. (18), who studied brain specimens from BACHD mice and found several mutant Htt proteins.
Mutant Htt interaction with Drp1
By co-IP and immunostaining analyses, using postmortem brain tissues from HD patients and brain tissues from BACHD mice, we demonstrated that Drp1 interacts with mutant Htt. Interestingly, mutant Htt-cleaved products of 82 and 40 kDa appeared to interact with Drp1 (Figs 3 and 5), and these abnormal interactions progressively increased in HD3 and HD4 patients. Very little interaction between Drp1 and WT Htt was observed in control subjects and non-transgenic, WT mice, indicating that this interaction is mutant Htt-specific. Further, it is interesting to note that mutant Htt and Drp1 interact in cortical tissues from 2-month-old BACHD mice. These observations, in particular Drp1 interaction with mutant in 2-month-old BACHD mice, indicate that the interaction between mutant Htt and Drp1 is an early event in the disease process, and that the polyQ repeat length and disease process are critical for this interaction.
Our findings are supported by a recent study of Song et al. (39), in which they sought to determine mutant Htt interaction with Drp1, using lysates from transgenic mice lines, YAC18 (control) and YAC128 (mutant Htt). They found increased interaction of mutant Htt with Drp1 in YAC128 mice and little interaction in YAC18, control mice. Similar to our findings, they also found increased mutant Htt interaction with Drp1 in HD brains relative to control subjects. However, their study did not define the specific bands of mutant Htt proteins' interaction with Drp1. Our results identified the two bands of mutant Htt (82 and 40 kDa) that specifically interact with Drp1 and that this interaction is polyQ-specific to Drp1.
Our extensive time-course double-labeling immunofluorescence analyses of Drp1 and mutant Htt-specific 1C2 antibodies revealed the formation of mutant Htt aggregates and that these aggregates interact with Drp1 in an age-dependent manner (Fig. 6). These formations of mutant Htt aggregates and Drp1 immunoreactivity in 14 DIV relative to 4 DIV further support the notion that these aggregates interact with Drp1 in an age-dependent manner.
Our findings, together with those from Song et al. (39), indicate that mutant Htt and Drp1 interact. These interactions may be involved in abnormal mitochondrial dynamics (increased fission and decreased fusion), may fragment mitochondria excessively and may ultimately cause neuronal damage.
Increased GTPase Drp1 enzymatic activity in HD brains and BACHD mice
In a previous study, we demonstrated increased mitochondrial fragmentation and decreased fusion in postmortem brains (36), but the cause of this increased fragmentation was not clear. In this study, using lysates from HD brains and control subjects and from BACHD and control WT mice, we found GTPase Drp1 enzymatic activity elevated, causing excessive mitochondrial fragmentation. Interestingly, we also found significantly increased Drp1 enzymatic activity in the cortex of HD patients relative to the control subjects, and in the striatum and cerebral cortex of BACHD mice relative to WT mice. We did not find increased Drp1 enzymatic activity in HD-unaffected region, cerebellum both HD patients and non-transgenic WT littermates of BACHD mice (data not shown). These observations confirmed our hypothesis that increased interaction between mutant Htt and Drp1 enhances Drp1 enzymatic activity, excessively fragments mitochondria and causes abnormal mitochondrial dynamics selectively in affected regions of HD brain.
Drp1 dominant mutation prevents mitochondrial fragmentation
Our findings that WT neurons transfected with WT-Drp1 show increased mitochondrial fragmentation in the cell body, and presence of less number of mitochondria in neurites and nerve terminals. On the contrary, elongated and uniformly distributed mitochondria were observed in WT neurons transfected with Drp1.K38A (dominant mutation). These observations further support that Drp1 is critical for mitochondrial fragmentation. Our findings concur with recent studies of Drp1 knockout mice (54,55) and cell culture studies of mutant Htt by Song et al. (39) and Wang et al. (37), all of which demonstrated that a lack of Drp1 or the presence of mutant Drp1 enhances mitochondrial fusion and decreases fission.
Further, recent studies of Drp1 knockout in mice revealed that double-knockouts are embryonic lethal (54,55) because of the presence of elongated mitochondria and decreased mitochondrial division in cells that caused developmental defects in embryos. However, primary neurons from Drp1 knockout mice (−/−) forebrain showed a decrease in the number of neurites and also the formation of defective synapses. These defects highlight the importance of Drp1-dependent decreased mitochondrial fission in Drp1−/− knockout neurons. Additional findings from studies that sought to determine mitochondrial fission and fusion in neurons also revealed that mitochondria formed extensive networks and elongated. These observations together with the findings of the present study (increased Drp1-negative mutation K38A caused excessive mitochondrial fusion and decreased fusion in neurons) suggest that a balanced or partial loss of Drp1 expression may protect neurons, particularly in a disease such as HD.
Defective mitochondrial transport and abnormal mitochondrial distribution in HD neurons
For the first time, we demonstrated significantly decreased total mitochondrial motility and anterograde-moving mitochondria in BACHD neurons relative to WT neurons. However, we found that the movement of retrograde mitochondria and the speed of moving mitochondria were unaffected in BACHD mice. Our observations agree with a previous study by Song et al. (39).
Our double-labeling analysis of BACHD primary cortical neurons with cDNAs of MitoDsRed and GFP revealed that increased numbers of mitochondria in the cell soma and reduced numbers of mitochondria in neuronal processes, neurite and terminals indicate that excessively fragmented mitochondria remain in the soma and are not able to transport to neuronal processes.
For the first time, we demonstrated that BACHD mice that express human full-length Htt with 97 polyQ repeats showed excessive fragmented mitochondria. Our observations were supported by findings from Song et al. (39). In a study in which they transfected exon 1 Htt with three different polyQ repeat lengths—16, 46 and 97—they reported that neurons with exogenously expressed exon 1 Htt with 17 polyQ repeats exhibited filamentous, normal and healthy mitochondria, whereas neurons expressing exon 1 Htt with 46 polyQ showed both elongated and round mitochondria. Neurons that exogenously expressed 97 polyQ repeats show mainly rounded and fragmented mitochondria. The findings of the present study together with the observations of Song et al. (39) suggest that mutant Htt with expanded polyQ repeats are responsible for mitochondrial fragmentation in HD neurons and that the fragmentation of mitochondria occurs along the length of polyQ and the stage of disease process and/or is age-dependent.
We recently reported on the increased interaction of amyloid beta (a toxic protein found in patients with Alzheimer's disease) with Drp1, which resulted in elevated Drp1 enzymatic activity, excessive mitochondrial fragmentation and altered mitochondrial distribution in neurons affected by the disease (44,56–58). We reported a similar finding in a study of HD, in which the interaction of mutant Htt had similar consequences: this interaction resulted in elevated Drp1 enzymatic activity, excessive mitochondrial fragmentation and altered mitochondrial distribution in HD neurons. These parallel findings for two different neurodegenerative diseases suggest a common pathway that may be involved in triggering abnormal mitochondrial dynamics and selective neuronal damage.
Synaptic and mitochondrial deficiencies in HD neurons
Synaptic deficiencies and mitochondrial ATP alterations likely contribute to the clinical symptoms in HD patients, such as chorea, dystonia and cognitive decline. We found significantly decreased mRNA levels of synaptic genes, synaptophysin and PSD95 in BACHD neurons, indicating that synaptic loss may be an early event in HD progression in BACHD mice. Our findings of increased levels of Drp1 and Fis1, CypD and Tomm40, and decreased levels of Mfn1 and Mfn2 in BACHD mice further support the presence of abnormal mitochondrial dynamics in BACHD mice. Our findings that mRNA levels of PGC1α and Sirt1 were decreased in BACHD mice not only agree with earlier research in HD (59–62), but also support the role of PGC1α and Sirt1 in promoting synaptic viability and neuronal protection.
We found significantly decreased immunoreactivity of synaptophysin in BACHD primary neurons relative to WT neurons, further supporting that synaptic deficiencies are present early on in the disease process.
Findings from our previous study of mutant Htt and mitochondria (36) together with those from the present study suggest that increased levels of mutant Htt cause abnormal mitochondrial dynamics and increased mitochondrial fragmentation in HD neurons. Mutant Htt-induced fragmented mitochondria are localized mainly in the cell body, and not able to transport to axons, dendrites and synapses, thus producing low mitochondrial ATP at synapses, leading to synaptic degeneration.
CONCLUSIONS
The purpose of our study was to understand how mutant Htt and mitochondria interact with each other and the consequences of this interaction. We studied this interaction and the effect of this interaction on mitochondrial damage and synaptic degeneration in HD. We found that mutant Htt interacts with the mitochondrial protein Drp1, which in turn enhances GTPase Drp1 enzymatic activity, and this enzymatic activity appears to cause excessive mitochondrial fragmentation and abnormal mitochondrial distribution in the HD neurons. These problems ultimately lead to selective synaptic degeneration in neurons affected by HD. With a better understanding of abnormal mitochondrial dynamics, and the role of mutant Htt interaction with Drp1 being clarified, therapeutic strategies can be developed to reduce the interaction between mutant Htt and Drp1, and to prohibit GTPase Drp1 enzymatic activity, thus preventing excessive mitochondrial fragmentation and abnormal mitochondrial distribution in HD neurons.
MATERIALS AND METHODS
Postmortem brains
Nine postmortem frozen brain specimens from the frontal cortex of HD patients and age-matched healthy subjects (controls) were obtained from the Harvard Tissue Resource Center. Three specimens were from patients with HD3 [graded according to VonSattel _et al_. (9)], three were from patients with HD4 and three were from the controls. Demographic details are described in a previous publication (36).
BACHD mice and primary neuronal cultures
To understand the mutant Htt effects in mitochondrial and synaptic dynamics, using brain tissues and primary neurons from the BACHD mice and controls (18), we investigated the expression of mutant Htt and its effects to mitochondrial dynamics and synaptic processes. The BACHD mice and non-transgenic littermates were housed at the Oregon National Primate Research Center of Oregon Health & Science University (OHSU). The OHSU Institutional Animal Care and Use Committee approved all procedures for animal care according to guidelines set forth by the National Institutes of Health. We genotyped for the HD transgene, using the DNA prepared from a tail biopsy of day-1 pups, following the protocol described in Gray et al. (18).
Primary neurons were prepared for our mutant Htt, mitochondrial and synaptic studies, as previously described (58,63). Briefly, day-1 mice were decapitated, and the brains were removed and maintained in room temperature with HABG (Hibernate A medium, Brain Bits, Springfield, IL, USA) supplemented with B-27 (Invitrogen, Carlsbad, CA, USA) and 0.5 mm GlutaMAX (Invitrogen). The cortex or hippocampus was then dissected and reserved for culture, and the cerebellum was used in this study for genotyping. The tissue was minced and then transferred to a solution of 2 mg/ml papain (Worthington Biochemical Corporation, Lakewood, NJ, USA), dissolved in Hibernate A without calcium (Brain Bits) and then supplemented with 0.5 mm GlutaMAX. Tissue was digested for 30 min at 30°C in a shaking water bath and then removed to 2 ml of HABG. Digested tissue was triturated 10 times with a fire-polished, siliconized, 9 in. glass pipette. Non-dissociated tissue was allowed to settle for 2 min, and then the supernatant was removed to a fresh tube. An additional 2 ml of HABG was added, and the process was repeated until 6 ml of dissociated cells were collected. Cells were centrifuged at 1000_g_ for 2 min and then washed with 2 ml of HABG. The pellets were resuspended in Neurobasal (Invitrogen) supplemented with B-27 and 0.5 mm GlutaMAX. Live cells were counted using the trypan blue exclusion method, and were plated at 500 cells/mm2 on poly-d-lysine-coated cover slips. Cells were moved to a 37°C, 5% CO2 incubator and allowed to adhere for 1 h. The medium was then changed to Neurobasal, supplemented with B-27 minus antioxidants and 0.5 mm GlutaMAX. One-half the growth medium was changed every 3 days.
Real time RT–PCR
Using RT–PCR, we measured mRNA expression levels of mitochondrial (Drp1, Fis1, Mfn1, CypD and Tomm40), neuroprotective (PGC1a and Sirt1) and synaptic (synaptophysin and PSF95) genes in the cortex of BACHD mice, as described in Shirendeb et al. (36), Manczak et al. (63) and Gutala and Reddy (64). Using the Primer Express software (Applied Biosystems), we designed the oligonucleotide primers for the housekeeping genes (β-actin, GAPDH, synaptic) and the mitochondrial genes. The oligonucleotide sequences for genes studied in this paper were: Drp1 forward primer 5′ GCGCTGATCCCGCGTCAT 3′ and reverse primer 5′ CCGCACCCACTGTGTTGA 3′; Fis1 forward primer 5′ GCCCCTGCTACTGGACCAT and reverse primer 5′ CCCTGAAAGCCTCACACTAAGG 3′; Mfn1 forward primer 5′ TCTCCAAGCCCAACATCTTCA 3′ and reverse primer 5′ ACTCCGGCTCCGAAGCA 3′; Mfn2 forward primer 5′ ACAGCCTCAGCCGACAGCAT 3′ and reverse primer 5′ TGCCGAAGGAGCAGACCTT 3′; Tomm40 forward primer 5′ CCTGCCCTCTGACCTTTCC and reverse primer 3′ GAGAGCTGGCAGCCAACAC 5′; CypD forward primer 5′ CAGCCAAGCCCTCCAACTC 3′ and reverse primer 5′ GCCGATGTCCACGTCAAAG 3′; synaptophysin forward primer 5′ CATTCAGGCTGCACCAAGTG 3′ and reverse primer 5′ TGGTAGTGCCCCCTTTAACG 3′; PSD95 forward primer 5′ GGACATTCAGGCGCACAAG and reverse primer 5′ TCCCGTAGAGGTGGCTGTTG 3′; PGC1α forward primer 5′ GGACAGTCTCCCCGTGGAT 3′ and reverse primer 5′ TCCATCTGTCAGTGCATCAAATG 3′; Sirt 1 forward primer 5′ GCCGCGGATAGGTCCATATA 3′ and reverse primer 5′ TCGAGGATCGGTGCCAAT 3′; β- actin forward primer 5′ ACGGCCAGGTCATCACTATTC 3′ and reverse primer 5′ AGGAAGGCTGGAAAAGAGCC 3′; GAPDH forward primer 5′ TTCCCGTTCAGCTCTGGG 3′ and reverse primer 5′ CCCTGCATCCACTGGTGC 3′.
Briefly, total RNA was isolated from neurons representing three independent cultures, in six-well plates, using TRIzol. Reverse transcription was performed with 2 μg of total RNA from each sample, using the Superscript III First Strand Synthesis System for RT–PCR (Invitrogen). RNA was combined with oligo-dT20, 1μl of oligo (dt) and 1μl of dNTPs (10 mm each) in a total volume of 12 μl; and then the mixture was heated to 65°C for 5 min. It was chilled on ice, and then 4 μl of 5× first-strand buffer, 2 μl of 0.1 m DTT and 1μl of RNAse out were added. Samples were incubated at 42°C for 2 min, and then 1μl of Superscript III (40 U/ml) was added. After a 50 min incubation at 42°C, the reaction was inactivated by heating the mixture to 70°C for 15 min.
Real-time quantitative PCR was performed using an ABI PRISM 7900HT Sequence Detection System (Applied Biosystems, Carlsbad, CA, USA) in a 25 μl volume. The reaction mixture for each primer comprised 1× PCR buffer, 2 mm MgCl2, 250 μm dNTPs, 0.3× SYBR Green, 3.12% DMSO, 0.015 U/μl GoldTaq, 50 ng cDNA and 200 nm primers. Both β-actin and GAPDH were measured as housekeeping genes that we used to normalize the gene expression data. However, we chose β-actin for normalization because this non-mitochondrial gene generates a less variable _C_T value between samples. _C_T values for gene products were normalized to β-actin _C_T values, and comparisons were made between experimental groups, using the Δ_C_T method. Briefly, the Δ_C_T value was calculated for each sample (_C_T gene of interest minus _C_T β-actin). Then the calibrator value was averaged (Δ_C_T) for the control samples. The calibrator was subtracted from the Δ_C_T for each control and from the experimental sample to derive the ΔΔ_C_T. The fold change was calculated as 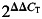 . Average fold change was calculated for each experimental group.
. Average fold change was calculated for each experimental group.
Western blot analysis
Using western blot analysis, we detected the mutant Htt protein expression using cortical protein lysates from BACHD and control mice. Protein concentrations were determined with the BCA protein assay (Pierce/Thermo Scientific). Ten percent polyacrylamide gels (Invitrogen) were loaded with 30 μg of protein per well as described in Manczak et al. (44). Using 3–8% Tris acetate gels, 30 μg of protein lysate was resolved to determine the mutant Htt proteins as described previously in Gray et al. (18). The resolved proteins were transferred to PVDF membranes using transfer buffer (Tris 25 mm, Glycine 190 mm, 20% methanol) (Millipore, San Diego, CA, USA) overnight at 15 V (Perkin Elmer). Membranes were then incubated for 1 h at room temperature, in a blocking buffer (5% dry milk dissolved in TBST). The nylon membranes were incubated overnight with the primary antibodies 1C2 (to identify expanded polyQ protein; 1:300; mouse monoclonal, Millipore) (65) and MAB 2166 (to identify both control and mutant Htt; 1:500; mouse monoclonal, Millipore). Following Calkins et al. (58), the membranes were washed with a TBST buffer three times at 10 min intervals and then incubated for 2 h with appropriate secondary antibodies, followed by three additional washes at 10 min intervals. Proteins were detected with the Supersignal West Pico chemiluminescent reagent (Thermo Scientific). Images of the exposed X-ray film were taken to visualize WT and mutant Htt proteins.
IP of Drp1 and mutant Htt
To determine the interaction between Drp1 and mutant Htt, we performed co-IP using affected and unaffected protein lysates from a large number of brains from HD3 and HD4 patients, from control subjects and from 2-month-old BACHD mice, as described in Manczak et al. (44). Briefly, we used a Dynabeads kit for IP (Invitrogen, Temicula, CA, USA). Fifty microliters of Dynabeads containing protein G was incubated with 10 µg of Drp1 antibodies for 1 h at room temperature, with rotation. The Dynabeads were then washed three times with a washing buffer and incubated with rotation overnight with 400 µg of lysate protein at 4°C. The incubated Dynabead–antigen/antibody complexes were washed again three times with a washing buffer, and an immunoprecipitant was eluted from the Dynabeads, using the NuPAGE LDS sample buffer. The IP elute was loaded onto a gel, followed by western blot analysis of Drp1 and mutant Htt (1C2) antibodies.
Mitochondrial motility
To determine whether mutant Htt impairs mitochondrial transport, we assessed mitochondrial motility in the primary neurons from BACHD and control mice, as previously described (58). Briefly, mitochondria were labeled by transfecting pDsRed2-mito (Clontech) and GFP (Clontech) into the hippocampal neurons at day 2 (DIV) with Lipofectamine 2000 (Invitrogen), according to the manufacturer's protocol. Mitochondrial motility was evaluated at DIV 12. Axonal processes were determined by morphological characteristics. Axons were identified as processes stemming from the soma if they were two to three times longer than other processes. Recordings were made on axonal segments, ∼20–100 µm from the soma. A series of time-lapse images was captured every 5 s, using a Leica SP5 AOBS confocal microscope with a heated (37°C), 5% CO2-controlled stage for 5 min. Z-stacks at each time point were collapsed to maximum projections, and the time series was archived as avi files. ImageJ software, with a mulitple kymograph plug-in, was used to analyze the avi files. Mitochondrial movements (direction and speed) were determined from the kymographic images. Mitochondria were considered stationary if they did not move >2 μm during the entire recording period. Each series of images was recorded for at least three randomly selected DsRed-mito-labeled cells per culture and for four cultures of independent pups.
Mitochondrial content in neurites
To determine the mitochondrial distribution in primary neurons from BACHD and WT mice, we assessed the mitochondria in neurites in the neuronal cultures from BACHD and control mice. Neuronal cultures were fixed with 4% paraformaldehyde (PFA) in PBS (PFA/PBS) for 10 min at room temperature and then washed with PBS. Images of cell bodies with neurites extending at least 100 mm were collected using a Leica SP5 AOBS confocal microscope with a 63× objective. GFP and DsRed were analyzed with the measurement tools in ImageJ, to determine the mitochondrial index of the neurites, the average mitochondrial length and the number of mitochondria per neurite length. Data were collected from at least four cells per culture and six cultures of independent pups per condition.
Immunocytochemistry of primary neurons
To determine the localization of mutant Htt, A11 oligomers and mitochondrial and synaptic proteins in BACHD neurons, we performed immunocytochemical analysis, following Calkins et al. (58). We used the following primary antibodies: Drp1 (1:200), 1C2 (1:200), A11 (rabbit polyclonal; 1:50; Invitrogen), MAP2 (1:300, rabbit polyclonal; Millipore) and synaptophysin (1:200, mouse monoclonal; Millipore). Briefly, we plated cells on 13 mm round cover slips coated with poly-d-lysine contained in a 24-well plate. After 14 DIV, the media were removed, and the cells were fixed with 4% PFA in PBS for 10–15 min at room temperature. Cover slips were washed with PBS, cell membranes were permeablized with 0.1% Triton X-100 in PBS for 5 min and then a blocking solution was applied (2% normal goat serum, 1% BSA in PBS). All incubations were carried out in a humidified environment. Samples were blocked for 2 h at room temperature and then incubated overnight with primary antibodies diluted in blocking solutions, at 4°C. After the primary antibodies were incubated, the cells were washed three times with PBS and then incubated with either goat-anti-rabbit-Alexa488 or goat-anti-mouse-Alexa568 (both 1:500, Invitrogen/Molecular Probes) for 2 h at room temperature. The cells were then washed three times again in PBS, and some sections were counterstained with DAPI (1:1000, KPL, Gaithersburg, MD, USA) (blue) for nuclear labeling. Then the cover slips were mounted on slides with a ProLong Gold antifade mounting reagent (Invitrogen). Cells were imaged using a Zeiss Axioskop 40 FL microscope.
Double-labeling analysis
To determine the interaction between Drp1 and mutant Htt, and A11 and mutant Htt, we performed double-labeling immunofluorescence analysis, using an anti-Drp1 antibody, 1C2 and A11, and primary cortical neurons as described in Manczak et al. (44) and Shirendeb et al. (36). For the first labeling, the slides were incubated overnight at 4°C with the anti-Drp1 antibody (1:200), under appropriate conditions and with appropriate reagents. For the second labeling, slides were incubated overnight at 4°C with 1C2 (1:200) or A11 (1:50 dilution, rabbit polyclonal). Next, the slides were incubated with appropriate secondary antibodies conjugated with Alexa 488 or 568 and then cover-slipped with ProLong Gold and photographed with a confocal microscope.
Drp1 and Drp1.K38A mutation transfections to WT neurons
To determine whether Drp1 dominant mutation, K38A, reduces mitochondrial fragmentation and enhances mitochondrial distribution in hippocampal neurons, we transfected WT neurons with pDsRed2-mito (Clontech) and Drp1 (WT) construct, and pDsRed2-mito (Clontech) and Drp1.K38A mutation constructs to day-2 WT hippocampal neurons separately with Lipofectamine 2000 (Invitrogen), according to the manufacturer's protocol. Neurons were harvested at DIV 12 and neuronal cultures were fixed with 4% PFA in PBS (PFA/PBS) for 5 min at room temperature and then washed with PBS. Neurons were processed for mitochondrial labeling and cover-slipped with ProLong Gold and photographed with a confocal microscope.
GTPase enzymatic activity of Drp1
Using a Novus Biological calorimetric kit (Littleton, CO, USA), we measured GTPase Drp1 enzymatic activity in frozen cortical tissues from HD patients, control subjects and 2-month-old BACHD mice. In a GTPase assay, Drp1 hydrolyzes GTP to GDP and inorganic phosphorous (Pi). We measured GTPase activity, based on the amount of inorganic phosphorous that the GTP produces. By adding the ColorLock Gold (orange) substrate to the inorganic phosphorous that is generated from GTP, we assessed GTP activity, based on the inorganic complex solution (green). Calorimetric measurements (green) were read in the wavelength range of 650 nm. We compared GTPase Drp1 activity in HD3 and HD4 patients and in control subjects. We also compared GTPase Drp1 enzymatic activity between BACHD mice and control mice.
Statistical analysis
All data were analyzed using the Prism3 software from GraphPad Software (San Diego, CA, USA). Results are expressed as mean ± SEM. The data were analyzed using an unpaired Student's _t_-test. The data from GTPase Drp1 enzymatic activity were retrieved from the cortex and striatum tissues and compared, to determine whether Drp1 activity is confined to HD-affected regions of the brain or whether Drp1 activity also occurs in unaffected regions. Statistical significance was attained at P ≤ 0.05.
FUNDING
This research was supported by NIH grants AG028072 (to P.H.R.), RR00163 (to P.H.R. and J.L.M., ONPRC) (P30-NS061800, Aicher, PI), NS31862 (Harvard Tissue Resource Center, Belmont, MA, USA) and Hereditary Disease Foundation (to J.L.M.) and Alzheimer Association grant IIRG-09-92429 (to P.H.R.).
ACKNOWLEDGEMENTS
We sincerely thank Dr van der Bliek, University of California, Los Angeles, for providing Drp1 cDNA constructs, and Dr Anda Cornea for her assistance with confocal imaging and mitochondrial transport assessment.
Conflict of Interest statement. Authors do not have conflict of interest in research presented in this paper.
REFERENCES
- 1.Folstein S.E. Huntington's Disease. Johns Hopkins University Press; 1990. [Google Scholar]
- 2.Montoya A., Price B.H., Menear M., Lepage M. Brain imaging and cognitive dysfunctions in Huntington's disease. J. Psychiatry Neurosci. 2006;31:21–29. [PMC free article] [PubMed] [Google Scholar]
- 3.Ross C.A., Tabrizi S.J. Huntington's disease: from molecular pathogenesis to clinical treatment. Lancet Neurol. 2011;10:83–98. doi: 10.1016/S1474-4422(10)70245-3. doi:10.1016/S1474-4422(10)70245-3. [DOI] [PubMed] [Google Scholar]
- 4.Kirkwood S.C., Su J.L., Conneally P., Foroud T. Progression of symptoms in the early and middle stages of Huntington disease. Arch. Neurol. 2001;58:273–278. doi: 10.1001/archneur.58.2.273. doi:10.1001/archneur.58.2.273. [DOI] [PubMed] [Google Scholar]
- 5.Mahant N., McCusker E.A., Byth K., Graham S. Huntington Study Group. Huntington's disease: clinical correlates of disability and progression. Neurology. 2003;61:1085–1092. doi: 10.1212/01.wnl.0000086373.32347.16. [DOI] [PubMed] [Google Scholar]
- 6.Hamilton J.M., Wolfson T., Peavy G.M., Jacobson M.W., Corey-Bloom J. Huntington Study Group. Rate and correlates of weight change in Huntington's disease. J. Neurol. Neurosurg. Psychiatry. 2004;75:209–212. doi: 10.1136/jnnp.2003.017822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Aziz N.A., van der Burg J.M., Landwehrmeyer G.B., Brundin P., Stijnen T. Roos R.A. EHDI Study Group. Weight loss in Huntington disease increases with higher CAG repeat number. Neurology. 2008;71:1506–1513. doi: 10.1212/01.wnl.0000334276.09729.0e. doi:10.1212/01.wnl.0000334276.09729.0e. [DOI] [PubMed] [Google Scholar]
- 8.Phan J., Hickey M.A., Zhang P., Chesselet M.F., Reue K. Adipose tissue dysfunction tracks disease progression in two Huntington's disease mouse models. Hum. Mol. Genet. 2009;18:1006–1016. doi: 10.1093/hmg/ddn428. doi:10.1093/hmg/ddn428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vonsattel J.P., Myers R.H., Stevens T.J., Ferrante R.J., Bird E.D., Richardson E.P. Neuropathological classification of Huntington's disease. J. Neuropathol. Exp Neurol. 1985;44:559–577. doi: 10.1097/00005072-198511000-00003. doi:10.1097/00005072-198511000-00003. [DOI] [PubMed] [Google Scholar]
- 10.Vonsattel J.P. Huntington disease models and human neuropathology: similarities and differences. Acta Neuropathol. 2008;115:55–69. doi: 10.1007/s00401-007-0306-6. doi:10.1007/s00401-007-0306-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Halliday G.M., McRitchie D.A., Macdonald V., Double K.L., Trent R.J., McCusker E. Regional specificity of brain atrophy in Huntington's disease. Exp. Neurol. 1998;154:663–672. doi: 10.1006/exnr.1998.6919. doi:10.1006/exnr.1998.6919. [DOI] [PubMed] [Google Scholar]
- 12.Byers R.K., Gilles F.H., Fung C. Huntington's disease in children: neuropathologic study of four cases. Neurology. 1973;23:561–569. doi: 10.1212/wnl.23.6.561. [DOI] [PubMed] [Google Scholar]
- 13.Spargo E., Everall I.P., Lantos P.L. Neuronal loss in the hippocampus in Huntington's disease: a comparison with HIV infection. J. Neurol. Neurosurg. Psychiatry. 1993;56:487–491. doi: 10.1136/jnnp.56.5.487. doi:10.1136/jnnp.56.5.487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Politis M., Pavese N., Tai Y.F., Tabrizi S.J., Barker R.A., Piccini P. Hypothalamic involvement in Huntington's disease: an in vivo PET study. Brain. 2008;131:2860–2869. doi: 10.1093/brain/awn244. doi:10.1093/brain/awn244. [DOI] [PubMed] [Google Scholar]
- 15.Soneson C., Fontes M., Zhou Y., Denisov V., Paulsen J.S., Kirik D., Petersén A. Huntington Study Group PREDICT-HD Investigators. Early changes in the hypothalamic region in prodromal Huntington disease revealed by MRI analysis. Neurobiol. Dis. 2010;40:531–543. doi: 10.1016/j.nbd.2010.07.013. doi:10.1016/j.nbd.2010.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.The Huntington's Disease Collaborative Research Group. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. Cell. 1993;72:971–983. doi: 10.1016/0092-8674(93)90585-e. doi:10.1016/0092-8674(93)90585-E. [DOI] [PubMed] [Google Scholar]
- 17.Reddy P.H., Mao P., Manczak M. Mitochondrial structural and functional dynamics in Huntington's disease. Brain. Res. Rev. 2009;61:33–48. doi: 10.1016/j.brainresrev.2009.04.001. doi:10.1016/j.brainresrev.2009.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gray M., Shirasaki D.I., Cepeda C., André V.M., Wilburn B., Lu X.H., Tao J., Yamazaki I., Li S.H., Sun Y.E., et al. Full-length human mutant huntingtin with a stable polyglutamine repeat can elicit progressive and selective neuropathogenesis in BACHD mice. J. Neurosci. 2008;28:6182–6195. doi: 10.1523/JNEUROSCI.0857-08.2008. doi:10.1523/JNEUROSCI.0857-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mangiarini L., Sathasivam K., Seller M., Cozens B., Harper A., Hetherington C., Lawton M., Trottier Y., Lehrach H., Davies S.W., Bates G.P. Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell. 2006;87:493–506. doi: 10.1016/s0092-8674(00)81369-0. doi:10.1016/S0092-8674(00)81369-0. [DOI] [PubMed] [Google Scholar]
- 20.DiFiglia M., Sapp E., Chase K.O., Davies S.W., Bates G.P., Vonsattel J.P., Aronin N. Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science. 1997;277:1990–1993. doi: 10.1126/science.277.5334.1990. doi:10.1126/science.277.5334.1990. [DOI] [PubMed] [Google Scholar]
- 21.Davies S.W., Turmaine M., Cozens B.A., DiFiglia M., Sharp A.H., Ross C.A., Scherzinger E., Wanker E.E., Mangiarini L., Bates G.P. Formation of neuronal intranuclear inclusions underlies the neurological dysfunction in mice transgenic for the HD mutation. Cell. 1997;90:537–548. doi: 10.1016/s0092-8674(00)80513-9. doi:10.1016/S0092-8674(00)80513-9. [DOI] [PubMed] [Google Scholar]
- 22.Reddy P.H., Williams M., Charles V., Garrett L., Pike-Buchanan L., Whetsell W.O., Jr., Miller G., Tagle D.A. Behavioural abnormalities and selective neuronal loss in HD transgenic mice expressing mutated full-length HD cDNA. Nat. Genet. 1998;20:198–202. doi: 10.1038/2510. doi:10.1038/2510. [DOI] [PubMed] [Google Scholar]
- 23.Schilling G., Becher M.W., Sharp A.H., Jinnah H.A., Duan K., Kotzuk J.A., Slunt H.H., Ratovitski T., Cooper J.K., Jenkins N.A., et al. Intranuclear inclusions and neuritic aggregates in transgenic mice expressing a mutant N-terminal fragment of huntingtin. Hum. Mol. Genet. 1999;8:397–407. doi: 10.1093/hmg/8.3.397. doi:10.1093/hmg/8.3.397. [DOI] [PubMed] [Google Scholar]
- 24.Hodgson J.G., Agopyan N., Gutekunst C.A., Leavitt B.R., LePiane F., Singaraja R., Smith D.J., Bissada N., McCutcheon K., Nasir J., et al. A YAC mouse model for Huntington's disease with full-length mutant huntingtin, cytoplasmic toxicity, and selective striatal neurodegeneration. Neuron. 1999;23:181–192. doi: 10.1016/s0896-6273(00)80764-3. doi:10.1016/S0896-6273(00)80764-3. [DOI] [PubMed] [Google Scholar]
- 25.Levine M.S., Klapstein G.J., Koppel A., Gruen E., Cepeda C., Vargas M.E., Jokel E.S., Carpenter E.M., Zanjani H., Hurst R.S., et al. Enhanced sensitivity to N-methyl-d-aspartate receptor activation in transgenic and knockin mouse models of Huntington's disease. J. Neurosci. Res. 1999;58:515–532. doi:10.1002/(SICI)1097-4547(19991115)58:4<515::AID-JNR5>3.0.CO;2-F. [PubMed] [Google Scholar]
- 26.Wheeler V.C., Auerbach W., White J.K., Srinidhi J., Auerbach A., Ryan A., Duyao M.P., Vrbanac V., Weaver M., Gusella J.F., Joyner A.L., MacDonald M.E. Length-dependent gametic CAG repeat instability in the Huntington's disease knock-in mouse. Hum. Mol. Genet. 1999;8:115–122. doi: 10.1093/hmg/8.1.115. doi:10.1093/hmg/8.1.115. [DOI] [PubMed] [Google Scholar]
- 27.Yamamoto A., Lucas J.J., Hen R. Reversal of neuropathology and motor dysfunction in a conditional model of Huntington's disease. Cell. 2000;101:57–66. doi: 10.1016/S0092-8674(00)80623-6. doi:10.1016/S0092-8674(00)80623-6. [DOI] [PubMed] [Google Scholar]
- 28.Kegel K.B., Meloni A.R., Yi Y., Kim Y.J., Doyle E., Cuiffo B.G., Sapp Y., Wang Y., Qin Z.H., Chen J.D., et al. Huntingtin is present in the nucleus, interacts with the transcriptional corepressor Cterminal binding protein, and represses transcription. J. Biol. Chem. 2002;277:7466–7476. doi: 10.1074/jbc.M103946200. doi:10.1074/jbc.M103946200. [DOI] [PubMed] [Google Scholar]
- 29.Kegel K.B., Sapp E., Yoder J., Cuiffo B., Sobin L., Kim Y.J., Qin Z.H., Hayden M.R., Aronin N., Scott D.L., et al. Huntingtin associates with acidic phospholipids at the plasma membrane. J. Biol. Chem. 2005;280:36464–36473. doi: 10.1074/jbc.M503672200. doi:10.1074/jbc.M503672200. [DOI] [PubMed] [Google Scholar]
- 30.Panov A.V., Gutekunst C.A., Leavitt B.R., Hayden M.R., Burke J.R., Strittmatter W.J., Greenamyre T. Early mitochondrial calcium defects in Huntington's disease are a direct effect of polyglutamines. Nat. Neurosci. 2002;5:731–736. doi: 10.1038/nn884. [DOI] [PubMed] [Google Scholar]
- 31.Choo Y.S., Johnson G.V., MacDonald M., Detloff P.J., Lesort M. Mutant huntingtin directly increases susceptibility of mitochondria to the calcium-induced permeability transition and cytochrome c release. Hum. Mol. Genet. 2004;13:1407–1420. doi: 10.1093/hmg/ddh162. [DOI] [PubMed] [Google Scholar]
- 32.Truant R., Atwal R., Burtnik A. Hypothesis: Huntingtin may function in membrane association and vesicular trafficking. Biochem. Cell Biol. 2006;84:912–917. doi: 10.1139/o06-181. doi:10.1139/o06-181. [DOI] [PubMed] [Google Scholar]
- 33.Strehlow A.N., Li J.Z., Myers R.M. Wild-type huntingtin participates in protein trafficking between the Golgi and the extracellular space. Hum. Mol. Genet. 2007;16:391–409. doi: 10.1093/hmg/ddl467. doi:10.1093/hmg/ddl467. [DOI] [PubMed] [Google Scholar]
- 34.Atwal R.S., Xia J., Pinchev D., Taylor J., Epand R.M., Truant R. Huntingtin has a membrane association signal that can modulate huntingtin aggregation, nuclear entry and toxicity. Hum. Mol. Genet. 2007;16:2600–2615. doi: 10.1093/hmg/ddm217. doi:10.1093/hmg/ddm217. [DOI] [PubMed] [Google Scholar]
- 35.Orr A.L., Li S., Wang C.E., Li H., Wang J., Rong J., Xu X., Mastroberardino P.G., Greenamyre J.T., Li X.L. N-terminal mutant huntingtin associates with mitochondria and impairs mitochondrial trafficking. J. Neurosci. 2008;28:2783–2792. doi: 10.1523/JNEUROSCI.0106-08.2008. doi:10.1523/JNEUROSCI.0106-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shirendeb U., Reddy A.P., Manczak M., Calkins M.J., Mao P., Tagle D.A., Reddy P.H. Abnormal mitochondrial dynamics, mitochondrial loss and mutant huntingtin oligomers in Huntington's disease: implications for selective neuronal damage. Hum. Mol. Genet. 2011;20:1438–1455. doi: 10.1093/hmg/ddr024. doi:10.1093/hmg/ddr024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang H., Lim P.J., Karbowski M., Monteiro M.J. Effects of overexpression of huntingtin proteins on mitochondrial integrity. Hum. Mol. Genet. 2009;18:737–752. doi: 10.1093/hmg/ddn404. doi:10.1093/hmg/ddn404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kim J., Moody J.P., Edgerly C.K., Bordiuk O.L., Cormier K., Smith K., Beal M.F., Ferrante R.J. Mitochondrial loss, dysfunction and altered dynamics in Huntington's disease. Hum. Mol. Genet. 2010;19:3919–3935. doi: 10.1093/hmg/ddq306. doi:10.1093/hmg/ddq306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Song W., Chen J., Petrilli A., Liot G., Klinglmayr E., Zhou Y., Poquiz P., Tjong J., Pouladi M.A., Hayden M.R., et al. Mutant huntingtin binds the mitochondrial fission GTPase dynamin-related protein-1 and increases its enzymatic activity. Nat. Med. 2011;17:377–382. doi: 10.1038/nm.2313. doi:10.1038/nm.2313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chan D.C. Mitochondrial fusion and fission in mammals. Annu. Rev. Cell Dev. Biol. 2006;22:79–99. doi: 10.1146/annurev.cellbio.22.010305.104638. doi:10.1146/annurev.cellbio.22.010305.104638. [DOI] [PubMed] [Google Scholar]
- 41.Reddy P.H., Reddy T.P., Manczak M., Calkins M.J., Shirendeb U., Mao P. Dynamin-related protein 1 and mitochondrial fragmentation in neurodegenerative diseases. Brain. Res. Rev. 2011;67:103–118. doi: 10.1016/j.brainresrev.2010.11.004. doi:10.1016/j.brainresrev.2010.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kageyama Y., Zhang Z., Sesaki H. Mitochondrial division: molecular machinery and physiological functions. Curr. Opin. Cell Biol. 2011;23:427–434. doi: 10.1016/j.ceb.2011.04.009. doi:10.1016/j.ceb.2011.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Moreira P.I., Zhu X., Wang X., Lee H.G., Nunomura A., Petersen R.B., Perry G., Smith M.A. Mitochondria: a therapeutic target in neurodegeneration. Biochim. Biophys. Acta. 2010;1802:212–220. doi: 10.1016/j.bbadis.2009.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Manczak M., Calkins M.J., Reddy P.H. Impaired mitochondrial dynamics and abnormal interaction of amyloid beta with mitochondrial protein Drp1 in neurons from patients with Alzheimer's disease: implications for neuronal damage. Hum. Mol. Genet. 2011;20:2495–2509. doi: 10.1093/hmg/ddr139. doi:10.1093/hmg/ddr139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Spampanato J., Gu X., Yang X.W., Mody I. Progressive synaptic pathology of motor cortical neurons in a BAC transgenic mouse model of Huntington's disease. Neuroscience. 2008;157:606–620. doi: 10.1016/j.neuroscience.2008.09.020. doi:10.1016/j.neuroscience.2008.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gu X., Greiner E.R., Mishra R., Kodali R., Osmand A., Finkbeiner S., Steffan J.S., Thompson L.M., Wetzel R., Yang X.W. Serines 13 and 16 are critical determinants of full-length human mutant huntingtin induced disease pathogenesis in HD mice. Neuron. 2009;64:828–840. doi: 10.1016/j.neuron.2009.11.020. doi:10.1016/j.neuron.2009.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Menalled L., El-Khodor B.F., Patry M., Suárez-Fariñas M., Orenstein S.J., Zahasky B., Leahy C., Wheeler V., Yang X.W., MacDonald M., et al. Systematic behavioral evaluation of Huntington's disease transgenic and knock-in mouse models. Neurobiol. Dis. 2009;35:319–336. doi: 10.1016/j.nbd.2009.05.007. doi:10.1016/j.nbd.2009.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Southwell A.L., Ko J., Patterson P.H. Intrabody gene therapy ameliorates motor, cognitive, and neuropathological symptoms in multiple mouse models of Huntington's disease. J. Neurosci. 2009;29:13589–13602. doi: 10.1523/JNEUROSCI.4286-09.2009. doi:10.1523/JNEUROSCI.4286-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pouladi M.A., Xie Y., Skotte N.H., Ehrnhoefer D.E., Graham R.K., Kim J.E., Bissada N., Yang X.W., Paganetti P., Friedlander R.M., Leavitt B.R., Hayden M.R. Full-length huntingtin levels modulate body weight by influencing insulin-like growth factor 1 expression. Hum. Mol. Genet. 2010;19:1528–1538. doi: 10.1093/hmg/ddq026. doi:10.1093/hmg/ddq026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kudo T., Schroeder A., Loh D.H., Kuljis D., Jordan M.C., Roos K.P., Colwell C.S. Dysfunctions in circadian behavior and physiology in mouse models of Huntington's disease. Exp. Neurol. 2011;228:80–90. doi: 10.1016/j.expneurol.2010.12.011. doi:10.1016/j.expneurol.2010.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.André V.M., Cepeda C., Fisher Y.E., Huynh M., Bardakjian N., Singh S., Yang X.W., Levine M.S. Differential electrophysiological changes in striatal output neurons in Huntington's disease. J. Neurosci. 2011;31:1170–1182. doi: 10.1523/JNEUROSCI.3539-10.2011. doi:10.1523/JNEUROSCI.3539-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hult S., Soylu R., Björklund T., Belgardt B.F., Mauer J., Brüning J.C., Kirik D., Petersén Å. Mutant huntingtin causes metabolic imbalance by disruption of hypothalamic neurocircuits. Cell Metab. 2011;13:428–439. doi: 10.1016/j.cmet.2011.02.013. doi:10.1016/j.cmet.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 53.Xiang Z., Valenza M., Cui L., Leoni V., Jeong H.K., Brilli E., Zhang J., Peng Q., Duan W., Reeves S.A., Cattaneo E., Krainc D. Peroxisome-proliferator-activated receptor gamma coactivator 1 α contributes to dysmyelination in experimental models of Huntington's disease. J. Neurosci. 2011;31:9544–9553. doi: 10.1523/JNEUROSCI.1291-11.2011. doi:10.1523/JNEUROSCI.1291-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wakabayashi J., Zhang Z., Wakabayashi N., Tamura Y., Fukaya M., Kensler T.W., Iijima M., Sesaki H. The dynamin-related GTPase Drp1 is required for embryonic and brain development in mice. J. Cell Biol. 2009;186:805–816. doi: 10.1083/jcb.200903065. doi:10.1083/jcb.200903065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ishihara N., Nomura M., Jofuku A., Kato H., Suzuki S.O., Masuda K., Otera H., Nakanishi Y., Nonaka I., Goto Y., et al. Mitochondrial fission factor Drp1 is essential for embryonic development and synapse formation in mice. Nat. Cell Biol. 2009;11:958–966. doi: 10.1038/ncb1907. doi:10.1038/ncb1907. [DOI] [PubMed] [Google Scholar]
- 56.Calkins M.J., Reddy P.H. Assessment of newly synthesized mitochondrial DNA using BrdU labeling in primary neurons from Alzheimer's disease mice: implications for impaired mitochondrial biogenesis and synaptic damage. Biochim. Biophys. Acta. 2011;1812:1182–1189. doi: 10.1016/j.bbadis.2011.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Calkins M.J., Reddy P.H. Amyloid beta impairs mitochondrial anterograde transport and degenerates synapses in Alzheimer's disease neurons. Biochim. Biophys. Acta. 2011;1812:507–513. doi: 10.1016/j.bbadis.2011.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Calkins M.J., Manczak M., Mao P., Shirendeb U., Reddy P.H. Impaired mitochondrial biogenesis, defective axonal transport of mitochondria, abnormal mitochondrial dynamics and synaptic degeneration in a mouse model of Alzheimer's disease. Hum. Mol. Genet. 2011 doi: 10.1093/hmg/ddr381. [Epub ahead of print 25 August 2011] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cui L., Jeong H., Borovecki F., Parkhurst C.N., Tanese N., Krainc D. Transcriptional repression of PGC-1alpha by mutant huntingtin leads to mitochondrial dysfunction and neurodegeneration. Cell. 2006;127:59–69. doi: 10.1016/j.cell.2006.09.015. doi:10.1016/j.cell.2006.09.015. [DOI] [PubMed] [Google Scholar]
- 60.Weydt P., Pineda V.V., Torrence A.E., Libby R.T., Satterfield T.F., Lazarowski E.R., Gilbert M.L., Morton G.J., Bammler T.K., Strand A.D., et al. Thermoregulatory and metabolic defects in Huntington's disease transgenic mice implicate PGC-1alpha in Huntington's disease neurodegeneration. Cell. Metab. 2006;4:349–362. doi: 10.1016/j.cmet.2006.10.004. [DOI] [PubMed] [Google Scholar]
- 61.St-Pierre J., Drori S., Uldry M., Silvaggi J.M., Rhee J., Jäger S., Handschin C., Zheng K., Lin J., Yang W., et al. Suppression of reactive oxygen species and neurodegeneration by the PGC-1 transcriptional coactivators. Cell. 2006;127:397–408. doi: 10.1016/j.cell.2006.09.024. doi:10.1016/j.cell.2006.09.024. [DOI] [PubMed] [Google Scholar]
- 62.Chaturvedi R.K., Adhihetty P., Shukla S., Hennessy T., Calingasan N., Yang L., Starkov A., Kiaei M., Cannella M., Sassone J., et al. Impaired PGC-1alpha function in muscle in Huntington's disease. Hum. Mol. Genet. 2009;18:3048–3065. doi: 10.1093/hmg/ddp243. doi:10.1093/hmg/ddp243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Manczak M., Mao P., Calkins M.J., Cornea A., Reddy A.P., Murphy M.P., Szeto H.H., Park B., Reddy P.H. Mitochondria-targeted antioxidants protect against amyloid-beta toxicity in Alzheimer's disease neurons. J. Alzheimers Dis. 2010;20(Suppl. 2):S609–S631. doi: 10.3233/JAD-2010-100564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gutala R.V., Reddy P.H. The use of real-time PCR analysis in a gene expression study of Alzheimer's disease post-mortem brains. J. Neurosci. Methods. 2004;132:101–107. doi: 10.1016/j.jneumeth.2003.09.005. doi:10.1016/j.jneumeth.2003.09.005. [DOI] [PubMed] [Google Scholar]
- 65.Trottier Y., Lutz Y., Stevanin G., Imbert G., Devys D., Cancel G., Saudou F., Weber C., David G., Tora L., et al. Polyglutamine expansion as a pathological epitope in Huntington's disease and four dominant cerebellar ataxias. Nature. 1995;378:403–436. doi: 10.1038/378403a0. doi:10.1038/378403a0. [DOI] [PubMed] [Google Scholar]