Ceramide Levels Regulated by Carnitine Palmitoyltransferase 1C Control Dendritic Spine Maturation and Cognition (original) (raw)
Background: CPT1C is highly expressed in hippocampus, but its cellular and physiological function is unknown.
Results: CPT1C overexpression increases ceramide levels, and CPT1C deficiency impairs dendritic spine morphology and spatial learning.
Conclusion: Regulation of ceramide levels by CPT1C is necessary for proper spine maturation.
Significance: We describe a new function of CPT1C in cognition.
Keywords: Ceramide, Ceramide Synthesis, Endoplasmic Reticulum (ER), Fatty Acid Transport, Neurons, Sphingolipid, Cognition, Dendritic Spines, Acyl-CoA, Spinogenesis
Abstract
The brain-specific isoform carnitine palmitoyltransferase 1C (CPT1C) has been implicated in the hypothalamic regulation of food intake and energy homeostasis. Nevertheless, its molecular function is not completely understood, and its role in other brain areas is unknown. We demonstrate that CPT1C is expressed in pyramidal neurons of the hippocampus and is located in the endoplasmic reticulum throughout the neuron, even inside dendritic spines. We used molecular, cellular, and behavioral approaches to determine CPT1C function. First, we analyzed the implication of CPT1C in ceramide metabolism. CPT1C overexpression in primary hippocampal cultured neurons increased ceramide levels, whereas in CPT1C-deficient neurons, ceramide levels were diminished. Correspondingly, CPT1C knock-out (KO) mice showed reduced ceramide levels in the hippocampus. At the cellular level, CPT1C deficiency altered dendritic spine morphology by increasing immature filopodia and reducing mature mushroom and stubby spines. Total protrusion density and spine head area in mature spines were unaffected. Treatment of cultured neurons with exogenous ceramide reverted the KO phenotype, as did ectopic overexpression of CPT1C, indicating that CPT1C regulation of spine maturation is mediated by ceramide. To study the repercussions of the KO phenotype on cognition, we performed the hippocampus-dependent Morris water maze test on mice. Results show that CPT1C deficiency strongly impairs spatial learning. All of these results demonstrate that CPT1C regulates the levels of ceramide in the endoplasmic reticulum of hippocampal neurons, and this is a relevant mechanism for the correct maturation of dendritic spines and for proper spatial learning.
Introduction
Carnitine palmitoyltransferase 1 (CPT1)5 enzymes catalyze the conversion of long-chain acyl-CoA to acyl-carnitines, thus facilitating the transport of long-chain fatty acids across intracellular membranes. There are three isoforms: the liver isoform CPT1A (1), the muscle isoform CPT1B (2), and the brain-specific isoform CPT1C (3). CPT1A and CPT1B are localized in the outer mitochondrial membrane and are rate-limiting enzymes in fatty acid β-oxidation.
The main isoform in brain, CPT1C, highly differs from the two other isozymes. Its C-terminal region is longer than that of the other CPTs (3) and is located in the endoplasmic reticulum (ER) of cells rather than in mitochondria (4). It has low CPT1 activity (4), but it binds the CPT1 physiological inhibitor malonyl-CoA with the same affinity as CPT1A (5). Finally, CPT1C is only present in mammals and appears to stem from a relatively recent cpt1a gene duplication (3). The other isozymes are expressed in such organisms as fish, reptiles, amphibians, or insects. This suggests a specific role for CPT1C in more evolved brains.
At the physiological level, CPT1C contributes to the control of food intake and energy homeostasis (5, 6). Two independent groups developed a CPT1C knock-out (KO) mouse, and both lines showed decreased food intake with respect to wild-type (WT) animals. However, when fed a high fat diet, they were more susceptible to obesity and diabetes, presenting lower rates of peripheral fatty acid oxidation. All of these effects were attributed to the hypothalamic function of CPT1C because ectopic overexpression of CPT1C in hypothalamus protected mice from adverse weight gain caused by a high fat diet (7). Moreover, the involvement of CPT1C in energy homeostasis has also been confirmed in transgenic animals overexpressing CPT1C specifically in the brain (8). At the molecular level, in collaboration with the group of Dr. Gary Lopaschuk, we showed that CPT1C is involved in the anorectic action of leptin, by modulating ceramide synthesis in the arcuate nucleus of the hypothalamus (9).
Interestingly, recent findings in tumor cells showed a new, unexpected role of CPT1C in the metabolic transformations reported in tumor cell growth (10). The authors demonstrated that CPT1C is frequently expressed in human lung tumors and protects cancerous cells from death induced by glucose deprivation or hypoxia. The results suggest that CPT1C might provide unidentified fatty acid-derived products that would be beneficial for cell survival under metabolic stress.
However, despite these recent findings about CPT1C, little is known about its physiological role during brain development and function. The finding that CPT1C is highly expressed in hippocampus (3) prompted us to look after other brain CPT1C functions beyond the control of energy homeostasis. Our results show that CPT1C is located in the ER of hippocampal neurons and regulates the maturation of dendritic spines by increasing ceramide levels. At the behavioral level, we demonstrate for the first time that CPT1C is involved in spatial learning.
EXPERIMENTAL PROCEDURES
Construction of Targeting Vector and Generation of KO Mice
A construct was generated using the pPNT vector (11). After correct recombination, this vector caused a 2.9-kb genomic deletion, including exons 12–15. The targeting construct was electroporated into 129/SvEv embryonic stem cells (ESC) by the Centre de Biotecnologia Animal i Teràpia Gènica at the Universitat Autonoma de Barcelona. Two positive ESC clones were expanded and verified for correct recombination by PCR amplification and Southern blot analysis. CPT1C+/− cells were injected into C57BL/6J blastocyst. Chimeric mice displaying >50% coat color chimerism were bred with C57BL/6J females to generate F1 offspring. The sixth backcrossed generation was used in all of the experiments.
Animal Housing
In behavioral studies, only males at 10–14 weeks of age were tested (n = 12). All of the behavioral testing was conducted by the same experimenters, blinded as to the genetic status of animals, in an isolated room and at the same time of day. All animal procedures met the guidelines of European Community Directive 86/609/EEC (EU directive 86/609, EU decree 2001-486) and Standards for Use of Laboratory Animals A5388-01 (National Institutes of Health) and were approved by the local ethics committee.
Morris Water Maze (MWM) Test
To test hippocampus-dependent spatial cognition, the MWM test was used, as described elsewhere (12). The water maze consisted of a circular pool (diameter 1.20 m, height 0.5 m). A white escape platform (15-cm diameter, height 24 cm) was located 1 cm below the water surface in a fixed position (northeast quadrant, 22 cm away the wall). All of the trials were recorded and traced with an image tracking system (SMART, Panlab, Spain) connected to a video camera. Escape latencies, length of the swimming paths, and swimming speed for each animal and trial were monitored and computed.
Cell Cultures and Plasmid Transfection
Hippocampal cultured neurons were obtained and cultured as described elsewhere (13). For plasmid transfection, neurons were grown for 14 days in vitro (DIV), transfected using the Effecten kit (Qiagen), and analyzed at 15 DIV. After transfection, neurons were fixed with 4% paraformaldehyde and 4% sucrose in PBS. Samples were mounted using Gel/Mount anti-fading medium (Invitrogen).
Virus Development and Cell Culture Infection
Two adeno-associated virus (AAV) vectors, serotype 1, AAV1-GFP, AAV1-CPT1C were constructed to drive cell expression of GFP and CPT1C, respectively. Vector plasmids carried the transgene expression cassette, including the cytomegalovirus promoter, the cDNA sequence of GFP and the rat CPT1C (3), the woodchuck posttranscriptional regulatory element (accession number AY468 486) to enhance transcription (14), and the bovine growth hormone polyadenosine transcription termination signal (bGH poly(A)) (bases 2326–2533, GenBankTM accession number M57764). The expression cassette was flanked by two inverted terminal repeats derived from AAV serotype 2. AAV1 vectors were produced in insect cells using baculovirus (15). The vector preparations used in this study had titers of 1 × 1012 and 2.5 × 1012 genome copies/ml for AAV1-GFP and AAV1-CPT1C, respectively.
AAV1-CPT1C virus infection was performed at 7 DIV in cells cultured in 6-well plates. Medium was removed and kept apart to be reused later. 0.5 ml of neurobasal medium without B27 and containing 0.5 mm glutamine and AAV at a concentration of 100,000 viruses/cell was added to each well and left to stand for 2 h. Then 1.5 ml of the preconditioned medium kept apart was added and left to stand for a further 7 days. Then cells were removed for analysis of CPT1C expression and ceramide levels. Myriocin (Sigma) treatment was performed 8 h before cell recollection.
Immunodetection in Brain Sections and Cultured Cells
Coronal sections (30 μm) from adult mouse forebrains were incubated with primary antibodies against glial fibrillary acidic protein (1:500; Chemicon MAB360) and CPT1C (1:100) overnight at 4 °C, washed three times in PBS (0.1 m), and incubated for 2 h with secondary antibodies coupled to fluorochromes Alexa 488 (for green fluorescence) and Alexa 568 (for red fluorescence) at a dilution of 1:500. In cultured neurons, anti-calreticulin polyclonal antibody (BD Biosciences) was used at a dilution of 1:50 for 1 h at 37 °C, and for the red fluorescence, the secondary antibody goat anti-mouse Alexafluor 546 (Molecular Probes) (1:500) was used. Sections and coverslips were mounted with Mowiol and observed using a Confocal Leica TCS SP2 (Leica Lasertechnik GmbH, Mannheim, Germany).
Image Analysis and Quantification of Dendrite Spine Density
Images were acquired using a digital camera (SpotRT, Diagnostic Instruments) attached to an epifluorescence microscope (Zeiss) equipped with a ×63 objective (Plan-Apochromat, Zeiss). All quantitative measurements were carried out using MetaMorph software (Molecular Devices). Approximately 100 dendrites from independent transfections were randomly selected for each construct to quantify the number of protrusions in proximal 50-mm sections of dendrites. Lengths of protrusions were determined by measuring the distance between the tip and the base.
Western Blot Analysis
Rabbit antibodies against the C-terminal region of mouse CPT1C and against CPT1A were as described elsewhere (4). Generally, 60 μg of protein extracts were subjected to SDS-PAGE. Dilutions of 1:500 and 1:1000 of anti-CPT1C and anti-CPT1A primary antibodies, respectively, were used. A 1:5000 dilution of secondary antibody was used. The blots were developed with the ECL Western blotting system (Amersham Biosciences).
Ceramide Quantification
Ceramides were extracted and analyzed via an LC-electrospray ionization-MS/MS system (API 3000 PE Sciex) in positive ionization as described elsewhere (16). Their concentrations were measured by MRM experiments using _N_-heptadecanoyl-d-_erythro_-sphingosine (C17-ceramide) as an internal standard (50 ng/ml). The method was linear over the range from 2 to 600 ng/ml.
Cell Feeding with Deuterated Serine
Hippocampal neurons at 14 DIV were treated with 4 mm dl-serine-_d_7 (CDN Isotopes, Cluzau Infolab) for different times. Ceramides were extracted, and two C18:0 deuterated ceramides were identified with an orbitrap mass spectrometer (Thermo Scientific). These ceramides were subsequently quantified using LC-electrospray ionization-MS/MS (API 3000 PE Sciex). The most abundant analyte corresponded to ceramide C18:0-_d_3, determined by a Q1 m/z = 569,567 and Q3 m/z = 267,287. Areas under the peak were measured and normalized with sample protein concentration.
Statistics
Data are expressed as means ± S.E. Statistical significance was determined by Student's t test for the difference between two normal groups, and the Mann-Whitney U test was used for non-normal distribution. One-way ANOVA with Bonferroni test for post hoc analysis was used for more than two groups. Performances in the MWM tests were compared using repeated measures ANOVA.
RESULTS
CPT1C Is Located throughout ER of Hippocampal Neurons, Even Penetrating into Dendritic Spines
It was previously described that CPT1C is highly expressed in the hippocampus (3). In order to determine the precise localization of the protein, we performed brain sections and incubated them with an anti-CPT1C antibody, kindly provided by the Wolfgang laboratory and previously used in the literature (4, 7). Fig. 1A clearly shows that CPT1C (in green) is expressed in pyramidal neurons of the hippocampus. Astrocytes were identified by glial fibrillary acidic protein (an astrocyte marker) antibody.
FIGURE 1.

CPT1C location in hippocampal neurons. A, CPT1C is present in neurons of the hippocampus, mainly pyramidal cells. Brain sections were double-immunodetected with anti-CPT1C antibody (green) and anti-glial fibrillary acidic protein antibody (red). B, hippocampal cultured neurons were double-transfected with pCPT1C-EGFP and pDS-Red at 11 DIV and visualized at 15 DIV. Images show that CPT1C is present in neuronal body, dendritic shaft, and spines (marked with arrows). pDs-Red transfection was performed to display the outline of the neuron. C, hippocampal cultured neurons were transfected with pDS-ER-Red to stain the ER. At 15 DIV, cells were immunodetected with anti-CPT1C antibodies (green). The merge image (yellow) demonstrates that CPT1C is localized to the ER membrane. D, Western blot analysis of CPT1C and CPT1A proteins in isolated microsomes and mitochondria from hippocampus of WT, heterozygous (HT), and KO mice.
To analyze the detailed localization of CPT1C in hippocampal neurons, we performed neuronal primary cultures and transfected them with pCPT1C-EGFP, a plasmid that encodes CPT1C fused to the N-terminal region of green fluorescence protein (EGFP) (4). Fig. 1B shows that CPT1C is located throughout the neuron, in neuronal bodies and dendrites. Detailed photographs of dendrites demonstrate that CPT1C is present mainly in shafts but also in spines (marked with arrows). The same cultures were transfected with pDS-Red (Clontech) that encodes the Discosoma sp. red fluorescent protein in the cytosol to display the outline of the cell.
To confirm that subcellular localization of endogenous CPT1C was in the ER, we transfected the cultured neurons with pDs-ER-Red2 (Clontech), which stains the ER red, and immunodetected endogenous CPT1C with anti-CPT1C antibody (in green). Fig. 1C shows that CPT1C is localized in the ER of cultured hippocampal neurons. Finally, Western blot experiments were also performed with different cellular fractions from mouse brain. Anti-CPT1A antibodies were used as a marker for mitochondria. Samples were retrieved from WT, CPT1C KO, and heterozygous mice developed in our laboratory (described under “Experimental Procedures”). Fig. 1D shows that CPT1C is present mainly in the microsomal fraction and that CPT1A is present mainly in mitochondria, confirming that CPT1C localizes to the ER membrane of cells.
CPT1C Regulates Levels of Ceramide in Cultured Neurons
Our group has recently reported that CPT1C regulates ceramide synthesis in arcuate nucleus of the hypothalamus as part of the signaling pathway of leptin (9). We wanted to examine whether CPT1C was also regulating ceramide levels in hippocampal neurons. We overexpressed CPT1C in primary hippocampal neurons using AAV1-CPT1C viruses. A 4-fold increase in CPT1C protein levels resulted in a 2-fold increase in ceramide levels with respect to control cells (cells infected with AAV1-GFP) (Fig. 2A). CPT1C overexpression mainly increased saturated ceramides (C16:0, C18:0, and C20:0). Ceramide C18:0 was the most abundant one in the hippocampal cultures, being 20 times more concentrated than the rest. We also measured ceramide levels in hippocampal cultured neurons from CPT1C KO mice. As shown in Fig. 2B, C18:0 ceramide levels were lower in KO cells than in WT cells, confirming that CPT1C regulates ceramide levels in hippocampal neurons.
FIGURE 2.
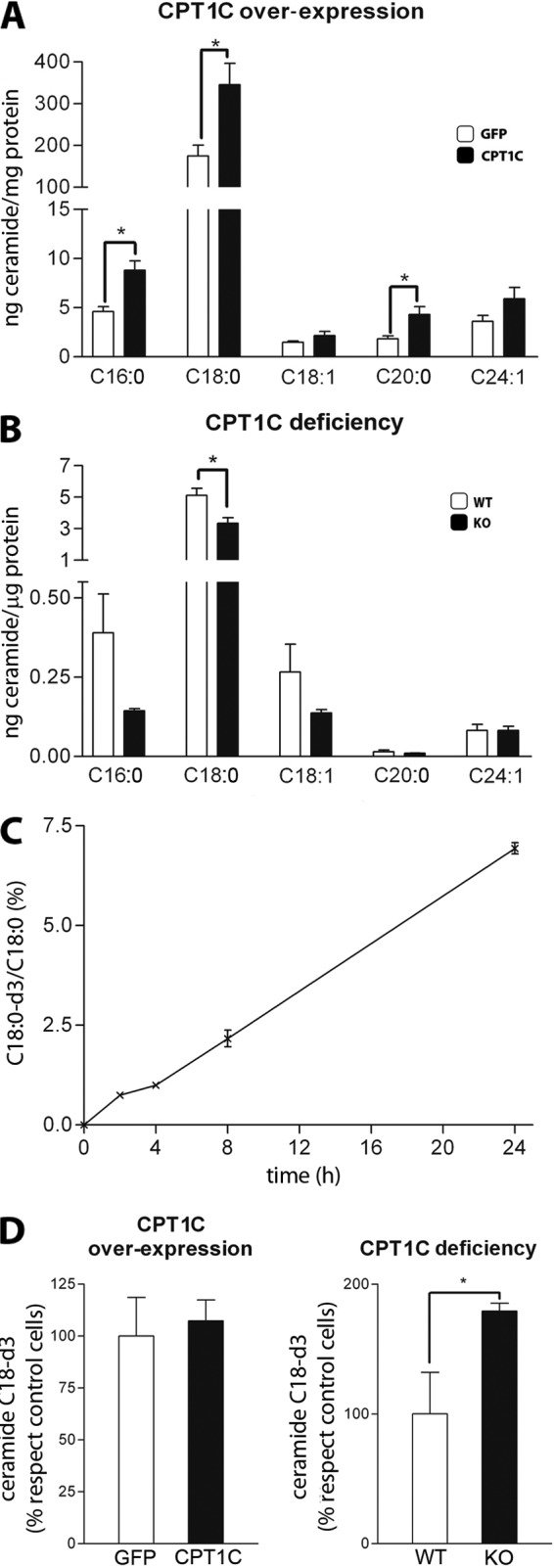
Regulation of ceramide levels by CPT1C. A, levels of ceramides in hippocampal neurons transduced with AAV1-GFP (as a control) or AAV1-CPT1C at 7 DIV. Cells were collected at 14 DIV. B, levels of ceramides in hippocampal neurons from WT and CPT1C KO mice. Cells were collected at 14 DIV. C, time course of incorporation of serine-_d_7 into ceramide C18:0-_d_3. Hippocampal cultured neurons from WT animals were treated with 4 mm serine-_d_7 at DIV14. Ceramides C18:0 and C18:0-_d_3 were analyzed at different times, and the percentage of incorporation is shown. D, effect of CPT1C overexpression and CPT1C deficiency on serine-_d_7 incorporation into ceramide C18:0-_d_3. Hippocampal cultured cells were transduced with AAV1-GFP (as a control) or AAV1-CPT1C at 7 DIV. Cells were treated with serine-_d_7 at DIV 14 and collected after 2.5 h of treatment. The percentage of variation in ceramide C18:0-_d_3 levels compared with the control cells is shown. Error bars, S.E.; n = 6; *, p < 0.05.
Because ceramide present in ER comes mainly from de novo synthesis, we examined whether CPT1C was activating this pathway. A 24-h pulse with 4 mm deuterated serine was first carried out in control neurons. The incorporation of deuterated serine into ceramide was linear during the first 24 h, at which point it reached a level of 7% (Fig. 2C). Because we were able to measure deuterated ceramide at the short time of 2 h, we decided to perform the next experiments at 2.5 h to minimize interference with other ceramide metabolic pathways. Fig. 2D shows that deuterated ceramide synthesis was not increased by CPT1C overexpression and was not decreased in CPT1C KO cells. These results clearly indicate that CPT1C does not activate the de novo synthesis of ceramide and suggest that CPT1C regulates ceramide levels by acting on another metabolic pathway. Surprisingly, the de novo synthesis of ceramide was even increased in CPT1C KO cells, suggesting that these neurons were activating this basal pathway to counterbalance the reduction in ceramide levels caused by CPT1C deficiency.
CPT1C KO Mice Have Reduced Ceramide Levels in Hippocampus
To examine whether CPT1C is involved in the regulation of hippocampal ceramide synthesis in adult mice, we measured ceramide levels in hippocampus from WT and CPT1C KO mice under ad libitum and fasted conditions. CPT1C KO mice showed lower ceramide levels in hippocampus than WT animals, mainly during fasting (Fig. 3). The most abundant ceramide found in the hippocampus was the C18:0 ceramide, being about 20 times more abundant than the rest (like in hippocampal cultures), which agrees with previous literature indicating that the major ceramide in brain neurons is C18:0 (17). Importantly, C18:0 ceramide levels were higher in WT than in KO mice during fasting, when it is known that the concentration of malonyl-CoA (the physiological inhibitor of CPT1 enzymes) is highly reduced (18), suggesting that CPT1C activity in the hippocampus is modulated by malonyl-CoA.
FIGURE 3.

Ceramide levels in hippocampus from ad libitum and fasted CPT1C KO and WT mice. Fasted mice were deprived of food for 15 h. Different ceramide species were measured: ceramide C16:0, ceramide C18:0, ceramide C18:1, ceramide C20:0, and ceramide C24:1. Error bars, S.E. n = 6; *, p < 0.05; **, p < 0.005; ***, p < 0.001, ANOVA test.
CPT1C Deficiency Increases Filopodia Density and Reduces Spine Maturation in Hippocampal Neurons
To examine the effects of CPT1C deficiency on dendritic spine density and morphology, we performed primary hippocampal cultures from CPT1C KO and WT mice, transfected the neurons with green fluorescent protein (GFP), and examined dendritic spines at 15 DIV. Neurons from CPT1C KO mice had the same protrusion density but larger protrusion length than WT neurons (Fig. 4, A–C). Morphological analysis revealed that CPT1C KO mice had a strong increase in filopodia number and a marked reduction of mature (mushroom and stubby) spines (Fig. 4, D–G). However, the spine head area in mature spines was the same in both genotypes (Fig. 4H). Overexpression of CPT1C on KO cultures reduced filopodia density and increased the percentage of mature spines to values similar to WT cultures (Fig. 5, B and D), confirming the requirement of CPT1C for efficient spine maturation.
FIGURE 4.

Dendritic spine density and morphology from CPT1C KO and WT hippocampal neurons. Hippocampal neurons were transfected (13 DIV) with pEGFP to visualize the outline of the cell. Protrusions were analyzed 2 days after transfection. Protrusion density (A) and protrusion length (B and C) were measured. Mature spines (A) and filopodia (B) are indicated. Spine morphology (D–F) was assayed by analysis of types of protrusions: filopodia (without head), mushroom (with head and neck), and stubby (with only head). G, percentage of mature spines (mushroom and stubby) relative to the total number of protrusions was also measured. H, spine head area was measured in mushroom and stubby spines. I, a representative image of dendritic spines from WT and CPT1C KO neurons. For the quantification of protrusion density, spine length, and morphology, ∼100 dendrites from independent transfections were selected randomly. Student's t tests were used to assess statistical significance of the differences. Error bars, S.E.; ***, p < 0.001.
FIGURE 5.

Rescue of CPT1C KO phenotype on spine morphology by CPT1C expression or ceramide treatment. A, hippocampal neurons treated with 1.5 μm C6-ceramide at 7 DIV and transfected with pEGFP (BD Biosciences) at 12 DIV, fixed, and analyzed for the morphology of dendritic protrusions at 15 DIV. B, hippocampal neurons were transfected with pIRES-CPT1C at 7 DIV and analyzed for spine morphology at 15 DIV. pIRES-CPT1C vector expresses both CPT1C and GFP proteins, which permits us to visualize in green the cells overexpressing CPT1C. C, hippocampal neurons at DIV9 were treated with 10 μm myriocin until 15 DIV. Cells were transfected with pEGFP at 12 DIV and analyzed for the morphology of dendritic protrusions at 15 DIV. D, a representative image showing dendritic spines from WT mice, KO mice, KO mice treated with C6-ceramide, KO mice transfected with pIRES-CPT1C, and WT mice treated with myriocin. For the quantification of spine morphology, ∼100 dendrites from independent transfections were selected randomly. Student's t tests and ANOVA post hoc were used to assess statistical significance of the differences. Error bars, S.E.; *, p < 0.05; ***, p < 0.001.
Ceramide Treatment Rescues CPT1C KO Phenotype on Spine Morphology
To corroborate that the reduction in ceramide synthesis caused by CPT1C deletion is the cause of the spine phenotype, we set up a rescue experiment in which CPT1C KO hippocampal cultures were incubated with 1.5 μm soluble C-6 ceramide for 7 days (from DIV 8 to 15). The ceramide dose used (<3 μm) does not induce neuronal apoptosis in hippocampal cultures (19). Exogenous ceramide treatment reversed the CPT1C KO phenotype by decreasing immature filopodia and restoring mature spine density to normal levels (Fig. 5, A and D). These results indicate that CPT1C regulation of spine maturation is mediated by ceramide.
Finally, to confirm that a reduction in ceramide levels is the cause of impaired spinogenesis, we treated hippocampal cultured neurons with myriocin, an inhibitor of ceramide biosynthesis, which has been described to reduce ceramide levels in cultured neurons (20). As shown in Fig. 5, C and D, myriocin treatment increased the density of filopodia and reduced the percentage of mature spines, a phenotype that completely resembles that observed in CPT1C KO cells.
CPT1C KO Mice Have Impaired Spatial Learning
To examine the spine maturation defects on cognition, we performed the MWM test. This test is usually used to measure hippocampus-dependent spatial navigation learning in mice. In the MWM, CPT1C KO showed significantly higher escape latency (delayed learning) during the 10 sessions of the acquisition phase (Fig. 6, A and B). The learning curves were significantly different from those of WT mice (repeated measures ANOVA F(1.22) = 6.726, p = 0.017) in the absence of swimming speed alteration, indicating pure learning impairment, with poorer performance not associated with motor deficits (Fig. 6C). Moreover, in the cued session, where the platform is visible (black flag), the escape latency of CPT1C KO mice was similar to that of the WT (Fig. 6A).
FIGURE 6.

Spatial learning and memory measured by MWM test. A, MWM performance of CPT1C KO and WT mice during the learning sessions as latency (s) to find the platform along the acquisition phase (A), removal (Rem), and cued sessions (Cue). PT, pretraining. B, visual pathway traced by all animals. The white round platform is located in the northeast (NE) quadrant. C, mean swimming speed along acquisition sessions. D, percentage of time spent in the target quadrant (NE) during the removal session; discontinuous lines represent the chance level in this session. E, percentage of permanence in quadrants during the reversal (Rev) session. Data are represented as mean ± S.E. (error bars); *, p < 0.05; **, p < 0.05; ***, p < 0.001, ANOVA test.
To test visuospatial memory, the platform was removed, and the time spent in each quadrant was measured. No significant differences between genotypes were detected in the preference for the trained quadrant, indicating that once the platform position was learned, it was equally retained in CPT1C KO and WT mice (Fig. 6D); the CPT1C KO deficits seem to be limited to the learning phase.
In the reversal test (Fig. 6E), which evaluates the ability of the mice to learn a new platform position (cognitive flexibility), no significant differences were observed between genotypes in the percentage of time spent in the previously trained quadrant (northeast; repeated measures ANOVA, F(1.22) = 0.086, p = 0.772). However, KO mice spent less time in the new goal quadrant (southwest; repeated measures ANOVA, F(1.22) = 8.676, p = 0.007), thus supporting the hypothesis of a hippocampus-dependent learning deficit in CPT1C KO mice.
DISCUSSION
Dendritic spine formation begins in the embryo and continues into early postnatal life but also occurs in the adult organism, where it contributes significantly to learning and memory formation. We demonstrate that the brain isoform CPT1C is present in dendritic spines and regulates the levels of ceramide in neurons, which is key to the transformation of dendritic filopodia into mature spines. This is the first time that CPT1C or ceramide levels have been directly involved in spine morphogenesis. At the physiological level, we show for the first time that CPT1C is involved in spatial learning.
CPT1C Regulates Ceramide Levels in Neurons
One of the relevant contributions of this study is the confirmation that CPT1C increases the levels of ceramide. We had previously described it in the arcuate nucleus of hypothalamus (9), and we now demonstrate it in hippocampal cultured neurons. In consequence, it may be a general phenomenon in neurons. We do not know the molecular mechanism by which CPT1C increases ceramide levels, but our results clearly demonstrate that it does not enhance de novo synthesis, as suggested previously (9). Although the de novo synthesis of ceramide is the main source of ceramide in ER, it can also be produced from the sphingosine pool (salvage pathway) or from the dephosphorylation of ceramide-1-phosphate. Therefore, CPT1C could be activating either of these two pathways. Another possibility is that CPT1C increases the levels of ceramide by inhibiting its elimination (by conversion to sphingosine, phosphorylation to ceramide-1-phosphate or incorporation into sphingomyelin). Further research is therefore required to determine the precise metabolic pathway in which CPT1C is involved.
Because CPT1C has low catalytic activity in vitro (4, 5), we hypothesize that CPT1C regulates the activity of this other enzyme involved in ceramide metabolism by protein-protein interaction. Therefore, under fasting conditions or reduction of malonyl-CoA levels, CPT1C might change its conformation and regulate this other enzyme, resulting in increased levels of ceramide.
CPT1C in Dendritic Spine Maturation
Our results implicate CPT1C in dendritic spine maturation. In absolute numbers, in cultured hippocampal neurons from CPT1C KO mice, the increase in filopodia corresponds with the decline in mature spine number, without altering the overall density of dendritic protrusions, which indicates that CPT1C is not necessary for the formation of new protrusions. However, it is necessary for the conversion of filopodia into mature spines. In addition, results show that the effect of CPT1C on dendritic spines is mediated by ceramide. The addition of ceramide to the cultured medium at low concentration reversed the CPT1C KO phenotype and induced spine maturation. A recent study demonstrates the presence of a new long-chain acyl-CoA synthetase (ACSL4) isoenzyme that localizes specifically in the ER of neurons. Its deficiency increases the percentage of filopodia and reduces the percentage of mature spines (21), in accordance with our results. This highlights the importance of fatty acid metabolism in spinogenesis and suggests that ACSL could provide the substrate necessary for ceramide synthesis in the ER of neurons.
There is only one study that correlates ceramide with the formation of dendritic spines (20). The authors report that coupled inhibition of cholesterol and ceramide synthesis causes alterations in the density and morphology of dendritic spines. Our work sheds light on the regulation of this process and identifies a role for CPT1C in the fine tuning of the modulation of ceramide synthesis, which is essential for the maturation of dendritic spines. The mechanism by which ceramides regulate spine maturation is unknown. However, ceramide binds to and regulates the activity of enzymes and signaling proteins, such as kinases, phosphatases, or membrane receptors (22). One example is protein phosphatase 1, which is activated by ceramide (23) and has been implicated in the conversion of filopodia into mature spines (24). In addition, ceramide is the building block of all cellular sphingolipids, which, in addition to cholesterol, are essential components of lipid rafts. These membrane microdomains are needed for the correct trafficking, anchorage, and activity of synaptic proteins and are preferred platforms for membrane-linked actin polymerization (25). All of these phenomena are necessary for synapse stability and maturation of dendritic spines. Therefore, the diminished ceramide levels found in CPT1C KO mice could alter the regulation of specific proteins or alter the formation of lipid rafts needed for synapse consolidation and spine maturation.
Physiologic Relevance of CPT1C
CPT1C-deficient mice present spatial learning impairment, with a clear delay in the acquisition phase, although they eventually learn and remember the location of the platform. It is important to emphasize that CPT1C deficiency does not affect swimming velocity or motivation and that longer acquisition times correspond to learning deficiencies. On the other hand, memory and cognitive flexibility (ability to modify behavior in an increasingly demanding cognitive task) are not altered in CPT1C KO mice. This indicates that CPT1C deficiency affects the process of consolidating new information but not retention or extinction. This phenotype could be directly related with the impaired dendritic spine maturation found in CPT1C KO mice and with the intact spine head area of mature spines found in both genotypes. In cognitive sciences, it is accepted that spine volume changes regulate new memory acquisition by enlarging and stabilizing smaller spines, whereas the existing memory persistence depends on changing volumes of larger spines (26). In addition, in human patients and most animal models of mental retardation, dendritic spines tend to be abnormally small and immature.
Results from our work show that CPT1C has other physiological roles apart from the regulation of food intake and energy homeostasis. We demonstrate that the molecular function of CPT1C is the fine tuning regulation of ceramide levels in neurons, which is needed for spine maturation during brain development. At the behavioral level, we demonstrate for the first time the involvement of CPT1C in learning, which opens the possibility that CPT1C mutations might be the cause of some human cognition disabilities of unknown etiology.
Acknowledgments
The editorial help of Robin Rycroft is gratefully acknowledged. We thank Josep Clotet for valuable discussions and Julia Geiger for technical support with cell cultures.
*
This work was supported by Ministerio de Educación y Ciencia, Spain, Grants SAF2007-61926, 2009SGR131, SAF2010-16427, CureFXSEU/FISPS09102673, and SAF2011-30520-C02-02 and by a grant from Fundació La Marató de TV3 (2007), Catalunya.
5
The abbreviations used are:
CPT1
carnitine palmitoyltransferase 1
ER
endoplasmic reticulum
MWM
Morris water maze
DIV
day(s) in vitro
AAV
adeno-associated virus
ANOVA
analysis of variance
EGFP
enhanced GFP.
REFERENCES
- 1.Esser V., Britton C. H., Weis B. C., Foster D. W., McGarry J. D. (1993) Cloning, sequencing, and expression of a cDNA encoding rat liver carnitine palmitoyltransferase I. Direct evidence that a single polypeptide is involved in inhibitor interaction and catalytic function. J. Biol. Chem. 268, 5817–5822 [PubMed] [Google Scholar]
- 2.Yamazaki N., Shinohara Y., Shima A., Terada H. (1995) High expression of a novel carnitine palmitoyltransferase I-like protein in rat brown adipose tissue and heart. Isolation and characterization of its cDNA clone. FEBS Lett. 363, 41–45 [DOI] [PubMed] [Google Scholar]
- 3.Price N., van der Leij F., Jackson V., Corstorphine C., Thomson R., Sorensen A., Zammit V. (2002) A novel brain-expressed protein related to carnitine palmitoyltransferase I. Genomics 80, 433–442 [DOI] [PubMed] [Google Scholar]
- 4.Sierra A. Y., Gratacós E., Carrasco P., Clotet J., Ureña J., Serra D., Asins G., Hegardt F. G., Casals N. (2008) CPT1c is localized in endoplasmic reticulum of neurons and has carnitine palmitoyltransferase activity. J. Biol. Chem. 283, 6878–6885 [DOI] [PubMed] [Google Scholar]
- 5.Wolfgang M. J., Kurama T., Dai Y., Suwa A., Asaumi M., Matsumoto S., Cha S. H., Shimokawa T., Lane M. D. (2006) The brain-specific carnitine palmitoyltransferase-1c regulates energy homeostasis. Proc. Natl. Acad. Sci. U.S.A. 103, 7282–7287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gao X. F., Chen W., Kong X. P., Xu A. M., Wang Z. G., Sweeney G., Wu D. (2009) Enhanced susceptibility of Cpt1c knockout mice to glucose intolerance induced by a high-fat diet involves elevated hepatic gluconeogenesis and decreased skeletal muscle glucose uptake. Diabetologia 52, 912–920 [DOI] [PubMed] [Google Scholar]
- 7.Dai Y., Wolfgang M. J., Cha S. H., Lane M. D. (2007) Localization and effect of ectopic expression of CPT1c in CNS feeding centers. Biochem. Biophys. Res. Commun. 359, 469–474 [DOI] [PubMed] [Google Scholar]
- 8.Reamy A. A., Wolfgang M. J. (2011) Carnitine palmitoyltransferase-1c gain-of-function in the brain results in postnatal microencephaly. J. Neurochem. 118, 388–398 [DOI] [PubMed] [Google Scholar]
- 9.Gao S., Zhu G., Gao X., Wu D., Carrasco P., Casals N., Hegardt F. G., Moran T. H., Lopaschuk G. D. (2011) Important roles of brain-specific carnitine palmitoyltransferase and ceramide metabolism in leptin hypothalamic control of feeding. Proc. Natl. Acad. Sci. U.S.A. 108, 9691–9696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zaugg K., Yao Y., Reilly P. T., Kannan K., Kiarash R., Mason J., Huang P., Sawyer S. K., Fuerth B., Faubert B., Kalliomäki T., Elia A., Luo X., Nadeem V., Bungard D., Yalavarthi S., Growney J. D., Wakeham A., Moolani Y., Silvester J., Ten A. Y., Bakker W., Tsuchihara K., Berger S. L., Hill R. P., Jones R. G., Tsao M., Robinson M. O., Thompson C. B., Pan G., Mak T. W. (2011) Carnitine palmitoyltransferase 1C promotes cell survival and tumor growth under conditions of metabolic stress. Genes Dev. 25, 1041–1051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tybulewicz V. L., Crawford C. E., Jackson P. K., Bronson R. T., Mulligan R. C. (1991) Neonatal lethality and lymphopenia in mice with a homozygous disruption of the c-abl proto-oncogene. Cell 65, 1153–1163 [DOI] [PubMed] [Google Scholar]
- 12.Arqué G., Fotaki V., Fernández D., Martínez de Lagrán M., Arbonés M. L., Dierssen M. (2008) Impaired spatial learning strategies and novel object recognition in mice haploinsufficient for the dual specificity tyrosine-regulated kinase-1A (Dyrk1A). PLoS One 3, e2575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Segura I., Essmann C. L., Weinges S., Acker-Palmer A. (2007) Grb4 and GIT1 transduce ephrinB reverse signals modulating spine morphogenesis and synapse formation. Nat. Neurosci. 10, 301–310 [DOI] [PubMed] [Google Scholar]
- 14.Grimm D., Kern A., Rittner K., Kleinschmidt J. A. (1998) Novel tools for production and purification of recombinant adenoassociated virus vectors. Hum. Gene Ther. 9, 2745–2760 [DOI] [PubMed] [Google Scholar]
- 15.Dentin R., Pégorier J. P., Benhamed F., Foufelle F., Ferré P., Fauveau V., Magnuson M. A., Girard J., Postic C. (2004) Hepatic glucokinase is required for the synergistic action of ChREBP and SREBP-1c on glycolytic and lipogenic gene expression. J. Biol. Chem. 279, 20314–20326 [DOI] [PubMed] [Google Scholar]
- 16.Merrill A. H., Jr., Sullards M. C., Allegood J. C., Kelly S., Wang E. (2005) Sphingolipidomics. High throughput, structure-specific, and quantitative analysis of sphingolipids by liquid chromatography tandem mass spectrometry. Methods 36, 207–224 [DOI] [PubMed] [Google Scholar]
- 17.Ben-David O., Futerman A. H. (2010) The role of the ceramide acyl chain length in neurodegeneration. Involvement of ceramide synthases. Neuromolecular Med. 12, 341–350 [DOI] [PubMed] [Google Scholar]
- 18.Tokutake Y., Onizawa N., Katoh H., Toyoda A., Chohnan S. (2010) Coenzyme A and its thioester pools in fasted and fed rat tissues. Biochem. Biophys. Res. Commun. 402, 158–162 [DOI] [PubMed] [Google Scholar]
- 19.Mitoma J., Ito M., Furuya S., Hirabayashi Y. (1998) Bipotential roles of ceramide in the growth of hippocampal neurons. Promotion of cell survival and dendritic outgrowth in dose- and developmental stage-dependent manners. J. Neurosci. Res. 51, 712–722 [DOI] [PubMed] [Google Scholar]
- 20.Hering H., Lin C. C., Sheng M. (2003) Lipid rafts in the maintenance of synapses, dendritic spines, and surface AMPA receptor stability. J. Neurosci. 23, 3262–3271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Meloni I., Parri V., De Filippis R., Ariani F., Artuso R., Bruttini M., Katzaki E., Longo I., Mari F., Bellan C., Dotti C. G., Renieri A. (2009) The XLMR gene ACSL4 plays a role in dendritic spine architecture. Neuroscience 159, 657–669 [DOI] [PubMed] [Google Scholar]
- 22.Breslow D. K., Weissman J. S. (2010) Membranes in balance. Mechanisms of sphingolipid homeostasis. Mol. Cell 40, 267–279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chalfant C. E., Kishikawa K., Mumby M. C., Kamibayashi C., Bielawska A., Hannun Y. A. (1999) Long chain ceramides activate protein phosphatase-1 and protein phosphatase-2A. Activation is stereospecific and regulated by phosphatidic acid. J. Biol. Chem. 274, 20313–20317 [DOI] [PubMed] [Google Scholar]
- 24.Terry-Lorenzo R. T., Roadcap D. W., Otsuka T., Blanpied T. A., Zamorano P. L., Garner C. C., Shenolikar S., Ehlers M. D. (2005) Neurabin/protein phosphatase-1 complex regulates dendritic spine morphogenesis and maturation. Mol. Biol. Cell 16, 2349–2362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Allen J. A., Halverson-Tamboli R. A., Rasenick M. M. (2007) Lipid raft microdomains and neurotransmitter signalling. Nat. Rev. Neurosci. 8, 128–140 [DOI] [PubMed] [Google Scholar]
- 26.Kasai H., Fukuda M., Watanabe S., Hayashi-Takagi A., Noguchi J. (2010) Structural dynamics of dendritic spines in memory and cognition. Trends Neurosci. 33, 121–129 [DOI] [PubMed] [Google Scholar]