Secondary Variants in Individuals Undergoing Exome Sequencing: Screening of 572 Individuals Identifies High-Penetrance Mutations in Cancer-Susceptibility Genes (original) (raw)
Abstract
Genome- and exome-sequencing costs are continuing to fall, and many individuals are undergoing these assessments as research participants and patients. The issue of secondary (so-called incidental) findings in exome analysis is controversial, and data are needed on methods of detection and their frequency. We piloted secondary variant detection by analyzing exomes for mutations in cancer-susceptibility syndromes in subjects ascertained for atherosclerosis phenotypes. We performed exome sequencing on 572 ClinSeq participants, and in 37 genes, we interpreted variants that cause high-penetrance cancer syndromes by using an algorithm that filtered results on the basis of mutation type, quality, and frequency and that filtered mutation-database entries on the basis of defined categories of causation. We identified 454 sequence variants that differed from the human reference. Exclusions were made on the basis of sequence quality (26 variants) and high frequency in the cohort (77 variants) or dbSNP (17 variants), leaving 334 variants of potential clinical importance. These were further filtered on the basis of curation of literature reports. Seven participants, four of whom were of Ashkenazi Jewish descent and three of whom did not meet family-history-based referral criteria, had deleterious BRCA1 or BRCA2 mutations. One participant had a deleterious SDHC mutation, which causes paragangliomas. Exome sequencing, coupled with multidisciplinary interpretation, detected clinically important mutations in cancer-susceptibility genes; four of such mutations were in individuals without a significant family history of disease. We conclude that secondary variants of high clinical importance will be detected at an appreciable frequency in exomes, and we suggest that priority be given to the development of more efficient modes of interpretation with trials in larger patient groups.
Introduction
High-throughput sequencing is effective for elucidating the cause of heritable disorders1 and for interrogating many genes in high-risk individuals.2 The number of research subjects undergoing exome or genome sequencing is rapidly increasing. Apart from the identification of the mutation causing the disorder for which the sequencing was performed (i.e., the primary variant), genome and exome sequencing have the potential to identify other clinically important results (i.e., secondary or so-called incidental variants). The number of clinically important secondary variants in human genomes is substantial. Each genome in the 1000 Genomes Project has 50–100 variants in disease-associated genes,3 and a screen4 of 104 exomes for 448 severe recessive diseases found an average carrier burden of 2.8 variants per person. Recommended interventions for highly penetrant conditions might include prophylactic cancer surgery and screening,5 implantation of a cardioverter defibrillator,6 and pharmacogenomic7 and reproductive decision making.8
The potential ability to identify secondary variants has led to controversy regarding whether these variants should be sought, and if they are sought or accidentally encountered, whether and how these variants should be returned to patients or research subjects.9–11 To shed light on this controversy, we set out to develop approaches to the analysis and return of whole-exome sequencing (WES) results for secondary (or so-called incidental) variants in genes associated with high-penetrance cancer-susceptibility syndromes in a cohort not ascertained for these conditions. We piloted these approaches on 572 ClinSeq12 participants and report the yield, utility, and limitations of this strategy. Finally, we discuss the implications of these findings for researchers generating these kinds of data and considering whether—and how—to return such results to study participants.
Subjects and Methods
Study Participants
ClinSeq participants were 45–65 years of age and gave consent for genome and exome sequencing and the return of results.12 They were selected for a range of atherosclerosis phenotypes, but not for personal or family histories of cancer. Family history, including ethnicity, was collected during the initial evaluation. The institutional review board (IRB) at the National Human Genome Research Institute reviewed and approved this study, and all subjects provided written informed consent.
Gene List
We developed a list (Table 1) comprising 27 cancer syndromes caused by mutations in 37 genes. The list was based on the “Concise handbook of familial cancer susceptibility syndromes—second edition” (2008).13 We added SDHAF2 (MIM 613019), implicated in paragangliomas 2 (MIM 601650), because that was positionally cloned after this chapter was written. The gene list was frozen as of the end of 2010. Seventeen primarily pediatric syndromes were excluded because these syndromes were unlikely to be clinically unrecognized in subjects older than 45 years. This curated list comprises mainly autosomal-dominant, high-penetrance syndromes except for familial adenomatous polyposis 2 (MIM 608456), an autosomal-recessive disorder, because it is similar in penetrance to some dominant, high-penetrance cancer syndromes.13
Table 1.
The 37 Cancer-Associated Genes Analyzed in this Study and a Summary of Variants Identified in Each Gene by Pathogenicity Score
| Gene | RefSeq Accession Number | Disease Associations | MIM Number | Total Variants | Pathogenicity Scorea | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| 0 | 1 | 2 | 3 | 4 | 5 | |||||
| APC | NG_008481.4 | familial adenomatous polyposis | 175100, 611731, and 135290 | 45 | 1 | 6 | − | 38 | − | − |
| BMPR1A | NG_009362.1 | familial juvenile polyposis | 174900 and 601299 | 4 | − | 1 | − | 3 | − | − |
| BRCA1 | NG_005905.2 | hereditary breast and ovarian cancer | 113705 | 40 | 2 | 13 | 3 | 20 | − | 2 |
| BRCA2 | NG_012772.1 | hereditary breast and ovarian cancer | 600185 | 52 | 1 | 11 | 9 | 28 | − | 3 |
| CDC73 (HPRT2) | NG_012691.1 | familial hyperparathyroidism and hereditary hyperparathyroidism-jaw tumor syndrome | 145000 and 145001 | 1 | − | − | − | 1 | − | − |
| CDH1 | NG_008021.1 | hereditary diffuse gastric cancer | 137215 and 192090 | 6 | − | 1 | − | 5 | − | − |
| CDKN2A | NG_007485.1 | hereditary multiple melanoma | 155601, 155755, 600160, and 606719 | 9 | − | 2 | − | 7 | − | − |
| FH | NG_012338.1 | hereditary renal cell carcinoma with multiple cutaneous and uterine leiomyomas | 605839, 136850, and 150800 | 5 | − | − | − | 5 | − | − |
| FLCN | NG_008001.1 | Birt-Hogg-Dubé syndrome | 135150 and 607273 | 10 | − | 2 | − | 8 | − | − |
| KIT | NG_007456.1 | gastrointestinal stromal tumor | 606764 and 164920 | 11 | 1 | 1 | − | 9 | − | − |
| MEN1 | NG_008929.1 | multiple endocrine neoplasia type 1 | 131100 | 6 | − | 2 | − | 4 | − | − |
| MET | NG_008996.1 | hereditary papillary renal cell carcinoma | 605074 and 164860 | 30 | 6 | 4 | − | 20 | − | − |
| MLH1 | NG_007109.1 | hereditary nonpolyposis colon cancer (Lynch syndrome) | 609310, 276300, 608089, 158320, 120436, and 120435 | 14 | − | 6 | − | 8 | − | − |
| MSH2 | NG_007110.1 | hereditary nonpolyposis colon cancer (Lynch syndrome) | 276300, 608089, 158320, 20435, and 609309 | 15 | − | 3 | − | 12 | − | − |
| MSH6 | NG_007111.1 | hereditary nonpolyposis colon cancer (Lynch syndrome) | 276300, 608089, 120435, and 600678 | 15 | 1 | 2 | − | 12 | − | − |
| MUTYH | NG_008189.1 | MYH-associated polyposis | 608456 and 604933 | 20 | 1 | 5 | − | 10 | 2 | 2 |
| NF1 | NG_009018.1 | neurofibromatosis type 1 | 162200, 162210, and 193520 | 14 | 1 | 2 | − | 11 | − | − |
| NF2 | NG_009057.1 | neurofibromatosis type 2 | 101000 and 607379 | 1 | − | − | − | 1 | − | − |
| PDGFRA | NG_009250.1 | gastrointestinal stromal tumor | 606764 and 173490 | 15 | 2 | 2 | − | 11 | − | − |
| PMS2 | NG_008466.1 | hereditary nonpolyposis colon cancer (Lynch syndrome) | 276300, 608089, 120435, and 600259 | 29 | 4 | 12 | − | 13 | − | − |
| PRKAR1A | NG_007093.2 | Carney complex type 1 | 160980, 188830, and 610489 | 0 | − | − | − | − | − | − |
| PTCH1 | NG_007664.1 | nevoid basal cell carcinoma syndrome | 109400 and 601309 | 20 | − | 4 | − | 16 | − | − |
| PTEN | NG_007466.1 | Cowden disease | 158350 and 601728 | 1 | − | − | − | 1 | − | − |
| RB1 | NG_009009.1 | hereditary retinoblastoma | 180200 | 4 | 1 | − | − | 3 | − | − |
| RET | NG_007489.1 | multiple endocrine neoplasia types 2A and 2B and familial medullary thyroid cancer | 171400, 155240, 162300, and 164761 | 15 | − | 4 | 1 | 10 | − | − |
| SDHAF2 | NG_023393.1 | hereditary paraganglioma | 601650 and 613019 | 2 | − | − | − | 2 | − | − |
| SDHB | NG_012340.1 | hereditary paraganglioma | 115310 and 185470 | 4 | − | 1 | − | 3 | − | − |
| SDHC | NG_012767.1 | hereditary paraganglioma | 605373 and 602413 | 9 | 4 | 2 | − | 2 | − | 1 |
| SDHD | NG_012337.1 | hereditary paraganglioma | 168000 and 602690 | 3 | − | 1 | − | 2 | − | − |
| SMAD4 | NG_013013.1 | familial juvenile polyposis | 174900, 175050, and 600993 | 2 | − | − | − | 2 | − | − |
| SMARCB1 | NG_009303.1 | schwannomatosis | 162091 and 601607 | 1 | − | − | − | 1 | − | − |
| STK11 | NG_007460.1 | Peutz-Jeghers syndrome | 175200 and 602216 | 0 | − | − | − | − | − | − |
| TP53 | NG_017013.1 | Li-Fraumeni syndrome | 151623, 191170, and 202300 | 2 | − | 1 | − | 1 | − | − |
| TSC1 | NG_012386.1 | tuberous sclerosis complex 1 | 191100 and 605284 | 17 | 1 | 2 | 4 | 10 | − | − |
| TSC2 | NG_005895.1 | tuberous sclerosis complex 2 | 191092 and 613254 | 28 | 1 | 4 | 11 | 12 | − | − |
| VHL | NG_008212.2 | von Hippel-Lindau syndrome | 193300 and 608537 | 1 | − | − | − | 1 | − | − |
| WT1 | NG_009272.1 | familial Wilms tumor 1 | 607102 and 194070 | 1 | − | − | − | 1 | − | − |
Next-Generation Sequencing and Variant Analysis
DNA was isolated from whole blood via the salting-out method (Qiagen, Valencia, CA). Solution-hybridization exome capture was performed with the SureSelect All Exon System (Agilent Technologies, Santa Clara, CA). The manufacturer's protocol version 1.0, compatible with Illumina paired-end sequencing, was used. Flow-cell preparation and sequencing of 101 bp paired-end reads were performed for the GAIIx sequencer14 (Illumina, San Diego, CA). Image analyses and base calling were performed as described.14 Reads were aligned to hg18 (NCBI build 36) with ELAND (Illumina). Uniquely aligned reads were grouped into ∼100 kb intervals, and unaligned reads were binned with their paired-end mates. Binned reads were aligned to their genomic sequence bin with cross_match and the use of parameters –minscore 21 and –masklevel 0. Typically, one or two 101 bp paired-end flow-cell lanes, or 4–8 Gb of sequence, were sufficient for the generation of ≥85% coverage of the targeted exome with high-quality variant detection (reported as a genotype at every callable position). Genotypes were called at high-quality sequence bases (Phred-like ≥Q20) with Most Probable Genotype14 (MPG). Filters were applied with the VarSifter Next-Gen variation analysis software.15
Our goal was to identify variations highly likely to be causative and to receive few false positives; therefore, sensitivity was sacrificed. We analyzed nonsense, frameshift, splice-site, and nonsynonymous variants in 37 cancer genes in 572 ClinSeq participants. Variants were filtered (Figure 1) on the basis of quality and frequency. Quality filters included a MPG score ≥10 and an MPG/read count ratio of >0.5. The most common of the syndromes analyzed was hereditary breast and ovarian cancer (HBOC, which includes BROVCA1 [MIM 604370] and BROVC2 [MIM 612555]), which had a frequency of ∼1/500 for BRCA1 (MIM 113075) and BRCA2 (MIM 600185) combined.16 We reasoned that an allele frequency of >0.5% in ClinSeq was appropriate for the exclusion of pathogenicity and that a SNP with a minor allele frequency of >0.015 (dbSNP17 build 132, minimum 120 chromosomes) was unlikely to cause a highly penetrant, rare, dominant disorder. Variants were graded from class 1 (almost certainly benign) to class 5 (definitely pathogenic) with a modified version of an established scale18. The modified classification scheme presented in Table 2 relies on qualitative rather than quantitative assessments of variant causation. Adequate data do not exist for the majority of variants to allow for quantitative assessments. Therefore, we modified the original scale. Variants that failed quality filters were assigned to class 0, and variants that were excluded as pathogenic on the basis of frequency data were assigned to class 1. For all other variants, the Human Gene Mutation Database (HGMD19) and locus-specific databases (LSDBs20,21) were consulted, and variants were assigned to pathogenicity classes according to the guidelines presented in Table 2. Missense variants or in frame indels not listed in HGMD or the consulted LSDBs (these variants are hereafter referred to as novel variants) were assigned to class 3. For novel nonsense, frameshift, and splice-site variants, the characteristics of the gene and the variant, as well as participant family history, were considered. When HGMD and/or an LSDB reported a variant as pathogenic, we reviewed relevant citations to determine whether the variant should be assigned to class 5. When the relevant citations supported causation, we analyzed family-history data. For missense alterations or in frame insertions/deletions, a single report was insufficient for the assignment of a variant to class 5. Variants not reported as pathogenic by HGMD and/or an LSDB were defined as variants of unknown significance (VUS). The VUSs were assigned to class 2 (highly likely to be benign), 4 (highly likely to be pathogenic), or 3 (a wide range from probably benign to possibly pathogenic). An initial data freeze was analyzed at 258 exomes. For the 258 exome dataset, relevant citations from HGMD and LSDBs were reviewed for all VUSs when they were being assigned to pathogenicity classes. It was determined that when HGMD or an LSDB did not identify a variant as pathogenic, a literature review did not result in any variant being assigned to class 5. For the 572 exome dataset, information available in HGMD and LSDBs was analyzed, but the primary literature was not reviewed for VUSs. Variants were assigned to class 2 if they had been reported multiple times as benign or if multiple pieces of evidence were presented against causation. Evidence against causation included presence in controls, co-occurrence with a known pathogenic mutation, and/or normal functional data. Class 4 was assigned when multiple primary reports defined the variant as pathogenic and evidence against causation was not presented. All other variants were assigned to class 3. Variants were scored by individual investigators and then reviewed by all authors. Probands with BRCA1 or BRCA2 class 5 variants were evaluated with BRCAPRO (CancerGene package version 5.1, Southwestern Medical Center at Dallas) and U.S. Preventive Services Task Force Guidelines22 (applicable only to unaffected women). The interpretations of individual variants, summarized in Table S1, available online, were submitted to ClinVar to be assigned a permanent accession. ClinVar is a new, centralized open-access database maintained by the National Institutes of Health National Center for Biotechnology Information (NCBI), which collects sequence-level and structural variant information, associated phenotypic data, and clinical assertions from numerous clinical laboratories and locus-specific databases. Although the ClinVar resource is under construction, data have been collected from several sources and are currently viewable through other active NCBI resources, such as Variation Viewer. We are pleased to have been the first to request ClinVar accessions before publication.
Figure 1.
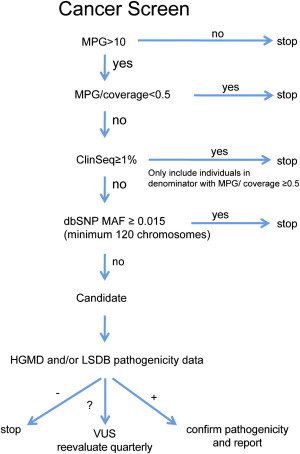
Filtering Criteria Used for Coding-Variant Interpretation
Variants were filtered for quality with MPG scores and coverage, frequency in ClinSeq and minor allele frequency in dbSNP, and data present in the Human Gene Mutation Database (HGMD) and locus specific databases (LSDBs) for each gene when available. Variants were determined to be benign, pathogenic, or of unknown significance (VUSs).
Table 2.
Sequence-Variant Pathogenicity Categorization
| Variants Not in Database | Pathogenic Variants | VUSs | Benign Variants | |||
|---|---|---|---|---|---|---|
| Missense Mutation | Nonsense, Frameshift, and Splice Mutations | Missense Mutation | Nonsense, Frameshift, and Splice Mutations | Any Mutation | Any Mutation | |
| Class 5 | consistent family historya and loss-of-function mutations known to cause disease | multiple primary reports as pathogenic and no evidence against causation | multiple or single primary report as pathogenic and no evidence against causation | |||
| Class 4 | equivocal family historya and loss-of-function mutations known to cause disease | multiple primary reports as pathogenic and evidence against causation or a single primary report as pathogenic with supporting evidence of causation | multiple primary reports as pathogenic and a single piece of evidence against causation | multiple primary reports as pathogenic | ||
| Class 3 | all novel missense | inconsistent family historya or loss-of-function mutations not known to cause disease | single primary report as pathogenic and no supporting evidence of causation | multiple primary reports as pathogenic and multiple pieces of evidence against causation or a single primary report as pathogenic and a single piece of evidence against causation | primary reports as a VUS | a single report as benign or primary reports as pathogenic |
| Class 2 | single primary report as pathogenic and multiple pieces of evidence against causation | single primary report as pathogenic with multiple evidence against causation | multiple pieces of evidence against causation | multiple primary reports as benign and no supporting evidence of causation or a single primary report as benign and multiple pieces of evidence against causation |
Return of Results
All class 5 variants predicted to cause an autosomal-dominant cancer syndrome were confirmed in our Clinical Laboratory Improvement Amendments (CLIA)-certified laboratory. The clinical test results were provided to the participants by a clinical geneticist. Participants with BRCA1 and BRCA2 variants were advised on cancer prevention and surveillance guidelines developed by the National Comprehensive Cancer Network (NCCN) and were referred to community clinical resources for ongoing surveillance. The participant with the SDHC (MIM 602413) variant was counseled regarding the paragangliomas 3 phenotype (MIM 605373) and its management according to published guidelines.23 Participants were encouraged to share their genetic results with their families. Participants with at-risk relatives were counseled to have those relatives receive mutation-specific clinical testing through a clinician and an approved clinical testing laboratory. Results containing monoallelic MUTYH (MIM 604933) class 5 variants implicated in recessive familial adenomatous polyposis 2 were not returned to participants as part of this study but will be validated and returned at a later time with other heterozygous-carrier results for disorders inherited in a recessive pattern.
Results
Participant Demographics
The dataset included 572 participants, of whom 92.2% were white and 97.7% were not of Hispanic or Latino background. Seventeen percent of the study population was of Ashkenazi ancestry. The median age was 58 years, and 46.5% were female and 53.5% were male.
Sequence Data
For the 572 exomes, 44.5 billion reads were generated, resulting in 3.84 trillion bp of sequence and 1,921,814 variants. Copy-number variants and indels greater than 10 bp were not assessed, which is a limitation of exome sequencing. A total of 181,736 variants were nonsynonymous, frameshift, nonsense, or splicing. The targeted coding sequences of the 37 genes comprised 101,235 bp. We achieved an overall coverage of 91.2%, and the per-gene coverage was between 22.3% (for STK11 [MIM 602216]) and 100% (for SDHD and SMAD4 [MIM 602690 and 600993, respectively]) (Figure 2). Narrowing the results to the 37 cancer genes identified 454 distinct potential nonsynonymous, frameshift, nonsense, or splice variants. Note that we counted variants once irrespective of whether they were present in one or more individuals. One variant was nonreference in all 572 participants and was excluded. An indel at the 3′ end of MLH1 (MIM 120436) did not alter the predicted protein sequence and was excluded. Two single-base variants were a single 2 bp indel and were combined. This left a total of 451 variants (Table S1).
Figure 2.
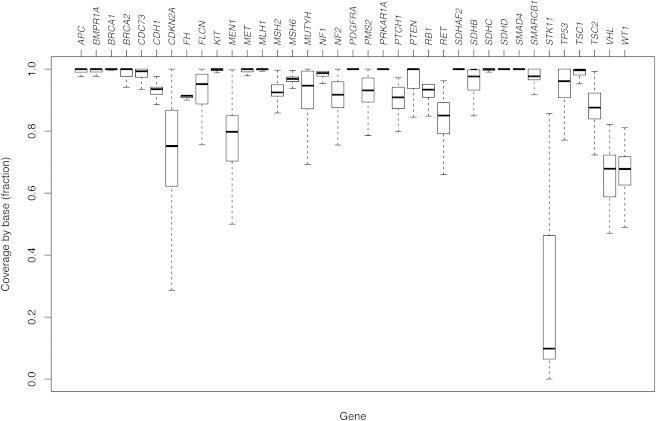
Box and Whisker Plots Showing Base Coverage for 37 Cancer-Associated Genes across a Cohort of 572 Probands
The MIM numbers for these genes are listed in Table 1.
Variant Filtering and Classification
The 451 variants were filtered for quality (n = 26, class 0), leaving 425. Ninety-four variants were defined as class 1 on the basis of ClinSeq frequency (n = 77) or dbSNP frequency (n = 17), leaving 331 (Figure 3). Of these 331 variants (classes 2–5), 186 were not listed in HGMD or the consulted LSDBs and were considered novel. All novel missense and single-amino-acid-deletion variants as well as alterations in non-HGMD transcripts were defined as class 3 (n = 186). Variants included in HGMD or the LSDBs, but not listed as pathogenic (n = 77), were classified on the basis of information available in the databases. Consideration was given to primary reports of pathogenicity as well as factors including but not limited to presence in controls and functional data. These 77 variants were defined as class 2 (n = 16) or class 3 (n = 61). The remaining 68 variants were listed as pathogenic in either HGMD (n = 59), the LSDBs (n = 4), or both (n = 5). When variants were listed as pathogenic in HGMD or LSDBs, relevant literature was reviewed. In cases where the literature suggested that a variant was likely to be pathogenic, family-history data were reviewed. Of these 68 variants, 5 were described as pathogenic for diseases other than cancer-susceptibility syndromes, leaving 63 variants for assessment. Fifty-three of these variants were defined as class 2 (n = 12) or class 3 (n = 41) on the basis of the literature review and/or participant family history, leaving ten variants.
Figure 3.
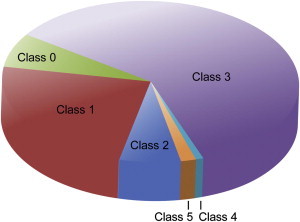
Characterization of 572 Variants by Pathogenicity Class
Variants were graded from 1 to 5 with a modified version of an established scale;18 1 is benign, and 5 is pathogenic. Variants that failed quality filters were defined as class 0. VUSs were defined as classes 2–4. Class 2 included variants highly likely to be benign, class 4 included variants highly likely to be pathogenic, and others were assigned to class 3.
These ten variants were assigned to classes 4 or 5 and included variants in MUTYH (class 4, n = 2; class 5, n = 2), BRCA1 (class 5, n = 2), BRCA2, (class 5, n = 3), and SDHC (class 5, n = 1). High-penetrance colon cancer is associated with biallelic MUTYH mutations; all variants identified in MUTYH were monoalleleic. A variant in SDHC (RefSeq NM_003001.3), c.43C>T (p.Arg15∗) (class 5), was identified in a single proband without a personal or family history of cancer. Mutations in SDHC predispose to head and neck paragangliomas.
The five remaining class 5 variants found among seven participants were in BRCA1 (n = 2) or BRCA2 (n = 3) (Table 3). One participant was heterozygous for BRCA1 (RefSeq NM_007294.3) c.68_69del (formerly described as c.del185AG) (p.Glu23Valfs∗17), an Ashkenazi founder mutation.24 His pedigree was not suggestive of HBOC given that he was assigned a prior probability of 0.3% for a BRCA1/2 mutation by the BRCAPRO algorithm25 (Figure 4A). Additionally, he did not meet NCCN guidelines for further risk evaluation. The second participant was heterozygous for BRCA1 c.547+2T>A, reported as clinically important in the Breast Cancer Information core database.26 This individual had a family history of breast and ovarian cancer and knew her mutation status before enrolling in our study.
Table 3.
Participant Information for Pathogenic Variants Identified in BRCA1 and BRCA2
| Gene | Mutations and RefSeq Accession Numbers | Sex | Age | Ethnicity | Mutation Results Prior to Study | BRCAPRO Score | Met NCCN Guidelines | Met USPSTF Referral Criteria (Women Only) |
|---|---|---|---|---|---|---|---|---|
| BRCA1 | c.547+2T>Aa (NM_007294.3) | female | 48 years | northern European | yes | N/A | yes | yes |
| BRCA1 | c.68_69del(p.Glu23Valfs∗17) (NM_007294.3) | male | 61 years | Ashkenazi Jewish | no | 0.3% | no | N/A |
| BRCA2 | c.5482_5486 del(p.Lys1828Valfs∗4) (NM_000059.3) | female | 56 years | Japanese | no | 0.0% | no | no |
| BRCA2 | c.5946del(p.Ser1982Argfs∗22) (NM_000059.3) | male | 57 years | Ashkenazi Jewish | no | 0.9% | yes | N/A |
| BRCA2 | c.5946del(p.Ser1982Argfs∗22) (NM_000059.3) | male | 60 years | Ashkenazi Jewish | no | 42.3% | yes | N/A |
| BRCA2 | c.5946del(p.Ser1982Argfs∗22) (NM_000059.3) | male | 55 years | Ashkenazi Jewish | yes | N/A | yes | N/A |
| BRCA2 | c.8297del(p.Thr2766Asnfs∗11) (NM_000059.3) | male | 59 years | Irish | no | 0.6% | no | N/A |
Figure 4.
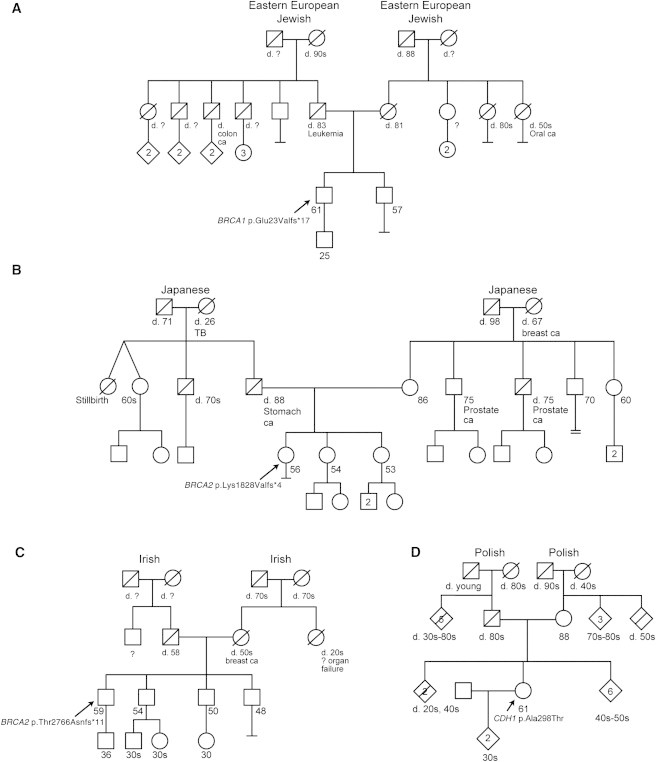
Family Histories for Selected Variants
The pathogenicity classes ascribed to variants detected in the probands of each family are as follows: class 5 for BRCA1 and BRCA2 variants in families A, B, and C and class 3 for the CDH1 variant in family D. The diamond symbol indicates relatives of probands in some families so that these families can remain anonymous. Of note, a first-degree relative and a second-degree relative of the proband in family D were diagnosed with prostate cancer; however, these individuals were from separate lineages, and prostate cancer is not thought to be a part of hereditary diffuse gastric cancer syndrome.
Three class 5 variants were identified among five participants in BRCA2. Three participants were heterozygous for BRCA2 (RefSeq NM_000059.3) c.5946del (formerly described as c.6174delT) (p.Ser1982Argfs∗22), an Ashkenazi founder mutation.24 All three of these probands reported pedigrees that met NCCN guidelines for further risk evaluation, but only one knew his mutation status before enrolling in our study. Two other frameshift mutations in BRCA2, c.5482_5486del (p.Lys1828Valfs∗4) and c.8297del (p.Thr2766Asnfs∗11), which are both known deleterious variants,27 were identified in our cohort. Neither of the participants with these mutations met NCCN guidelines for further risk evaluation given that they were assigned prior probabilities of 0 and 0.6% for a BRCA1/2 mutation by the BRCAPRO algorithm25,28 (Figures 4B and 4C).
Discussion
Our goal was to pilot an analytic method to identify clinically important secondary variants on a reasonable set of genes from a large set of exome-sequence data. We focused our analysis on 37 genes for 27 cancer-predisposing syndromes (Table 1) well represented by LSDBs and published data. Although this is not an exhaustive gene list, it includes most of the well-curated cancer-susceptibility loci and provides an appropriate starting point for this type of analysis. This analysis has the potential to reveal generalizable conclusions about the analytic approach necessary for identifying such variants and the relative frequency of clinically important variants. With respect to the current debate surrounding secondary variants,29,30 these results provide data on the analysis and frequency of secondary variants in a research cohort. These data prove that medically important variants occur at an appreciable frequency and that ignoring such variants could be detrimental to the well-being of research participants.
The data illustrate a range of results from pathogenic to benign. We detected a total of five class 5 variants in genes associated with breast and ovarian cancer among seven probands in the ClinSeq cohort. These included two BRCA1 class 5 variants (each in a single subject) and three BRCA2 class 5 variants (one in three subjects and two in single subjects). Our detection of these pathogenic variants in seven probands illustrates the ability of exome sequencing to identify secondary variants that are of high potential medical impact; in the cases of BRCA1 and BRCA2, the identification of these variants can substantially reduce mortality in females.5,31 An important question to ask is whether the research participants with these variants should, or could, have been readily identified or diagnosed by other means. Of those with BRCA1 and BRCA2 variants, four probands reported family histories that met NCCN guidelines for further risk assessment for hereditary breast and ovarian cancer; three did not. Of the four probands who met NCCN guidelines, two had already undergone testing for BRCA1 and BRCA2. The other two individuals, both males, had not done so at the time of enrollment and might not have done so outside of this study (Table 3). The fact that three (one with a BRCA1 mutation and two with BRCA2 mutations) out of seven probands with these high-penetrance mutations were not predicted by standard approaches highlights the limitations of a family-history-based approach to the detection of hereditary cancer risk. Familial risk assessment of breast- and ovarian-cancer-susceptibility syndromes can be limited by small family size or a paucity of females.32 Population-based series have shown that about half of BRCA1 and BRCA2 heterozygotes with incident cancers lack a family history of breast or ovarian cancer.33
An additional class 5 variant was the SDHC variant c.43C>T (p.Arg15∗). Mutations in SDHC typically cause benign, unifocal paragangliomas, which might cause cranial nerve damage leading to considerable morbidity.34,35 The c.43C>T variant has been detected in the probands of several studies, including one report with no individual clinical data,36 one report of a patient with paraganglioma and a gastrointestinal stromal tumor and an unknown family history,37 and one report of a patient with a glomus tumor but no family data.38 No symptoms of this disorder were reported by the subjects upon enrollment in these studies, and the family history did not identify any affected individuals. Clinical evaluation of this research subject is underway.
In addition to these class 5 variants in BRCA1, BRCA2, and SDHC, we classified two variants in MUTYH (RefSeq NM_001048171.1) as class 5. These two variants, c.494A>G (p.Tyr165Cys) and c.1145G>A (p.Gly382Asp), are present in the general population at a combined frequency of approximately 0.8% and account for the majority of mutations in MUTYH (dbSNP). One of these variants was detected in two subjects, and the other was detected in five subjects. MUTYH mutations cause high-penetrance colon-cancer susceptibility inherited in an autosomal-recessive pattern. Pathogenic variants in this gene might modestly increase the risk of colon cancer in a heterozygous state. Because none of the variants were found in the biallelic state, we elected to treat these variants as autosomal-recessive carrier variants.
We classified two variants as class 4 pathogenicity. Both of these variants were in MUTYH and will be handled similarly to the MUTYH variants described above.
We found a total of 293 class 3 and 28 class 2 variants. One example of a class 3 variant for which the literature review and analysis of our data led to the downgrading of its pathogenicity is a variant in CDH1 (MIM 192090), c.892G>A (RefSeq NM_004360.3) (p.Ala298Thr), in two unrelated ClinSeq probands with negative family histories. Pathogenic CDH1 mutations cause hereditary diffuse gastric cancer (MIM 137215), which is rare and highly penetrant.39 The c. 892A>G (p.Ala298Thr) variant was reported in a single family,40 and in vitro studies showed abnormal results in a functional assay.40,41 On the basis of these data, the CDH1 variant was listed as causative in HGMD; however, our identification of two probands (2/572) with this variant and without a family history of gastric or breast cancer (a representative family is shown in Figure 4D) argues against this variant causing a highly penetrant cancer syndrome. We conclude that this variant is of unknown pathogenicity, and we therefore defined it as class 3. Clarifying its pathogenicity will require further research. Additional efforts to clarify pathogenicity would be impractical for all class 3 and class 2 variants. As noted above, the prior probability of disease in a group that is unselected for these phenotypes is small. These data suggest that exome interrogation for these 27 genes in a population not ascertained for cancer generates many more class 2 or 3 variants (n = 321) than class 4 or 5 variants (n = 10). This burden of ambiguous variants is a significant issue and should be considered by clinicians, researchers, and IRBs when designing WES studies.
Our experience is emblematic of challenges in interpreting high-throughput data. HGMD and many LSDBs have limited curation resources and significant misclassification rates.4,42 In addition, the pathogenicity determination of variants can vary among LSDBs and HGMD, making interpretation of these data challenging. Of the 451 variants identified in this study, nine that were identified as pathogenic in HGMD were reclassified as benign on the basis of frequency in controls. Fifty-nine other variants identified as pathogenic in HGMD were assigned pathogenicity scores between 2 and 4 on the basis of co-occurrence with known pathogenic mutations, limited information on causality, or association to a disease other than a high-penetrance cancer syndrome. Two variants (c.1145G>A [p.Gly382Asp] and c.494A>G [p.Tyr165Cys] in MUTYH) assessed here as class 5 were identified in HGMD as disease-associated polymorphisms rather than disease-causing mutations possibly on the basis of their high allele frequency in the general population. Both of these variants were also included in dbSNP as rs36053993 and rs34612342. dbSNP includes both pathogenic and nonpathogenic variants. We observed that the pathogenicity determination of a given variant was generally more conservative in LSDBs than in HGMD. Of the nine variants previously classified as pathogenic in an LSDB, we assigned eight of those nine variants pathogenicity scores of 5. Furthermore, no variant that was not identified as pathogenic in the corresponding LSDB was assigned a pathogenicity score of 5. The 1000 Genomes dataset3 does not include phenotypic data, so deriving conclusions of causation from those data is difficult. These complexities of determining causality are not novel and have bedeviled single-gene testing laboratories for years. However, the scale of WES greatly magnifies these issues and argues for increased efforts to improve mutation databases before genomic screening of healthy individuals moves into the clinical realm. Cohorts such as ClinSeq, which have robust phenotypic data and the ability to perform iterative clinical research, should be useful for the assessment of the pathogenicity of variants and the improvement of these databases.
We detected eight cases of a high-penetrance autosomal-dominant tumor or cancer syndrome among 572 persons undergoing WES. The high rate of breast and ovarian cancer susceptibility is attributable in part to the large proportion (∼17%) of Ashkenazi Jewish participants in ClinSeq and the 2.5% prevalence of BRCA1 or BRCA2 founder mutations among that group. However, three participants with BRCA1 or BRCA2 mutations were not of Ashkenazi Jewish heritage, and two of these individuals did not meet NCCN guidelines for further risk assessment. Additionally, the individual with the SDHC mutation did not have a family history of paraganglioma. In all, at least three of the seven individuals with class 5 variants had no indication from family history that they were at an increased risk of developing familial cancer.
This study has a number of limitations. Like the early adopters of genetic testing in specialty clinics or those undergoing direct-to-consumer genetic testing, ClinSeq subjects are not representative of the general population. The cohort has a high average income and educational level and exhibits a high degree of curiosity and motivation regarding genetic testing and research.43 Another potential source of bias is that some ClinSeq participants enrolled on the basis of family history. This effect is evident in at least one other rare disease in the ClinSeq cohort (Biesecker et al., unpublished data). It should not be concluded that the overall prevalence of pathogenic mutations in this cohort is representative of the general population. A further complication might be the average age of our cohort. The average age of 58 years might cause a reduced number of high-penetrance cancer alleles in the sample because many of these variants cause morbidity and mortality at younger ages. Although 572 exomes is a prodigious amount of data, we cannot measure clinical utility. We do not have cost-effectiveness data because neither the sequencing costs nor the costs of the downstream medical evaluations can be readily measured. We did not measure the resources required to annotate these exomes, but we estimate that analyzing these 37 genes required 1–2 hours of time per sample. This time requirement is falling as our experience accumulates. We can now analyze additional exomes in less than 1 hr. Irrespective of this downward trend, scaling this manual curation approach to the entire genome is impractical, and these data highlight the urgent need for improved analytic algorithms and mutation databases for the automation of these processes. To address this issue, we submitted a description of our study and interpretations to ClinVar. The ClinVar infrastructure and accessioning of submitted data is designed to facilitate curation of variants at multiple levels (e.g., uncurated, single-source curation, expert-level curation, or practice guidelines). The goal of this resource is to provide a freely available archive of reported human variation and the evidence used for the generation of the interpretation of that variation with the viewpoint of providing infrastructure for future reinterpretation.
Overall, the data argue for the potential medical utility of the interpretation and return of secondary variants because the number of identified individuals with clinically important results is substantial. In addition, these data suggest that it might be possible to implement a clinical screen for rare cancer-susceptibility syndromes with the use of WES data and that cases that would otherwise go undiagnosed until family members manifest cancer might be detected with a genomics-first approach. Genetic testing for cancer susceptibility identified by an atypical disease in the proband and/or by positive family history has been widely embraced, suggesting that oncology might be one of the earliest specialties to benefit from genomic screening. Studies of other highly penetrant, clinically important, adult-onset disorders will help clarify the potential utility of screening with the use of genomics. Further work is needed for the expansion of these efforts into larger, more varied cohorts, the expansion of the gene target list, the automation of these processes, and the determination of their efficacy.
Acknowledgments
The authors are grateful for the contributions of the staff of the National Institutes of Health (NIH) Intramural Sequencing Center and the NIH Clinical Center; specifically, we thank the clinical support and nursing staff for their help with the clinical aspects of this study. Donna Maglott facilitated the inclusion of ClinVar accession numbers. This study was funded by the Intramural Research Program of the NIH National Human Genome Research Institute.
Supplemental Data
Document S1. Table S1
Web Resources
The URLs for data presented herein are as follows:
- Breast Cancer Information Core database, http://research.nhgri.nih.gov/bic/
- ClinVar, http://www.ncbi.nlm.nih.gov/clinvar/
- dbSNP, http://www.ncbi.nlm.nih.gov/projects/SNP/
- The Human Gene Mutation Database, http://www.hgmd.org/
- Leiden Open Variation Database, http://www.lovd.nl/2.0/index_list.php
- NCCN Guidelines Version 1.2011, http://www.nccn.org/professionals/physician_gls/pdf/genetics_screening.pdf
- Phrap/Cross_match/Swat, http://www.phrap.org/phredphrapconsed.html#block_phrap
- VarSifter, http://research.nhgri.nih.gov/software/VarSifter/
Accession Numbers
The ClinVar accession numbers for the variants identified in the 572 ClinSeq participants are listed in Table S1.
References
- 1.Ng S.B., Buckingham K.J., Lee C., Bigham A.W., Tabor H.K., Dent K.M., Huff C.D., Shannon P.T., Jabs E.W., Nickerson D.A. Exome sequencing identifies the cause of a mendelian disorder. Nat. Genet. 2010;42:30–35. doi: 10.1038/ng.499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Walsh T., Lee M.K., Casadei S., Thornton A.M., Stray S.M., Pennil C., Nord A.S., Mandell J.B., Swisher E.M., King M.C. Detection of inherited mutations for breast and ovarian cancer using genomic capture and massively parallel sequencing. Proc. Natl. Acad. Sci. USA. 2010;107:12629–12633. doi: 10.1073/pnas.1007983107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.1000 Genomes Project Consortium A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bell C.J., Dinwiddie D.L., Miller N.A., Hateley S.L., Ganusova E.E., Mudge J., Langley R.J., Zhang L., Lee C.C., Schilkey F.D. Carrier testing for severe childhood recessive diseases by next-generation sequencing. Sci. Transl. Med. 2011;3:ra4. doi: 10.1126/scitranslmed.3001756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Domchek S.M., Friebel T.M., Singer C.F., Evans D.G., Lynch H.T., Isaacs C., Garber J.E., Neuhausen S.L., Matloff E., Eeles R. Association of risk-reducing surgery in BRCA1 or BRCA2 mutation carriers with cancer risk and mortality. JAMA. 2010;304:967–975. doi: 10.1001/jama.2010.1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schwartz P.J., Spazzolini C., Priori S.G., Crotti L., Vicentini A., Landolina M., Gasparini M., Wilde A.A., Knops R.E., Denjoy I. Who are the long-QT syndrome patients who receive an implantable cardioverter-defibrillator and what happens to them? Data from the European Long-QT Syndrome Implantable Cardioverter-Defibrillator (LQTS ICD) Registry. Circulation. 2010;122:1272–1282. doi: 10.1161/CIRCULATIONAHA.110.950147. [DOI] [PubMed] [Google Scholar]
- 7.Ashley E.A., Butte A.J., Wheeler M.T., Chen R., Klein T.E., Dewey F.E., Dudley J.T., Ormond K.E., Pavlovic A., Morgan A.A. Clinical assessment incorporating a personal genome. Lancet. 2010;375:1525–1535. doi: 10.1016/S0140-6736(10)60452-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lebo R.V., Grody W.W. Testing and reporting ACMG cystic fibrosis mutation panel results. Genet. Test. 2007;11:11–31. doi: 10.1089/gte.2006.9996. [DOI] [PubMed] [Google Scholar]
- 9.Caulfield T., McGuire A.L., Cho M., Buchanan J.A., Burgess M.M., Danilczyk U., Diaz C.M., Fryer-Edwards K., Green S.K., Hodosh M.A. Research ethics recommendations for whole-genome research: Consensus statement. PLoS Biol. 2008;6:e73. doi: 10.1371/journal.pbio.0060073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wolf S.M., Lawrenz F.P., Nelson C.A., Kahn J.P., Cho M.K., Clayton E.W., Fletcher J.G., Georgieff M.K., Hammerschmidt D., Hudson K. Managing incidental findings in human subjects research: Analysis and recommendations. J. Law Med. Ethics. 2008;36:219–248. doi: 10.1111/j.1748-720X.2008.00266.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bredenoord A.L., Kroes H.Y., Cuppen E., Parker M., van Delden J.J. Disclosure of individual genetic data to research participants: The debate reconsidered. Trends Genet. 2011;27:41–47. doi: 10.1016/j.tig.2010.11.004. [DOI] [PubMed] [Google Scholar]
- 12.Biesecker L.G., Mullikin J.C., Facio F.M., Turner C., Cherukuri P.F., Blakesley R.W., Bouffard G.G., Chines P.S., Cruz P., Hansen N.F., NISC Comparative Sequencing Program The ClinSeq Project: Piloting large-scale genome sequencing for research in genomic medicine. Genome Res. 2009;19:1665–1674. doi: 10.1101/gr.092841.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lindor N.M., McMaster M.L., Lindor C.J., Greene M.H. Concise handbook of familial cancer susceptibility syndromes - second Edition. J Natl Cancer Inst Monogr. 2008;2008:1–93. doi: 10.1093/jncimonographs/lgn001. [DOI] [PubMed] [Google Scholar]
- 14.Teer J.K., Bonnycastle L.L., Chines P.S., Hansen N.F., Aoyama N., Swift A.J., Abaan H.O., Albert T.J., Margulies E.H., Green E.D., NISC Comparative Sequencing Program Systematic comparison of three genomic enrichment methods for massively parallel DNA sequencing. Genome Res. 2010;20:1420–1431. doi: 10.1101/gr.106716.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Teer J.K., Green E.D., Mullikin J.C., Biesecker L.G. VarSifter: Visualizing and analyzing exome-scale sequence variation data on a desktop computer. Bioinformatics. 2012;28:599–600. doi: 10.1093/bioinformatics/btr711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stratton M.R., Rahman N. The emerging landscape of breast cancer susceptibility. Nat. Genet. 2008;40:17–22. doi: 10.1038/ng.2007.53. [DOI] [PubMed] [Google Scholar]
- 17.Sherry S.T., Ward M.H., Kholodov M., Baker J., Phan L., Smigielski E.M., Sirotkin K. dbSNP: The NCBI database of genetic variation. Nucleic Acids Res. 2001;29:308–311. doi: 10.1093/nar/29.1.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Plon S.E., Eccles D.M., Easton D., Foulkes W.D., Genuardi M., Greenblatt M.S., Hogervorst F.B., Hoogerbrugge N., Spurdle A.B., Tavtigian S.V., IARC Unclassified Genetic Variants Working Group Sequence variant classification and reporting: Recommendations for improving the interpretation of cancer susceptibility genetic test results. Hum. Mutat. 2008;29:1282–1291. doi: 10.1002/humu.20880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stenson P.D., Ball E.V., Mort M., Phillips A.D., Shiel J.A., Thomas N.S., Abeysinghe S., Krawczak M., Cooper D.N. Human Gene Mutation Database (HGMD): 2003 update. Hum. Mutat. 2003;21:577–581. doi: 10.1002/humu.10212. [DOI] [PubMed] [Google Scholar]
- 20.Fokkema I.F., den Dunnen J.T., Taschner P.E. LOVD: Easy creation of a locus-specific sequence variation database using an “LSDB-in-a-box” approach. Hum. Mutat. 2005;26:63–68. doi: 10.1002/humu.20201. [DOI] [PubMed] [Google Scholar]
- 21.Greenblatt M.S., Brody L.C., Foulkes W.D., Genuardi M., Hofstra R.M., Olivier M., Plon S.E., Sijmons R.H., Sinilnikova O., Spurdle A.B., IARC Unclassified Genetic Variants Working Group Locus-specific databases and recommendations to strengthen their contribution to the classification of variants in cancer susceptibility genes. Hum. Mutat. 2008;29:1273–1281. doi: 10.1002/humu.20889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.U.S. Preventive Services Task Force Genetic risk assessment and BRCA mutation testing for breast and ovarian cancer susceptibility: Recommendation statement. Ann. Intern. Med. 2005;143:355–361. doi: 10.7326/0003-4819-143-5-200509060-00011. [DOI] [PubMed] [Google Scholar]
- 23.Klein R.D., Lloyd R.V., Young W.F. Hereditary Paraganglioma-Pheochromocytoma Syndromes. In: Pagon R.A., Bird T.D., Dolan C.R., Stephens K., editors. GeneReviews. University of Washington; 2009. [Google Scholar]
- 24.Neuhausen S., Gilewski T., Norton L., Tran T., McGuire P., Swensen J., Hampel H., Borgen P., Brown K., Skolnick M. Recurrent BRCA2 6174delT mutations in Ashkenazi Jewish women affected by breast cancer. Nat. Genet. 1996;13:126–128. doi: 10.1038/ng0596-126. [DOI] [PubMed] [Google Scholar]
- 25.Parmigiani G., Berry D., Aguilar O. Determining carrier probabilities for breast cancer-susceptibility genes BRCA1 and BRCA2. Am. J. Hum. Genet. 1998;62:145–158. doi: 10.1086/301670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Szabo C., Masiello A., Ryan J.F., Brody L.C. The breast cancer information core: Database design, structure, and scope. Hum. Mutat. 2000;16:123–131. doi: 10.1002/1098-1004(200008)16:2<123::AID-HUMU4>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 27.Edwards S.M., Kote-Jarai Z., Meitz J., Hamoudi R., Hope Q., Osin P., Jackson R., Southgate C., Singh R., Falconer A., Cancer Research UK/Bristish Prostate Group UK Familial Prostate Cancer Study Collaborators. British Association of Urological Surgeons Section of Oncology Two percent of men with early-onset prostate cancer harbor germline mutations in the BRCA2 gene. Am. J. Hum. Genet. 2003;72:1–12. doi: 10.1086/345310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Berry D.A., Iversen E.S., Jr., Gudbjartsson D.F., Hiller E.H., Garber J.E., Peshkin B.N., Lerman C., Watson P., Lynch H.T., Hilsenbeck S.G. BRCAPRO validation, sensitivity of genetic testing of BRCA1/BRCA2, and prevalence of other breast cancer susceptibility genes. J. Clin. Oncol. 2002;20:2701–2712. doi: 10.1200/JCO.2002.05.121. [DOI] [PubMed] [Google Scholar]
- 29.Wolf S.M., Crock B.N., Van Ness B., Lawrenz F., Kahn J.P., Beskow L.M., Cho M.K., Christman M.F., Green R.C., Hall R. Managing incidental findings and research results in genomic research involving biobanks and archived data sets. Genet. Med. 2012;14:361–384. doi: 10.1038/gim.2012.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kohane I.S., Hsing M., Kong S.W. Taxonomizing, sizing, and overcoming the incidentalome. Genet. Med. 2012;14:399–404. doi: 10.1038/gim.2011.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mitra A.V., Bancroft E.K., Barbachano Y., Page E.C., Foster C.S., Jameson C., Mitchell G., Lindeman G.J., Stapleton A., Suthers G., IMPACT Study Collaborators Targeted prostate cancer screening in men with mutations in BRCA1 and BRCA2 detects aggressive prostate cancer: Preliminary analysis of the results of the IMPACT study. BJU Int. 2011;107:28–39. doi: 10.1111/j.1464-410X.2010.09648.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Weitzel J.N., Lagos V.I., Cullinane C.A., Gambol P.J., Culver J.O., Blazer K.R., Palomares M.R., Lowstuter K.J., MacDonald D.J. Limited family structure and BRCA gene mutation status in single cases of breast cancer. JAMA. 2007;297:2587–2595. doi: 10.1001/jama.297.23.2587. [DOI] [PubMed] [Google Scholar]
- 33.Rubinstein W.S., Jiang H., Dellefave L., Rademaker A.W. Cost-effectiveness of population-based BRCA1/2 testing and ovarian cancer prevention for Ashkenazi Jews: A call for dialogue. Genet. Med. 2009;11:629–639. doi: 10.1097/GIM.0b013e3181afd322. [DOI] [PubMed] [Google Scholar]
- 34.Schiavi F., Boedeker C.C., Bausch B., Peçzkowska M., Gomez C.F., Strassburg T., Pawlu C., Buchta M., Salzmann M., Hoffmann M.M., European-American Paraganglioma Study Group Predictors and prevalence of paraganglioma syndrome associated with mutations of the SDHC gene. JAMA. 2005;294:2057–2063. doi: 10.1001/jama.294.16.2057. [DOI] [PubMed] [Google Scholar]
- 35.Bardella C., Pollard P.J., Tomlinson I. SDH mutations in cancer. Biochim. Biophys. Acta. 2011;1807:1432–1443. doi: 10.1016/j.bbabio.2011.07.003. [DOI] [PubMed] [Google Scholar]
- 36.Burnichon N., Rohmer V., Amar L., Herman P., Leboulleux S., Darrouzet V., Niccoli P., Gaillard D., Chabrier G., Chabolle F., PGL.NET network The succinate dehydrogenase genetic testing in a large prospective series of patients with paragangliomas. J. Clin. Endocrinol. Metab. 2009;94:2817–2827. doi: 10.1210/jc.2008-2504. [DOI] [PubMed] [Google Scholar]
- 37.Pasini B., McWhinney S.R., Bei T., Matyakhina L., Stergiopoulos S., Muchow M., Boikos S.A., Ferrando B., Pacak K., Assie G. Clinical and molecular genetics of patients with the Carney-Stratakis syndrome and germline mutations of the genes coding for the succinate dehydrogenase subunits SDHB, SDHC, and SDHD. Eur. J. Hum. Genet. 2008;16:79–88. doi: 10.1038/sj.ejhg.5201904. [DOI] [PubMed] [Google Scholar]
- 38.Neumann H.P., Erlic Z., Boedeker C.C., Rybicki L.A., Robledo M., Hermsen M., Schiavi F., Falcioni M., Kwok P., Bauters C. Clinical predictors for germline mutations in head and neck paraganglioma patients: Cost reduction strategy in genetic diagnostic process as fall-out. Cancer Res. 2009;69:3650–3656. doi: 10.1158/0008-5472.CAN-08-4057. [DOI] [PubMed] [Google Scholar]
- 39.Fitzgerald R.C., Hardwick R., Huntsman D., Carneiro F., Guilford P., Blair V., Chung D.C., Norton J., Ragunath K., Van Krieken J.H., International Gastric Cancer Linkage Consortium Hereditary diffuse gastric cancer: Updated consensus guidelines for clinical management and directions for future research. J. Med. Genet. 2010;47:436–444. doi: 10.1136/jmg.2009.074237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Brooks-Wilson A.R., Kaurah P., Suriano G., Leach S., Senz J., Grehan N., Butterfield Y.S., Jeyes J., Schinas J., Bacani J. Germline E-cadherin mutations in hereditary diffuse gastric cancer: assessment of 42 new families and review of genetic screening criteria. J. Med. Genet. 2004;41:508–517. doi: 10.1136/jmg.2004.018275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mateus A.R., Simões-Correia J., Figueiredo J., Heindl S., Alves C.C., Suriano G., Luber B., Seruca R. E-cadherin mutations and cell motility: A genotype-phenotype correlation. Exp. Cell Res. 2009;315:1393–1402. doi: 10.1016/j.yexcr.2009.02.020. [DOI] [PubMed] [Google Scholar]
- 42.Cotton R.G., Auerbach A.D., Beckmann J.S., Blumenfeld O.O., Brookes A.J., Brown A.F., Carrera P., Cox D.W., Gottlieb B., Greenblatt M.S. Recommendations for locus-specific databases and their curation. Hum. Mutat. 2008;29:2–5. doi: 10.1002/humu.20650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Facio F.M., Brooks S., Loewenstein J., Green S., Biesecker L.G., Biesecker B.B. Motivators for participation in a whole-genome sequencing study: Implications for translational genomics research. Eur. J. Hum. Genet. 2011;19:1213–1217. doi: 10.1038/ejhg.2011.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Document S1. Table S1