Immunotherapy for Prostate Cancer: Biology and Therapeutic Approaches (original) (raw)
Abstract
Although prostate cancer was not historically considered to be a particularly immune-responsive cancer, recent clinical trials have demonstrated that immunotherapy for prostate cancer can lead to improvements in overall survival (OS). These studies include randomized controlled trials with sipuleucel-T and another with PROSTVAC-VF, both of which rely on stimulating the immune system to target prostate proteins. This review discusses the most promising developments over the past year in immune-based therapy for prostate cancer and the opportunities that lie ahead. Recent randomized immunotherapy trials in prostate cancer have demonstrated improvements in OS but without the concomitant improvements in progression-free survival. This uncoupling of survival from clinical response poses challenges to clinical management, because conventional measures of objective response cannot be used to identify patients benefiting from treatment. There is a significant need to identify immunologic or clinical surrogates for survival so that clinical benefit can be assessed in a timely manner. Immunotherapy is now an established treatment approach for prostate cancer, with multiple clinical trials demonstrating improvements in OS. Significant challenges to this modality remain, including determining best clinical setting for immunotherapy, identifying patients who benefit, and defining relevant clinical and immunologic end points. Nevertheless, the broader availability of novel immunotherapies will provide opportunities not only to target different components of the immune system but also to combine immunotherapies with other treatments for improved clinical efficacy.
INTRODUCTION
With the US Food and Drug Administration (FDA) approval of sipuleucel-T (Provenge; Dendreon, Seattle, WA), immunotherapy is now an established treatment modality for prostate cancer. Although conventional treatments for prostate cancer have relied on androgen disruption and cytotoxicity, immunotherapy relies on activating the host immune system to specifically target tumors. With each promising step in prostate cancer immunotherapy, general themes in the clinical pattern of response have emerged, which may have an impact on clinical practice: length of time required to initiate antitumor immunity, initial progression of disease, subsequent improvement in overall survival (OS), and lack of definitive biomarkers to know when immune therapies are working. In this review, we discuss the biology of an antitumor immune response and focus on the key approaches and challenges in immunotherapy that have translated into promising, and now established, treatments in prostate cancer.
PROSTATE CANCER IMMUNOLOGY
An adaptive immune response develops through a sequence of events: first, activation of antigen-presenting cells (APCs) in the presence of a target antigen; second, presentation of the antigen to T cells; third, targeting of antigen by activated T cells; and fourth, downregulation of T-cell response (Fig 1). The goal of immunotherapy is to promote this effector response against cancerous cells. During APC activation, immature APCs take up tumor antigens from the environment and process them into peptides that are displayed on the cell surface by major histocompatibility complexes (MHCs). Once mature and activated, APCs display these MHC-peptide complexes along with costimulatory molecules (B7 ligands) on the cell surface, thereby becoming proficient presenters of tumor antigens to T cells. Of the APCs that stimulate T cells, dendritic cells (DCs) are considered the most important, because they can sensitize naive T cells to novel antigens and establish helper T-cell responses polarized to support antitumor response. APC maturation and activation can be enhanced by cytokines, such as granulocyte-macrophage colony-stimulating factor (GM-CSF), which promote the production and differentiation of mature monocytes and DCs from hematopoietic progenitor cells,1 and by stimulation of the Toll-like family of receptors (TLRs), which recognize molecular patterns normally associated with pathogens.2 TLRs are already being exploited for cancer treatment (eg, Bacillus Calmette-Guérin mycobacteria for superficial bladder carcinoma and imiquimod for superficial basal carcinoma). The next step involves effectively presenting antigen to effector T cells, an event that requires at least two signals. Recognition of antigen by its cognate T-cell receptor provides one of these signals. Costimulation by the B7 family of ligands on APCs interacting with the CD28 receptor on T cells delivers the second requisite signal. Both are essential for T-cell activation; otherwise, T cells are rendered tolerant, or unresponsive, to the antigen being targeted. Activated APCs also produce cytokines to drive naive T-cell activation and differentiation into effector T cells. These T cells in turn produce additional cytokines necessary for expansion and survival, such as interleukin-2 (IL-2) and interferon gamma (IFN-γ). These lymphocytes subsequently infiltrate tumor sites where the target antigen is present and mediate tumor cell lysis. T-cell activation, however, also turns on a number of inhibitory pathways that can blunt effector T-cell responses. These innate immune checkpoints are important in winding down immune responses after infections, but in the setting of malignancy, which can generate an immunosuppressive microenvironment, they can abort antitumor responses. Cytotoxic T lymphocyte–associated receptor 4 (CTLA4)3 and programmed death 1 (PD-1)4 are members of the CD28 family of coreceptors that negatively regulate T-cell responses.
Fig 1.
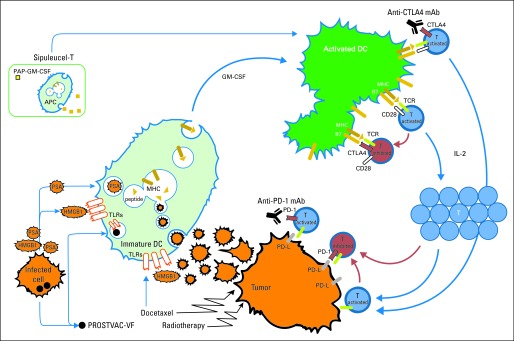
Overview of tumor-specific immune response and components targeted by individual immunotherapies. Dendritic cell (DC) activation normally requires uptake, processing, and presentation of tumor antigens by immature DCs. Triggering of Toll-like receptors (TLRs) on DCs also leads to DC activation. When T cells receive two signals—first, from binding of antigen–major histocompatibility complex (MHC) to T-cell receptor (TCR), and second, from B7 binding to CD28—T cells proliferate in interleukin-2 (IL-2) –dependent manner, migrate to tumor, and directly lyse tumor cells. PROSTVAC-VF is a poxvirus vaccine that delivers prostate-specific antigen (PSA). It activates immature DCs with uptake of encoded tumor antigens released from infected cells and has three costimulation signals encoded in virus to enhance DC-to–T-cell interaction. Sipuleucel-T consists of harvested antigen-presenting cells (APCs) cultured with fused protein consisting of prostatic acid phosphatase (PAP) and granulocyte-macrophage colony-stimulating factor (GM-CSF). This product is reinfused into patients. In vitro manipulation of cells as well as GM-CSF presumably enhances antigen presentation. Anti–cytotoxic T lymphocyte–associated receptor 4 (CTLA4) monoclonal antibodies (anti-CTLA4 mAb) target CTLA4 coreceptors, which negatively regulates T-cell activation. Anti–programmed death 1 (PD-1) monoclonal antibodies (anti–PD-1 mAb) target PD-1 receptors, which negatively regulates function of memory T cells. PD-1 can be expressed on tumor infiltrating lymphocytes, whereas ligands for PD-1 (PD-L) are often expressed by tumors. Cytotoxic effects of radiation therapy and docetaxel can also modulate immune responses with release of proinflammatory signals from dying cells (eg, high-mobility group protein B1 [HMGB1] is a TLR4 agonist). In addition, docetaxel can also act as a TLR agonist.
In prostate cancer, there are inherent barriers in each process. Antigens relevant in prostate cancer, such as prostate-specific antigen (PSA), prostate-specific membrane antigen, prostate stem-cell antigen, and prostatic acid phosphatase (PAP), were selected based on their nearly exclusive expression to prostate tissue, but being self-proteins, they are not inherently immunogenic.5 Lymphocytes that can recognize self-proteins such as these are usually eliminated during development to prevent autoimmunity, and the rare reactive lymphocytes that survive this selection process are subject to a series of regulatory mechanisms (eg, immunosuppressive cytokines such as transforming growth factor β or regulatory T cells [Tregs])6,7 to further maintain immune tolerance. Moreover, tumors have evolved mechanisms to co-opt these processes to evade the immune system. Tumor cells can impede the maturation of DCs or prevent expression of costimulatory molecules necessary for T-cell activation. MHC expression and peptide processing can be downregulated to block recognition by cytotoxic T cells.8,9 The tumor microenvironment can also support the expansion of Tregs that can inhibit effector T-cell function.10,11 Immune responses can also be inhibited by innate immune checkpoints. Both CTLA4 and PD-1 are upregulated with T-cell activation, and the ligands for PD-1 (PD-L1, PD-L2) are often expressed by tumors. Thus, the most successful therapies are designed to overcome these immunosuppressive mechanisms.
APC-ACTIVATING IMMUNOTHERAPY
A number of cytokines (GM-CSF) and agents (TLR agonists, Bacillus Calmette-Guérin) can enhance antigen presentation by APCs. GM-CSF recruits and promotes the maturation of monocytes and granulocytes from stem-cell precursors and potently activates macrophages and DCs. As a single agent, GM-CSF has modest activity, modulating PSA responses in patients with castration-resistant prostate cancer (CRPC).12 Long-term delay in disease progression can be seen in up to 15% of patients with serologic progression,13 and GM-CSF may have activity in other cancers.14,15 Neoadjuvant GM-CSF may increase the numbers of prostate tumor–resident DCs that carry an activated phenotype.16 Because of its nonspecific stimulatory properties, GM-CSF is now frequently used as an immune adjuvant in many immunotherapy trials.
ANTIGEN-TARGETED IMMUNOTHERAPY
One advantage with prostate cancer is that primary definitive treatment involves surgical resection or ablative radiotherapy, mitigating the concern for raising autoimmunity against residual prostate tissue. A number of methods have been devised to deliver tumor-associated antigens in a way that promotes tumor-specific T-cell responses (eg, peptide vaccines, virally packaged antigens, and DNA-based antigen-expressing vectors). DCs have also been loaded ex vivo with tumor antigens or whole tumor lysates to induce antitumor immune responses.17–20 The most notable developments in antigen-targeted prostate cancer immunotherapy are summarized in Table 1.
Table 1.
Selected Randomized Clinical Trials of Immunotherapy in Prostate Cancer
| Immunotherapy | Trial Design | Phase | Target Population | Antigen | No. of Patients | Median OS | P | Reference |
|---|---|---|---|---|---|---|---|---|
| Sipuleucel-T | IMPACT (D9902B) comparing sipuleucel-T v placebo; random assignment 2:1; crossover allowed | III | Asymptomatic metastatic CRPC | PAP/GM-CSF | 512 | 25.8 v 21.7 months | .03 | Kantoff et al21 |
| Sipuleucel-T | D9902A comparing sipuleucel-T v placebo; random assignment 2:1; crossover allowed | III | Asymptomatic metastatic CRPC | PAP/GM-CSF | 98 | 19.0 v 15.3 months | .331 | Higano et al22 |
| Sipuleucel-T | D9901 comparing sipuleucel-T v placebo; random assignment 2:1; crossover allowed | III | Asymptomatic metastatic CRPC | PAP/GM-CSF | 127 | 25.9 v 21.4 months | .010 | Small et al23 |
| PROSTVAC-VF | TBC-PRO-002 comparing PROSTVAC-VF v placebo; random assignment 2:1; crossover allowed | II | Asymptomatic metastatic CRPC | PSA | 125 | 25.1 v 16.6 months | .006 | Kantoff et al24 |
| PROSTVAC-VF | Comparing PROSTVAC-VF v PROSTVAC-VF plus GM-CSF | II | Metastatic CRPC (chemotherapy naive) | PSA | 32 | 26.6 months (results reported as one overall group; no added benefit with GM-CSF) | NS | Gulley et al25 |
| PSA-poxvirus vaccine | Comparing PSA-poxvirus vaccine v nilutamide; crossover allowed | II | Nonmetastatic CRPC | PSA | 42 | 5.1 v 3.4 years; subgroup analysis of crossover patients (6.2 v 3.7 years) favored men initially treated with vaccine followed by vaccine plus hormone therapy | .13 (subgroup, .045) | Madan et al26 |
| Prostate GVAX | VITAL-1 comparing prostate GVAX v docetaxel plus prednisone | III | Asymptomatic metastatic CRPC | Multiple | 626 | 20.7 v 21.7 months (closed after interim analysis showed futility) | .78 | Higano et al27 |
| Prostate GVAX | VITAL-2 comparing prostate GVAX plus docetaxel v docetaxel plus prednisone | III | Symptomatic metastatic CRPC | Multiple | 408 of 600 planned | 12.2 v 14.1 months (halted for imbalance of deaths) | .0076 | Small et al28 |
Sipuleucel-T
Sipuleucel-T is a personalized cellular therapy that uses ex vivo antigen presentation to induce an antitumor immune response. The product is derived from peripheral blood mononuclear blood cells harvested by leukapheresis that are enriched for APCs with density-gradient centrifugation. The cells are cultured ex vivo for 36 to 44 hours with a fusion protein (PA2024) linking PAP and GM-CSF.29 PAP was selected based on the evidence that immunization can drive T cell–mediated immune responses in both rats and humans.30,31 PAP is thought to be targeted to immature DCs by GM-CSF within this cellular product. GM-CSF presumably would also enhance DC maturation, although a comparison of GM-CSF–treated leukapheresed cells with sipuleucel-T showed significant increases in cytokine levels (IL-2, IFN-γ) and T-cell activation from the sipuleucel-T product.32 After 2 days of culture, antigen-loaded APCs and the other immune cells (including T cells) contained in the culture become activated and are then infused back into patients. These antigen-loaded APCs would presumably stimulate T cells to target PAP-expressing prostate cancer cells. Treatment involves three rounds of apheresis and intravenous infusions administered every 2 weeks.
The vaccine underwent three randomized controlled trials to confirm its clinical benefit. In these studies, patients in the placebo arms underwent leukapheresis and received infusions of cells that were cocultured without PA2024, amounting to less than one third of the original apheresis product. Progressing patients in the placebo arm who could cross over received a cellular product derived from the coculture of PA2024 with the remaining apheresed cells that were cryopreserved at the time of placebo preparation (APC8015F). The first randomized, placebo-controlled phase III trial for sipuleucel-T (D9901) enrolled 127 men with asymptomatic metastatic CRPC randomly assigned at a ratio of two to one.23 The study did not meet the primary end point of time to disease progression (TTP; 11.7 v 10.0 weeks). However, median OS was improved by 4 months over placebo (25.9 v 21.4 months; P = .01). A second similarly designed trial (D9902A) did not meet its primary end point of TTP, but it showed prolonged OS that did not reach statistical significance (19.0 v 15.3 months; P = .33).22 The lack of a significant survival benefit may reflect the premature discontinuation of the study at 98 patients to launch the subsequent trial; thus, it was likely underpowered to detect a difference. The IMPACT trial (Immunotherapy for Prostate Adenocarcinoma Treatment; D9902B), a randomized, double-blind, placebo-controlled phase III trial that enrolled 512 men at a ratio of two to one, was designed with OS as the primary end point. Patients enrolled onto this trial were similar to those in the previous two studies (Eastern Cooperative Oncology Group performance status of 0 to 1, minimally or asymptomatic, no pathologic fractures, no visceral metastases, and no recent chemotherapy within 3 months or ≤ two prior chemotherapy regimens), but it also included men with any Gleason score (GS). The study recapitulated the results of D9901, showing a 4.1-month improvement in median OS (25.8 v 21.7 months) with no effect on TTP (14.6 v 14.4 weeks).21 These registration trials were notable for showing a survival benefit despite a crossover design for placebo-treated patients, and this improvement in survival remained after adjustment for post-therapy docetaxel use. This effect was also observed consistently among patient subgroups (independent of GS, number of bone metastases, PSA level, lactate dehydrogenase level, performance status, prior chemotherapy, and bisphosphonate use).21 Patients who crossed over from the placebo arm also had better survival with frozen APC8015F than those who did not. An exploratory review of all patients treated with APC8015F after receiving placebo versus those who were not revealed a median OS of 20.0 versus 9.8 months (hazard ratio, 0.52; P < .001), even after adjustment for baseline characteristics.33 Toxicities were limited to infusion-related chills (54%), nausea (28%), fever (29%), headache (16%), and fatigue (39%) within the first few days of treatment, although a trend toward increased but infrequent cerebrovascular events (2.4% v 1.8%; P = 1.0) was observed. On the basis of these results, the FDA approved sipuleucel-T in April 2010 for the treatment of asymptomatic or minimally symptomatic metastatic CRPC.
Despite this approval, questions remain as to how sipuleucel-T should be used. First, the trials showed that patients would continue to have disease progression, making it difficult to determine who would derive a clinical benefit. As a result, after treatment with sipuleucel-T, patients presumably will move onto other treatment, but the appropriate timing for this is unclear. Second, the target population of these trials was limited to patients with CRPC with minimal or no symptoms related to prostate cancer, yet a trend toward benefit could be seen independent of many risk factors, including GS, suggesting broader applicability. Although a majority of patients were chemotherapy naive, those who had received prior chemotherapy (18.2%) also seemed to benefit from treatment. Nevertheless, sipuleucel-T relies on an intact immune system to work, so this treatment is likely not appropriate for patients who have been heavily pretreated with chemotherapy or are receiving systemic corticosteroids. Moreover, because sipuleucel-T does not affect disease progression, this treatment should be administered soon after the development of CRPC with nonvisceral metastases. If sipuleucel-T treatment is administered after multiple secondary hormonal manipulations and/or docetaxel, then the patient may have a relatively short time to develop an immune response before having to embark on subsequent treatment that will likely include steroids and/or chemotherapy. Nevertheless, when patients manifest disease progression after sipuleucel-T treatment, they should proceed immediately to the next appropriate treatment rather than wait for a clinical response, because these occur infrequently.
Future studies should investigate whether patients with earlier-stage disease (eg, nonmetastatic CRPC) would benefit. The capacity for antitumor response presumably degrades during the course of disease, and providing treatment early to leverage a more competent immune system may improve clinical outcome. The probabilities of survival at 3 years (31.7% with sipuleucel-T v 23% with placebo) also indicate that a small proportion of patients significantly benefited from treatment. Whether the survival impact could be extended if sipuleucel-T were administered at an earlier stage of disease, and whether a treatment population could be better defined by immunologic assessments, remain to be determined. Therefore, developing predictive biomarkers could help guide patient selection for this treatment.
PROSTVAC-VF
PROSTVAC-VF (National Cancer Institute/BN ImmunoTherapeutics, Mountain View, CA) is a viral vaccine that consists of a combination of recombinant vaccinia and fowlpox viruses that encode PSA and a triad of T-cell costimulatory molecules composed of lymphocyte function–associated antigen 3, intercellular adhesion molecule 1, and B7-1 (collectively labeled as TRICOM).34 This multiviral combination promotes tumor immunity in a number of ways: first, vaccinia as a vector is immunogenic; second, infected cells undergo necrosis, releasing PSA that is then processed by immature DCs; and third, pro-inflammatory danger signals that are also released by cell necrosis can further activate DCs. Because neutralizing antibody responses are induced against vaccinia virus, which could potentially limit further treatment with vaccinia plus PSA, the poxvirus-based vaccination was refined to use a heterologous prime/boost strategy in which patients are first immunized with the vaccinia platform and subsequently treated with the fowlpox virus.35 Early-phase trials demonstrated safety of the vectors, induction of PSA-specific immune responses, and reduction in PSA velocity,35–37 leading to a randomized, double-blinded phase II trial with PROSTVAC-VF in men with asymptomatic CRPC.24 This trial did not meet the primary end point of progression-free survival (PFS; 3.7 months in control arm v 3.8 months in treatment arm), but OS greatly favored patients who received PROSTVAC-VF (25.1 v 16.6 months), with a 43% reduction in death and 8.5-month improvement in median OS at 3 years poststudy.24 Such advantages are striking in light of the crossover design and allowance for post-treatment chemotherapy use, but one troubling concern is the low OS seen in the placebo arm, despite a lower-risk target population (those with GS > 7 were excluded) than that in the IMPACT trial (21.7-month median OS in IMPACT placebo arm) and with 19 of 40 placebo-treated patients crossing over. Subsequent treatments after disease progression in this trial were not tracked, so potential imbalances between the two arms (eg, treatment with docetaxel) could also have affected OS. A phase III clinical trial in minimally symptomatic CRPC is planned for late 2011.
For both sipuleucel-T and PROSTVAC-VF, the target population was limited to men with an Eastern Cooperative Oncology Group status of 0 or 1, without visceral disease or opioid analgesic use for pain. For PROSTVAC-VF, men with a GS of more than 7 were also excluded. Patients with these characteristics were selected so that they could presumably live long enough to allow for benefit from the activity of vaccines, which may not be appreciated until 1 year after treatment. Moreover, evidence of immune regulation (presence of regulatory T cells, myeloid-derived suppressor cells, immunosuppressive cytokines such as transforming growth factor β),6,38,39 both peripherally and within the tumor microenvironment, suggests that vaccination must surmount multiple challenges to activate an effective antitumor response. Studies in which these challenges are minimized (eg, in earlier disease states) could reveal improved clinical benefit.
DNA Vaccines
DNA-based vaccines rely on injecting expression plasmids that encode full-length tumor antigens to allow for endogenous processing and presentation by DCs. These offer the advantage of facile recombinant engineering to employ any target antigen and rapid scalability. Pro-inflammatory agents that can activate DCs such as TLR agonists or recruit DCs such as GM-CSF have thus been engineered into DNA vectors to provide a backbone of immune activating signals. An early-phase clinical trial with a DNA vaccine has revealed the potential to apply this approach in humans. The study showed that men with biochemical recurrence of disease (stage D0) could be safely immunized with a DNA vaccine encoding PAP plus copies of a TLR agonist (CpG).40 PAP-specific T-cell responses were detected in a proportion of study patients.41 The design is also notable for treating men at an earlier stage of disease, presumably targeting a more appropriate population for immunotherapy. Unfortunately, modulation of PSA velocity cannot serve as a surrogate for OS. Without reliable biochemical and immunologic surrogates for survival, detecting clinical benefit in a population with early disease may require long follow-up intervals to detect meaningful clinical events.
Whole Tumor Cell Vaccines
Single antigen–specific vaccines often battle both immunoselective and suppressive pressures that can render the antigen invisible or tolerogenic to the immune response. The argument for vaccinating multiple antigens, then, is to provide a number of targets to avoid a one-or-none response. Genetically modified tumor cells represent one such approach. Among the most studied in prostate cancer is GVAX (BioSante Pharmaceuticals, Lincolnshire, IL), which consists of two allogeneic prostate cancer cell lines (LNCaP and PC3) engineered to express GM-CSF.42 This approach would presumably deliver multiple antigens, some of which are known, but others may be unknown. Nevertheless, two large phase III trials, one with GVAX alone and a second in combination with docetaxel, failed to show improvements in OS in patients treated with docetaxel plus prednisone.27,28 The reasons for failure are not clear, but in hindsight, there were many variables that had not been addressed at the phase II level. Although there is preclinical evidence to suggest that chemotherapy can induce immunomodulatory effects and therefore be additive to immunotherapy,43 how GVAX should be combined with docetaxel was not addressed before the phase III trial. Finally, use of docetaxel in the control arms of both trials may not have been appropriate given the different kinetics of response seen with immunotherapies.
IMMUNE CHECKPOINT BLOCKADE
Checkpoints exist to dampen the immune response, and blocking these receptors can potently maintain T-cell activation systemically. Unlike sipuleucel-T and PROSTVAC-VF, this approach relies on enhancing endogenous immune responses. CTLA4 is a receptor on activated T cells that normally serves to inhibit further T-cell activation (Fig 2). Ipilimumab (Yervoy; Bristol-Myers Squibb, New York, NY) is a humanized immunoglobulin G1 kappa monoclonal antibody that targets CTLA4. In a randomized phase III trial in patients with previously treated unresectable or metastatic melanoma, ipilimumab at a dose of 3 mg/kg, with and without a melanoma gp100 peptide vaccine, was shown to improve OS by 4 months compared with gp100 vaccine alone (10.0, 10.1, and 6.4 months, respectively).44 Not unlike sipuleucel-T and PROSTVAC-VF for CRPC, PFS in this trial was low and not affected (2.76, 2.86, and 2.76 months, respectively). On the basis of this trial, ipilimumab was approved by the FDA in March 2011 for the treatment of metastatic melanoma.
Fig 2.

Overview of cytotoxic T lymphocyte–associated antigen 4 (CTLA4) blockade. After T-cell activation, CTLA4 receptors are recruited to the T-cell surface and compete with CD28 for binding to B7. (A) When CTLA4 binds to B7, it inactivates T cells, resulting in downregulation of T-cell immune response. (B) Anti-CTLA4 antibodies (anti-CTLA4 mAb) can block this interaction by binding to CTLA4, resulting in augmented T-cell activation. MHC II, major histocompatibility complex II; TCR, T-cell receptor.
To our knowledge, the first in-human clinical trial with ipilimumab was performed in patients with CRPC.45 Ipilimumab monotherapy (3 mg/kg) induced clinical responses in two patients, who experienced PSA declines of more than 50% lasting 60 and 135 days, respectively. An additional eight of the 14 treated patients experienced declines in PSA of less than 50%. One patient experienced a grade 3 corticosteroid-responsive rash. Because CTLA-4 blockade upregulates T-cell activity, there has been interest in focusing responses toward tumor antigens by combining antibody treatment with other immunologic agents or with cytotoxic treatments to release antigens from dying tumor cells. Ipilimumab has been combined with GM-CSF, demonstrating PSA responses as well as objective tumor responses in CRPC.46 Ipilimumab has also been combined with docetaxel,47 PROSTVAC-VF,48 and GVAX,49 with clinical responses seen in each of these trials. So far, the combination of ipilimumab with a single dose of docetaxel in a phase II study revealed no advantage over docetaxel alone, although glucocorticoids were administered with chemotherapy and may have dampened immune responses.47 However, with PROSTVAC-VF, combined treatment in men with CRPC may have an additive effect on OS (31.8 months).48 When combined with GVAX, PSA declines of more than 50% were reported in a significant number of patients receiving combination therapy.49 Two phase III trials are currently under way in prostate cancer. The first trial randomly assigns patients with docetaxel-refractory CRPC to placebo versus ipilimumab after limited radiotherapy to a metastatic site (eg, bone metastasis), which may potentially release tumor antigens for APCs to present. The second phase III trial randomly assigns chemotherapy-naive patients with CRPC to receive ipilimumab or placebo.
Immune-related adverse events are common with CTLA4 blockade and can be life threatening. Upward to 60% of patients experience some autoimmune toxicity, and severe grade 3 to 4 events can be seen in approximately 10% to 20%.50–52 The most common toxicities are GI (diarrhea, colitis), skin (pruritus, rash), and liver (transaminitis) events. Prompt initiation of high-dose steroids is imperative to treat grade 3 to 4 immune-mediated adverse events and has made these toxicities largely manageable. Clinical responses have also manifested after the initiation of steroid treatment, so clinical efficacy can be uncoupled from the toxicities. Prophylactic steroid treatment such as with budesonide has not been shown to prevent diarrhea.53 In the initial phase I and II trials in CRPC, a significant proportion of clinical responders to ipilimumab also developed endocrinopathies, including pan-hypopituitarism, hypothyroidism, and adrenal insufficiency. Although treatment-associated adrenal insufficiency could potentially confound results by indirectly promoting antitumor activity, clinical responses have also been seen in the absence of measureable changes in adrenal hormones, indicating that antitumor immune responses are also induced directly with treatment. At present, we cannot predict which patients will develop these toxicities, which will likely preclude its use in patients with hormone-sensitive prostate cancer.
Antibodies to PD-1 have also entered phase I clinical trials, demonstrating efficacy in a number of malignancies, including prostate cancer. PD-1 represents another coinhibitory receptor that can be expressed on memory cells and is thought to be a marker of exhausted cells. Antibodies that block PD-1 can enhance tumor-specific T-cell responses.54,55 Interestingly, CD8+ T cells that infiltrate prostate and melanoma tumors express high levels of PD-1 and have impaired effector functions, suggesting that reversal of PD-1 signaling in those cells can have direct effects on the tumor landscape.56,57 This was suggested in a phase I trial showing objective responses in a number of tumors with PD-1 antagonists (MDX-1106; Bristol-Myers Squibb).58 A partial response was seen in one (6.7%) of 15 patients, and stable disease (> 4 months) was seen in three (20%) of 15 patients with CRPC.59 Although immune-related toxicities seem similar to those with ipilimumab, the frequency of these may be lower in these preliminary studies.
CONVENTIONAL THERAPIES AS IMMUNE MODULATORS
The deleterious effects of cytotoxic treatment (eg, chemotherapy and radiation therapy) on hematopoietic cells have traditionally precluded their combination with immune therapies. Several lines of evidence, however, suggest that cytotoxic treatments may not only provide additional benefit in combination but may also mediate their own efficacy through immunomodulatory effects. In animal models, tumor-cell turnover induced by chemotherapy triggers the release of ATP and endogenous TLR4 agonists (HMGB1) to activated DCs (through both purinergic receptor P2RX7 and NLRP3 inflammasome, respectively).60 By recognizing danger signals released from dying cells, these in turn promote the maturation of DCs and priming of CD8+ effector T cells through and IL-1β– and caspase (caspase 1) –dependent pathways.61 Similarly, tumor regression with radiation treatment may also be dependent on the immune recognition of danger signals released by dying cells.62,63 Furthermore, taxanes can function as TLR agonists, thereby directly activating DCs, and can also negatively modulate the frequency of regulatory T cells.64 Thus, cytotoxic treatments could be combined with immunotherapies, including immune checkpoint blockade. Combination trials with ipilimumab include the phase III trial combining local radiation with ipilimumab. In a phase II experience with combination radiotherapy and ipilimumab, 10 of 45 patients exhibited PSA declines of more than 50%, but 11 developed grade 3 or higher toxicities.65 Radiation with PROSTVAC-VF therapy seemed to have a more favorable toxicity profile and incurred significant PSA-specific T-cell responses, whereas none were induced with radiation alone.66 Cytotoxic treatments should be presumably administered before or concurrently with these immunotherapies, and use of steroids (eg, as premedication) should be minimized to avoid steroid-induced immunosuppression.
Androgen deprivation in prostate cancer can also affect the levels of T cells in peripheral lymphoid tissues, increase T-cell sensitivity to antigen-specific stimulation, and decrease time of lymphocyte recovery after chemotherapy-induced lymphocyte depletion.67,68 Moreover, androgen disruption can induce T-cell infiltration into the prostate.69 Thus, there exists the biologic rationale of combining these treatments with immunotherapy. A randomized phase II trial comparing nilutamide, a PSA-poxvirus vaccine, and combination (at crossover) in men with nonmetastatic CRPC suggested that vaccination before second-line hormone therapy may provide long-term survival advantages over receiving nilutamide upfront, raising the hypothesis that the order of treatment may improve survival at indolent stages of disease.26 Issues that need to be addressed in future studies include the optimal dose and schedule of combination regimens. With immunotherapies that lack significant toxicities, administering these treatments when androgen deprivation is being initiated (eg, with biochemically relapsed prostate cancer) may be a potential approach to maximize the immunogenicity of these treatments.
IMMUNOLOGIC BIOMARKERS
Identifying immunologic end points to immunotherapy would thus help guide the development of immunotherapy trials. Tools being studied to monitor systemic immune activation include immunophenotyping by flow cytometry and assessment of serum cytokine responses. There is emerging evidence to suggest that the amount of immune reserve can be prognostic; a total lymphocyte count greater than 1,000 per μl was associated with improved OS in patients receiving ipilimumab for advanced melanoma,70 and levels of CRP are currently being used as a biomarker to evaluate response to CTLA4 blockade. Persistently elevated levels of inducible costimulator–positive CD4 T cells have also been associated with improved clinical outcomes in melanoma.71 Changes in the balance of effector T cells and Tregs may also reflect treatment activity and are currently being investigated in combination ipilimumab and GM-CSF trials.46
Assays to assess for antigen-specific responses are also being developed. These include MHC-peptide tetramer staining, T-cell proliferation, cytotoxicity assays, delayed-type hypersensitivity, enzyme-linked immunosorbent assay for antigen-specific antibodies, and enzyme-linked immunosorbent spot assays that detect IFN-γ or granzyme-producing T cells.72 These assays are being studied in different clinical trials.73 Induction of antigen-specific antibody responses detected by enzyme-linked immunosorbent assay was associated with improved clinical outcome with sipuleucel-T treatment.21 The induction of enhanced T-cell immune responses to PSA detected by IFN-γ enzyme-linked immunosorbent spot assay was also associated with prolonged OS with PROSTVAC-VF treatment.25 These assays will need to be validated prospectively to establish their clinical utility.
DEVELOPING PROSTATE CANCER IMMUNOTHERAPY
The long natural history of prostate cancer provides both unique challenges and opportunities for developing immunotherapies. Measurements of efficacy in early-phase studies often assess PSA response, objective responses, and TTP, but none of these end points can serve as surrogates for OS.74,75 Once safety is established, phase II clinical trials can be performed in the different states of prostate cancer to establish immunologic efficacy and help refine treatments for further study. In addition to patients with CRPC, patients with earlier stages of disease can provide additional opportunities to demonstrate clinical and immune responses, particularly because many of these treatments do not have significant adverse effects. In the setting of recurrent prostate cancer after definitive therapy, immunotherapies have been administered before initiation of androgen deprivation to assess for immune effects. Neoadjuvant trials (ie, immunotherapy before radical prostatectomy) are also now being used to examine immune responses in tissues as well as in blood. Determining responses in these clinical settings, however, is complicated by the lack of measurable disease and validated PSA end points for survival. Nevertheless, determining TTP or PFS as primary end points may not translate into improved OS, the definitive end point for phase III studies. As demonstrated by sipuleucel-T, PROSTVAC-VF, and ipilimumab in melanoma, no statistically significant differences in PFS were seen, yet therapies conferred statistically significant advantages in OS. This uncoupling between PFS and OS could reflect the time that is required for vaccines to establish an immunologic effect. Thus, early measurements may capture clinical progression before antitumor immunity is established. Clinical benefit could therefore manifest as prolonged stabilization of disease. This is supported by the delayed separation of Kaplan-Meier curves, which only becomes evident beyond 1 year after treatment with either sipuleucel-T or PROSTVAC-VF.21,24 Without surrogate end points for OS, phase III trials for prostate cancer immunotherapy may be relegated to metastatic CRPC, which may not reflect the best clinical setting for these therapies.
SUMMARY
In summary, immunotherapy is now a part of the armamentarium for prostate cancer, but there still remains room for improvement. Trial design continues to evolve in light of the biologic properties of immunologic agents: the delayed survival benefit, potential durability of response, and inadequacy of both standard biochemical and radiographic criteria to evaluate treatment response. Application of guidelines that have been established to evaluate immune-mediated tumor responses radiographically (immune-related response criteria) and standardize cellular immune response assays across multiple centers may improve trial design and implementation.73,76 Given that a subset of patients have achieved durable responses, mechanistic studies should be performed to understand the basis of successful treatment. Patient selection will also be important given age-related immunosenescence77 and tumor-mediated immune suppression.78 Moreover, as more agents become available to treat advanced prostate disease, the best clinical setting to administer immunotherapy will need to be investigated. For now, sipuleucel-T and PROSTVAC-VF have been primarily evaluated in patients with CRPC, yet immunotherapies should be more immunologically efficacious in earlier stages of disease. Biochemical relapse after definitive therapy represents one setting in which these therapies could be tested. Alternatively, the neoadjuvant setting could provide opportunities to study immune responses not just in the blood but in tissues as well. Unfortunately, until surrogates for OS are developed, clinical efficacy for immunotherapies will likely have to be validated in CRPC so that OS can be assessed in a timely fashion.
Footnotes
Supported by Grant No. R01CA136753 from the National Cancer Institute.
Authors' disclosures of potential conflicts of interest and author contributions are found at the end of this article.
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
Although all authors completed the disclosure declaration, the following author(s) indicated a financial or other interest that is relevant to the subject matter under consideration in this article. Certain relationships marked with a “U” are those for which no compensation was received; those relationships marked with a “C” were compensated. For a detailed description of the disclosure categories, or for more information about ASCO's conflict of interest policy, please refer to the Author Disclosure Declaration and the Disclosures of Potential Conflicts of Interest section in Information for Contributors.
Employment or Leadership Position: None Consultant or Advisory Role: Lawrence Fong, Pfizer (C) Stock Ownership: None Honoraria: None Research Funding: Lawrence Fong, Dendreon Expert Testimony: None Other Remuneration: None
AUTHOR CONTRIBUTIONS
Conception and design: All authors
Collection and assembly of data: All authors
Data analysis and interpretation: All authors
Manuscript writing: All authors
Final approval of manuscript: All authors
REFERENCES
- 1.Markowicz S, Engleman EG. Granulocyte-macrophage colony-stimulating factor promotes differentiation and survival of human peripheral blood dendritic cells in vitro. J Clin Invest. 1990;85:955–961. doi: 10.1172/JCI114525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Iwasaki A, Medzhitov R. Regulation of adaptive immunity by the innate immune system. Science. 2010;327:291–295. doi: 10.1126/science.1183021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fong L, Small EJ. Anti-cytotoxic T-lymphocyte antigen-4 antibody: The first in an emerging class of immunomodulatory antibodies for cancer treatment. J Clin Oncol. 2008;26:5275–5283. doi: 10.1200/JCO.2008.17.8954. [DOI] [PubMed] [Google Scholar]
- 4.Brown JA, Dorfman DM, Ma FR, et al. Blockade of programmed death-1 ligands on dendritic cells enhances T cell activation and cytokine production. J Immunol. 2003;170:1257–1266. doi: 10.4049/jimmunol.170.3.1257. [DOI] [PubMed] [Google Scholar]
- 5.Fong L, Small EJ. Immunotherapy for prostate cancer. Curr Urol Rep. 2006;7:239–246. doi: 10.1007/s11934-006-0027-8. [DOI] [PubMed] [Google Scholar]
- 6.Shariat SF, Shalev M, Menesses-Diaz A, et al. Preoperative plasma levels of transforming growth factor beta(1) (TGF-beta(1)) strongly predict progression in patients undergoing radical prostatectomy. J Clin Oncol. 2001;19:2856–2864. doi: 10.1200/JCO.2001.19.11.2856. [DOI] [PubMed] [Google Scholar]
- 7.Miller AM, Lundberg K, Ozenci V, et al. CD4+CD25high T cells are enriched in the tumor and peripheral blood of prostate cancer patients. J Immunol. 2006;177:7398–7405. doi: 10.4049/jimmunol.177.10.7398. [DOI] [PubMed] [Google Scholar]
- 8.Chang CC, Ogino T, Mullins DW, et al. Defective human leukocyte antigen class I-associated antigen presentation caused by a novel beta2-microglobulin loss-of-function in melanoma cells. J Biol Chem. 2006;281:18763–18773. doi: 10.1074/jbc.M511525200. [DOI] [PubMed] [Google Scholar]
- 9.Chang CC, Campoli M, Restifo NP, et al. Immune selection of hot-spot beta 2-microglobulin gene mutations, HLA-A2 allospecificity loss, and antigen-processing machinery component down-regulation in melanoma cells derived from recurrent metastases following immunotherapy. J Immunol. 2005;174:1462–1471. doi: 10.4049/jimmunol.174.3.1462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Getnet D, Maris CH, Hipkiss EL, et al. Tumor recognition and self-recognition induce distinct transcriptional profiles in antigen-specific CD4 T cells. J Immunol. 2009;182:4675–4685. doi: 10.4049/jimmunol.0803400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shafer-Weaver KA, Anderson MJ, Stagliano K, et al. Cutting edge: Tumor-specific CD8+ T cells infiltrating prostatic tumors are induced to become suppressor cells. J Immunol. 2009;183:4848–4852. doi: 10.4049/jimmunol.0900848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Small EJ, Reese DM, Um B, et al. Therapy of advanced prostate cancer with granulocyte macrophage colony-stimulating factor. Clin Cancer Res. 1999;5:1738–1744. [PubMed] [Google Scholar]
- 13.Rini BI, Weinberg V, Bok R, et al. Prostate-specific antigen kinetics as a measure of the biologic effect of granulocyte-macrophage colony-stimulating factor in patients with serologic progression of prostate cancer. J Clin Oncol. 2003;21:99–105. doi: 10.1200/JCO.2003.04.163. [DOI] [PubMed] [Google Scholar]
- 14.Rini BI, Stadler WM, Spielberger RT, et al. Granulocyte-macrophage–colony stimulating factor in metastatic renal cell carcinoma: A phase II trial. Cancer. 1998;82:1352–1358. doi: 10.1002/(sici)1097-0142(19980401)82:7<1352::aid-cncr19>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- 15.Spitler LE, Grossbard ML, Ernstoff MS, et al. Adjuvant therapy of stage III and IV malignant melanoma using granulocyte-macrophage colony-stimulating factor. J Clin Oncol. 2000;18:1614–1621. doi: 10.1200/JCO.2000.18.8.1614. [DOI] [PubMed] [Google Scholar]
- 16.Fong L, Dao V, O'Brien S, et al. Neoadjuvant immunotherapy for prostate cancer with GM-CSF and tumor infiltration by antigen presenting cells. J Clin Oncol. 2008;26(suppl):147s. abstr 3063. [Google Scholar]
- 17.Heiser A, Coleman D, Dannull J, et al. Autologous dendritic cells transfected with prostate-specific antigen RNA stimulate CTL responses against metastatic prostate tumors. J Clin Invest. 2002;109:409–417. doi: 10.1172/JCI14364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fong L, Brockstedt D, Benike C, et al. Dendritic cell-based xenoantigen vaccination for prostate cancer immunotherapy. J Immunol. 2001;167:7150–7156. doi: 10.4049/jimmunol.167.12.7150. [DOI] [PubMed] [Google Scholar]
- 19.Fong L, Brockstedt D, Benike C, et al. Dendritic cells injected via different routes induce immunity in cancer patients. J Immunol. 2001;166:4254–4259. doi: 10.4049/jimmunol.166.6.4254. [DOI] [PubMed] [Google Scholar]
- 20.Pandha HS, John RJ, Hutchinson J, et al. Dendritic cell immunotherapy for urological cancers using cryopreserved allogeneic tumour lysate-pulsed cells: A phase I/II study. BJU Int. 2004;94:412–418. doi: 10.1111/j.1464-410X.2004.04922.x. [DOI] [PubMed] [Google Scholar]
- 21.Kantoff PW, Higano CS, Shore ND, et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N Engl J Med. 2010;363:411–422. doi: 10.1056/NEJMoa1001294. [DOI] [PubMed] [Google Scholar]
- 22.Higano CS, Schellhammer PF, Small EJ, et al. Integrated data from 2 randomized, double-blind, placebo-controlled, phase 3 trials of active cellular immunotherapy with sipuleucel-T in advanced prostate cancer. Cancer. 2009;115:3670–3679. doi: 10.1002/cncr.24429. [DOI] [PubMed] [Google Scholar]
- 23.Small EJ, Schellhammer PF, Higano CS, et al. Placebo-controlled phase III trial of immunologic therapy with sipuleucel-T (APC8015) in patients with metastatic, asymptomatic hormone refractory prostate cancer. J Clin Oncol. 2006;24:3089–3094. doi: 10.1200/JCO.2005.04.5252. [DOI] [PubMed] [Google Scholar]
- 24.Kantoff PW, Schuetz TJ, Blumenstein BA, et al. Overall survival analysis of a phase II randomized controlled trial of a poxviral-based PSA-targeted immunotherapy in metastatic castration-resistant prostate cancer. J Clin Oncol. 2010;28:1099–1105. doi: 10.1200/JCO.2009.25.0597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gulley JL, Arlen PM, Madan RA, et al. Immunologic and prognostic factors associated with overall survival employing a poxviral-based PSA vaccine in metastatic castrate-resistant prostate cancer. Cancer Immunol Immunother. 2010;59:663–674. doi: 10.1007/s00262-009-0782-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Madan RA, Gulley JL, Schlom J, et al. Analysis of overall survival in patients with nonmetastatic castration-resistant prostate cancer treated with vaccine, nilutamide, and combination therapy. Clin Cancer Res. 2008;14:4526–4531. doi: 10.1158/1078-0432.CCR-07-5048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Higano C, Saad F, Somer B, et al. A phase III trial of GVAX immunotherapy for prostate cancer versus docetaxel plus prednisone in asymptomatic, castration-resistant prostate cancer (CRPC). Presented at the Genitourinary Cancers Symposium; February 26-28, 2009; Orlando, FL. abstr LBA150. [Google Scholar]
- 28.Small E, Demkow T, Gerritsen W, et al. A phase III trial of GVAX immunotherapy for prostate cancer in combination with docetaxel versus docetaxel plus prednisone in symptomatic, castration-resistant prostate cancer (CRPC). Presented at the 2009 Genitourinary Cancers Symposium; February 26-28, 2009; Orlando, FL. abstr 7. [Google Scholar]
- 29.Small EJ, Fratesi P, Reese DM, et al. Immunotherapy of hormone-refractory prostate cancer with antigen-loaded dendritic cells. J Clin Oncol. 2000;18:3894–3903. doi: 10.1200/JCO.2000.18.23.3894. [DOI] [PubMed] [Google Scholar]
- 30.Fong L, Ruegg CL, Brockstedt D, et al. Induction of tissue-specific autoimmune prostatitis with prostatic acid phosphatase immunization: Implications for immunotherapy of prostate cancer. J Immunol. 1997;159:3113–3117. [PubMed] [Google Scholar]
- 31.McNeel DG, Nguyen LD, Disis ML. Identification of T helper epitopes from prostatic acid phosphatase. Cancer Res. 2001;61:5161–5167. [PubMed] [Google Scholar]
- 32.Sheikh NA, Wesley JD, Chadwick E, et al. Characterization of antigen-specific T-cell activation and cytokine expression induced by sipuleucel-T. Presented at the 2011 Genitourinary Cancers Symposium; February 17-19, 2011; Orlando FL. abstr 155. [Google Scholar]
- 33.George DJ, Nabhan C, Gomella LG, et al. Subsequent treatment with APC8015F and its effect on survival in the control arm of phase III sipuleucel-t studies. Presented at the 2011 Genitourinary Cancers Symposium; February 17-19, 2011; Orlando, FL. abstr 139. [Google Scholar]
- 34.Madan RA, Arlen PM, Mohebtash M, et al. Prostvac-VF: A vector-based vaccine targeting PSA in prostate cancer. Expert Opin Investig Drugs. 2009;18:1001–1011. doi: 10.1517/13543780902997928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kaufman HL, Wang W, Manola J, et al. Phase II randomized study of vaccine treatment of advanced prostate cancer (E7897): A trial of the eastern cooperative oncology group. J Clin Oncol. 2004;22:2122–2132. doi: 10.1200/JCO.2004.08.083. [DOI] [PubMed] [Google Scholar]
- 36.DiPaola RS, Plante M, Kaufman H, et al. A phase I trial of pox PSA vaccines (PROSTVAC-VF) with B7-1, ICAM-1, and LFA-3 co-stimulatory molecules (TRICOM) in patients with prostate cancer. J Transl Med. 2006;4:1. doi: 10.1186/1479-5876-4-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Arlen PM, Skarupa L, Pazdur M, et al. Clinical safety of a viral vector based prostate cancer vaccine strategy. J Urol. 2007;178:1515–1520. doi: 10.1016/j.juro.2007.05.117. [DOI] [PubMed] [Google Scholar]
- 38.Vergati M, Cereda V, Madan RA, et al. Analysis of circulating regulatory T cells in patients with metastatic prostate cancer pre- versus post-vaccination. Cancer Immunol Immunother. 2011;60:197–206. doi: 10.1007/s00262-010-0927-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. 2009;9:162–174. doi: 10.1038/nri2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McNeel DG, Dunphy EJ, Davies JG, et al. Safety and immunological efficacy of a DNA vaccine encoding prostatic acid phosphatase in patients with stage D0 prostate cancer. J Clin Oncol. 2009;27:4047–4054. doi: 10.1200/JCO.2008.19.9968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Becker JT, Olson BM, Johnson LE, et al. DNA vaccine encoding prostatic acid phosphatase (PAP) elicits long-term T-cell responses in patients with recurrent prostate cancer. J Immunother. 2010;33:639–647. doi: 10.1097/CJI.0b013e3181dda23e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ward JE, McNeel DG. GVAX: An allogeneic, whole-cell, GM-CSF-secreting cellular immunotherapy for the treatment of prostate cancer. Expert Opin Biol Ther. 2007;7:1893–1902. doi: 10.1517/14712598.7.12.1893. [DOI] [PubMed] [Google Scholar]
- 43.Wada S, Yoshimura K, Hipkiss EL, et al. Cyclophosphamide augments antitumor immunity: Studies in an autochthonous prostate cancer model. Cancer Res. 2009;69:4309–4318. doi: 10.1158/0008-5472.CAN-08-4102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hodi FS, O'Day SJ, McDermott DF, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363:711–723. doi: 10.1056/NEJMoa1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Small EJ, Tchekmedyian NS, Rini BI, et al. A pilot trial of CTLA-4 blockade with human anti-CTLA-4 in patients with hormone-refractory prostate cancer. Clin Cancer Res. 2007;13:1810–1815. doi: 10.1158/1078-0432.CCR-06-2318. [DOI] [PubMed] [Google Scholar]
- 46.Fong L, Kwek SS, O'Brien S, et al. Potentiating endogenous antitumor immunity to prostate cancer through combination immunotherapy with CTLA4 blockade and GM-CSF. Cancer Res. 2009;69:609–615. doi: 10.1158/0008-5472.CAN-08-3529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Small E, Higano C, Tchekmedyian N, et al. Randomized phase II study comparing 4 monthly doses of ipilimumab (MDX-010) as a single agent or in combination with a single dose of docetaxel in patients with hormone-refractory prostate cancer. J Clin Oncol. 2006;24(suppl):243s. abstr 4609. [Google Scholar]
- 48.Madan RA, Mohebtash M, Arlen PM, et al. Overall survival (OS) analysis of a phase l trial of a vector-based vaccine (PSA-TRICOM) and ipilimumab (ipi) in the treatment of metastatic castration-resistant prostate cancer (mCRPC) J Clin Oncol. 2010;28(suppl):216s. abstr 2550. [Google Scholar]
- 49.Gerritsen W, van den Eertwegh AJ, de Gruijl T, et al. Expanded phase I combination trial of GVAX immunotherapy for prostate cancer and ipilimumab in patients with metastatic hormone-refractory prostate cancer (mHPRC) J Clin Oncol. 2008;26(suppl):285s. abstr 5146. [Google Scholar]
- 50.Hodi FS. Overcoming immunological tolerance to melanoma: Targeting CTLA-4. Asia Pac J Clin Oncol. 2010;6(suppl 1):S16–S23. doi: 10.1111/j.1743-7563.2010.01271.x. [DOI] [PubMed] [Google Scholar]
- 51.Hersh EM, O'Day SJ, Powderly J, et al. A phase II multicenter study of ipilimumab with or without dacarbazine in chemotherapy-naive patients with advanced melanoma. Invest New Drugs. 2010;29:489–498. doi: 10.1007/s10637-009-9376-8. [DOI] [PubMed] [Google Scholar]
- 52.O'Day SJ, Maio M, Chiarion-Sileni V, et al. Efficacy and safety of ipilimumab monotherapy in patients with pretreated advanced melanoma: A multicenter single-arm phase II study. Ann Oncol. 2010;21:1712–1717. doi: 10.1093/annonc/mdq013. [DOI] [PubMed] [Google Scholar]
- 53.Weber J, Thompson JA, Hamid O, et al. A randomized, double-blind, placebo-controlled, phase II study comparing the tolerability and efficacy of ipilimumab administered with or without prophylactic budesonide in patients with unresectable stage III or IV melanoma. Clin Cancer Res. 2009;15:5591–5598. doi: 10.1158/1078-0432.CCR-09-1024. [DOI] [PubMed] [Google Scholar]
- 54.Hirano F, Kaneko K, Tamura H, et al. Blockade of B7–H1 and PD-1 by monoclonal antibodies potentiates cancer therapeutic immunity. Cancer Res. 2005;65:1089–1096. [PubMed] [Google Scholar]
- 55.Iwai Y, Terawaki S, Honjo T. PD-1 blockade inhibits hematogenous spread of poorly immunogenic tumor cells by enhanced recruitment of effector T cells. Int Immunol. 2005;17:133–144. doi: 10.1093/intimm/dxh194. [DOI] [PubMed] [Google Scholar]
- 56.Sfanos KS, Bruno TC, Meeker AK, et al. Human prostate-infiltrating CD8+ T lymphocytes are oligoclonal and PD-1+ Prostate. 2009;69:1694–1703. doi: 10.1002/pros.21020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ahmadzadeh M, Johnson LA, Heemskerk B, et al. Tumor antigen-specific CD8 T cells infiltrating the tumor express high levels of PD-1 and are functionally impaired. Blood. 2009;114:1537–1544. doi: 10.1182/blood-2008-12-195792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Brahmer JR, Drake CG, Wollner I, et al. Phase I study of single-agent anti-programmed death-1 (MDX-1106) in refractory solid tumors: Safety, clinical activity, pharmacodynamics, and immunologic correlates. J Clin Oncol. 2010;28:3167–3175. doi: 10.1200/JCO.2009.26.7609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.McDermott DF, Drake CG, Sznol M, et al. A phase I study to evaluate safety and antitumor activity of biweekly BMS-936558 (anti-PD-1, MDX-1106/ONO-4538) in patients with RCC and other advanced refractory malignancies. Presented at the 2011 Genitourinary Cancers Symposium; February 17-19, 2011; Orlando, FL. [Google Scholar]
- 60.Ghiringhelli F, Apetoh L, Tesniere A, et al. Activation of the NLRP3 inflammasome in dendritic cells induces IL-1beta-dependent adaptive immunity against tumors. Nat Med. 2009;15:1170–1178. doi: 10.1038/nm.2028. [DOI] [PubMed] [Google Scholar]
- 61.Green DR, Ferguson T, Zitvogel L, et al. Immunogenic and tolerogenic cell death. Nat Rev Immunol. 2009;9:353–363. doi: 10.1038/nri2545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Apetoh L, Ghiringhelli F, Tesniere A, et al. Toll-like receptor 4-dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat Med. 2007;13:1050–1059. doi: 10.1038/nm1622. [DOI] [PubMed] [Google Scholar]
- 63.Apetoh L, Ghiringhelli F, Tesniere A, et al. The interaction between HMGB1 and TLR4 dictates the outcome of anticancer chemotherapy and radiotherapy. Immunol Rev. 2007;220:47–59. doi: 10.1111/j.1600-065X.2007.00573.x. [DOI] [PubMed] [Google Scholar]
- 64.Vicari AP, Luu R, Zhang N, et al. Paclitaxel reduces regulatory T cell numbers and inhibitory function and enhances the anti-tumor effects of the TLR9 agonist PF-3512676 in the mouse. Cancer Immunol Immunother. 2009;58:615–628. doi: 10.1007/s00262-008-0586-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Slovin SF, Beer TM, Higano CS, et al. Initial phase II experience of ipilimumab (IPI) alone and in combination with radiotherapy (XRT) in patients with metastatic castration-resistant prostate cancer (mCRPC) J Clin Oncol. 2009;27(suppl):268s. abstr 5138. [Google Scholar]
- 66.Gulley JL, Arlen PM, Bastian A, et al. Combining a recombinant cancer vaccine with standard definitive radiotherapy in patients with localized prostate cancer. Clin Cancer Res. 2005;11:3353–3362. doi: 10.1158/1078-0432.CCR-04-2062. [DOI] [PubMed] [Google Scholar]
- 67.Sutherland JS, Goldberg GL, Hammett MV, et al. Activation of thymic regeneration in mice and humans following androgen blockade. J Immunol. 2005;175:2741–2753. doi: 10.4049/jimmunol.175.4.2741. [DOI] [PubMed] [Google Scholar]
- 68.Roden AC, Moser MT, Tri SD, et al. Augmentation of T cell levels and responses induced by androgen deprivation. J Immunol. 2004;173:6098–6108. doi: 10.4049/jimmunol.173.10.6098. [DOI] [PubMed] [Google Scholar]
- 69.Mercader M, Bodner BK, Moser MT, et al. T cell infiltration of the prostate induced by androgen withdrawal in patients with prostate cancer. Proc Natl Acad Sci U S A. 2001;98:14565–14570. doi: 10.1073/pnas.251140998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ku GY, Yuan J, Page DB, et al. Single-institution experience with ipilimumab in advanced melanoma patients in the compassionate use setting: Lymphocyte count after 2 doses correlates with survival. Cancer. 2010;116:1767–1775. doi: 10.1002/cncr.24951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bogunovic D, O'Neill DW, Belitskaya-Levy I, et al. Immune profile and mitotic index of metastatic melanoma lesions enhance clinical staging in predicting patient survival. Proc Natl Acad Sci U S A. 2009;106:20429–20434. doi: 10.1073/pnas.0905139106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Fong L, Small EJ. Immunotherapy for prostate cancer. Curr Oncol Rep. 2007;9:226–233. doi: 10.1007/s11912-007-0026-z. [DOI] [PubMed] [Google Scholar]
- 73.Hoos A, Eggermont AM, Janetzki S, et al. Improved endpoints for cancer immunotherapy trials. J Natl Cancer Inst. 2010;102:1388–1397. doi: 10.1093/jnci/djq310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Collette L, Burzykowski T, Schröder FH. Prostate-specific antigen (PSA) alone is not an appropriate surrogate marker of long-term therapeutic benefit in prostate cancer trials. Eur J Cancer. 2006;42:1344–1350. doi: 10.1016/j.ejca.2006.02.011. [DOI] [PubMed] [Google Scholar]
- 75.Scher HI, Jia X, de Bono JS, et al. Circulating tumour cells as prognostic markers in progressive, castration-resistant prostate cancer: A reanalysis of IMMC38 trial data. Lancet Oncol. 2009;10:233–239. doi: 10.1016/S1470-2045(08)70340-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wolchok JD, Hoos A, O'Day S, et al. Guidelines for the evaluation of immune therapy activity in solid tumors: Immune-related response criteria. Clin Cancer Res. 2009;15:7412–7420. doi: 10.1158/1078-0432.CCR-09-1624. [DOI] [PubMed] [Google Scholar]
- 77.Shimatani K, Nakashima Y, Hattori M, et al. PD-1+ memory phenotype CD4+ T cells expressing C/EBPalpha underlie T cell immunodepression in senescence and leukemia. Proc Natl Acad Sci U S A. 2009;106:15807–15812. doi: 10.1073/pnas.0908805106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Willimsky G, Blankenstein T. Sporadic immunogenic tumours avoid destruction by inducing T-cell tolerance. Nature. 2005;437:141–146. doi: 10.1038/nature03954. [DOI] [PubMed] [Google Scholar]