Loss-of-function mutations in MGME1 impair mtDNA replication and cause multi-systemic mitochondrial disease (original) (raw)
. Author manuscript; available in PMC: 2013 Aug 1.
Published in final edited form as: Nat Genet. 2013 Jan 13;45(2):214–219. doi: 10.1038/ng.2501
Abstract
Known disease mechanisms in mitochondrial DNA (mtDNA) maintenance disorders alter either the mitochondrial replication machinery (POLG1, POLG22 and C10orf23) or the biosynthesis pathways of deoxyribonucleoside 5′-triphosphates for mtDNA synthesis4–11. However, in many of these disorders, the underlying genetic defect has not yet been discovered. Here, we identified homozygous nonsense and missense mutations in the orphan gene C20orf72 in three families with a mitochondrial syndrome characterized by external ophthalmoplegia, emaciation, and respiratory failure. Muscle biopsies showed mtDNA depletion and multiple mtDNA deletions. C20orf72, hereafter MGME1 (mitochondrial genome maintenance exonuclease 1), encodes a mitochondrial RecB-type exonuclease belonging to the PD-(D/E)XK nuclease superfamily. We demonstrate that MGME1 cleaves single-stranded DNA and processes DNA flap substrates. Upon chemically induced mtDNA depletion, patient fibroblasts fail to repopulate. They also accumulate intermediates of stalled replication and show increased levels of 7S DNA, as do MGME1-depleted cells. Hence, we show that MGME1-mediated mtDNA processing is essential for mitochondrial genome maintenance.
At present, only three nuclear genes directly involved in mtDNA replication have been associated with mitochondrial disease1–3. Pathogenic mutations in these genes result in two molecular phenotypes, multiple mtDNA deletions (“multiple mtDNA deletion disorders”) and/or reduction of mtDNA copy number (“mtDNA depletion syndromes”, MDS). Multiple mtDNA deletion disorders and MDS are clinically and genetically overlapping entities, known collectively as mtDNA maintenance disorders. Their clinical manifestations range from severe multi-organ involvement in early childhood to tissue-specific manifestations, e.g. myopathies, in late adulthood. Progressive external ophthalmoplegia (PEO) is commonly associated with these disorders, particularly the late-onset phenotypes.
We identified a Lebanese family (Family I) with three children affected by a severe multi-systemic mitochondrial disorder (Supplementary Fig. 1a). Initial symptoms were ptosis in childhood, followed by mild PEO, diffuse skeletal muscle wasting and weakness, profound emaciation, and respiratory distress. Skeletal muscle biopsies showed scattered cytochrome-_c_-oxidase (COX)-negative fibers and ragged-red fibers (RRF) in all patients. Respiratory chain enzyme activities were decreased in skeletal muscle (Supplementary Table 1). Mutations in mtDNA and nuclear genes associated with PEO were excluded by Sanger sequencing. To find the underlying genetic cause, we captured the exome from patient P1976 and performed massively parallel sequencing12. We applied several filtering steps excluding intronic variants at non-canonical splice sites, synonymous variants as well as variants present in 816 exomes from patients with unrelated diseases (Supplementary Table 2). Assuming an autosomal recessive mode of inheritance and focusing on genes predicted to encode mitochondrial proteins13, this approach selected two genes, SPTLC3 and C20orf72. Unlike SPTLC3, C20orf72 harbored a homozygous nonsense mutation (c.456G>A, p.Trp152*), found in all affected but not in healthy siblings, while both parents were heterozygous carriers (Fig. 1).
Figure 1. Protein sequence, domain architecture, mutations and subcellular localization of MGME1.

(a) Top: Domain architecture of MGME1 indicating the mutations indentified in the patients. The key motifs of the PD-(D/E)XK nuclease superfamily are indicated in Roman numerals and the sequence details of the motifs characteristic for the RecB-type subgroup are shown as color boxes below the schematic structure (“h” indicates a hydrophobic residue). “MTS” indicates the predicted mitochondrial targeting signal. Bottom: Alignment of protein sequences of the orthologues of MGME1 and RecB-type nucleases. The c.698A>G, p.Tyr233Cys mutation is highlighted in black. Colons denote chemical similarity between the sequences, asterisks indicate identical residues. GenBank accession numbers of sequences used in the alignment are as follows: Human, NP_443097; Mouse, NP_083260; Chicken, XP_415017; Green anole lizard, XP_003219931, Xenopus, NP_001120532; Zebrafish, NP_0001008640; Lancelet, XP_002610511; Sea squirt, XP_002119404; Cyanobacteria, YP_002377694; AddA Bacillus subtilis, YP_004207075; RecB Escherichia coli, EGB85545. (b) The predicted active site structural motif of MGME1 based on the known crystal structure of the homologous RecB-type nucleases (left). Structure prediction was made using the 3D-Jury algorithm and modelled using the MODELLER software. The active site structure of E.coli RecB is provided for comparison (right). (c) Western blot showing protein level of MGME1 in P1976 fibroblasts (FB1976) and control fibroblasts. β-actin was used as a loading control. (d) Cellular localization of the GFP-tagged variant of MGME1 (green) in human fibroblasts. mtSSBP was used as a mitochondrial marker (red). Nuclei were stained with DAPI (blue). Co-localization of the green and red signal appears yellow on digitally overlaid images (overlay). Scale bars indicate 10μm.
In parallel, we applied targeted MitoExome sequencing14,15 and identified two additional patients from an Italian family (Family II) with the same homozygous nonsense mutation in C20orf72 (Supplementary Fig. 1b). Haplotype analysis revealed a homozygous region of 1.1 Mb shared by both families, suggesting that the mutation derives from a common founder (Supplementary Table 3). In addition, we identified a homozygous c.698A>G, p.Tyr233Cys mutation in a sporadic case from Germany. All patients presented with strikingly similar clinical and histological phenotypes (Table 1, Supplementary Note, Supplementary Fig. 2).
Table 1.
Clinical overview of patients harboring MGME1 mutations
| Family I (Lebanese) | Family II (Italian) | Sporadic case (German) | ||||
|---|---|---|---|---|---|---|
| P1976 | P2061 | P3737 | P4050 | P4052 | P931 | |
| Sex | male | female | male | male | male | female |
| Age at first visit (y) | 10 | 17 | ~14 | 31 | 36 | 35 |
| Current age (y) | 26 | 33 | death at 26 (cardiac failure) | 53 | 42 | death at 73 (respiratory failure) |
| MGME1 mutation | c.456G>A, p.Trp152* | c.456G>A, p.Trp152* | c.456G>A, p.Trp152* | c.456G>A, p.Trp152* | c.456G>A, p.Trp152* | c.698A>G, p.Tyr233Cys |
| Clinical phenotype (major clinical findings) | PEO with proximal weakness and generalized muscle wasting, profound emaciation, respiratory failure (non- invasive ventilation) | PEO with exercise intolerance and generalized muscle wasting, profound emaciation, dyspnea | PEO with proximal weakness and generalized muscle wasting, profound emaciation, respiratory failure (non-invasive ventilation) | PEO with proximal weakness, respiratory failure (non-invasive ventilation), emaciation in advanced stage | ||
| BMI | 11.8 | 12.7 | 12.3 | n.a. | 17.1 | n.a. |
| Secondary clinical symptoms | mental retardation, gastrointestinal symptoms, renal colics, spinal deformities | mental retardation, gastrointestinal symptoms, spinal deformities, mild ataxia | mental retardation, gastrointestinal symptoms, spinal deformities, dilated cardiomyopathy | mild spinal deformities | renal colics, mild spinal deformities | cardiac arrhythmias, depressive episodes, memory deficits |
| Brain imaging (y) | cerebellar atrophy on MRI and cranial CT (10 and 26) | cerebellar atrophy on MRI (17) | cerebellar atrophy on MRI (23) | n.a. | mild cerebral atrophy on cranial CT (57) | |
| Skeletal muscle biopsy (y) | vastus lateralis (10): few RRF, few scattered COX- negative fibers (0.4%) | biceps brachii (17): RRF, few scattered COX- negative fibers (0.6%) | vastus lateralis (23): RRF, scattered COX- negative fibers (6.2%) | biceps brachii (31): few RRF, scattered COX-negative fibers (1.4–1.7%) | biceps brachii (37), triceps brachii (41): few RRF, scattered COX-negative fibers (1.5%, 2%) | vastus lateralis (57): RRF, scattered COX- negative fibers (3.6%) |
| Respiratory chain enzyme activities | combined complex I/IV-deficiency | complex I deficiency | normal | combined complex I/IV-deficiency | n.a. | |
| long-range PCR | multiple mitochondrial DNA deletions (present in all muscle biopsies) | |||||
| NMDAS (0–145 Pts.) | 48 Pts. (age 26) | 37 Pts. (age 33) | n.a. |
Analysis of the primary structure of the protein encoded by C20orf72, renamed MGME1, revealed that it belongs to the PD-(D/E)XK nuclease superfamily16, a large and diverse group of enzymes having various nucleic acid cleavage activities. In addition to the principal PD-(D/E)XK motifs, I, II and III (Fig. 1a, green, yellow, blue), MGME1 contains a QhxxY motif characteristic of the RecB-type family17 (Fig. 1a, red). 3D homology modeling of the MGME1 active site based on the crystal structure of RecB18 indicated a very similar arrangement of the key catalytic residues (Fig. 1b). The c.456G>A, p.Trp152* mutation is upstream of the PD-(D/E)XK nuclease motif, so that a truncated product is very likely to be non-functional. Even so, we did not observe any detectable levels of either the full-length or the truncated MGME1 in P1976 fibroblasts (Fig. 1c), the latter most likely owing to nonsense-mediated mRNA decay. The c.698A>G, p.Tyr233Cys mutation found in the sporadic case affects a conserved amino acid residue (Fig. 1a). MGME1 contains a predicted N-terminal mitochondrial targeting signal and immunohistochemical subcellular localization of GFP-tagged MGME1 in human fibroblasts (Fig. 1d) and cell fractionation (Supplementary Fig. 3) revealed that MGME1 in fact localizes to mitochondria.
To clarify the molecular consequences of MGME1 mutations, we analyzed available tissue samples for the presence of mtDNA deletions or mtDNA depletion. Long-range PCR revealed the presence of multiple mtDNA deletions in the muscle biopsies from all patients and from blood and urine samples of Family I (Supplementary Fig. 4). The deletions were unusually large in comparison with those observed in POLG patients. Quantitative assessment of deletions by single-molecule PCR revealed total loads between 0.04% and 1.5% in skeletal muscle (Supplementary Table 4), which is similar to the levels observed in patients with POLG mutations (Supplementary Table 4). MtDNA copy number analysis detected substantial depletion in all adult patients (P2061, P3737, P4050, P4052 and P931) (Fig. 2a, gray bars). Only the muscle biopsy from patient P1976, which was taken when he was 10 years old and only mildly symptomatic, had normal mtDNA copy numbers. The finding of mtDNA depletion and mtDNA deletions in skeletal muscle from the MGME1 patients suggested perturbed mtDNA maintenance as the primary cause of the mitochondrial disease.
Figure 2. Loss of MGME1 leads to mtDNA depletion and elevated levels of 7S DNA.
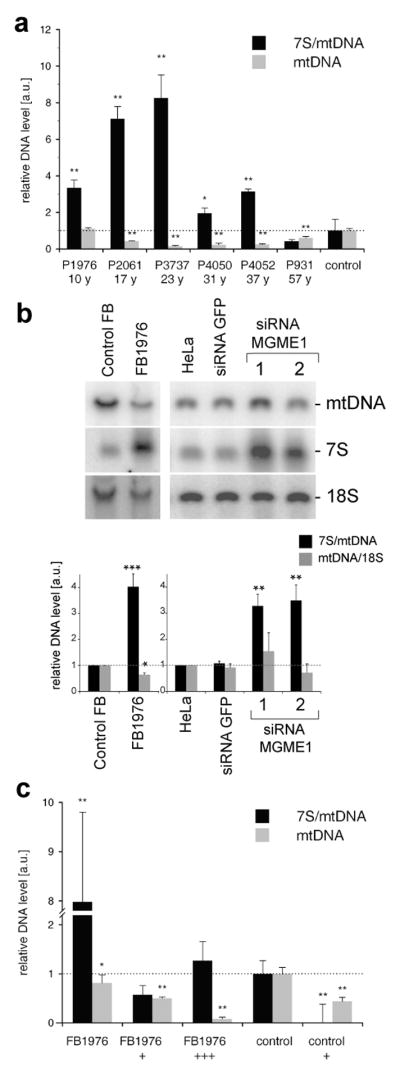
(a) Relative mtDNA copy numbers (gray bars) and 7S/mtDNA ratios (black bars) in skeletal muscle from patients with MGME1 mutations. The data are shown as averages ± standard deviations (SD) of three independent qPCR determinations and normalized to the corresponding control values. The control values are averages ± SD from eleven skeletal muscle biopsies samples, age range 18 – 33 years, mtDNA copy number range 8,146–11,416 molecules per nucleus. * P<0.05, ** P<0.01; two-tailed Student’s t- test. (b) Total DNA from control or patient (FB1976) fibroblasts (cell passage 15–21) and from untransfected HeLa cells (HeLa), cells transfected with siRNA to GFP or siRNA to MGME1 for 6 days analyzed by 1D Southern blotting with a radioactive probe specific for the non-coding region (NCR) in human mtDNA (mtDNA region: 14,258–4,121) followed by a probe specific for the 18S rDNA. The values of the relative DNA level (black bars, 7S DNA/mtDNA; grey bars, mtDNA/18S rDNA) obtained by quantifying PhosphorImager scans of Southern blots using ImageQuant software and normalized for the values obtained for control fibroblasts or untransfected HeLa cells. * P<0.05, ** P<0.01, *** P<0.001; two-tailed Student’s t- test; n=3, error bars=1 SD. Age of biopsy indicated. (c) Relative mtDNA copy numbers (grey bars) and 7S/mtDNA ratios (black bars) in fibroblasts of a patient with a _MGME1_-null mutation (P1976, cell passage 6–10) and a control. When indicated, the fibroblasts were transduced with low (“+”) or high (“+++”) titer of a lentiviral MGME1 construct. * P<0.05, ** P<0.01; two-tailed Student’s t- test; n=3, error bars=1 SD.
In addition to mtDNA depletion, we observed a relative increase of 7S DNA levels. 7S DNA is the single-stranded component of the mtDNA displacement loop (D-loop), and is postulated to be an intermediate of prematurely terminated mtDNA replication. The ratios of 7S/mtDNA were up to 8-fold increased in 5 of 6 investigated muscle tissues (Fig. 2a, black bars) and the fibroblasts from patient P1976 (Fig. 2b, black bars). This phenomenon was also detected in cells subjected to siRNA down-regulation of MGME1 (Fig. 2b and Supplementary Fig. 5a), suggesting a direct involvement of MGME1 in the turnover of 7S DNA.
To rescue the phenotype of the MGME1 mutation, P1976 fibroblasts were transfected with a lentiviral MGME1wt expression construct. Overexpression of MGME1wt for 2 weeks substantially decreased the 7S DNA level, but it also caused a more general mtDNA depletion (Fig. 2c and Supplementary Fig. 5b). Depletion of mtDNA was also detectable in control fibroblasts after lentiviral MGME1wt overexpression. These findings imply that an optimal level of MGME1 is critical for the proper maintenance of the mitochondrial genome.
To investigate the enzymatic properties of MGME1, the recombinant protein was isolated from human mitochondria by affinity purification (Supplementary Fig. 6a). MGME1 cleaved ssDNA, but not ssRNA nor RNA/DNA hybrids. The enzyme required free nucleic acid ends for catalysis, as it was unable to degrade circular ssDNA (Fig. 3a) or dsDNA substrates containing a nick or ssDNA gap of 12 nt (Supplementary Fig. 7). The enzyme had a strong preference for ssDNA over dsDNA (Fig. 3a). Blocking of the 5′-end, but not the 3′-end, of ssDNA with a biotin-streptavidin moiety reduced the cutting rate to about 10 – 20% as compared to the unblocked control at low enzyme concentrations (Supplementary Fig. 8). The recombinant MGME1 processed DNA 5′ and 3′ splayed arm and flap-like substrates by removing the ssDNA segment, but stopped at the ssDNA-dsDNA junction (Fig. 3b–c). However, the flap-like substrates with displaced 5′ DNA end were processed more efficiently than ones with a displaced 3′ end (Fig. 3d). In addition, MGME1 processed RNA-DNA chimaeric oligonucleotides by initiating DNA degradation at a position 2–5 nucleotides downstream from the RNA-DNA junction (Fig. 3e). Notably, the enzyme processed splayed arm and flap-like substrates, where the displaced 5′ arm contained RNA (Fig. 3e). A point mutation of the conserved Lys residue of the predicted PD-(D/E)XK motif III (p.Lys253Ala) completely abolished all nucleolytic activities tested (Fig. 3 and Supplementary Fig. 6b). We were unable to test the enzymatic activity of the enzyme variant found in the sporadic case (p.Tyr233Cys) most likely owing to impaired folding (Online Methods and Supplementary Fig. 6c). These in vitro results confirmed the computational prediction that MGME1 was a PD-(D/E)XK-type nuclease. The requirement of nucleic acid ends with preference for the DNA 5′-end for catalysis, the ability to process DNA flap structures as well as flap structures containing RNA at the 5′-end make MGME1 a good candidate enzyme for the processing of displaced DNA containing Okazaki fragments during RNA-primed DNA synthesis on the lagging strand19 or of DNA flaps during long-patch base excision repair20.
Figure 3. Characterization of the enzymatic activity of MGME1.

(a) DNase activity of MGME1 and the preference towards ssDNA vs. dsDNA. 1 pmol of radioactively labeled substrates (asterisks indicate the label) was incubated for 30 min with increasing concentrations (0.125, 0.25, 0.5, 1, 2, 4 pmoles) of purified MGME1 or 4 pmols of the p.Lys253Ala mutant (K253A). Reaction products were subjected to denaturing PAGE and autoradiography; were quantified, and the intensity was plotted against the enzyme concentration (right). (b) and (c) MGME1 activity on 5′ displaced (b) and 3′ displaced (c) splayed arm and flap-like DNA structures. Reaction conditions were as per (a). Asterisks indicate positions of radioactive label. (d) Comparison of the MGME1 processing efficiency on the 5′ displaced (5′-flap) and 3′ displaced (3′-flap) flap-like DNA substrates with mapping of the radioactive products. Signal intensities of processed and partially processed reaction product were quantified and plotted as in a. (e) MGME1 activity on RNA-DNA chimeric substrates that resemble Okazaki fragments. Increasing concentrations of purified MGME1 were incubated with a 5′ radioactively end-labelled RNA-DNA substrates as schematically indicated in the reaction conditions as described in (a). Numbers on the top indicate oligonucleotide lengths. SA, splayed-arm structure. Quantification of the reactions upon PAGE and autoradiography is shown to the right.
To investigate the influence of the MGME1 nonsense mutation on mtDNA replication capability, we performed mtDNA depletion and repopulation experiments in fibroblast cultures from patient P1976. Upon treatment with the nucleotide analogue ddC21, control and patient fibroblasts developed mtDNA depletion, with mtDNA falling to <10% of the untreated steady state level within 12 days exposure (Fig. 4a). This occurs because ddC acts both as a competitive inhibitor of mtDNA polymerase and as a terminator of nascent strand elongation22. After ddC withdrawal (Fig. 4a, arrow), mtDNA repopulation was severely impaired in MGME1-null cells (only 8% of control mtDNA copy number was reached after 16 days of culture without ddC). This clearly indicates perturbed mtDNA replication, similar to that observed in patients harbouring pathogenic POLG mutations22. Interestingly, the mtDNA depletion rate after exposure to ddC was considerably slower in the MGME1-null cells than in controls, suggesting impaired mtDNA breakdown22.
Figure 4. Perturbed mitochondrial replication in patient fibroblasts and MGME1-depleted cells.
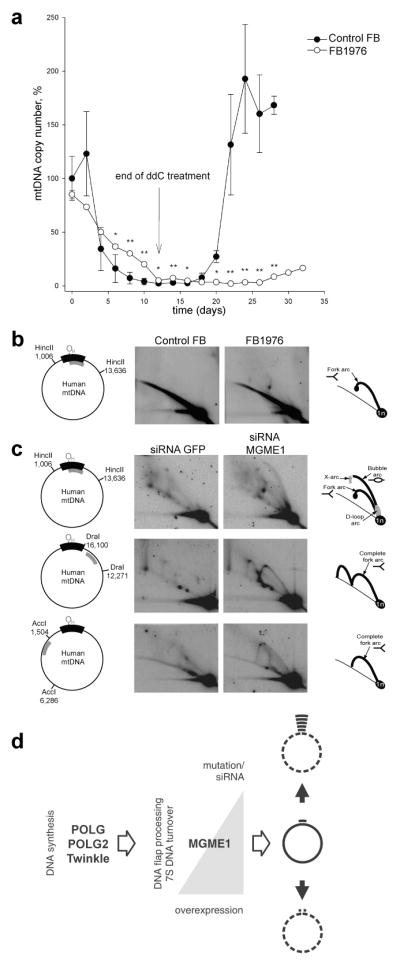
(a) MtDNA copy number during induced depletion and repopulation of human control fibroblasts (filled circles) and MGME1-null fibroblasts (FB1976 open circles). MtDNA copy numbers were determined by qPCR. Depletion of mtDNA was achieved by addition of 20 μM ddC to the culture medium for 12 days. MtDNA copy numbers were determined by qPCR using the mitochondrial primers MT3922F25 and MT4036R26. Data points for FB1976 represent the mean of three determinations ± SD values. The data for control fibroblasts are averages ± SD of mtDNA depletion-repopulation experiments with two separate cell lines. * P<0.05, ** P<0.01; two-tailed Student’s t- test. (b) and (c) Mitochondrial DNA replication in MGME1-null fibroblasts and MGME1-depleted cells. Total DNA from control or FB1976 fibroblasts (b) and HeLa cells transfected with siRNA to GFP or siRNA to MGME1 for 6 days (c) was subjected to two-dimensional neutral–neutral agarose-gel electrophoresis (2DNAGE) followed by Southern blotting. Restriction enzymes and probes used (grey bars) are indicated in the far left column of the figure. The nucleotide positions of the probes were as follows: _Hinc_II: nt 16,341–151, _Dra_I: nt 13,202–13,800 and _Acc_I: nt 3,587–4,161. Black bar indicates the NCR; OH, origin of H-strand replication. The interpretation based on23,28 is provided in the far right column of the figure. 1n, – non-replicating fragment. (d) Scheme showing the proposed involvement of MGME1 in mtDNA maintenance. Mutation or siRNA knockdown of MGME1 results in accumulation of replication intermediates followed by mtDNA depletion and 7S DNA accumulation (right black arrow up). In contrast, overexpression of MGME1 causes promiscuous degradation of mtDNA (including 7S DNA) resulting in mtDNA depletion (right black arrow down).
To further examine the consequence of the MGME1 nonsense mutation on mtDNA replication, we analyzed the pattern of mitochondrial replication intermediates in patient fibroblasts using two-dimensional agarose gel electrophoresis (2D-AGE). We observed pronounced replication stalling and accumulation of replication intermediates in the P1976 fibroblasts but not in control fibroblasts (Fig. 4b). This increase in stalled mtDNA replication intermediates was also detected in HeLa cells subjected to MGME1 siRNA, and replication stalling persisted throughout the entire mtDNA molecule (Fig. 4c). A similar mtDNA replication defect in 2D-AGE has been reported for mutations in C10orf2 (Twinkle) and POLG23.
In conclusion, our findings imply that mtDNA processing by MGME1 is essential for effective mtDNA synthesis. Most importantly, we show that mutations in this mitochondrial exonuclease affect proper mtDNA maintenance (Fig. 4d) and are a clinically relevant cause of mtDNA disorders. Thus far, only two nuclear nucleases also present in mitochondria, FEN120 and DNA224–26, and one mitochondrial enzyme, ExoG27, have been proposed to be involved in mtDNA flap processing. Studies on mitochondrial forms of FEN1 and DNA2 as well as ExoG have focused on their role in long-patch excision repair in mitochondria and nothing is known about their contribution to mtDNA replication. Although, in principle MGME1 might play a redundant role in mtDNA repair, it is the first identified mitochondrial exonuclease shown to be involved in mtDNA replication, likely through the processing of displaced DNA or RNA-DNA in the flap-like structures during mtDNA synthesis.
ONLINE METHODS
Methods and any associated references are available in the online version of the paper.
Mutation screening
Exome sequencing of the index patient of Family I (P1976) was performed on a Genome Analyzer IIx system (Illumina) after in-solution enrichment of exonic sequences (SureSelect Human All Exon 38 Mb kit, Agilent). The sample was sequenced using two lanes of a flowcell as 54 bp paired-end runs. Read alignment was performed with BWA (version 0.5.8) to the human genome assembly hg19. Single-nucleotide variants and small insertions and deletions were detected with SAMtools (v 0.1.7). Variant annotation was performed with custom scripts. Dealing with a mitochondrial disorder, we focused on genes encoding proteins with a confirmed or predicted mitochondrial localization corresponding to a MitoP2-score >0.513. Sequence analysis by Sanger sequencing confirmed the c.456G>A mutation in the homozygous state in all three affected siblings from Family I.
The homozygous C20orf72 nonsense mutation (c.456G>A, p.Trp152*) in patient P4052 from Family II was detected by a MitoExome sequencing approach described previously15. Briefly, DNA was extracted from blood, and hybrid selection (Agilent SureSelect) was used to select for the mtDNA and coding exons of over 1000 nuclear genes associated with mitochondrial function15, which were sequenced on an Illumina HiSeq with paired 76bp reads. Reads were aligned to the reference human genome assembly hg19, and variants were detected using the Genome Analysis Toolkit29. All variants previously associated with disease (MITOMAP: A Human Mitochondrial Genome Database, and HGMD: Human Gene Mutation Database) were prioritized as well as all rare variants predicted to be deleterious and consistent with recessive mode of pathogenesis29. The homozygous variant c.456G>A; p.Trp152* in C20orf72 was the only candidate mutation meeting these stringent prioritization criteria.
The homozygous c.698A>G, p.Tyr233Cys mutation in patient P931 was detected in a screening of 50 German PEO patients by Sanger sequencing.
Quantitative PCR
Copy numbers of mtDNA were determined by quantitative PCR essentially as described previously30. Briefly, a segment of the mtDNA outside the 7S region was amplified using primers MT3922F25 and MT4036R26 (first number, 5′-nucleotide of primer; second number, length of primer; F, forward primer; R, reverse primer; numbering according to reference sequence NC_012920). Primers MT16520F24, and MT35R24 were used together with TaqMan probe MT16557F25 to amplify a short segment inside the 7S region of the mtDNA. CT values were defined at the inflection points of fitted sigmoid curves (4-parameter Chapman curves), and were compared to those of the single copy nuclear gene KCNJ10 amplified by primers KIR835F19 and KIR903R19 and TaqMan probe KIR857F27 (numbering according to sequence U52155). Relative 7S/mtDNA ratios were calculated by subtracting outside-7S copy numbers from inside-7S copy numbers, divided by outside-7S copy numbers, and subsequently normalizing to the control value. Reactions were performed on a MyiQ qPCR system (Bio-Rad, Munich, Germany) using either TaqMan probes, or iQ SYBR Green Supermix (Bio-Rad, Munich, Germany) under the following conditions: 95°C for 3 min, and 45 cycles of 95°C for 15 s and 60°C for 1 min.
Detection of mtDNA deletions
Large-scale deletions of the mitochondrial genome were detected by long-range PCR using either primer pair MT5462F28 and MT45R22, or the primer pair MT15974F23 and MT15623R20 (Supplementary Table 5). For the amplification, TaKaRa LA Taq Hot Start DNA polymerase (Takara Bio Inc., Otsu, Japan) was used under the following conditions: 95°C for 2.5 min, 10 cycles of 92°C for 20 s and 68°C for 14 min, 20 cycles of 92°C for 25 s and 68°C for 16 min, and 72°C for 10 min. To quantify deletion levels, a single-molecule PCR approach was used as described previously30. Briefly, single mtDNA deletions were amplified using the same conditions as in long-range PCR, but in 42 cycles of PCR. To estimate the amounts of total mtDNA, primers MT16520F24 and MT4833R24 were used. Total template DNA was diluted to degrees, where only a part of multiple identical reactions resulted in amplification products (ideally less than 50 %). It is reasonable to assume that, under these conditions, each positive reaction originated from a single mtDNA molecule or a single deleted mtDNA molecule dividing the degree of dilution for single mtDNA molecules by the degree of dilution for single deleted mtDNA molecules (e.g. 6 positive reactions out of 15 at dilution 10−5 for total mtDNA and 3 positive reactions out of 15 at dilution 10−3 for deleted mtDNA gives a deletion ratio of 5×10−3).
Cell culture, transduction and transfection
HeLa cells were cultured in Dulbecco’s Modified Eagle Medium (DMEM, Gibco) supplemented with 10% foetal calf serum (Thermo Scientific), 10 U/ml penicillin and 10 μg/ml streptomycin (Gibco). Fibroblast cell lines cells were maintained in high-glucose Dulbecco’s modified Eagle’s medium (DMEM, Invitrogen) supplemented with 10% fetalbovine serum (Invitrogen), 200 μM uridine (Sigma) and 1% penicillin/streptomycin (Invitrogen), at 37°C in a humidified atmosphere of 5% CO2.
The C20orf72 ORF was cloned into the lentiviral expression vector pLENTI6.3. The p.Lenti6.3/V5-TOPO vector system (Invitrogen) was utilized for lentiviral mediated expression of wild type (wt) cDNA in skin fibroblast cell lines31.
Transfected HEK293 cells were maintained in DMEM containing 10% tetracycline-free foetal calf serum (Biochrom AG), 10 U/ml penicillin and 10 μg/ml streptomycin (Gibco), 50 μg/ml hygromycin (Invitrogen) and 15 μg/ml blasticidin (Invivogen).
For siRNA experiments, 100,000 cells were transfected with siRNA against MGME1 or negative control (Invitrogen Stealth) using Lipofectamine RNAiMAX (Invitrogen) for three days, then retransfected for a further three days. siRNA sequences are given in Supplementary Table 5.
Protein purification
The full-length cDNA clone for MGME1 was obtained from MRC Geneservice and cloned in-frame with a FLAG.STREP2 tag into the expression vector pcDNA5/FRT/TO (Invitrogen). The p.Tyr233Cys and p.Lys253Ala variants of MGME1 were created by oligonucleotide-directed mutagenesis (primer sequences are given in Supplementary Table 5). The MGME1 wild-type, p.Tyr233Cys and p.Lys253Ala constructs were transfected into HEK Flp-In T-Rex 293 cells (Invitrogen) using Cell Line Nucleofector (Lonza), applying programme A-23. Transfected cells were induced to express recombinant MGME1 or its mutants with 50 ng/ml doxycycline for 48 hours, after which mitochondria were isolated by hypotonic lysis and differential centrifugation as described previously32. Recombinant MGME1 protein was isolated using a gravity flow Strep-Tactin column (IBA) as described previously32. In order to assess the purity of the recombinant enzymes Strep-Tactin purified proteins were separated by SDS–PAGE, Coomassie-stained and all protein bands were excised from gels and identified by mass spectrometry. Typically, for the wild-type and p.Lys253Ala variants the procedure yielded ~150 μg of recombinant enzyme that was ~95 % pure, starting from 4 – 4.5 g of cells (Supplementary Fig. 6a–b). In the same conditions the p.Tyr233Cys variant gave only about 1/30 of this yield and preparations were contaminated (~90 %) with mitochondrial chaperone HSP70, suggestive for impaired folding of the recombinant p.Tyr233Cys protein (Supplementary Fig. 6c).
In vitro nuclease assays
1 nmol of labelled substrates were incubated with the indicated amounts of purified MGME1 protein in a reaction buffer containing 10 mM Tris pH 7.6, 20 mM MgCl2, 1 mM DTT and 0.1 mg/ml BSA at 37 °C for 30 min. Reactions were then snap frozen on dry ice and separated on 10% denaturing polyacrylamide gels, dried, and imaged using a PhosphorImager (GE Healthcare). For streptavidin end-blocking of biotinylated oligonucleotides, reactions also contained 15 mg/ml streptavidin (New England Biolabs).
DNA isolation, gel separation and hybridization
For one-dimensional gels, 3 μg of total genomic DNA was restricted according to manufacturer’s instructions (New England Biolabs), separated on 0.6% agarose gels and blotted overnight onto nylon membrane (GE Magnaprobe). Membranes were hybridised with appropriate radiolabelled probes overnight at 65 °C in 7% SDS and 0.25 M sodium phosphate buffer (pH 7.6), then washed with 1 x SSC with 0.1% SDS three times for 20 min at 65 °C and imaged using a Phosphorimager (GE Healthcare). For two-dimensional gels, total DNA was isolated from cells by sequential phenol-chloroform extraction according to established protocols33. 10–20 μg of DNA was restricted according to manufacturer’s instructions (New England Biolabs) and separated on agarose gels according to protocols described in detail elsewhere33. Gels were then blotted and hybridised as above, followed by washing with 1 × SSC three times for 20 min, then 1 × SSC with 0.1% SDS three times for 20 min. Primer sequences used to produce the probes are given in Supplementary Table 5.
Antibodies
Western blotting was performed on cell lysates and cell fractions using primary antibodies to MGME1 (Sigma HPA040913, 1:500 dilution), β-actin (Sigma A2228, 1:100,000), TFAM (a kind gift from Prof. R. Wiesner, University of Cologne, 1:40,000), Histone H4 (Abcam ab10158, 1:5,000), GAPDH (Abcam ab9482, 1:1,000) and TOM20 (Santa Cruz sc-11415, 1:1,000). Secondary antibodies used were goat anti-rabbit HRP (Promega W401B, 1:2,000) and goat anti-mouse HRP (Promega W402B, 1:2,000).
Supplementary Material
SuppInfoTablesFigs
Acknowledgments
T.J.N. and M.M. are grateful to Stuart Wood and Ian Holt for stimulating discussions during the course of this work. We are grateful to Susanne Beyer, Karin Kappes-Horn, Manuela Stepien-Mering, Evelyn Botz and Rosi Hellinger for technical assistance.
This work was supported by the Medical Research Council (T.J.N., J.R., M.Minczuk), the German Bundesministerium für Bildung und Forschung (BMBF) through funding of the Systems Biology of Metabotypes grant (SysMBo #0315494A), the E-Rare project GENOMIT (#01GM1207) and of the German Network for mitochondrial disorders (mitoNET) including C.K., T.K. (mitoNET 01GM0862), T.M., H.P. (mitoNET 01GM0867), and W.S.K. (mitoNET 01GM0868). W.S.K was funded by the Deutsche Forschungsgemeinschaft (SFB TR3 A11 and D12). V.K.M. was supported by grants from the National Institutes of Health (GM077465 and GM097136).
Footnotes
Data access
The individuals did not provide consent for making exome data available to public databases or database with managed access. We will submit the disease causing variants to ClinVar. We have consent to share the data with cooperation partners. Within a research cooperation, variant data are available on request.
AUTHOR CONTRIBUTIONS
C.K. identified, clinically characterized, collected samples, histochemically analyzed skeletal muscle biopsies of Family I and the sporadic case, and obtained fibroblasts of P1976; M.S., D.R., G.P.C., M.Moggio, C.M.Q., S.D. identified, clinically characterized, collected samples, histochemically analyzed skeletal muscle biopsies of Family II and obtained fibroblasts of P4050 and P4052; T.B.H., T.W., T.M.S., T.M., H.P. performed exome sequencing and analysis of Family I; S.E.C., V.K.M. performed targeted mitochondrial exome sequencing and analysis of Family II; T.J.N., G.Z., M.Minczuk performed the computational analysis; T.J.N. analyzed protein levels, performed subcellular localization, purified and characterized recombinant MGME1, analyzed the siRNA-knockdown cells and P1976 fibroblasts; T.B.H., K.D., A.I., H.P. performed subcellular localization and complementation experiments; S. S. performed the mtDNA repopulation experiments; V.P. performed the copy number and deletion quantification; K.H. screened PEO samples for MGME1 mutations and identified P931; J.R. contributed to the characterization of P1976 fibroblasts; T.K., T.M. provided samples and coordinate the German network of mitochondrial disorders; C.K., G.Z., M.Minczuk, W.S.K. and H.P. planned the project and wrote the manuscript.
References
- 1.Van Goethem G, et al. Mutation of POLG is associated with progressive external ophthalmoplegia characterized by mtDNA deletions. Nat Genet. 2001;28:211–212. doi: 10.1038/90034. [DOI] [PubMed] [Google Scholar]
- 2.Longley MJ, et al. Mutant POLG2 disrupts DNA polymerase gamma subunits and causes progressive external ophthalmoplegia. Am J Hum Genet. 2006;78:1026–1034. doi: 10.1086/504303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Spelbrink JN, et al. Human mitochondrial DNA deletions associated with mutations in the gene encoding Twinkle, a phage T7 gene 4-like protein localized in mitochondria. Nat Genet. 2001;28:223–231. doi: 10.1038/90058. [DOI] [PubMed] [Google Scholar]
- 4.Nishino I, Spinazzola A, Hirano M. Thymidine phosphorylase gene mutations in MNGIE, a human mitochondrial disorder. Science. 1999;283:689–692. doi: 10.1126/science.283.5402.689. [DOI] [PubMed] [Google Scholar]
- 5.Kaukonen J, et al. Role of adenine nucleotide translocator 1 in mtDNA maintenance. Science. 2000;289:782–785. doi: 10.1126/science.289.5480.782. [DOI] [PubMed] [Google Scholar]
- 6.Mandel H, et al. The deoxyguanosine kinase gene is mutated in individuals with depleted hepatocerebral mitochondrial DNA. Nat Genet. 2001;29:337–341. doi: 10.1038/ng746. [DOI] [PubMed] [Google Scholar]
- 7.Saada A, et al. Mutant mitochondrial thymidine kinase in mitochondrial DNA depletion myopathy. Nat Genet. 2001;29:342–344. doi: 10.1038/ng751. [DOI] [PubMed] [Google Scholar]
- 8.Elpeleg O, et al. Deficiency of the ADP-forming succinyl-CoA synthase activity is associated with encephalomyopathy and mitochondrial DNA depletion. Am J Hum Genet. 2005;76:1081–1086. doi: 10.1086/430843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Spinazzola A, et al. MPV17 encodes an inner mitochondrial membrane protein and is mutated in infantile hepatic mitochondrial DNA depletion. Nat Genet. 2006;38:570–575. doi: 10.1038/ng1765. [DOI] [PubMed] [Google Scholar]
- 10.Bourdon A, et al. Mutation of RRM2B, encoding p53-controlled ribonucleotide reductase (p53R2), causes severe mitochondrial DNA depletion. Nat Genet. 2007;39:776–780. doi: 10.1038/ng2040. [DOI] [PubMed] [Google Scholar]
- 11.Ostergaard E, et al. Deficiency of the alpha subunit of succinate-coenzyme A ligase causes fatal infantile lactic acidosis with mitochondrial DNA depletion. Am J Hum Genet. 2007;81:383–387. doi: 10.1086/519222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Haack TB, et al. Molecular diagnosis in mitochondrial complex I deficiency using exome sequencing. J Med Genet. 2012;49:277–283. doi: 10.1136/jmedgenet-2012-100846. [DOI] [PubMed] [Google Scholar]
- 13.Elstner M, et al. MitoP2: an integrative tool for the analysis of the mitochondrial proteome. Mol Biotechnol. 2008;40:306–315. doi: 10.1007/s12033-008-9100-5. [DOI] [PubMed] [Google Scholar]
- 14.Calvo SE, et al. Molecular diagnosis of infantile mitochondrial disease with targeted next-generation sequencing. Sci Transl Med. 2012;4:118ra10. doi: 10.1126/scitranslmed.3003310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pagliarini DJ, et al. A mitochondrial protein compendium elucidates complex I disease biology. Cell. 2008;134:112–123. doi: 10.1016/j.cell.2008.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Steczkiewicz K, et al. Sequence, structure and functional diversity of PD-(D/E)XK phosphodiesterase superfamily. Nucleic Acids Res. 2012;40:7016–45. doi: 10.1093/nar/gks382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Aravind L, et al. Holliday junction resolvases and related nucleases: identification of new families, phyletic distribution and evolutionary trajectories. Nucleic Acids Res. 2000;28:3417–32. doi: 10.1093/nar/28.18.3417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Singleton MR, et al. Crystal structure of RecBCD enzyme reveals a machine for processing DNA breaks. Nature. 2004;432:187–93. doi: 10.1038/nature02988. [DOI] [PubMed] [Google Scholar]
- 19.Holt IJ, et al. Coupled leading- and lagging-strand synthesis of mammalian mitochondrial DNA. Cell. 2000;100:515–524. doi: 10.1016/s0092-8674(00)80688-1. [DOI] [PubMed] [Google Scholar]
- 20.Liu P, et al. Removal of oxidative DNA damage via FEN1-dependent long-patch base excision repair in human cell mitochondria. Mol Cell Biol. 2008;28:4975–4987. doi: 10.1128/MCB.00457-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brown TA, Clayton DA. Release of replication termination controls mitochondrial DNA copy number after depletion with 2′,3′-dideoxycytidine. Nucleic Acids Res. 2002;30:2004–2010. doi: 10.1093/nar/30.9.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stewart JD, et al. POLG mutations cause decreased mitochondrial DNA repopulation rates following induced depletion in human fibroblasts. Biochim Biophys Acta. 2011;1812:321–325. doi: 10.1016/j.bbadis.2010.11.012. [DOI] [PubMed] [Google Scholar]
- 23.Wanrooij S, et al. Expression of catalytic mutants of the mtDNA helicase Twinkle and polymerase POLG causes distinct replication stalling phenotypes. Nucleic Acids Res. 2007;35:3238–3251. doi: 10.1093/nar/gkm215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Copeland WC, Longley MJ. DNA2 resolves expanding flap in mitochondrial base excision repair. Mol Cell. 2008;32:457–458. doi: 10.1016/j.molcel.2008.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zheng L, et al. Human DNA2 is a mitochondrial nuclease/helicase for efficient processing of DNA replication and repair intermediates. Mol Cell. 2008;32:325–336. doi: 10.1016/j.molcel.2008.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Duxin JP, et al. Human Dna2 is a nuclear and mitochondrial DNA maintenance protein. Mol Cell Biol. 2009;29:4274–4282. doi: 10.1128/MCB.01834-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tann AW, et al. Apoptosis induced by persistent single-strand breaks in mitochondrial genome: critical role of EXOG (5′-EXO/endonuclease) in their repair. J Biol Chem. 2011;286:31975–31983. doi: 10.1074/jbc.M110.215715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pohjoismäki JL, et al. Mammalian mitochondrial DNA replication intermediates are essentially duplex but contain extensive tracts of RNA/DNA hybrid. J Mol Biol. 2010;397:1144–1155. doi: 10.1016/j.jmb.2010.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McKenna A, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zsurka G, et al. Clonally expanded mitochondrial DNA mutations in epileptic individuals with mutated DNA polymerase gamma. J Neuropathol Exp Neurol. 2008;67:857–866. doi: 10.1097/NEN.0b013e3181839b2d. [DOI] [PubMed] [Google Scholar]
- 31.Danhauser K, et al. Cellular rescue-assay aids verification of causative DNA-variants in mitochondrial complex I deficiency. Mol Genet Metab. 2011;103:161–166. doi: 10.1016/j.ymgme.2011.03.004. [DOI] [PubMed] [Google Scholar]
- 32.Rorbach J, et al. PDE12 removes mitochondrial RNA poly(A) tails and controls translation in human mitochondria. Nucleic Acids Res. 2011;39:7750–7763. doi: 10.1093/nar/gkr470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Reyes A, et al. Analysis of replicating mitochondrial DNA by two-dimensional agarose gel electrophoresis. Methods Mol Biol. 2007;372:219–232. doi: 10.1007/978-1-59745-365-3_16. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
SuppInfoTablesFigs