Bleach Gel: A Simple Agarose Gel for Analyzing RNA Quality (original) (raw)
. Author manuscript; available in PMC: 2013 Jul 2.
Published in final edited form as: Electrophoresis. 2012 Jan;33(2):366–369. doi: 10.1002/elps.201100335
Abstract
RNA-based applications requiring high quality, non-degraded RNA are a foundational element of many research studies. As such, it is paramount that the integrity of experimental RNA is validated prior to cDNA synthesis or other downstream applications. In the absence of expensive equipment such as microfluidic electrophoretic devices, and as an alternative to the costly and time-consuming standard formaldehyde gel, RNA quality can be quickly analyzed by adding small amounts of commercial bleach to TAE buffer-based agarose gels prior to electrophoresis. In the presence of low concentrations of bleach, the secondary structure of RNA is denatured and potential contaminating RNases are destroyed. Because of this, the ‘bleach gel’ is a functional approach that addresses the need for an inexpensive and safe way to evaluate RNA integrity and will improve the ability of researchers to rapidly analyze RNA quality.
Keywords: Agarose, Bleach, Denaturing gel, Electrophoresis, RNA quality
One of the major issues affecting the integrity of RNA is the ubiquitous presence of ribonucleases (RNases). These enzymes are present throughout the phylogenetic trees of both prokaryotes [1] and eukaryotes [2-4] and even appear in some viruses [5,6]. Because these enzymes are found in microorganisms, many of which are dispersed throughout the air, they can easily contaminate laboratory samples. In addition, RNases are secreted from the skin [7] and are found in various fluids produced by the human body including tears, saliva, mucus, and sweat. RNases rapidly degrade RNA, some at a rate of 39.2 nmol/min per mg [8]. RNases are also resilient and resist a vast number of chemical insults. For example, RNase A remains active in conditions such as pH ranges between 2 and 10 [9,10], solutions of up to 8 M urea [11], extreme temperatures (15-80°C) [10,11], and boiling for 30 minutes [12]. If a research sample containing RNA becomes compromised by RNases, subsequent analyses utilizing that sample may produce unreliable results. Therefore, laboratory precautions must always be taken in preparing samples to inhibit degradation of RNA.
Once RNA samples have been prepared, denaturing gel electrophoresis is frequently used to visually assess the quality of RNA. The denaturing gel is a time-intensive procedure requiring toxic reagents. Denaturing gels for RNA analysis usually contain formaldehyde [13], formamide [13], or urea [14,15], but other compounds have also been employed including glyoxal/DMSO [16], mercuric hydroxide [17], guanidine thiocyanate [18], and SDS [19]. A TAE-based gel for RNA has been previously been described, although it requires sample preparation with hot formamide [20]. All of these reagents disrupt the secondary structure of RNA, allowing for proper analysis of the sample during gel electrophoresis. However, most of the procedures require multiple washing steps, the use of special running buffers that increase the length of the procedure, and special precautions due to the use of toxic reagents.
In the past several years, microfluidics-based technologies, such as the 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA), ScreenTape® R6K (Lab901, now a part of Agilent Technologies, Santa Clara, CA), and Experion (Bio-Rad Laboratories, Hercules, CA) have become commercially available for quantitating and analyzing RNA concentration and integrity. An attractive feature of these electrophoresis systems is their reliability. For instance, the 2100 Bioanalyzer and ScreenTape® R6K platforms parallel each other in their capacity to evaluate RNA samples [21]. Indeed, these instruments are versatile tools, despite their dependency on RNA integrity in order to perform accurate RNA quantification [22]. Ultimately, the primary drawback of these advanced technologies is their cost, and as a consequence, their accessibility to many researchers.
For quick analysis of RNA integrity, our lab has often replaced formaldehyde gel electrophoresis with the use of standard TAE-based agarose gels normally used in analysis of DNA. It is unclear whether the degraded RNA in these gels was due to poor quality RNA preparation or because RNases were present on the apparatus and degraded the sample while the gel was running. We therefore addressed this concern by incorporating common household bleach (6% sodium hypochlorite) into a TAE and agarose mixture prior to melting the agarose. As previous studies have shown that protein denaturation and damage by oxidation occurs after exposure to hypochlorite [23], we postulate that the 6% sodium hypochlorite present in commercial bleach denatures the secondary structure of RNA by destroying hydrogen bonds [24] and destroys any RNases present in the gel [23].
It is the aim of this paper to introduce an alternative approach for analyzing RNA quality by gel electrophoresis. By incorporating commercial chlorine bleach (6% sodium hypochlorite; Clorox®, Oakland, CA) into a standard TAE agarose gel, we show that we can quickly visualize the condition of an RNA sample. Figure 1 illustrates the ability of increasing concentrations of bleach to protect RNA from degradation by RNase A. Each 1% agarose gel solution was initially spiked with 20 ng/ml of RNase A in order to demonstrate the effect of bleach to inhibit the enzyme. Although it is unlikely that this degree of contamination would occur naturally, the use of this large amount of RNase A demonstrates the strength of bleach towards inactivating RNases. Increasing amounts (0% to 5% v/v) of commercial bleach were added to the agarose mixtures [for each 50 ml agarose gel, 0 to 2.5 ml of undiluted bleach (6% sodium hypochlorite) was added], and the solutions were incubated at room temperature for 5-10 minutes [though the addition of bleach with no incubation was equally efficacious (data not shown)]. The agarose gels were then heated to melt the agarose and cooled. Three μl of 10 mg/ml ethidium bromide were added to each gel, and the gels were poured into molds and allowed to solidify.
Figure 1. Protective effect of bleach on RNA quality in RNase-containing gels.
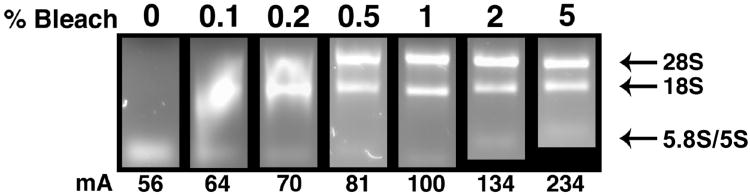
Standard 1% TAE agarose gels were made containing 20 ng/ml of RNase A along with various concentrations of household bleach (0% to 5.0% v/v Clorox®). Each gel was loaded with 10 μL 1× DNA Loading Buffer containing 1 μg of total RNA isolated from 4T1.2 mouse mammary carcinoma cells and run for ∼35 minutes at a constant 100 V. The presence of 28S, 18S, and 5.8S/5S ribosomal RNA (rRNA) bands are absent in the gel containing 0% bleach. Increasing rRNA band integrity occurs with bleach concentrations of 0.1 to 0.5% (a 0.5% bleach has 250 μl bleach/50 ml gel). The 28S, 18S, and 5.8S/5S rRNA bands are intact in gels with bleach concentrations of 1.0 to 5.0% (indicated by arrows). An increase in bleach concentration also results in a linear increase in amperage. These results suggest that a 1% TAE agarose ‘bleach gel’ is best run at a concentration of 0.5% to 1% v/v bleach.
The gels were placed in mini-gel electrophoresis apparatuses and submerged completely with 1× TAE buffer. Each gel was loaded with samples consisting of 1× DNA Loading buffer, prepared from a 10× DNA Loading buffer stock (1.9 mM xylene cyanol, 1.5 mM bromophenol blue, 25% glycerol in sterile dH20) and 1 μg of total RNA isolated from 4T1.2 mouse mammary carcinoma cells using STAT-60 (Tel-Test Inc., Friendswood, TX). The gels were run for ∼35 minutes with constant voltage (100 V) prior to imaging under UV transillumination. In the presence of 20 mg/ml RNase A and the absence of bleach, the entire RNA sample was degraded (Fig. 1). With the addition of bleach at concentrations of 0.5% v/v (250 μl of household bleach per 50 ml gel) and above, degradation of RNA by RNase A did not occur (Fig.1).
When assessing the quality of RNA by gel electrophoresis, the presence of three distinct bands suggests high quality RNA. For eukaryotic RNA, the top band represents 28S ribosomal RNA (rRNA), which runs at ∼4.8 kb; the middle band represents 18S rRNA at ∼2.0 kb; and the third band represents 5.8S (154 nt) and 5S (117 nt) RNA. Transfer RNAs (73-93 nt) may or may not be visible [25-28]. Smeared bands or a 28S:18S band intensity ratio below 2:1 indicate poor RNA quality. It is clear from Figure 1 that we obtained high quality RNA, based on this criterion, and were able to analyze the RNA using the ‘bleach gel’. In addition, bands of a 1 kb DNA ladder (New England BioLabs, Inc., Ipswich, MA) were intact (data not shown), suggesting that DNA integrity is maintained in the ‘bleach gel’.
With increasing bleach concentrations, the electrical resistance through the gel was decreased, resulting in a linear increase in milliamps (mA). At very high concentrations of bleach, increased amperage was needed to reach the desired voltage (Fig. 1). Although only Clorox® bleach was used in our ‘bleach gels’, bleach as old as three years was tested and performed as well as Clorox® bleach less than a year old (data not shown). Whether other brands of bleach may be effective remains unknown. Moreover, if the bleach were added after the agarose was melted, the bleach could interfere with the setting of the gel and have deleterious effects on the ethidium bromide used to stain the RNA (data not shown). By adding the bleach prior to melting the agarose suspension, the aforementioned conflicts were circumvented. As an alternative to TAE-based agarose gels, TBE buffer-based gels are commonly used in DNA electrophoresis. In a ‘bleach gel’ made with TBE, RNase inactivation and RNA denaturation were comparable to that of a TAE-based ‘bleach gel’ (data not shown). Finally, a cost comparison was performed and the outcome demonstrated that a 50 ml ‘bleach gel’ costs approximately 7-fold less than a standard RNA analysis gel containing formaldehyde (Table 1). The simplicity of preparing the ‘bleach gel’ parallels that of an agarose gel for analyzing DNA. For these reasons, our short protocol (Table 2) is an economical and convenient option for analyzing RNA integrity.
Table 1.
Cost comparison between the ‘bleach gel’ and a standard formaldehyde gel.
| Gel (50 mL)a) | 1% ‘Bleach Gel’ | 1% Standard RNA Gel |
|---|---|---|
| Agarose, 0.5 g | 0.573∣0.573 | 0.573∣0.573 |
| 10× TAE, 5 mL | $0.181 | - |
| Bleach, 500 μL | $0.00026254 | - |
| EtBr (10 mg/mL), 2 μL | $0.00848 | - |
| 10× MESA, 5 mL | - | $0.7725 |
| 37% Formaldehyde, 8 mL | - | $6.688 |
| Loading Dye (10 μL per sample)b) | ||
| EtBr (10 mg/mL), 0.1 μL | - | $0.000424 |
| 10× DNA Loading Buffer, 2 μL | 0.00608∣0.00608 | 0.00608∣0.00608 |
| Formamide, 5 μL | - | $0.00195 |
| 37% Formaldehyde, 1.75 μL | - | $0.001463 |
| Running Buffer (300 mL) | ||
| 10× TAE, 30 mL | $1.08 | - |
| 10× MESA, 30 mL | - | $4.635 |
| Total Cost per Gelc) | $1.84882254 | $12.678417 |
Table 2.
‘Bleach gel’ protocol.
| Step | ‘Bleach Gel’ Preparation |
|---|---|
| 1 | Add 1.0% w/v agarosea) to 1× TAE bufferb), e.g. 0.5 g in 50 mL. |
| 2 | Add 1.0% v/v Clorox® bleachc), e.g. 500 μL in 50 mL, and incubate at room temperature for 5 minutes, with occasional swirling. |
| 3 | Heat the suspension to melt the agarose. |
| 4 | Allow solution to cool before adding ethidium bromided) to a final concentration of about 0.5 μg/mL. |
| 5 | Pour solution into gel mold and allow the ‘bleach gel’ to solidify. |
| Load and Run RNA Samples | |
| 6 | Load RNA sample mixed with 10× DNA loading buffere) to a final concentration of 1×. Also, load DNA ladder. |
| 7 | Electrophorese gel in 1× TAE buffer at 100 V for approximately 35 minutes, before visualizing by UV transillumination. |
While using readily available and cost-effective reagents, this new procedure minimizes preparation time, is simple to execute, and reduces the amount of toxic agents used. In conclusion, we believe the simple ‘bleach gel’ is an effective, safe, affordable, and rapid way to evaluate RNA quality.
Acknowledgments
We are grateful to NIH (#P20 RR016454 and #R15 CA106274), the American Cancer Society (#RSG-09-276-01-CSM), the Komen for the Cure (#KG100513), NASA (#NNX10AN29A), and Mountain States Tumor and Medical Research Institute (MSTMRI) for funding this work.
Abbreviations
TAE
Tris-acetate-EDTA
TBE
Tris-borate-EDTA
DEPC
diethylpyrocarbonate
MESA
MOPS-EDTA sodium acetate
EtBr
ethidium bromide
Footnotes
The authors declare no competing interests.
References
- 1.Kazantsev AV, Pace NR. Nat Rev Microbiol. 2006;4:729–740. doi: 10.1038/nrmicro1491. [DOI] [PubMed] [Google Scholar]
- 2.Jin L, Kryukov K, Suzuki Y, Imanishi T, Ikeo K, Gojobori T. Gene. 2009;435:1–8. doi: 10.1016/j.gene.2008.12.022. [DOI] [PubMed] [Google Scholar]
- 3.Vieira J, Fonseca NA, Vieira CP. J Mol Evol. 2009;69:32–41. doi: 10.1007/s00239-009-9249-y. [DOI] [PubMed] [Google Scholar]
- 4.Barber GN. RNA Biol. 2009;6:35–39. doi: 10.4161/rna.6.1.7565. [DOI] [PubMed] [Google Scholar]
- 5.Taddeo B, Sciortino MT, Zhang W, Roizman B. Proc Natl Acad Sci U S A. 2007;104:12163–12168. doi: 10.1073/pnas.0705245104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lim D, Gregorio GG, Bingman C, Martinez-Hackert E, Hendrickson WA, Goff SP. J Virol. 2006;80:8379–8389. doi: 10.1128/JVI.00750-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Harder J, Schroder JM. J Biol Chem. 2002;277:46779–46784. doi: 10.1074/jbc.M207587200. [DOI] [PubMed] [Google Scholar]
- 8.Crooke ST, Lemonidis KM, Neilson L, Griffey R, Lesnik EA, Monia BP. Biochem J. 1995;312(Pt 2):599–608. doi: 10.1042/bj3120599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bartholeyns J, Baudhuin P. Biochem J. 1977;164:675–683. doi: 10.1042/bj1640675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dalaly BK, Eitenmiller RR, Friend BA, Shahani KM. Biochim Biophys Acta. 1980;615:381–391. doi: 10.1016/0005-2744(80)90505-7. [DOI] [PubMed] [Google Scholar]
- 11.Kalnitsky G, Resnick H. J Biol Chem. 1959;234:1714–1717. [PubMed] [Google Scholar]
- 12.Kunita M. J Gen Physiol. 1940;24:15–32. doi: 10.1085/jgp.24.1.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rave N, Crkvenjakov R, Boedtker H. Nucleic Acids Res. 1979;6:3559–3567. doi: 10.1093/nar/6.11.3559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Donis-Keller H, Maxam AM, Gilbert W. Nucleic Acids Res. 1977;4:2527–2538. doi: 10.1093/nar/4.8.2527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stoeckle MY, Guan L. BioTechniques. 1993;15(227):230–221. [PubMed] [Google Scholar]
- 16.McMaster GK, Carmichael GG. Proc Natl Acad Sci U S A. 1977;74:4835–4838. doi: 10.1073/pnas.74.11.4835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Alwine JC, Kemp DJ, Stark GR. Proc Natl Acad Sci U S A. 1977;74:5350–5354. doi: 10.1073/pnas.74.12.5350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Goda SK, Minton NP. Nucleic Acids Res. 1995;23:3357–3358. doi: 10.1093/nar/23.16.3357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Virca GD, Northemann W, Shiels BR, Widera G, Broome S. Biotechniques. 1990;8:370–371. [PubMed] [Google Scholar]
- 20.Masek, Tomas, Vopalensky Vaclav, Suchomelova Petra, Pospisek Martin. Anal Biochem. 2005;336:46–50. doi: 10.1016/j.ab.2004.09.010. [DOI] [PubMed] [Google Scholar]
- 21.Wilkes TM, Devonshire AS, Ellison SLR, Foy CA. BMC Res Notes. 2010;3:1–7. doi: 10.1186/1756-0500-3-89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Becker C, Hammerle-Fickinger A, Riedmaier I, Pfaffl MW. Methods. 2010;50:237–243. doi: 10.1016/j.ymeth.2010.01.010. [DOI] [PubMed] [Google Scholar]
- 23.Hawkins CL, Davies MJ. Biochem J. 1998;332(Pt 3):617–625. doi: 10.1042/bj3320617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Guo-Dong Kang, JG C, Wei-Dong Chen, Xing-Ming Jie, Yi-Ming Cao, Quan Yuana. J Memb Sci. 2007;300:165–171. [Google Scholar]
- 25.Imbeaud S, Graudens E, Boulanger V, Barlet X, Zaborski P, Eveno E, Mueller O, Schroeder A, Auffray C. Nucleic Acids Res. 2005;33:e56. doi: 10.1093/nar/gni054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schroeder A, Mueller O, Stocker S, Salowsky R, Leiber M, Gassmann M, Lightfoot S, Menzel W, Granzow M, Ragg T. BMC Mol Biol. 2006;7:3. doi: 10.1186/1471-2199-7-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sambrook J, Karlen A, E F, Maniatis T. Molecular Cloning, a laboratory manual. Cold Spring Harbor Laboratory Press; New York: 1989. [Google Scholar]
- 28.Davoren PA, McNeill RE, Lowery AJ, Kerin MJ, Miller N. BMC Mol Biol. 2008;9:76. doi: 10.1186/1471-2199-9-76. [DOI] [PMC free article] [PubMed] [Google Scholar]