DNA topology, not DNA sequence, is a critical determinant for Drosophila ORC–DNA binding (original) (raw)
Abstract
Drosophila origin recognition complex (ORC) localizes to defined positions on chromosomes, and in follicle cells the chorion gene amplification loci are well-studied examples. However, the mechanism of specific localization is not known. We have studied the DNA binding of DmORC to investigate the _cis_-requirements for DmORC:DNA interaction. DmORC displays at best six-fold differences in the relative affinities to DNA from the third chorion locus and to random fragments in vitro, and chemical probing and DNase1 protection experiments did not identify a discrete binding site for DmORC on any of these fragments. The intrinsic DNA-binding specificity of DmORC is therefore insufficient to target DmORC to origins of replication in vivo. However, the topological state of the DNA significantly influences the affinity of DmORC to DNA. We found that the affinity of DmORC for negatively supercoiled DNA is about 30-fold higher than for either relaxed or linear DNA. These data provide biochemical evidence for the notion that origin specification in metazoa likely involves mechanisms other than simple replicator–initiator interactions and that in vivo other proteins must determine ORC's localization.
Keywords: DNA binding, Drosophila ORC, replication, topology
Introduction
In a eukaryotic cell committed to duplication, chromosomal DNA replication initiates at many sites called origins of DNA replication. The process that determines origin selection is understood in some depth for fungal genomes, but in multicellular organisms there is still considerable mystery as to how origin specification is achieved (DePamphilis, 1999; Gilbert, 2001). In Saccharomyces cerevisiae, origins contain multiple functional modules within ∼150 bp of DNA, including the highly conserved and essential ARS consensus sequence (ACS), which serves as the binding site for the hexameric origin recognition complex (ORC) (Bell and Stillman, 1992). Following binding to the origin, ORC nucleates the assembly of all other replication proteins, demonstrating that specific DNA binding by ORC is the first essential step in the initiation process (Bell, 2002).
In contrast to budding yeast, replication origins in Schizosaccharomyces pombe are generally longer (500–1500 bp), do not contain conserved consensus sequences analogous to the ACS, and are characterized by a high degree of redundancy. S. pombe ORC (SpORC) binds to AT-rich origin sequences via an AT-hook DNA-binding domain on subunit SpORC4p, a unique feature of SpORC, demonstrating that various modes of origin recognition by ORC have evolved in eukaryotes (Chuang and Kelly, 1999; Moon et al, 1999; Kong and DePamphilis, 2001; Lee et al, 2001).
Physical mapping techniques have identified specific origin regions at a number of loci in various metazoans, and in some instances large deletions affect the function in transgenic constructs. The problem, however, is further complicated because origin selection is developmentally regulated and seems to change, as proscribed by a well-defined tissue and temporally regulated program. In the early embryonic stages of Drosophila and Xenopus, origin site selection appears to be promiscuous with respect to DNA sequence (although not necessarily random; Blow et al, 2001; Hyrien et al, 2003), indicating that specific ‘replicator' sequences are dispensable (Harland and Laskey, 1980; Mechali and Kearsey, 1984; Spradling and Orr-Weaver, 1987; Smith and Calos, 1995). Specific origin usage occurs later in development, showing that some mechanism(s) for selection of initiation zones or sites must exist (Hyrien et al, 1995; Sasaki et al, 1999).
The discovery that ORC exists in metazoans and is essential for DNA replication provides an obvious approach for defining origins by studying how ORC associates with DNA and by uncovering the rules for how ORC finds the appropriate _cis_-acting sites in vivo. One of the most tractable systems for studying origin choice and ORC localization in metazoans is provided by the DNA amplicons in the follicle cells surrounding the developing oocyte in Drosophila melanogaster (Calvi and Spradling, 1999). In these somatic cells, the chorion genes on the third and X chromosome undergo site-specific DNA amplification to allow for a rapid increase in the number of templates for later transcription of the egg shell genes. The _cis_-elements controlling third chromosome chorion gene amplification have been determined and include ACE-3, a 300 base pair fragment that is sufficient to localize DmORC in follicle cells, and ori-β, the primary origin located within this amplicon that also localizes DmORC (Austin et al, 1999).
Even though ORC is bound specifically at chromosomal regions containing origins of replication in both differentiated insect and human somatic cells, little is known about how the ORC complex finds these origins (Austin et al, 1999; Beall et al, 2002; Keller et al, 2002). ORC localization and origin selection may involve an elaborate pathway with many regulators intervening both upstream and downstream of ORC–chromatin association (Walter and Newport, 1997; Okuno et al, 2001; DePamphilis, 2003). In any case, a quantitative analysis of DmORC's preference for DNA sequences has not been reported, nor has a specific DNA-binding sequence for a metazoan ORC been revealed.
We have demonstrated that recombinant DmORC (rDmORC) can substitute for embryonic ORC in a cell-free replication system (Chesnokov et al, 1999). Furthermore, we showed that rDmORC binds both DNA and chromatin in an ATP-dependent manner in vitro. Similar to ScORC, ATP-binding, but not –ATP-hydrolysis, is required for ATP-dependent DNA binding, and DNA binding slows the kinetics of ATP hydrolysis (Chesnokov et al, 2001). In this study, we examine the DNA-binding properties of DmORC by a variety of methods. We have monitored DNA binding by quantitative gel-shift assays employing competitor DNAs to yield relative binding constants for DmORC for DNA fragments derived from the third chorion locus and from heterologous sources. While small preferences for some DNAs were indeed found, our conclusion is that DmORC cannot solely rely upon its intrinsic DNA-binding specificity to guide it to replication origins. Consistent with this finding, attempts to define a specific DNA sequence to which DmORC binds were unsuccessful. Together with other results these data led us to speculate that the binding differences we detected may reflect differences in the particular DNA structure that exists in the preferred test fragments.
ORC subunits 1, 4, and 5 are members of the AAA+ family and are thus structurally related to the prokaryote initiator protein DnaA, suggesting that common mechanisms might exist for both prokaryotic and eukaryotic DNA replication initiation (Davey et al, 2002; Erzberger et al, 2002). Specific binding of DnaA is dependent upon negatively ‘(−)' supercoiled DNA (Fuller and Kornberg, 1983; Bramhill and Kornberg, 1988). A similar requirement for origin function in bacteriophage λ also suggests that (−) supercoiled DNA may be broadly important for origin function (Schnos et al, 1988). We tested this parameter for the DmORC–DNA complex and found that (−) supercoiling dramatically increases DmORC affinity but not specificity to DNA. The implication of these studies to the general usefulness of the replicon hypothesis in its simple form is discussed.
Results
DmORC binds to DNA fragments of variable sequence composition
Our initial goal was to determine a high-affinity DmORC-binding site in ACE-3 and ori-β using DNase 1 protection. We were unable to detect a discrete binding site with embryonic, tissue culture, or rDmORC. Instead, we found that with increasing amounts of DmORC the entire fragment was protected from DNase 1 digestion (supplementary Figure 1). One interpretation of these data is that DmORC might have affinity for many sites within the test fragment, all of which have similar binding constants. Among these sites, one or a few might be preferred, but to achieve a significant DNase 1 protection for such a putative site close to 100% occupancy would be required, and at that point protection of the other sites would also be observed. In such situations, gel-shift assays are more sensitive, and we thus focused our efforts on this protocol.
Detection of an ATP-dependent DmORC–DNA complex using EMSA requires excess carrier DNA. However, by varying the amount of carrier DNA, multiple additional ATP-dependent DmORC–DNA complexes of decreased mobility were detected (Figure 1A, lanes 2–4). At 30 μg/ml competitor, only one complex was detected (lane 5). Conversely, when the concentration of rDmORC was increased versus a constant concentration of carrier DNA, only one complex was visible at lower DmORC concentrations (Figure 1B, lanes 4 and 5), while a series of complexes was detected at higher DmORC concentrations (Figure 1B, lanes 6 and 7). These data are consistent with the suggestion that the test fragment contains multiple sites for DmORC occupancy.
Figure 1.
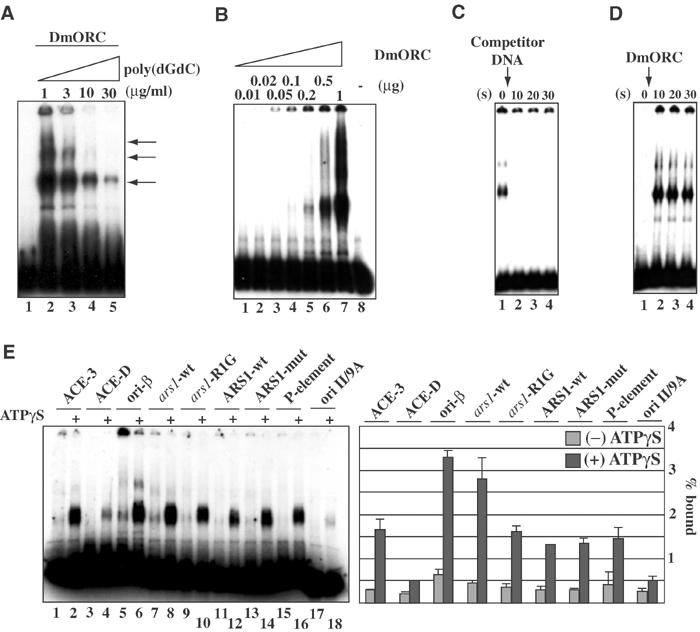
rDmORC binds to various DNA sequences. (A) EMSA showing the ATP-dependent binding of rDmORC to a radiolabeled ACE-3 fragment at increasing poly(dGdC)·poly(dGdC) competitor concentration as indicated. Lane 1, no protein. (B) EMSA showing the ATP-dependent binding of increasing amounts of rDmORC to ACE-3 at 50 μg/ml poly(dGdC)·poly(dGdC) competitor. (C) rDmORC–ACE-3 complex at equilibrium (lane 1) was chased with excess cold competitor DNA. Aliquots were analyzed by EMSA in intervals as indicated (lanes 2–4). *(s)=seconds (D) rDmORC binding to a radiolabeled ACE-3 fragment was monitored over time as indicated (lanes 2–4). Lane 1, no DmORC. (E) EMSA of rDmORC binding to various radiolabeled DNA fragments with or without 0.5 mM ATPγS as indicated. The bar diagram depicts binding efficiencies as a fraction of the bound probe. The averages and standard deviations for three independent experiments are shown.
To further define the rDmORC–DNA interaction, we examined the kinetics of binding to and dissociation from DNA. To measure an off-rate, rDmORC–ACE-3 complexes were allowed to equilibrate before adding excess sheared salmon sperm DNA just prior to EMSA. Surprisingly, an off-rate was too rapid to be measured by this technique, as no complexes were detected after as short as a 10-s incubation with competitor (Figure 1C). This is in contrast to ScORC, which has an extremely low and unmeasured off-rate (Mizushima et al, 2000; SP Bell, personal communication). The on-rate was also too fast to be determined by EMSA, with maximum binding reached within 10 s (Figure 1D). These studies establish that rDmORC–DNA binding is dynamic, despite a nanomolar affinity constant for the ATP-dependent rDmORC–DNA complex.
We employed a variety of chemical modification interference and missing contact assays to discern specific nucleotides required for the observed gel-shift complexes. End-labeled DNA fragments were treated with formic acid, hydrazine, _N_-ethyl-nitroso-urea (ENU), or diethyl-pyrocarbonate (DEPC), and subsequently subjected to EMSA using rDmORC. The hydrolysis patterns were identical for both shifted and nonshifted DNAs, indicating that DmORC–DNA complex formation does not require any single specific or critical base pair contacts (supplementary Figure 2). In addition, DMS footprinting did not reveal regions within ACE-3 or ori-β that were protected by DmORC (not shown). These findings are consistent with the hypothesis that DmORC binds to a large number of sites within the test fragments and that differences in affinities between the most preferred to the weakest site are small.
As we were unable to detect a specific binding site for DmORC on ACE-3 or ori-β, we examined the binding of DmORC to unrelated ∼300 base pair DNA fragments to try to find commonalities for sequence recognition. DmORC is a phosphoprotein in vivo and dephosphorylation increases the avidity of DmORC for DNA in vitro, while hyperphosphorylation disrupts ATP-dependent DNA binding by DmORC (D Remus and M Botchan, unpublished). Strikingly, either λ-protein phosphatase-treated (Figure 1E) or untreated (not shown) rDmORC bound to all fragments with similar affinity. The fragment _ars1_-R1G contains a linker substitution in an essential 30 bp A/T-rich region of S. pombe ars1 (Clyne and Kelly, 1995). Only an ∼2-fold reduction in affinity was found between both S. pombe fragments, with an overall affinity similar to that of ACE-3 (Figure 1E, lanes 8 and 10). The S. cerevisiae ARS1 element has critical recognition elements for ScORC, and replacement of the A/T-rich ACS by a G/C-rich sequence (ARS/858–865) destroys ScORC binding and replication activity (Marahrens and Stillman, 1992). DmORC bound both S. cerevisiae fragments with equivalent affinities (Figure 1E, lanes 12 and 14), demonstrating that DmORC does not share DNA sequence specificity with ScORC. We were surprised to find that DmORC bound to a P-element end-containing fragment as well as to ACE-3 (Figure 1E, lane 16). As summarized by the bar graph in Figure 1E, DmORC displays at most a seven-fold reproducible difference in affinity between fragments (e.g. compare ACE-D and ori-β), demonstrating that DmORC has at best a weak intrinsic sequence discrimination.
We used EMSA to test quantitatively the relative affinity of DmORC to various regions derived from the third chromosome chorion gene cluster. The gene cluster was divided into 11 ∼300–350 bp regions (see the illustrations in Figures 2B and C) to provide both specific and nonspecific DNA substrates. DmORC binding to a radiolabeled ACE-3 fragment was assayed in the presence of increasing amounts of each respective competitor DNA. As a measure of relative binding affinity, the competitor concentration at which DmORC binding to the ACE-3 probe was reduced by 50% ([C]1/2) was determined. We first compared the affinities of rDmORC and embryonic DmORC to ACE-3 and ACE-D (Figure 2A). Both rDmORC and embryonic DmORC exhibited a five- to six-fold preference for ACE-3 over ACE-D, showing that rDmORC does not differ from embryonic DmORC in its DNA-binding specificity. We then tested the relative affinities of rDmORC for all fragments in two to three independent competition experiments (Figure 2B). The results are summarized in Figure 2C, in which [C]1/2 was normalized to ACE-3. The relative affinities of rDmORC for the various fragments varied from one- to six-fold, with the weakest relative affinities occurring for fragments ACE-D, s18-3, and s15-2, and an ∼2.5-fold reduced affinity for fragments s18-1, s18-2, and s15-1. Binding to ori-β, ori-D, ACE-U2, and ACE-U1 was essentially indistinguishable from ACE-3. Strikingly, rDmORC bound with only an ∼2-fold reduced efficiency to a random vector sequence. These data demonstrate that the intrinsic sequence specificity of DmORC is very low. DNA binding to ‘nonspecific' sequences is only 1–6-fold reduced compared to ‘specific' sequences. Clearly, the important genetic replicator determinants do not contain DNA sequences to which DmORC binds with the specificity that might have been anticipated by simple models.
Figure 2.
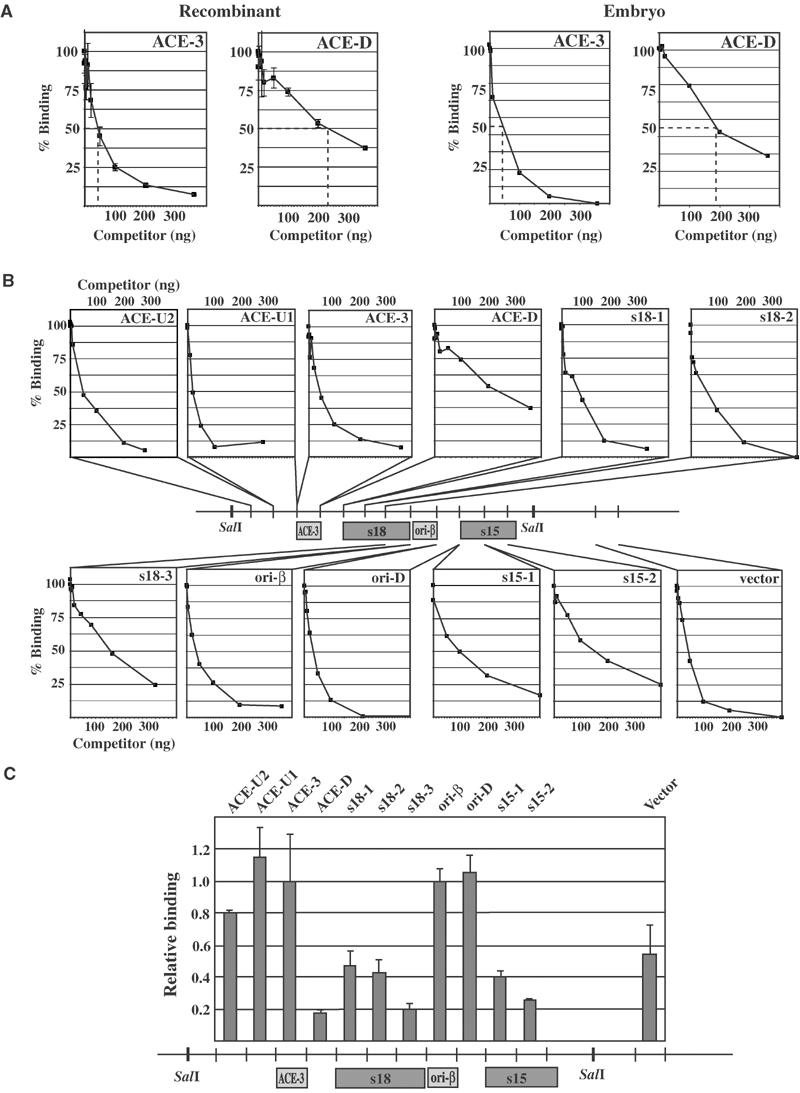
rDmORC binds with similar affinity to fragments of the third chromosome chorion gene cluster. ATP-dependent binding of rDmORC to a radiolabeled ACE-3 fragment was assayed by EMSA in the presence of competitor fragment DNA indicated in the upper right corner of each plot. % binding is relative to the fraction of ACE-3 bound in the absence of competitor. (A) Relative binding efficiencies of rDmORC (left two panels) and embryonic (0–12 h) DmORC to ACE-3 in the presence of increasing amounts of ACE-3 and ACE-D competitor. Dashed lines indicate [C]1/2. (B) Binding plots of all fragments tested. The relative position of each fragment in the chorion gene cluster is schematized in the center. (C) Summary plot depicting [C]1/2 for each fragment normalized to ACE-3. The average and standard deviation for two to three independent experiments are shown.
We have previously shown that the ATPase activity of DmORC is inhibited by ACE-3- or ori-β-containing DNA (Chesnokov et al, 2001). We therefore asked whether this inhibition was dependent on the DNA sequence. We found that all DNA fragments tested similarly inhibit DmORC's ATPase activity, regardless of the fragment concentration (Figure 3A). The maximum inhibition for all fragments tested was ∼2-fold. Various DNAs, including single- and double-stranded M13, single-stranded ΦX-174, and pBluescript plasmid, also result in an ∼2-fold inhibition of the ATPase activity of rDmORC (data not shown). Interestingly, maximum inhibition of rDmORC's ATPase activity was reached at a molar rDmORC:DNA ratio of 2–4. In addition, no significant difference in inhibition was detected in time-course experiments between ACE-3 and ACE-D, when either fragment was used at a saturating ∼3-fold molar excess (Figure 3B). Thus, ATPase inhibition does not appear to require a conformational change in rDmORC that is dependent on a specific DNA sequence.
Figure 3.
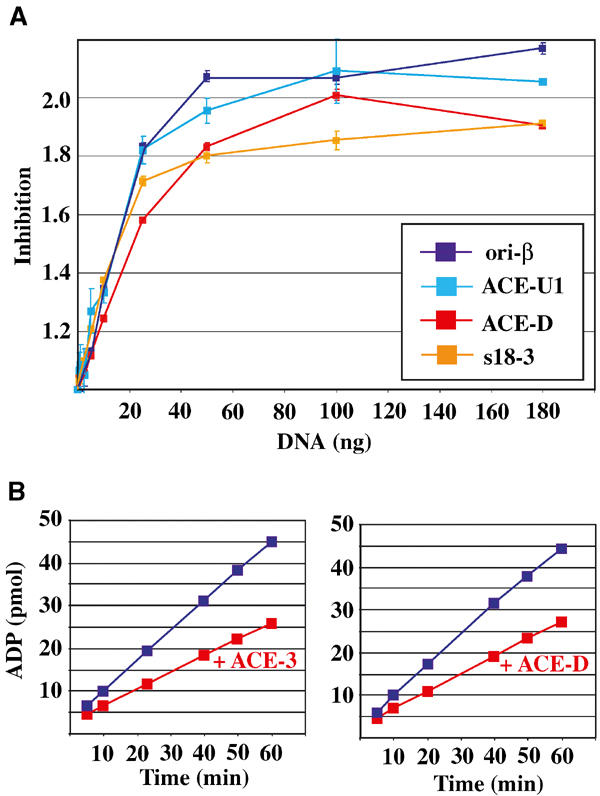
Inhibition of rDmORC's ATPase activity by DNA does not require a specific DNA sequence. (A) ATP hydrolysis by DmORC in the presence of increasing amounts of ori-β (dark blue line), ACE-U1 (light blue), ACE-D (red), and s18-3 (orange). The factor of inhibition is plotted as a function of fragment concentration. Averages and standard deviations for two independent experiments are shown. (B) Time course of the amount of ADP produced by rDmORC in the absence (blue lines) or presence (red lines) of ACE-3- (left panel) or ACE-D- (right panel) containing DNA.
DmORC binds preferentially to negatively supercoiled DNA
Given the absolute requirements for supercoiling and specific DNA binding for the prokaryote initiators, it was important to ask whether the topology of a DNA template affects rDmORC–DNA binding. A filter-binding assay was developed to monitor rDmORC binding to a 3H-thymidine-labeled plasmid DNA. We characterized the binding of rDmORC to pBluescript (pBS) or derivatives that contained either the ori-β or the ACE-3 fragment. As shown in Figure 4A, retention of pBS/ori-β on the nitrocellulose filter was dependent upon rDmORC, and titration of rDmORC at a constant plasmid DNA concentration revealed a simple binding curve without any indication of cooperativity. However, binding of rDmORC to plasmid DNA was stimulated about three-fold in the presence of ATP (Figure 4B). The concentration of ATP required for maximal binding is 10–20 μM, similar to the ATP-dependent rDmORC binding to linear DNA fragments using EMSA (Chesnokov et al, 2001). However, a significant fraction of rDmORC–DNA complexes occurs independently of ATP, consistent with earlier reports for DmORC binding to linear fragments (Austin et al, 1999; Chesnokov et al, 2001).
Figure 4.
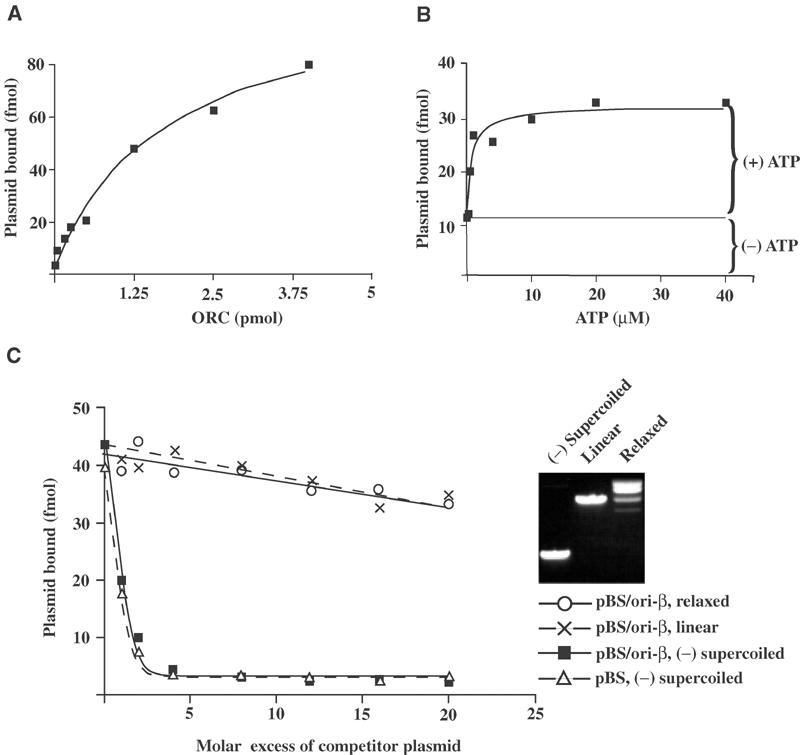
DmORC binds preferentially to (−) supercoiled DNA. rDmORC binding to 125 fmol 3H-labeled pBS/ori-β was assayed by nitrocellulose filter binding. (A) Titration of rDmORC with 0.5 mM ATP. (B) Titration of ATP with 1 pmol rDmORC. (C) rDmORC binding to 3H-labeled (−) supercoiled pBS/ori-β in the presence of increasing amounts of unlabeled (−) supercoiled pBS/ori-β (▪), pBS/ori-β relaxed with _Topo_I (○), pBS/ori-β linearized with _Bam_HI (×), or (−) supercoiled pBluescript (▵). Inset panel: Ethidium-bromide-stained agarose gel showing (−) supercoiled, linearized, and relaxed pBS/ori-β.
Next, the relative affinity of rDmORC for linear, relaxed, and (−) supercoiled plasmid DNA was assayed using the filter-binding assay. We performed competition experiments, monitoring the binding of rDmORC to 3H-thymidine-labeled (−) supercoiled plasmid DNA in the presence of increasing amounts of unlabeled plasmid DNA of various topologies. (−) supercoiled plasmid DNA competed efficiently for rDmORC binding; however, this binding was not dependent on the presence of ori-β (or ACE-3, data not shown) since pBluescript competed with similar efficiency (Figure 4C). Strikingly, linear and relaxed plasmid DNAs were about 26- and 28-fold, respectively, less efficient for competition as determined by linear regression of the data in Figure 4C. A 25–30-fold preferred binding of rDmORC to (−) supercoiled DNA versus linear or relaxed DNA implies that DNA topology has a significantly greater impact upon DmORC–DNA complex formation than the DNA sequence (∼6-fold).
Competition experiments using the filter-binding assay require fairly large amounts of template DNA and do not readily allow the quantitation of DmORC bound per DNA molecule. A glycerol gradient-based assay system provides a quantitative way to analyze the binding of rDmORC to plasmid DNAs of variable topology. Differential labeling of supercoiled and linear plasmid DNA with 3H and 32P, respectively, allows for quantitative and distinct monitoring of DmORC binding to DNA of variable topology in one reaction using liquid-scintillation counting.
Figure 5A illustrates the mobility of linear, nicked circular, and (−) supercoiled DNA in a glycerol gradient. Plasmid topologies were independently confirmed by probing the fractions by Southern blotting. With these conditions, rDmORC alone sediments in a peak slightly above the position where plasmid DNAs would be found (Figure 5B). A significant fraction (∼30%) precipitated due to the low-salt (100 mM KCl) DNA-binding conditions. When rDmORC (2.5-fold molar excess) was assayed in a binding reaction that contained both 32P-labeled linear plasmid DNA and 3H-labeled (−) supercoiled plasmid at equimolar concentrations, a significant fraction of the (−) supercoiled plasmid was bound by rDmORC, while the linear plasmid remained essentially unbound (Figure 5C). Binding to (−) supercoiled DNA was characterized by a significant decrease of free (−) supercoiled DNA and by a concomitant shift in (−) supercoiled DNA to positions of higher mobility between 2.4 and 3.8 ml. The 2.4–3.8 ml fractions also contained all of the soluble rDmORC. In all, 58% or 274 fmol of the (−) supercoiled DNA co-fractionated with rDmORC and 13% or 61 fmol of (−) supercoiled plasmid pelleted at the bottom of the gradient. An estimated 40–50% or 1–1.2 pmol of rDmORC co-fractionated with (−) supercoiled DNA, corresponding to approximately 3–4 rDmORC molecules per molecule of (−) supercoiled plasmid in the soluble fractions. The heterogeneous mobility of rDmORC-bound supercoiled plasmid may thus be explained by variable stoichiometries of rDmORC–plasmid complexes.
Figure 5.
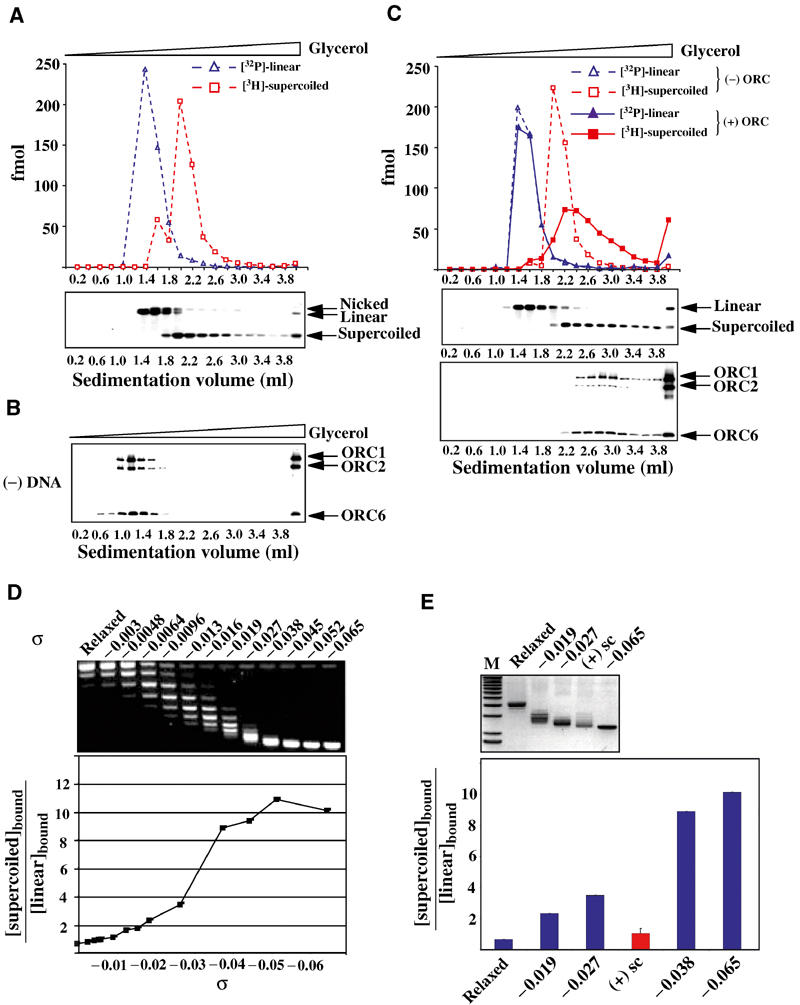
Superhelical density determines the preference for DmORC–DNA binding. Binding reactions containing equimolar 32P-labeled linear and 3H-labeled (−) supercoiled DNA were fractionated by glycerol gradient sedimentation and assayed for each radiolabel by liquid scintillation counting, analyzed by Southern blotting for pBS/ori-β, and by Western blotting for rDmORC subunits 1, 2, and 6. (A) Upper panel: sedimentation profile of linear (blue dashed line) and (−) supercoiled (red dashed line) DNA in the absence of rDmORC. Lower panel: Southern blot of the same fractions. (B) Western blot: sedimentation of rDmORC without DNA. (C) Sedimentation profile of linear (blue) and (−) supercoiled (red) DNA in the absence (dashed lines) and presence (solid lines) of rDmORC. Middle panel: Southern blot analysis of binding reaction; lower panel: Western blot, DmORC sedimentation in binding reaction. (D) rDmORC binding to (−) supercoiled DNA of variable σ. An ethidium-bromide-stained gel of the tested DNAs is shown on top of a graph depicting the ratios of bound supercoiled DNA to bound linear DNA as a function of σ of input supercoiled DNA. (E) Relative binding efficiency of (+) supercoiled DNA (red column) compared to (−) supercoiled DNA of bracketing superhelical densities. An ethidium-bromide-stained gel of relaxed, (−) supercoiled, and (+) supercoiled DNA is shown on top. M=1 kb ladder.
Next, we examined whether the preference for (−) supercoiled DNA required a specific degree of superhelical density. To this end, an array of variably (−) supercoiled 3H-labeled plasmid DNAs was generated and each plasmid was assayed for its relative binding efficiency to rDmORC compared to 32P-labeled linear plasmid DNA using the glycerol gradient-based assay described above. The results are summarized in Figure 5D, in which the ratio of bound supercoiled DNA to bound linear DNA was plotted over σ. Relaxed DNA bound slightly less efficiently (0.8-fold) to rDmORC than linear plasmid DNA. Equal binding was observed at σ values of up to −0.01. The sigmoidal curve observed for DmORC:DNA binding likely reflects a cooperative change in template DNA structure associated with increased affinity by DmORC to the constrained DNA. This transition for increased DNA binding occurs around a σ value of −0.03 to −0.04. The binding preference increased to a factor of about 10, which did not significantly increase further up to a σ value of −0.065. The marked preference for supercoiled DNA has a positive temperature dependence (data not shown), and the effects are thus greater in the prior filter-binding assays executed at 25°C than for the sedimentation assays at 4°C. In summary, these data demonstrate that the degree of superhelicity determines the relative affinity of rDmORC for a DNA template, and that a discrete change in superhelicity promotes maximum binding. ScORC binds single-stranded DNA with higher affinity than duplex DNA in the absence of ATP (Lee et al, 2000). Although we did not observe a preference for single-stranded DNA in the presence or absence of ATP for the DmORC, DNA structure in general is likely to influence ORC's affinity to DNA.
Superhelical DNA is characterized by crossovers of the duplex (writhe) and by a slightly underwound state for negative supercoils (twist). To discriminate between these two features, which could either or both be responsible for the preferential binding of rDmORC, we generated positively ‘(+)' supercoiled plasmid DNA by relaxing (−) supercoiled plasmid with _Topo_I in the presence of recombinant archaeal histone HMfB. We obtained (+) supercoiled plasmids with writhe comparable to (−) supercoiled plasmids between σ −0.027 and −0.038 (Figure 5E and data not shown). When examined for its relative binding efficiency to rDmORC, no preference for (+) supercoiled plasmid over linearized plasmid by rDmORC was observed (Figure 5E). In contrast, (−) supercoiled DNAs of σ −0.027 and −0.038 were preferentially bound by factors of 3.5 and 9, respectively. Even (−) supercoiled DNA of σ −0.019, which is significantly reduced in superhelical density compared to the (+) supercoiled template, exhibited a 2.3-fold preference over linearized plasmid, demonstrating that (−) supercoiled DNA is preferentially bound by rDmORC compared to (+) supercoiled DNA.
Simple DNA duplex crossovers thus appeared not to be responsible for the increased binding efficiency of rDmORC. Preference for DmORC binding to (−) supercoiled DNA may be caused by two nonmutually exclusive mechanisms. (−) supercoiled DNA can promote wrapping of the DNA around a protein core, if a particular chirality to the wrap is important. Alternatively, the underwound state of (−) supercoiled DNA energetically favors strand separation, and such an unwound final state may be optimal for the DmORC–DNA interaction. Thus, rDmORC might locally unwind the DNA, as has been reported for many other initiator proteins, including DnaA. In either case, why did we not observe a progression of weaker affinities comparing negative to relaxed to positive supercoiled DNA? As DmORC binds to positive and relaxed DNA with about equal affinity, we suggest that the complexes formed with these templates are not equivalent to those formed upon negative supercoils. For example, multiple protein subunits may contact (−) supercoiled DNA, leading to a final state not mimicked by these other templates where only a weakly bound DNA:protein complex is achieved.
Both wrapping and unwinding can be detected by relaxation with _Topo_I. We asked whether addition of rDmORC to plasmid DNA led to a linking number change in the plasmid relative to plasmid unbound by DmORC. Increasing amounts of rDmORC were incubated with a constant amount of plasmid DNA, _Topo_I was added to remove any negative supercoils from the plasmids, and purified DNA topoisomers were resolved by agarose gel electrophoresis in the presence of chloroquine and visualized by Southern blotting (Figure 6). Starting at molar rDmORC:plasmid ratios of 1.5–2 (0.15–0.2 μg ORC), (−) supercoiled topoisomers that were absent when rDmORC was excluded from the reaction could be detected. At higher rDmORC:plasmid ratios, a wide distribution of negative topoisomers was visible. This pattern was reproducible over a range of _Topo_I concentrations and therefore is not due to a partial activity of the topoisomerase enzyme. The lower panel depicts a merged scanning profile of lane 2 (− ORC, red) and lane 8 (+ ORC, blue). Subtracting the unbound pattern intensities (lane 2) from the bound pattern (lane 8) yields a difference map as shown in the inset, and from these data the supercoiled species peak with a linking number change (Δ_L_K) of −3 to −4 (topoisomers E–F). From the glycerol gradient-binding experiment, we estimate that under these conditions approximately 10–20% of the supercoils are bound by DmORC, a number consistent with the smear of topoisomers resistant to topoisomerase activity (∼14%). Thus, from our estimates of the number of DmORC molecules bound to supercoiled DNA from the experiment shown in Figure 5 (3–4 rDmORC/supercoiled DNA molecule), we estimate that each complex introduces a Δ_L_K equivalent to −1. In order to test whether the plasmids were unwound in the presence of DmORC, we probed the DmORC-bound plasmid DNAs for single-stranded regions using P1 nuclease. In a second approach, dimethyl sulfate (DMS), which modifies unwound DNA and thereby prevents re-annealing at such sites, was added to rDmORC–plasmid-binding reactions and the DNA was subsequently probed for single-stranded regions using S1 nuclease. In neither case were we able to detect rDmORC-dependent unwinding of the (−) supercoiled plasmid DNA (data not shown).
Figure 6.
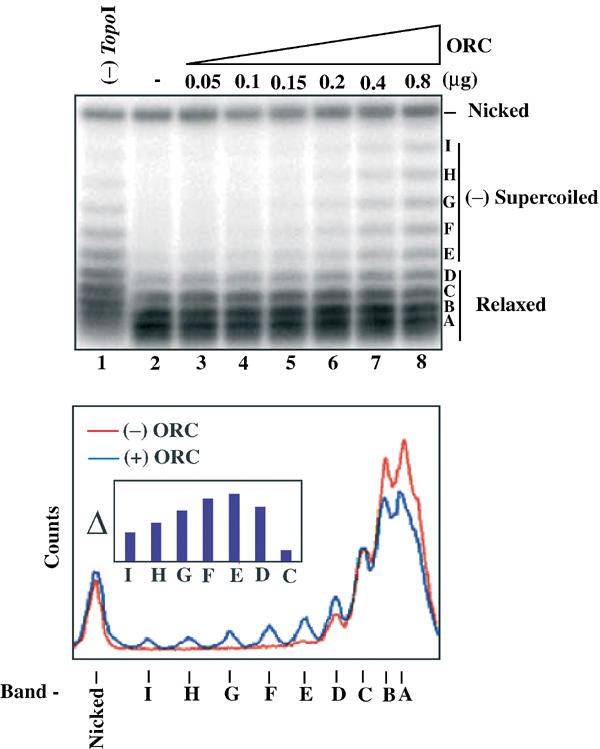
rDmORC binding to (−) supercoiled DNA induces a topology change in the DNA. A measure of 250 fmol of (−) supercoiled pBS/ori-β was incubated with increasing amounts of rDmORC in the presence of _Topo_I, the DNA repurified and analyzed by Southern blotting (upper panel). Lane 1, no _Topo_I; Lane 2, no rDmORC. Lower panel: lane scan of lanes −ORC (red) and +0.8 μg ORC (blue). Inset: Difference plot of topoisomer band intensities from Southern blot lanes 8 and 2. The letters denote topoisomers.
Discussion
At the start of this work, our anticipation was that DmORC would bind with some specificity to sequences within ACE-3 and ori-β. The expectation arose because ACE-3 is necessary and sufficient for DmORC's localization in follicle cells and many DmORC activities are similar to those of ScORC. We focused upon the ATP-dependent DNA–protein interactions that resist competitor DNA, in the hope that additional DNA contacts made by the DmORC–ATP complex would enhance the specificity. Attempts to detect a specific footprint by DNase 1 protection were not successful, nor did we uncover specific bases whose modification or removal interfered with the ATP-dependent gel shift. These negative results alone cannot rule out the possibility that under some binding conditions, DmORC will display sequence specificity. However, other experiments reported here are most consistent with the notion that DmORC does not recognize specific sequence motifs.
We measured the relative binding affinities for a wide variety of DNA fragments with different base compositions and found that DmORC did not bind ACE-3 more avidly than a pBluescript fragment or a fragment from the Drosophila P-element transposon. DmORC bound to DNA fragments spanning the third chromosome amplification locus with only 2–6-fold differences in binding affinity, which included ACE-3, ori-β, and flanking sequences. Our results are consistent with those of Austin et al, who did not quantitatively address the issue of DmORC–DNA-binding specificity. It is instructive to compare DmORC's range of sequence preferences to those of other DNA-binding proteins. For the lac repressor, a protein with exquisite specific to nonspecific discrimination, this ratio is ∼106-fold, while at the other end for such specificity the Drosophila homeodomain proteins show 102–103-fold preferences for recognition sites over random DNA (Riggs et al, 1972; Affolter et al, 1990). Thus, DmORC seems more a general DNA-binding factor with specificities superimposed by DNA structure, just as histone octamers have preferences for certain DNAs. While our manuscript was in preparation, a report (Vashee et al, 2003) appeared which came to a somewhat similar conclusion with regard to human ORC binding to DNA. Our data showing a very consequential effect of (−) supercoiling on DmORC affinity for DNA suggest that some feature of the DNA structure may underlie preferential binding to some DNA fragments over others. These results led us to speculate that DmORC is intrinsically promiscuous for DNA binding, which is reflected by the relative sequence independence for origin usage as it occurs in the early Drosophila embryo. The important question as to how DmORC finds its way specifically to the ACE-3 locus in follicle cells remains unresolved. Some of the biochemical results reported here provide new ideas for speculation.
The topological structure of the DNA significantly affects DmORC association with DNA. At physiologically relevant conditions, (−) supercoiled DNA is a ∼30-fold more avid target for DmORC–DNA binding than linear or relaxed DNA. This structural sensitivity provides an order of magnitude to DmORC's affinity to DNA over the sequence information alone. Transient local alterations in chromosomal DNA topology may result from the activity of chromatin remodeling complexes following nucleosome displacement. A local loss of a nucleosome will at least transiently produce a region of (−) supercoiled DNA. Creation of supercoiled domains within chromatin might provide a regulatory role for remodeling complexes in DmORC localization and origin regulation. Such a mechanism requires DmORC binding to supercoiled DNA in fierce competition with cellular topoisomerases, which are known to resolve globally plectonemic DNA supercoils within minutes in yeast (Saavedra and Huberman, 1986; Pederson and Morse, 1990). The rapid on and off-rates of DmORC on DNA as observed here, or the possible association of DmORC with chromatin remodeling factors, might circumvent the counteractive effect of topoisomerases in vivo. Preferential binding requires a certain degree of negative supercoiling, and DmORC binds (+) supercoiled DNA less efficiently than (−) supercoiled DNA, indicating that DmORC does not simply recognize DNA crossovers in the supercoiled template. (−) supercoiled DNA generally promotes left-handed wrapping of the DNA around a protein core, and the underwound state of (−) supercoiled DNA promotes DNA unwinding. Both mechanisms have roles in the function of the DnaA initiator of Escherichia coli. In this context, we also note that the chromosome around ARS1 is under torsional stress when bound by ScORC in vivo (Diffley and Cocker, 1992).
Initiator proteins from all the well-characterized prokaryotic and eukaryotic systems have multiple critical roles in the DNA replication process, including DNA site targeting, local DNA duplex remodeling and melting, replication protein assembly at origins, and replication initiation rate control by a variety of regulatory pathways. At our present level of understanding, it is too early to summarize firmly what might be conserved across all kingdoms with regard to all roles for the respective DnaA-, and ORC-related proteins. Very likely, ORC participates in early DNA remodeling, perhaps in complex with Cdc6 and other proteins. Further studies on the DmORC:supercoiled DNA complex should clarify the physical basis for ORC's preference for binding such DNA. It is striking that the structures of the core AAA+ domains of DnaA and ORC subunits 1, 4, and 5 are remarkably similar, and that melting of DNA by DnaA is promoted by ATP binding, multimerization, and supercoiling. We did not find an increase in nuclease sensitivity on DNAs bound by DmORC. If regions of DNA melting were protected by DmORC or limited to only a few base pairs, such melting might have escaped detection by the methods that we used. We emphasize that the inability to focus upon a particular DNA sequence in these studies eliminates our ability to use more sensitive methods to detect local DNA conformational changes.
DnaA targets origin sequences by a domain resembling the tryptophan repressor DNA-binding domain (Erzberger et al, 2002), whereas the ORC:DNA recognition appears to be highly divergent even within the few species analyzed to date. In the context of the replicon hypothesis, ORC is an initiator. However, the puzzle still remains as to how replicator DNA sequences as proposed by Jacob et al (1963) should be viewed in metazoans. The original replicon model, based in large part on the replication behaviors of different episomes and the host chromosome in E. coli, posits that the initiator activates replication through site-specific binding to a DNA sequence. The following points raise important issues with regard to the usefulness of the replicon model in metazoans. DmORC itself likely relies at least sometimes upon other proteins for origin targeting, and some of these targeting proteins may work indirectly or directly. At other times, specific replicator sequences may simply not exist. Targeting is expected to follow a complex pathway involving repressors, chromatin structure, co-activators, and epigenetic signals. Origin selection will then likely be exemplified by many special cases, as has been observed for numerous promoter:gene activation mechanisms. In such a model, ORC, Cdc6, and other initiation factors might serve general functions in origin activation, rather than play a determining role in origin specification. The question then is: How many DNA sequence elements will actually function as truly dedicated ‘replicators', distinct from organizers of chromosomal domains controlling general DNA metabolic functions including transcription?
Materials and methods
Electrophoretic mobility shift assay
rDmORC was purified as described (Chesnokov et al, 2001). Reactions were carried out in 15 μl buffer A/100 mM KCl/5 mM MgCl2/0.12 mg/ml BSA/20 μg/ml poly(dGdC)·poly(dGdC) as described (Chesnokov et al, 2001), with modifications described in the figure legends. For off-rate determination, the reaction volume was increased to 150 μl; ATP was 0.5 mM. The reaction was allowed to equilibrate for 30 min at 25°C. At 30 min, 30 μg (3 μl) of sheared salmon sperm DNA (Sigma) was added to the reaction and 10 μl aliquots of the reaction were applied onto a running gel in time intervals as indicated. For on-rate determination, the reaction volume was increased to 150 μl; ATP was 0.5 mM. Following DmORC addition, 10 μl aliquots were loaded on a running gel at intervals as indicated. DNA fragments corresponding to Drosophila ACE-3, ori-β and ACE-D, S. cerevisiae ARS1-wt and ARS1-mut, S. pombe ars1-wt and ars1-R1G, the P-element ends, and S. coprophila oriII/9A were PCR-amplified from plasmids pT2, pARS1/WT and pARS1/858–865, pRC20 and pRCR1G, pPI25.1, and an oriII/9A PCR fragment, and subcloned into pBLUESCRIPT II KS(+), respectively. Binding was quantitated on a PhosphoImager (Fuji). For competitive gel-shift experiments, DNA fragments were PCR-amplified from pT2. Third chorion fragment positions and corresponding primer pairs are provided as supplementary materials.
ATPase assays
Reactions were carried out at 25°C in 25 μl of buffer A/100 mM KCl/5 mM MgCl2/0.12 mg/ml BSA containing 50 μM ATP (incl. 1 μCi α[32P]ATP) and 80 nM purified DmORC. For the DNA titration experiments, reactions were incubated for 60 min at 25°C and stopped by spotting 1 μl of each reaction on a PEI-cellulose TLC plate (Sigma). For the time-course experiments, the concentration of DNA was 160 nM and time points were taken by spotting 1 μl of the reaction on a PEI-cellulose TLC plate as indicated. The plates were developed in 0.6 M Na2HPO4/NaH2PO4, pH 3.5, and quantitated on a PhosphoImager (Fuji).
Plasmid binding
Metabolic labeling of plasmid DNA with [methyl-3H]-thymidine (ICN) was performed in E. coli DH5α as described (Julin et al, 1986). Plasmid DNAs of varying (−) superhelicity were generated as described (Keller, 1975). ΔLks were determined by the band-counting method (Keller, 1975) and σ values were calculated from σ=(Lk−Lk0)/Lk0=ΔLk/Lk0 (Lk0 for pBS/ori-β=313.43). (+) supercoiled plasmid DNA was prepared from 3H-labeled (−) supercoiled plasmid DNA as described (LaMarr et al, 1997). Filter-binding reactions were carried out in 50 μl of buffer A/100 mM KCl/5 mM MgCl2/50 μg/ml BSA containing 250 ng (2.4 nM) 3H-labeled DNA and DmORC, ATP, and cold competitor DNA, as indicated in the figure. Reactions were incubated at 25°C for 30 min, filtered at ∼0.3 ml/min through a 0.45 μm nitrocellulose membrane using a 96-well mini-vacuum manifold (both Schleicher and Schuell), and retained radiolabel quantitated by liquid-scintillation counting. For glycerol gradient plasmid-binding experiments, 25 nM DmORC was incubated for 30 min at 25°C in 100 μl buffer A/100 mM KCl/5 mM MgCl2/0.12 mg/ml BSA/500 μM ATP containing 5 nM 32P-end-labeled linear and 5 nM 3H-labeled circular-closed DNA. Reactions were fractionated by centrifugation at 4°C, 42 000 r.p.m., for 8 h on a 4 ml 15–35% glycerol gradient containing buffer A/100 mM KCl. Fractions (200 μl) were collected from the top and analyzed by liquid scintillation counting, Southern blot, and Western blot analysis.
Topology assay
Reactions were carried out in 20 μl of buffer A/75 mM KCl/25 mM NaCl/50 μg/ml BSA/2 mM MgCl2 incl. 25 ng/μl (8 nM) plasmid DNA, 500 μM ATP, and DmORC, as indicated in the figure. Reactions were incubated for 30 min at 25°C. Subsequently, 1 U _Topo_I was added to the reaction and incubated for another 30 min at 25°C. The reaction was stopped by adding 0.5% SDS/10 mM EDTA, and DNA was purified by phenol/chloroform extraction and EtOH precipitation. A measure of 50 ng DNA of each reaction was analyzed by agarose gel-electrophoresis in 1 μg/ml chloroquine, followed by Southern blotting using a 32P-labeled probe against pBluescript. Topoisomer band intensities were quantitated on a PhosphoImager (Pharmacia).
Supplementary Material
Supplementary Figure 1
Supplementary Figure 2
Supplementary Materials
Acknowledgments
Nick Cozzarelli and Nancy Crisona (University of California, Berkeley) gave helpful insights for experimental protocols and ideas. Kathleen Sandman (OSU, Columbus), Tom Kelly (MSKCC, New York), Don Rio (UC Berkeley), Bruce Stillman (CSHL, Cold Spring Harbor), Anja-Katrin Bielinsky (University of Minnesota, Minneapolis), and Alan Spradling (Carnegie Institution of Washington, Baltimore) provided plasmids. Peter Lewis purified the embryonic DmORC. This work was supported by the National Institutes of Health Grant CA-30490.
References
- Affolter M, Percival-Smith A, Muller M, Leupin W, Gehring WJ (1990) DNA binding properties of the purified Antennapedia homeodomain. Proc Natl Acad Sci USA 87: 4093–4097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Austin RJ, Orr-Weaver TL, Bell SP (1999) Drosophila ORC specifically binds to ACE3, an origin of DNA replication control element. Genes Dev 13: 2639–2649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beall EL, Manak JR, Zhou S, Bell M, Lipsick JS, Botchan MR (2002) Role for a Drosophila Myb-containing protein complex in site-specific DNA replication. Nature 420: 833–837 [DOI] [PubMed] [Google Scholar]
- Bell SP (2002) The origin recognition complex: from simple origins to complex functions. Genes Dev 16: 659–672 [DOI] [PubMed] [Google Scholar]
- Bell SP, Stillman B (1992) ATP-dependent recognition of eukaryotic origins of DNA replication by a multiprotein complex. Nature 357: 128–134 [DOI] [PubMed] [Google Scholar]
- Blow JJ, Gillespie PJ, Francis D, Jackson DA (2001) Replication origins in Xenopus egg extract Are 5-15 kilobases apart and are activated in clusters that fire at different times. J Cell Biol 152: 15–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bramhill D, Kornberg A (1988) Duplex opening by dnaA protein at novel sequences in initiation of replication at the origin of the E. coli chromosome. Cell 52: 743–755 [DOI] [PubMed] [Google Scholar]
- Calvi BR, Spradling AC (1999) Chorion gene amplification in Drosophila: a model for metazoan origins of DNA replication and S-phase control. Methods 18: 407–417 [DOI] [PubMed] [Google Scholar]
- Chesnokov I, Gossen M, Remus D, Botchan M (1999) Assembly of functionally active Drosophila origin recognition complex from recombinant proteins. Genes Dev 13: 1289–1296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chesnokov I, Remus D, Botchan M (2001) Functional analysis of mutant and wild-type Drosophila origin recognition complex. Proc Natl Acad Sci USA 98: 11997–12002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuang RY, Kelly TJ (1999) The fission yeast homologue of Orc4p binds to replication origin DNA via multiple AT-hooks. Proc Natl Acad Sci USA 96: 2656–2661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clyne RK, Kelly TJ (1995) Genetic analysis of an ARS element from the fission yeast Schizosaccharomyces pombe. EMBO J 14: 6348–6357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davey MJ, Jeruzalmi D, Kuriyan J, O'Donnell M (2002) Motors and switches: AAA+ machines within the replisome. Nat Rev Mol Cell Biol 3: 826–835 [DOI] [PubMed] [Google Scholar]
- DePamphilis ML (1999) Replication origins in metazoan chromosomes: fact or fiction? BioEssays 21: 5–16 [DOI] [PubMed] [Google Scholar]
- DePamphilis ML (2003) Eukaryotic DNA replication origins: reconciling disparate data. Cell 114: 274–275 [DOI] [PubMed] [Google Scholar]
- Diffley JF, Cocker JH (1992) Protein–DNA interactions at a yeast replication origin. Nature 357: 169–172 [DOI] [PubMed] [Google Scholar]
- Erzberger JP, Pirruccello MM, Berger JM (2002) The structure of bacterial DnaA: implications for general mechanisms underlying DNA replication initiation. EMBO J 21: 4763–4773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuller RS, Kornberg A (1983) Purified dnaA protein in initiation of replication at the Escherichia coli chromosomal origin of replication. Proc Natl Acad Sci USA 80: 5817–5821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert DM (2001) Making sense of eukaryotic DNA replication origins. Science 294: 96–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harland RM, Laskey RA (1980) Regulated replication of DNA microinjected into eggs of Xenopus laevis. Cell 21: 761–771 [DOI] [PubMed] [Google Scholar]
- Hyrien O, Marheineke K, Goldar A (2003) Paradoxes of eukaryotic DNA replication: MCM proteins and the random completion problem. BioEssays 25: 116–125 [DOI] [PubMed] [Google Scholar]
- Hyrien O, Maric C, Mechali M (1995) Transition in specification of embryonic metazoan DNA replication origins. Science 270: 994–997 [DOI] [PubMed] [Google Scholar]
- Jacob F, Brenner S, Cuzin F (1963) On the regulation of DNA replication in bacteria. Cold Spring Harb Symp Quant Biol 28: 329–348 [Google Scholar]
- Julin DA, Riddles PW, Lehman IR (1986) On the mechanism of pairing of single- and double-stranded DNA molecules by the recA and single-stranded DNA-binding proteins of Escherichia coli. J Biol Chem 261: 1025–1030 [PubMed] [Google Scholar]
- Keller C, Ladenburger EM, Kremer M, Knippers R (2002) The origin recognition complex marks a replication origin in the human TOP1 gene promoter. J Biol Chem 277: 31430–31440 [DOI] [PubMed] [Google Scholar]
- Keller W (1975) Determination of the number of superhelical turns in simian virus 40 DNA by gel electrophoresis. Proc Natl Acad Sci USA 72: 4876–4880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong D, DePamphilis ML (2001) Site-specific DNA binding of the Schizosaccharomyces pombe origin recognition complex is determined by the Orc4 subunit. Mol Cell Biol 21: 8095–8103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaMarr WA, Sandman KM, Reeve JN, Dedon PC (1997) Large scale preparation of positively supercoiled DNA using the archaeal histone HMf. Nucleic Acids Res 25: 1660–1661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee DG, Makhov AM, Klemm RD, Griffith JD, Bell SP (2000) Regulation of origin recognition complex conformation and ATPase activity: differential effects of single-stranded and double-stranded DNA binding. EMBO J 19: 4774–4782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JK, Moon KY, Jiang Y, Hurwitz J (2001) The Schizosaccharomyces pombe origin recognition complex interacts with multiple AT-rich regions of the replication origin DNA by means of the AT-hook domains of the spOrc4 protein. Proc Natl Acad Sci USA 98: 13589–13594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marahrens Y, Stillman B (1992) A yeast chromosomal origin of DNA replication defined by multiple functional elements. Science 255: 817–823 [DOI] [PubMed] [Google Scholar]
- Mechali M, Kearsey S (1984) Lack of specific sequence requirement for DNA replication in Xenopus eggs compared with high sequence specificity in yeast. Cell 38: 55–64 [DOI] [PubMed] [Google Scholar]
- Mizushima T, Takahashi N, Stillman B (2000) Cdc6p modulates the structure and DNA binding activity of the origin recognition complex in vitro. Genes Dev 14: 1631–1641 [PMC free article] [PubMed] [Google Scholar]
- Moon KY, Kong D, Lee JK, Raychaudhuri S, Hurwitz J (1999) Identification and reconstitution of the origin recognition complex from Schizosaccharomyces pombe. Proc Natl Acad Sci USA 96: 12367–12372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okuno Y, McNairn AJ, den Elzen N, Pines J, Gilbert DM (2001) Stability, chromatin association and functional activity of mammalian pre-replication complex proteins during the cell cycle. EMBO J 20: 4263–4277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pederson DS, Morse RH (1990) Effect of transcription of yeast chromatin on DNA topology in vivo. EMBO J 9: 1873–1881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riggs AD, Lin S, Wells RD (1972) Lac repressor binding to synthetic DNAs of defined nucleotide sequence. Proc Natl Acad Sci USA 69: 761–764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saavedra RA, Huberman JA (1986) Both DNA topoisomerases I and II relax 2 micron plasmid DNA in living yeast cells. Cell 45: 65–70 [DOI] [PubMed] [Google Scholar]
- Sasaki T, Sawado T, Yamaguchi M, Shinomiya T (1999) Specification of regions of DNA replication initiation during embryogenesis in the 65-kilobase DNApolalpha-dE2F locus of Drosophila melanogaster. Mol Cell Biol 19: 547–555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnos M, Zahn K, Inman RB, Blattner FR (1988) Initiation protein induced helix destabilization at the lambda origin: a prepriming step in DNA replication. Cell 52: 385–395 [DOI] [PubMed] [Google Scholar]
- Smith JG, Calos MP (1995) Autonomous replication in Drosophila melanogaster tissue culture cells. Chromosoma 103: 597–605 [DOI] [PubMed] [Google Scholar]
- Spradling A, Orr-Weaver T (1987) Regulation of DNA replication during Drosophila development. Annu Rev Genet 21: 373–403 [DOI] [PubMed] [Google Scholar]
- Vashee S, Cvetic C, Lu W, Simancek P, Kelly TJ, Walter JC (2003) Sequence-independent DNA binding and replication initiation by the human origin recognition complex. Genes Dev 17: 1894–1908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter J, Newport JW (1997) Regulation of replicon size in Xenopus egg extracts. Science 275: 993–995 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1
Supplementary Figure 2
Supplementary Materials