STAT-3: A Molecular Hub for Signaling Pathways in Gliomas (original) (raw)
. Author manuscript; available in PMC: 2014 Jan 9.
Published in final edited form as: Mol Cancer Res. 2008 May;6(5):10.1158/1541-7786.MCR-07-2180. doi: 10.1158/1541-7786.MCR-07-2180
Abstract
Glioblastoma (GBM) is the most common and severe primary brain tumor in adults. Its aggressive and infiltrative nature renders the current therapeutics of surgical resection, radiation, and chemotherapy relatively ineffective. Accordingly, recent research has focused on the elucidation of various signal transduction pathways in GBM, particularly aberrant activation. This review focuses on the STAT-3 signal transduction pathway in the context of this devastating tumor. STAT-3 is aberrantly activated in human GBM tissues, and this activation is implicated in controlling critical cellular events thought to be involved in gliomagenesis such as cell cycle progression, apoptosis, angiogenesis, and immune evasion. There are no reports of gain of function mutations in GBM; rather, the activation of STAT-3 is thought to be a consequence of either dysregulation of upstream kinases or loss of endogenous inhibitors. This review provides detailed insight into the multiple mechanisms of STAT-3 activation in GBM, as well as describing endogenous and chemical inhibitors of this pathway and their clinical significance. In GBM, STAT-3 acts a “molecular hub” to link extracellular signals to transcriptional control of proliferation, cell cycle progression, and immune evasion. Because STAT-3 plays this central role in GBM signal transduction, it has significant potential as a therapeutic target.
Keywords: STATs, Gliomas, Signal Transduction
Introduction
Malignant gliomas, the most common type of primary brain tumors, are highly aggressive, infiltrative and destructive. The most common and severe form of malignant glioma, the World Health Organization (WHO) grade IV astrocytic glioblastoma (GBM) (1, 2), affects approximately 13,000 people each year, with men more often affected than women. The infiltrative and aggressive nature of GBM renders current treatments such as surgical resection, radiation and chemotherapy relatively ineffective (1, 3). Median survival after treatment is 14 months, and despite advances in the basic understanding of cancer biology, this poor prognosis has not improved for several decades (1, 4). For this reason, recent studies have focused on understanding the molecular signaling pathways implicated in GBM progression.
GBM progression presents molecular biologists with a challenge because components of several different signaling pathways, such as Phosphoinositide-3 Kinase (PI3K), AKT, Ras, and Mitogen Activated Protein Kinases (MAPK), and receptor tyrosine kinases including the epidermal growth factor receptor (EGFR) and the vascular endothelial growth factor receptor (VEGFR), all appear to contribute strongly to the growth and promotion of GBM (5, 6). We now know, however, that these diverse signaling pathways converge at specific transcription factors, including Signal Transducers and Activator of Transcription-3 (STAT-3). STAT-3 gene targets affect proliferation, growth, and apoptosis, and aberrant activation of STAT-3 has been identified not only in GBM, but also in a number of other human cancers including breast, lung, ovarian, pancreatic, skin, and prostate cancers, and Hodgkins lymphoma, myeloma, and acute myeloid leukemia (7). This review describes how STAT-3 activation and resulting downstream effects play a role in GBM progression, and how therapeutic strategies to inhibit STAT-3 signaling provide new opportunities for GBM treatment.
STAT-3 and its Activation Pathways
The STAT family of cytoplasmic latent transcription factors consists of seven members: STAT-1-4, STAT-5a, STAT-5b, and STAT-6 (8). STAT-3 activation results in expression of genes that control cell proliferation, survival, differentiation and development. Like all STAT proteins, STAT-3 is activated by tyrosine phosphorylation in response to stimulation by cytokines and growth factors. Specifically, STAT-3 activation is downstream of receptor engagement by members of the Interleukin-6 (IL-6) cytokine family, including IL-6, Oncostatin M (OSM), and Leukemia Inhibitory Factor (LIF), and by growth factors such as PDGF, FGF and EGF (9). In addition to initial activation by tyrosine phosphorylation, phosphorylation of STAT-3 on serine residue 727 maximally activates its transcriptional activity (10).
STAT-3 is tyrosine phosphorylated by three types of kinases: receptor tyrosine kinases such as EGFR, FGFR or PDGFR, Janus Kinase (JAK) family members, which are constitutively bound to the cytoplasmic tails of cytokine receptors, or non-receptor associated tyrosine kinases including Ret, Src or the Bcl-Abl fusion protein (Figure 1A). After tyrosine phosphorylation, STAT-3 homo-dimerizes or hetero-dimerizes with STAT-1, translocates to the nucleus, and binds to consensus DNA sequences, such as the TTN4-5AA sequence (where N refers to any nucleotide base), within promoters of its target genes (8) (Figure 1A). STAT proteins then cooperate with other transcription factors to regulate expression of numerous genes, including bcl-2, bcl-xL, mcl-1, p21WAF1/CIP1, and cyclin D1 (11, 12).
Figure 1. The STAT-3 Signaling Pathway.
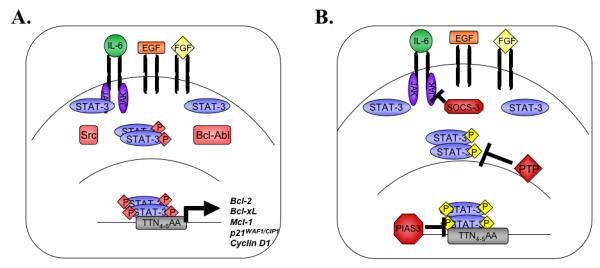
A. Cytokines such as IL-6, or growth factors including EGF and FGF, initiate STAT-3 signal transduction when they bind to their receptors and activate intracellular kinases. JAK proteins or receptor tyrosine kinases recruit inactive STAT-3 monomers and phosphorylate them on tyrosine 705. STAT-3 is also activated directly through interaction with the oncogenic kinases Src and Bcl-Abl. Tyrosine phosphorylated STAT-3 dimerizes and translocates to the nucleus, where STAT-3 transcriptional regulation is further modulated by serine phosphorylation. Active STAT-3 dimers bind to consensus sequences in the promoters of genes that regulate cell growth and anti-apoptotic behavior. B. Multiple STAT-3 endogenous inhibitors attenuate STAT-3 signaling. SOCS-3 inhibits JAK activation and subsequent signal transduction in the cytoplasm, while PIAS3 inhibits STAT-3 DNA binding in the nucleus. Protein tyrosine phosphatases such as SHP-1 and SHP-2 dephosphorylate active STAT-3 complexes.
Regulation of STAT-3 Activity
Because STAT-3 affects transcription of genes involved in apoptosis and cell cycle, tight control of STAT-3 activity is imperative to prevent malignant transformation of cells. Following induction of target gene expression, endogenous negative regulators attenuate STAT protein activity on a number of levels. Suppressors of Cytokine Signaling (SOCS) proteins downregulate the upstream kinase activity responsible for STAT-3 phosphorylation (13), while the Protein Inhibitors of Activated STATs (PIAS) proteins and protein tyrosine phosphatases (PTPs) target STAT proteins directly (Figure 1B) (14, 15).
STAT-3 Inhibition by SOCS-3
SOCS-3 is a member of the SOCS protein family, which is comprised of seven SOCS proteins (SOCS-1 through SOCS-7) and Cytokine Inducible SH2 Domain Containing Protein (CIS) (16). The cytoplasmic, inducibly expressed SOCS proteins attenuate STAT activity by inhibiting upstream JAK activation in a classic negative feedback loop (Figure 1B and 2) (16). Activated STAT-3 induces the expression of SOCS-3 (17, 18), which subsequently inhibits STAT-3 signaling by binding to and attenuating the signal transduction of gp130-related cytokine receptors and their associated JAK kinases (13).
Figure 2. IL-6 Cytokine Signaling Pathways.
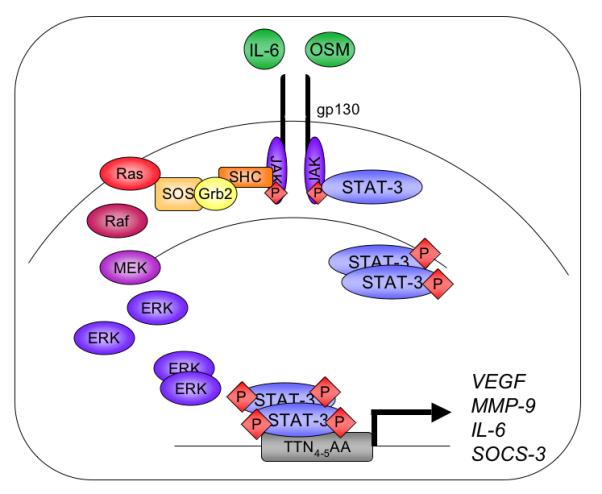
Members of the IL-6 cytokine family bind to a common receptor subunit, gp130, which either homo-or hetero-dimerizes upon ligand binding. JAK proteins are auto- and trans-phosphorylated and phosphorylate one of four tyrosine residues on the intracellular portion of the gp130 subunit. JAK protein activation results in downstream signal transduction through both the MAPK and STAT-3 pathways. Receptor interactions with the adaptor proteins SHC and Grb2 activate the MEK-ERK pathway. Alternatively, STAT-3 monomers are recruited to the receptor, become tyrosine phosphorylated and dimerize through their SH2 domains. STAT-3 dimers translocate to the nucleus and bind to consensus sequences in the promoters of genes including VEGF, MMP-9, IL-6, and SOCS-3.
STAT-3 Inhibition by PIAS Proteins and Protein Tyrosine Phosphatases
The highly conserved PIAS family of proteins includes PIAS1, PIAS3, PIASx, and PIASy. Except for PIAS1, each protein has two isoforms, and PIAS3 regulates STAT-3. In contrast to the inducibly-expressed SOCS proteins, PIAS proteins are constitutively expressed in the nucleus, and mediate transcriptional repression by directly interfering with the binding of STAT proteins and other transcription factors to their target DNA sequences (19). All PIAS proteins contain a zinc ring finger domain, an N-terminal LXXLL motif, C-terminal acidic domain, a serine/threonine rich domain, and a recently discovered PINIT motif that is involved in nuclear retention (19, 20) (Figure 3). PIAS proteins inhibit STAT transcriptional activity by interfering with DNA binding via their N-terminal domains (14). PIAS proteins also recruit transcriptional co-repressors such as histone deacetylases to target gene promoters to inhibit transcription (21, 22). Furthermore, PIAS proteins influence the activation status of transcription factors by directly modifying the proteins themselves. Some PIAS proteins, including PIAS3, exhibit E3-SUMO (Small Ubiquitin-like MOdifier) ligase activity and SUMOylate a variety of transcription factors, including p53, c-Jun, and c-Myb (19, 23, 24). SUMO modification affects their transcriptional ability, either by activation or inhibition (25).
Figure 3. Structural Domains of the PIAS3 Protein.
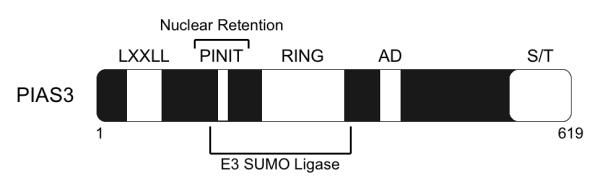
PIAS3 is a 619 amino acid protein that shares 40% homology to the other PIAS proteins. PIAS3 contain an N-terminal LXXLL motif, a PINIT motif, which is involved in nuclear retention, a zinc ring finger domain (RING), a C-terminal acidic domain (AD) and a serine/threonine (S/T) rich domain. The PINIT motif and the RING finger domain are involved in the E3 SUMO ligase activity exhibited by PIAS3.
PTPs act in both the cytoplasm and nucleus to inhibit target protein phosphorylation events. STAT-3 signaling is dampened by PTPs, such as the SH2-domain containing tyrosine phosphatase family (SHP-1 and SHP-2), which down-regulate STAT-3 activation directly by dephosphorylating active STAT-3 complexes (15).
The Role of STAT-3 in Oncogenesis and Immune Evasion
In mice, the STAT-3−/− phenotype is embryonic lethal at E6.5-7 (26), demonstrating that STAT-3 is essential to normal cellular functions. Conditional STAT-3 knock-out mice and other deletion models have revealed a role for STAT-3 in wound healing, T-cell development, mammary gland development, cell cycle progression, apoptosis, and proliferation (26-28). A number of these basic cellular events are involved in tumor development; consequently, STAT-3 has been implicated in oncogenesis by promoting abnormal cell cycle progression, angiogenesis, apoptosis, tissue invasion, and immune evasion (29). Many tumor-derived cell lines require STAT proteins, particularly STAT-3, to maintain a transformed phenotype (7), and STAT-3 is constitutively activated in 50% to 90% of diverse human cancers (7, 30). In experimental settings, introduction of a constitutively active STAT-3 mutant, STAT-3C, was sufficient to transform cells, and these cells could form tumors in nude mice (31). Furthermore, a dominant-negative mutant of STAT-3 blocked transformation by v-src (32). These observations suggest that many types of oncogenesis may be dependent on STAT-3 activity.
In addition to promoting oncogenesis, constitutively active STAT-3 also perpetuates tumor growth by undermining tumor recognition pathways in the immune system (33). In several experimental systems, STAT-3 promoted tumor immune evasion on multiple fronts by inhibiting pro-inflammatory cytokine signaling, blocking the anti-tumor activity of immune cells themselves, and promoting tolerogenesis by amplifying regulatory T (TReg) cells. Blocking STAT-3 activity in tumor cells, with either dominant-negative or anti-sense STAT-3, resulted in elevated expression of pro-inflammatory mediators, including interferon-β (IFN-β), tumor necrosis factor-α (TNF-α), IL-6, and the chemokines RANTES and IP-10. Conversely, activating STAT-3 inhibited expression of both IL-6 and RANTES in normal fibroblasts (34).
In addition to blocking pro-inflammatory cytokine signals, STAT-3 activation blunts the immunogenic response of immune cells themselves, further tipping the balance to a tolerogenic response. Animal and patient studies support this idea; in tumor-bearing mice, inhibiting STAT-3 increased the anti-tumor activity of T cells, natural killer cells, and neutrophils (35). Bone-marrow derived dendritic cells, which are thought to promote immune tolerance, are immature and dysfunctional in both humans and animals with cancer (36, 37), but inhibition of STAT-3 enhanced dendritic cell maturation in tumor-bearing mice. Additionally, mice that lack STAT-3 in hematopoietic cells had elevated levels of major histocompatibilty complex (MHC) class II, CD80, and CD86, compared to mice with intact STAT-3 expression (35). Taken together, these results demonstrate that STAT-3 is involved in maintaining the immature dendritic cell phenotype and promoting tumor immune tolerance.
More specifically, in gliomas, activation of STAT-3 has been implicated in inhibiting the T-cell response. Glioma-infiltrating CD8+ T cells were characterized as neither activated nor proliferating (CD8+CD25−), a hallmark of the immunosuppressive tumor environment (38). One route to reversing immune tolerance is by targeting CD8+CD25− T cells themselves. When STAT-3 activity in monocytes from glioma patients was inhibited, the expression of co-stimulatory molecules such as CD80 and CD86 was upregulated, an event which presumably enhanced T cell activation and is a critical step in reversing immune evasion (39). These data demonstrate that inhibiting STAT-3 affects the activation of glioma-infiltrating CD8+ T cells, which serve as one of many potential targets in overcoming immune suppression.
STAT-3 activity was shown to be elevated in tumor-associated TReg cells that maintain tumor immune evasion (35, 40-42). In these TReg cells, elevated activation of STAT-3 increased proliferation and promoted expression of forkhead box P3 (FOXP3), transforming growth factor-beta (TGF-β), and interleukin 10 (IL-10), all of which inhibit CD8+ T cell differentiation and dendritic cell maturation (33, 40, 41, 43). TReg cells have been demonstrated in the blood and within the tumor microenvironment of GBM patients, and are thought to contribute to the lack of effective immune responsiveness against GBMs (38, 44). The STAT-3 activation status of the TReg cells was not examined in these studies. Interestingly, TReg cells have been reported to be deficient in SOCS-3 expression, while SOCS-3 overexpression in these cells decreased their proliferation and suppressive function (45). The observed activation of STAT-3 in tumor-associated TReg cells may be attributable to the associated absence of the endogenous inhibitor SOCS-3. Taken together, these data show that STAT-3 activation promoted not only oncogenesis but also immune evasion, specifically by inhibiting expression of pro-inflammatory mediators, suppressing both dendritic cell maturation and CD8+ T cell activation, and promoting proliferation of TReg cells. These results demonstrate how two separate and equally important steps in oncogenesis, transformation and immune evasion, intersect at the point of STAT-3 activation.
STAT-3 Activation in Gliomas
It is clear that STAT-3 activation is critically involved in tumorigenesis, but in vitro, GBM cells have shown varying levels of constitutive STAT-3 activation. Using electrophoretic mobility shift assays (EMSAs), immunofluorescence, and immunoblotting, we and others have demonstrated that numerous GBM cell lines express very little activated STAT-3 under basal, unstimulated conditions (46, 47)1. However, using similar techniques, others have reported that some GBM cells exhibited constitutive STAT-3 activation, as well as constitutively high levels of anti-apoptotic proteins such as Bcl-xL, Bcl-2, Mcl-1, and c-Myc (48, 49). Several studies examining human GBM tissues observed constitutive activation of STAT-3, as assessed by tyrosine phosphorylation (48, 50, 51)1. Additionally, we detected elevated levels of serinephosphorylated STAT-3 in human GBM tissues1, demonstrating for the first time the presence of the maximally-activated form of STAT-3 in GBM. Immunohistochemistry studies showed that tyrosine phosphorylated STAT-3 localized to tumor endothelial cells (50), suggesting a role for STAT-3 in angiogenesis. As well, tyrosine phosphorylated STAT-3 has been localized to tumor cells (51). Taken together, these observations implicate a role for STAT-3 in GBM pathology.
Upstream Mediators of Aberrant STAT-3 Activation
Missense mutations of STAT-3 have been reported in patients with hyper IgE syndrome (52, 53). In human cancers, however, aberrant STAT-3 activity stems from dysregulation of upstream tyrosine kinases or loss of negative feedback mechanisms. For example, stably transforming cells with the oncogenic Src tyrosine kinase was sufficient to induce constitutive activation of STAT-3 (54) and downstream C-reactive protein and c-fos promoter activities (55). In order to understand how STAT-3 is improperly activated in GBM and other cancers, research has focused on unraveling the upstream mechanisms by which STAT-3 is activated, and understanding how regulation of these mechanisms fail in the context of GBM. Below we discuss how, in the context of GBM, members of the IL-6 cytokine family or growth factors initiate the STAT-3 activation pathway by interacting with their receptors.
STAT-3 Activation by IL-6 Cytokines
The IL-6 cytokine family consists of a number of structurally related proteins, such as IL-6, OSM, Ciliary Neurotrophic Factor (CNTF), and LIF. These pleiotropic cytokines mediate signal transduction through the MAPK or the JAK-STAT-3 pathways, and initiate these signaling pathways by binding to their receptors. This binding step leads to the homodimerization of gp130 or heterodimerization of gp130 with other gp130-related receptor subunits, such as the OSM receptor (9, 27). JAK proteins then phosphorylate the gp130 receptor subunit on one of four specific tyrosine residues, which recruits and activates the STAT-3 protein (9) (Figure 2). IL-6 expression is regulated by multiple stimuli, examples of which include hypoxia, pro-inflammatory mediators, and IL-6 cytokines themselves, including both IL-6 and OSM. These many factors are known to be upregulated in various diseases of the CNS, including GBM (56).
Substantial data gathered from cell lines, mouse models, and patient samples support a role for IL-6 proteins in GBM-associated STAT-3 activation. Several groups observed constitutive expression of IL-6 and OSM in human GBM cells, both in vivo and in vitro (57-59). The elevated expression of IL-6 cytokines is thought to be responsible, at least in part, for both constitutive and induced activation of STAT-3 in GBM. In some GBM cell lines, autocrine IL-6 expression resulted in constitutive activation of STAT-3, and neutralizing antibodies to IL-6 reduced STAT-3 activation, inhibited cell proliferation, and induced apoptosis (48). In a number of other human brain tumor cell lines, stimulation with IL-6, LIF, CNTF, or OSM resulted in induced STAT-3 activation (46), and treatment of human astroglioma cells with OSM increased the STAT-3-dependent expression and activation of MMP-9 and VEGF (59, 60).
In a GBM mouse model, development of gliomas required the presence of IL-6; mice that lacked IL-6 failed to develop tumors. To further explore this IL-6 dependence, heterozygous GFAP-v-src transgenic mice were utilized to compare astrocytic tumor incidence between animals that express IL-6 (v-src+/−/IL-6+/+) and those that lack IL-6 (vsrc+/−/IL-6−/−). While 21% of the v-src+/−/IL-6+/+ mice (12/56) developed tumors of varying grades, only 2.8% of the v-src+/−/IL-6−/− mice (1/35) developed tumors (61). These results suggest that loss of IL-6 in predisposed mouse models suppressed glioma formation.
Human GBM tissues also amplify expression of the IL-6 gene (57, 62). Immunohistochemistry studies on patient samples revealed that IL-6 was localized to GBM tumor cells, and IL-6 activity was detected in the cerebrospinal fluid and tumor cysts of GBM patients (57). Additionally, expression of pro-survival effectors downstream of STAT-3, such as MMP-9 and VEGF, was elevated in human GBM tissues and implicated in tumor angiogenesis (63). It is clear from these studies that IL-6 promotes glioma development in vivo, at least in part through the action of STAT-3.
STAT-3 Activation by EGFR
The transmembrane receptor tyrosine kinase EGFR is amplified in approximately 50% of GBMs. In approximately 50% of these cases, the GBMs express a mutant EGFR that lacks a portion of the extracellular ligand-binding domain (EGFRvIII). In studies comparing EGFRvIII to wild-type EGFR, the mutant was persistently auto-phosphorylated at low levels (64) and could not be down-regulated. Auto-phosphorylation of EGFRvIII resulted in inefficient EGF signal attenuation and persistent activation of downstream kinase pathways, including those involving STAT-3, Ras/MAPK and AKT (5). In fact, the expression of tyrosine phosphorylated STAT-3 correlated significantly with the expression of EGFRvIII in human GBM tissues (51).
D54-MG glioma cells expressing EGFRvIII showed higher motility than their wild-type counterparts (65), which suggests that expression of EGFRvIII enhanced glioblastoma cell invasion. EGFRvIII expression also promotes tumorigenesis in vivo; when implanted subcutaneously or intracerebrally into nude mice, EGFRvIII-expressing U87-MG cells produced larger and faster-growing tumors than those formed by wild type EGFR-expressing cells (66). Current evidence suggests that the mechanism behind the increased tumor size and growth rate in EGFRvIII-expressing cells lies in the activation of downstream effectors such as STAT-3 and AKT. Recent work in lung cancer cells showed that STAT-3 cooperates with AKT in EGFR signal transduction, and that the cooperative activation of these signaling intermediates contributed to changes in expression of the cell cycle intermediates cyclin D1 and p27 (67). In support of this observation, tumors formed by a variety of glioblastoma cells expressing EGFRvIII had larger volumes than tumors formed by wild-type EGFR-expressing cells, due to higher levels of proliferation as measured by Ki67 immunoreactivity (68). To determine the mechanism behind the increased proliferation, the EGFRvIII-expressing cells were examined for expression of cell cycle-related proteins. Compared to wild-type controls, EGFRvIII-expressing glioblastoma cells had lower levels of the cyclin-dependent kinase inhibitor, p27, higher CDK2-Cyclin A activity, and constitutively high levels of hyperphosphorylated RB proteins. Importantly, this pro-growth phenotype was dependent on the PI3K/AKT pathway, because inhibition of AKT resulted in decreased tumor volume and increased p27 expression (68). Taken together, these data illustrate that expression of EGFRvIII in glioblastoma cells enhances gliomagenesis by promoting cooperative activation of multiple downstream effectors such as STAT-3 and AKT.
STAT-3 Activation by FGFR
Signaling initiated by FGF through the FGFR is another mechanism by which a growth factor promotes the transcriptional activation of STAT-3 (69). Glioma cell growth was dependent on a functional FGFR pathway in C6 glioma cells; introduction of a dominant-negative FGFR attenuated FGF signaling, which resulted in reduced anchorage-dependent growth rates in these cells (70). When cells expressing the mutant FGFR were xenografted into immunodeficient mice or transplanted into rat brain, onset of tumor development was delayed and tumor volume was reduced compared to wild-type FGFR controls. Tumors formed from FGFR dominant-negative expressing cells exhibited lower levels of VEGF compared to tumors from wild-type counterparts (70), which helps to explain why the dominant-negative FGFR-expressing tumors displayed slower growth rates. Interestingly, STAT-3 is a downstream transcriptional target of FGFR (69), and activation of STAT-3 results in expression of VEGF (59). These observations suggest that STAT-3 was the critical link between FGFR signaling and VEGF-mediated tumor growth in C6 glioma cells. In summary, although multiple cytokines and growth factors may independently contribute to GBM pathology, STAT-3 appears to be a critical “molecular hub” that links these pathways together.
Mechanisms of STAT-3 Inhibition in Gliomas
STAT-3 is a promising target for GBM therapy, not only because it is a convergence point for several signaling pathways that promote glioma growth and maintenance, but also because aberrant STAT-3 activation results from upstream dysregulation, not constitutively active STAT-3 mutations. STAT-3 should therefore retain its ability to respond to direct regulatory stimuli or external inhibition, independent of upstream signaling. Several current therapeutics target STAT-3 activity in cancers other than GBM (Figure 4). Recent work has approached STAT-3 inhibition from two fronts: 1) through RNA interference or chemical inhibitors, and 2) through modulation of endogenous regulators such as PIAS3 and SOCS-3.
Figure 4. Inhibition of STAT-3 Signal Transduction.
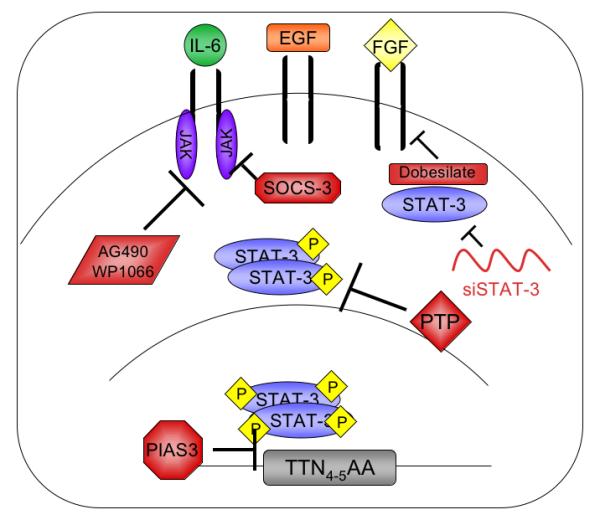
A variety of endogenous and pharmacologic inhibitors can attenuate STAT-3 signaling. SOCS-3, PIAS3, and various protein tyrosine phosphatases (PTP), inhibit STAT-3 activity endogenously. STAT-3 specific siRNA degrades STAT-3 mRNA. Pharmacologic inhibition of JAK activity by AG490 and WP1066 dampens the signals that result in STAT-3 activation. Attenuation of FGF signaling by dobesilate also inhibits STAT-3 mediated gene expression by attenuating kinase signals upstream of STAT-3 activation.
Direct STAT-3 Inhibition
Although not yet applied to GBM, several compounds attenuate STAT-3 signaling by targeting the STAT-3 protein directly. Researchers demonstrated that platinum compounds interfere with STAT-3 activation and abrogate signaling mediated by constitutively active STAT-3, presumably by direct action. This block of STAT-3 activity was accompanied by inhibition of cell growth and induction of apoptosis in breast, lung, and prostate cancer cell lines (71). Decoy oligonucleotides, or G-quartets, are competitive inhibitory structures comprised of guanine-rich oligonucleotides. By competitively binding activated STAT-3, G-quartets inhibited STAT-3 binding to endogenous gene promoters and therefore attenuated STAT-3-induced gene expression (72, 73). Another chemical inhibitor of STAT-3, S31-201 (NSC 74859), mediated its anti-tumor activity by inhibiting expression of pro-survival genes and attenuating the growth of human breast tumors in vivo (74). S31-201 was found to inhibit STAT-3 homodimer formation, STAT-3 DNA binding, and the resulting transcriptional activity, suggesting that the anti-tumor activity of this compound was mediated in part by a direct block of STAT-3 signal transduction (74). Using GBM cell lines, direct inhibition of STAT-3 activity using RNA interference (RNAi) triggered apoptosis and inhibited survival (75). RNAi-mediated downregulation of STAT-3 in A172 and U251-MG glioma cell lowered levels of pro-survival proteins such as Survivin and Bcl-xL and increased levels of apoptosis, as measured by cleaved caspase 3 levels and annexin V staining (75). This link between STAT-3 activity and GBM cell survival warrants testing the clinical effectiveness of existing STAT-3 inhibitors on GBM progression.
Inhibition of Upstream Kinases
Because aberrant STAT-3 activation is usually the result of the overactivity of upstream kinases, several pharmacological interventions that act by inhibiting growth factor receptors or other upstream kinases have been very effective at abrogating STAT-3 activity. Inhibition of EGFR with gefitinib (Iressa, AstraZeneca, USA) resulted in inhibition of STAT-3 tyrosine phosphorylation (76). Because the activation of STAT-3 depends on its direct phosphorylation by tyrosine kinases (27), several pharmacological JAK (AG490, WP1066, and JSI-124 (cucurbitacin I)) and Src (PD180970) inhibitors showed promising STAT-3 inhibition in vitro, and are in various early stages of experimental testing (48, 49, 77). The JAK inhibitor AG490 blocked constitutively active STAT-3 in the astroglioma cell line U251-MG (48), which decreased cell survival and increased cleavage of the apoptotic marker PARP. AG490-treated cells also displayed a dose-dependent inhibition in expression of pro-survival proteins, including Bcl-xL, Bcl-2, and Mcl-1 (48). These proteins are all gene targets of STAT-3, suggesting that the apoptotic response was mediated by STAT-3 inhibition.
Although AG490 is very effective in vitro, it did not consistently offer the same anti-tumor results in vivo; therefore, the structure of AG490 was modified to produce the more potent and active WP1066 compound (49). Preliminary in vivo studies showed that WP1066 successfully crossed the blood-brain-barrier, a key feature in drug design for glioma patients (39), and that it significantly inhibited the growth of glioma xenografts compared to untreated controls (49). Importantly, the effects of WP1066 are tumor specific. This compound specifically inhibited GBM cell viability without affecting the viability of normal human astrocytes (49).
The mechanism behind WP1066-mediated GBM cell growth attenuation appears to be rooted in its potent inhibition of STAT-3. In initial studies, WP1066 blocked tyrosine phosphorylation of STAT-3 by JAK proteins, which resulted in a failure of STAT-3 to translocate to the nucleus and mediate its transcriptional effects. WP1066 treatment of U87-MG and U373-MG cells decreased STAT-3 mediated expression of Bcl-xL, Mcl-1, and c-Myc, which resulted in increased apoptosis and reduced GBM cell viability (49). WP1066 also reversed immune tolerance in GBM patients, presumably by blocking STAT-3 mediated immune suppression and tolerogenesis. WP1066 stimulated proliferation of T cells from GBM patients and enhanced immunogenic responses in glioma-infiltrating microglia, macrophages, and peripheral blood monocytes (39). WP1066 also promoted immunogenic responses by upregulating the costimulatory molecules CD80 and CD86 and the T cell effector cytokines IL-2, IL-4, IL-12, and IL-15 (39). Because pharmacologic inhibitors of JAK proteins attenuate STAT-3-mediated GBM cell growth, glioma cell viability, and immune evasion in GBM, the JAK proteins have emerged as attractive targets for subsequent STAT-3 downregulation.
Dihydroxy-2,5 benzenesulphonate (Dobesilate) is thought to inhibit activation of STAT-3 by attenuating the upstream FGF signaling pathway. Dobesilate is currently used to treat diabetic retinopathy and chronic venous insufficiency, because it has been shown to block FGF driven neovascularization (78, 79). Treatment of C6 glioma cells with dobesilate in vitro triggered apoptosis and growth arrest (80). Further studies in glioma cells showed that dobesilate significantly inhibited constitutive expression of tyrosine phosphorylated STAT-3 (81), activation of the MAPK Extracellular Signal Related Protein Kinases1/2 (ERK1/2) (82), and expression of the pro-survival proteins Bcl-xL and cyclin D1 (81). These results support the idea that dobesilate increased apoptosis and decreased cell growth and survival in part by blocking STAT-3 activation.
The observed effects of AG490, WP1066, and dobesilate collectively illustrate that pharmacological inhibitors of individual kinases that operate upstream of STAT-3 have significant potential as GBM chemotherapeutic agents. Furthermore, inhibition of these various upstream kinases in combination with direct STAT-3 inhibitors may also be effective in GBM therapy. Recent work has demonstrated that inhibition of a single tyrosine kinase pathway provides little benefit in reducing glioma cell survival and growth; however, a combinatorial strategy to inhibit multiple upstream tyrosine kinases, such as EGFR and PDGFR, as well as PI3-K, proved to significantly reduce intracellular signaling, survival, and anchorage-independent growth of glioma cells (83). This combinatorial strategy provided novel evidence that inhibition of aberrant signaling pathways by multiple mechanisms may be effective for treatment of GBM. In addition, it supports the notion that STAT-3 inhibition by direct, indirect, or a combinatorial approach using existing pharmacological inhibitors, has promise as a clinical target in GBM.
Endogenous Negative Regulation by PIAS3
In addition to targeting STAT-3 for pharmacological inhibition, recent studies have focused on enhancing endogenous cellular mechanisms for modulating STAT-3 activity; namely, the inhibitory pathways involving PIAS3, SOCS-3 and PTPs (15).
Because of its ability to inhibit STAT-3 transcriptional activation at multiple levels, PIAS3 has recently become a focus of cancer biologists. In support of this, we showed that inhibiting constitutive PIAS3 expression with siRNA significantly increased proliferation of glioblastoma cells1. In contrast, PIAS3 overexpression in these cells attenuated proliferation and inhibited OSM-mediated STAT-3 promoter activity, suggesting that ectopic expression of PIAS3 could inhibit STAT-3 transcriptional activity in GBM1. Importantly, we recently showed that expression of the PIAS3 protein was largely absent in human GBM tissues, despite unchanged PIAS3 mRNA levels1, suggesting that the PIAS3 protein was rapidly degraded in GBM tissues. It was also reported that PIAS3 expression was diminished in human ALK+ T cell lymphoma samples, which was associated with STAT-3 dysregulation (84). Taken together, these observations indicate that downregulation of PIAS3 may cause or exacerbate gliomagenesis, and that re-introduction of PIAS3 could potentially inhibit GBM progression.
Although PIAS3 was first described as a negative regulator of STAT-3 transcriptional activity, it is now known to also modulate NF-κB, PI3-K, and TGF-β signaling pathways, all of which are associated with poor prognosis in GBM patients (51, 85-90). In vitro, PIAS3 downregulated the NF-κB pathway by interacting with p65 and repressing its transcriptional activity (91). PIAS3 also modulated the PI3-K pathway by interacting with AKT and suppressing its phosphorylation and activation (92). Studies in various cell lines showed that PIAS3 overexpression affected AKT signaling in both prostate and lung cancer cells, with subsequent effects on cell growth and apoptosis (92, 93). PIAS proteins also regulated transcriptional activity of SMAD proteins, the downstream targets of TGF-β signaling (25). These results strongly suggest that enhancing PIAS3 expression may inhibit multiple signaling pathways, such as STAT-3, NF-κB, PI3-K and TGF-β, that are overactive in human GBM tissues.
Involvement of SOCS-3 in Gliomas
Because SOCS-3 is an endogenous inhibitor of STAT-3 signaling (16), as well as a STAT-3 transcriptional target (13, 17), the evidence correlating SOCS-3 expression and STAT-3 activity is conflicting, and somewhat confusing. SOCS-3 works in a negative-feedback loop to suppress STAT-3 signaling; therefore, it is reasonable to suggest that loss of SOCS-3 may contribute to STAT-3 activation and tumor progression. Indeed, reports describing SOCS-3 hypermethylation and subsequent loss of expression in a variety of cancers support the idea that SOCS-3 may have a tumor-suppressing function (94-97). In contrast, elevated SOCS-3 expression was reported in human breast cancer and melanoma tissues, as well as in a subset of classical Hodgkin lymphoma cell lines and primary lymphoma cells (98-104). In several studies, SOCS-3 promoted cell growth and proliferation in a number of cancers (100, 103, 105), and elevated levels of SOCS-3 were reported to confer a growth advantage to a melanoma cell line (103). Lymphoma cells with low levels of endogenous SOCS-3 expression succumbed to proliferation arrest while cells that endogenously expressed higher levels of SOCS-3 did not undergo arrest (100).
SOCS-3 overexpression has been attributed to constitutive STAT-3 activation in various cancers (101, 106). However, there are few reports describing SOCS-3 expression in GBM. Our studies showed that human GBM tissues expressed higher levels of SOCS-3 than did control brain tissues, and that SOCS-3 promoter activity and mRNA expression is inhibited by ectopic PIAS3 expression1. Moreover, constitutive expression of SOCS-3 in human GBM tissues correlated with enhanced cell survival and radioresistance in vitro (105). This resistance was dependent on SOCS-3 expression, as evidenced by marked sensitivity to ionizing radiation in SOCS-3−/− mouse embryonic fibroblasts (MEFs) compared to wild-type MEFs (105). Interestingly, U87-MG glioma cells which constitutively express SOCS-3 displayed significant radioresistance that was attenuated by expression of a dominant-negative STAT-3 construct (105). Current results collectively demonstrate that constitutive expression of SOCS-3 and the associated radioresistance were both mediated by STAT-3, adding another aspect of tumorigenesis mediated by activated STAT-3, i.e. radioresistance.
Interestingly, a unique characteristic of glioblastoma cells that are resistant to ionizing radiation is the expression of the cell surface marker, CD133, a hallmark of neural precursor cells. CD133+ cells have recently been identified as the tumor initiating subset of glioblastoma cells (107). Upon treatment with ionizing radiation, growth of glioblastoma cells grown in vitro or as grafts in mice was largely inhibited, but the proportion of CD133+ cells was greater. In addition, heightened DNA repair responses were detected in CD133+ glioblastoma cells (107). Because the expression of SOCS-3 in GBM cells was also shown to correlate with resistance to ionizing radiation (105), it is tempting to speculate that CD133+ cells might also express elevated levels of SOCS-3, as well as activated STAT-3. Experiments to examine this parameter of STAT-3 signaling are currently being explored.
Although SOCS-3 is a negative regulator of STAT-3, it is also a target gene of STAT-3 (13); therefore, the simultaneous overexpression/activation of both of these proteins in GBM is reasonable. It is possible that as a transcriptional target of STAT-3, SOCS-3 is involved in promoting cell growth, cell proliferation, and resistance to radiation. Importantly, however, studies examining the expression and function of SOCS-3 in GBM are limited in number. Future experiments should be aimed at confirming the elevated expression of SOCS-3 in GBMs, identifying the cell types expressing SOCS-3, as well as elucidating the role of SOCS-3 in GBM.
Conclusions
In summary, aberrant expression and activation of the STAT-3 transcription factor has been implicated in GBM pathology both in vivo and in vitro. Aberrant STAT-3 activation was often associated with signaling initiated by IL-6 cytokine family members or growth factors such as EGF and FGF. These signaling pathways converge at the molecular hub STAT-3, which links various extracellular signals to transcriptional control of cell proliferation, cell cycle, and apoptosis. The central role STAT-3 plays in GBM-associated cell signaling makes it an attractive therapeutic target in the ongoing search for relevant GBM therapies, and studies thus far have revealed significant clinical potential in inhibiting STAT-3.
Acknowledgements
We acknowledge Dr. Rebecca Goldstein for excellent editorial assistance.
Grant Support: This work was supported in part by National Institutes of Health grants P50 CA-97247 and R01 NS-54158 (E.N.B.).
Footnotes
1
Brantley, EC, Nabors, LB, Gillespie, GY, Choi, Y-H, Palmer, CA, Harrison, K, Roarty, K, Benveniste, EN. Loss of PIAS3 in Glioblastoma Multiforme tumors: Implications for STAT-3 activation and gene expression. Submitted 2008.
References
- 1.Ohgaki H, Kleihues P. Epidemiology and etiology of gliomas. Acta Neuropathol (Berl) 2005;109:93–108. doi: 10.1007/s00401-005-0991-y. [DOI] [PubMed] [Google Scholar]
- 2.Louis DN, Ohgaki H, Wiestler OD, et al. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol (Berl) 2007;114:97–109. doi: 10.1007/s00401-007-0243-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schwartzbaum JA, Fisher JL, Aldape KD, Wrensch M. Epidemiology and molecular pathology of glioma. Nat Clin Pract Neurol. 2006;2:494–503. doi: 10.1038/ncpneuro0289. [DOI] [PubMed] [Google Scholar]
- 4.Stupp R, Mason WP, van den Bent MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352:987–96. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 5.Rao RD, James CD. Altered molecular pathways in gliomas: an overview of clinically relevant issues. Semin Oncol. 2004;31:595–604. doi: 10.1053/j.seminoncol.2004.07.002. [DOI] [PubMed] [Google Scholar]
- 6.Guha A, Mukherjee J. Advances in the biology of astrocytomas. Curr Opin Neurol. 2004;17:655–62. doi: 10.1097/00019052-200412000-00004. [DOI] [PubMed] [Google Scholar]
- 7.Bromberg J. Stat proteins and oncogenesis. J Clin Invest. 2002;109:1139–42. doi: 10.1172/JCI15617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Reich NC, Liu L. Tracking STAT nuclear traffic. Nat Rev Immunol. 2006;6:602–12. doi: 10.1038/nri1885. [DOI] [PubMed] [Google Scholar]
- 9.Heinrich PC, Behrmann I, Haan S, Hermanns HM, Muller-Newen G, Schaper F. Principles of interleukin (IL)-6-type cytokine signalling and its regulation. Biochem J. 2003;374:1–20. doi: 10.1042/BJ20030407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wen Z, Zhong Z, Darnell JE., Jr Maximal activation of transcription by Stat1 and Stat3 requires both tyrosine and serine phosphorylation. Cell. 1995;82:241–50. doi: 10.1016/0092-8674(95)90311-9. [DOI] [PubMed] [Google Scholar]
- 11.Germain D, Frank DA. Targeting the cytoplasmic and nuclear functions of signal transducers and activators of transcription 3 for cancer therapy. Clin Cancer Res. 2007;13:5665–9. doi: 10.1158/1078-0432.CCR-06-2491. [DOI] [PubMed] [Google Scholar]
- 12.Bowman T, Garcia R, Turkson J, Jove R. STATs in oncogenesis. Oncogene. 2000;19:2474–88. doi: 10.1038/sj.onc.1203527. [DOI] [PubMed] [Google Scholar]
- 13.Starr R, Willson TA, Viney EM, et al. A family of cytokine-inducible inhibitors of signalling. Nature. 1997;387:917–21. doi: 10.1038/43206. [DOI] [PubMed] [Google Scholar]
- 14.Chung CD, Liao J, Liu B, et al. Specific inhibition of Stat3 signal transduction by PIAS3. Science. 1997;278:1803–5. doi: 10.1126/science.278.5344.1803. [DOI] [PubMed] [Google Scholar]
- 15.Rakesh K, Agrawal DK. Controlling cytokine signaling by constitutive inhibitors. Biochem Pharmacol. 2005;70:649–57. doi: 10.1016/j.bcp.2005.04.042. [DOI] [PubMed] [Google Scholar]
- 16.Yoshimura A, Naka T, Kubo M. SOCS proteins, cytokine signalling and immune regulation. Nat Rev Immunol. 2007;7:454–65. doi: 10.1038/nri2093. [DOI] [PubMed] [Google Scholar]
- 17.Qin H, Roberts KL, Niyongere SA, Cong Y, Elson CO, Benveniste EN. Molecular mechanism of lipopolysaccharide-induced SOCS-3 gene expression in macrophages and microglia. J Immunol. 2007;179:5966–76. doi: 10.4049/jimmunol.179.9.5966. [DOI] [PubMed] [Google Scholar]
- 18.Ramana CV, Kumar A, Enelow R. Stat1-independent induction of SOCS-3 by interferon-gamma is mediated by sustained activation of Stat3 in mouse embryonic fibroblasts. Biochem Biophys Res Commun. 2005;327:727–33. doi: 10.1016/j.bbrc.2004.12.074. [DOI] [PubMed] [Google Scholar]
- 19.Shuai K. Regulation of cytokine signaling pathways by PIAS proteins. Cell Res. 2006;16:196–202. doi: 10.1038/sj.cr.7310027. [DOI] [PubMed] [Google Scholar]
- 20.Duval D, Duval G, Kedinger C, Poch O, Boeuf H. The ‘PINIT’ motif, of a newly identified conserved domain of the PIAS protein family, is essential for nuclear retention of PIAS3L. FEBS Lett. 2003;554:111–8. doi: 10.1016/s0014-5793(03)01116-5. [DOI] [PubMed] [Google Scholar]
- 21.Imoto S, Sugiyama K, Muromoto R, Sato N, Yamamoto T, Matsuda T. Regulation of transforming growth factor-beta signaling by protein inhibitor of activated STAT, PIASy through Smad3. J Biol Chem. 2003;278:34253–8. doi: 10.1074/jbc.M304961200. [DOI] [PubMed] [Google Scholar]
- 22.Long J, Matsuura I, He D, Wang G, Shuai K, Liu F. Repression of Smad transcriptional activity by PIASy, an inhibitor of activated STAT. Proc Natl Acad Sci U S A. 2003;100:9791–6. doi: 10.1073/pnas.1733973100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schmidt D, Muller S. Members of the PIAS family act as SUMO ligases for c-Jun and p53 and repress p53 activity. Proc Natl Acad Sci U S A. 2002;99:2872–7. doi: 10.1073/pnas.052559499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sharrocks AD. PIAS proteins and transcriptional regulation--more than just SUMO E3 ligases? Genes & Dev. 2006;20:754–8. doi: 10.1101/gad.1421006. [DOI] [PubMed] [Google Scholar]
- 25.Shuai K, Liu B. Regulation of gene-activation pathways by PIAS proteins in the immune system. Nat Rev Immunol. 2005;5:593–605. doi: 10.1038/nri1667. [DOI] [PubMed] [Google Scholar]
- 26.Akira S. Roles of STAT3 defined by tissue-specific gene targeting. Oncogene. 2000;19:2607–11. doi: 10.1038/sj.onc.1203478. [DOI] [PubMed] [Google Scholar]
- 27.Hirano T, Ishihara K, Hibi M. Roles of STAT3 in mediating the cell growth, differentiation and survival signals relayed through the IL-6 family of cytokine receptors. Oncogene. 2000;19:2548–56. doi: 10.1038/sj.onc.1203551. [DOI] [PubMed] [Google Scholar]
- 28.Harris TJ, Grosso JF, Yen HR, et al. Cutting edge: An in vivo requirement for STAT3 signaling in TH17 development and TH17-dependent autoimmunity. J Immunol. 2007;179:4313–7. doi: 10.4049/jimmunol.179.7.4313. [DOI] [PubMed] [Google Scholar]
- 29.Yu H, Jove R. The STATs of cancer--new molecular targets come of age. Nat Rev Cancer. 2004;4:97–105. doi: 10.1038/nrc1275. [DOI] [PubMed] [Google Scholar]
- 30.Buettner R, Mora LB, Jove R. Activated STAT signaling in human tumors provides novel molecular targets for therapeutic intervention. Clin Cancer Res. 2002;8:945–54. [PubMed] [Google Scholar]
- 31.Bromberg JF, Wrzeszczynska MH, Devgan G, et al. Stat3 as an oncogene. Cell. 1999;98:295–303. doi: 10.1016/s0092-8674(00)81959-5. [DOI] [PubMed] [Google Scholar]
- 32.Bromberg JF, Horvath CM, Besser D, Lathem WW, Darnell JE., Jr Stat3 activation is required for cellular transformation by v-src. Mol Cell Biol. 1998;18:2553–8. doi: 10.1128/mcb.18.5.2553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yu H, Kortylewski M, Pardoll D. Crosstalk between cancer and immune cells: role of STAT3 in the tumour microenvironment. Nat Rev Immunol. 2007;7:41–51. doi: 10.1038/nri1995. [DOI] [PubMed] [Google Scholar]
- 34.Wang T, Niu G, Kortylewski M, et al. Regulation of the innate and adaptive immune responses by Stat-3 signaling in tumor cells. Nat Med. 2004;10:48–54. doi: 10.1038/nm976. [DOI] [PubMed] [Google Scholar]
- 35.Kortylewski M, Kujawski M, Wang T, et al. Inhibiting Stat3 signaling in the hematopoietic system elicits multicomponent antitumor immunity. Nat Med. 2005;11:1314–21. doi: 10.1038/nm1325. [DOI] [PubMed] [Google Scholar]
- 36.Almand B, Resser JR, Lindman B, et al. Clinical significance of defective dendritic cell differentiation in cancer. Clin Cancer Res. 2000;6:1755–66. [PubMed] [Google Scholar]
- 37.Vicari AP, Caux C, Trinchieri G. Tumour escape from immune surveillance through dendritic cell inactivation. Semin Cancer Biol. 2002;12:33–42. doi: 10.1006/scbi.2001.0400. [DOI] [PubMed] [Google Scholar]
- 38.Hussain SF, Yang D, Suki D, Aldape K, Grimm E, Heimberger AB. The role of human glioma-infiltrating microglia/macrophages in mediating antitumor immune responses. Neuro Oncol. 2006:261–79. doi: 10.1215/15228517-2006-008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hussain SF, Kong LY, Jordan J, et al. A novel small molecule inhibitor of signal transducers and activators of transcription 3 reverses immune tolerance in malignant glioma patients. Cancer Res. 2007;67:9630–6. doi: 10.1158/0008-5472.CAN-07-1243. [DOI] [PubMed] [Google Scholar]
- 40.Kasprzycka M, Marzec M, Liu X, Zhang Q, Wasik MA. Nucleophosmin/anaplastic lymphoma kinase (NPM/ALK) oncoprotein induces the T regulatory cell phenotype by activating STAT3. Proc Natl Acad Sci U S A. 2006;103:9964–9. doi: 10.1073/pnas.0603507103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zou W. Immunosuppressive networks in the tumour environment and their therapeutic relevance. Nat Rev Cancer. 2005;5:263–74. doi: 10.1038/nrc1586. [DOI] [PubMed] [Google Scholar]
- 42.Doganci A, Eigenbrod T, Krug N, et al. The IL-6R alpha chain controls lung CD4+CD25+ Treg development and function during allergic airway inflammation in vivo. J Clin Invest. 2005;115:313–25. doi: 10.1172/JCI22433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kinjyo I, Inoue H, Hamano S, et al. Loss of SOCS3 in T helper cells resulted in reduced immune responses and hyperproduction of interleukin 10 and transforming growth factor-beta 1. J Exp Med. 2006;203:1021–31. doi: 10.1084/jem.20052333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jordan JT, Sun W, Hussain SF, Deangulo G, Prabhu SS, Heimberger AB. Preferential migration of regulatory T cells mediated by glioma-secreted chemokines can be blocked with chemotherapy. Cancer Immunol Immunother. 2008;57:123–31. doi: 10.1007/s00262-007-0336-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pillemer BB, Xu H, Oriss TB, Qi Z, Ray A. Deficient SOCS3 expression in CD4+CD25+FoxP3+ regulatory T cells and SOCS3-mediated suppression of Treg function. Eur J Immunol. 2007;37:2082–89. doi: 10.1002/eji.200737193. [DOI] [PubMed] [Google Scholar]
- 46.Schaefer LK, Menter DG, Schaefer TS. Activation of stat3 and stat1 DNA binding and transcriptional activity in human brain tumour cell lines by gp130 cytokines. Cell Signal. 2000;12:143–51. doi: 10.1016/s0898-6568(99)00077-7. [DOI] [PubMed] [Google Scholar]
- 47.Van Wagoner NJ, Choi C, Repovic P, Benveniste EN. Oncostatin M regulation of interleukin-6 expression in astrocytes: biphasic regulation involving the mitogen-activated protein kinases ERK1/2 and p38. J Neurochem. 2000;75:563–75. doi: 10.1046/j.1471-4159.2000.0750563.x. [DOI] [PubMed] [Google Scholar]
- 48.Rahaman SO, Harbor PC, Chernova O, Barnett GH, Vogelbaum MA, Haque SJ. Inhibition of constitutively active Stat3 suppresses proliferation and induces apoptosis in glioblastoma multiforme cells. Oncogene. 2002;21:8404–13. doi: 10.1038/sj.onc.1206047. [DOI] [PubMed] [Google Scholar]
- 49.Iwamaru A, Szymanski S, Iwado E, et al. A novel inhibitor of the STAT3 pathway induces apoptosis in malignant glioma cells both in vitro and in vivo. Oncogene. 2007;26:2435–44. doi: 10.1038/sj.onc.1210031. [DOI] [PubMed] [Google Scholar]
- 50.Schaefer LK, Ren Z, Fuller GN, Schaefer TS. Constitutive activation of Stat3alpha in brain tumors: localization to tumor endothelial cells and activation by the endothelial tyrosine kinase receptor (VEGFR-2) Oncogene. 2002;21:2058–65. doi: 10.1038/sj.onc.1205263. [DOI] [PubMed] [Google Scholar]
- 51.Mizoguchi M, Betensky RA, Batchelor TT, Bernay DC, Louis DN, Nutt CL. Activation of STAT3, MAPK, and AKT in malignant astrocytic gliomas: correlation with EGFR status, tumor grade, and survival. J Neuropathol Exp Neurol. 2006;65:1181–8. doi: 10.1097/01.jnen.0000248549.14962.b2. [DOI] [PubMed] [Google Scholar]
- 52.Holland SM, DeLeo FR, Elloumi HZ, et al. STAT3 mutations in the hyper-IgE syndrome. N Engl J Med. 2007;357:1608–19. doi: 10.1056/NEJMoa073687. [DOI] [PubMed] [Google Scholar]
- 53.Minegishi Y, Saito M, Tsuchiya S, et al. Dominant-negative mutations in the DNA-binding domain of STAT3 cause hyper-IgE syndrome. Nature. 2007;448:1058–62. doi: 10.1038/nature06096. [DOI] [PubMed] [Google Scholar]
- 54.Yu CL, Meyer DJ, Campbell GS, et al. Enhanced DNA-binding activity of a Stat3-related protein in cells transformed by the Src oncoprotein. Science. 1995;269:81–3. doi: 10.1126/science.7541555. [DOI] [PubMed] [Google Scholar]
- 55.Turkson J, Bowman T, Garcia R, Caldenhoven E, De Groot RP, Jove R. Stat3 activation by Src induces specific gene regulation and is required for cell transformation. Mol Cell Biol. 1998;18:2545–52. doi: 10.1128/mcb.18.5.2545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Van Wagoner NJ, Benveniste EN. Interleukin-6 expression and regulation in astrocytes. J Neuroimmunol. 1999;100:124–39. doi: 10.1016/s0165-5728(99)00187-3. [DOI] [PubMed] [Google Scholar]
- 57.Van Meir E, Sawamura Y, Diserens AC, Hamou MF, de Tribolet N. Human glioblastoma cells release interleukin 6 in vivo and in vitro. Cancer Res. 1990;50:6683–8. [PubMed] [Google Scholar]
- 58.Van Wagoner NJ, Oh JW, Repovic P, Benveniste EN. Interleukin-6 (IL-6) production by astrocytes: autocrine regulation by IL-6 and the soluble IL-6 receptor. J Neurosci. 1999;19:5236–44. doi: 10.1523/JNEUROSCI.19-13-05236.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Repovic P, Fears CY, Gladson CL, Benveniste EN. Oncostatin-M induction of vascular endothelial growth factor expression in astroglioma cells. Oncogene. 2003;22:8117–24. doi: 10.1038/sj.onc.1206922. [DOI] [PubMed] [Google Scholar]
- 60.Chen SH, Gillespie GY, Benveniste EN. Divergent effects of oncostatin M on astroglioma cells: influence on cell proliferation, invasion, and expression of matrix metalloproteinases. Glia. 2006;53:191–200. doi: 10.1002/glia.20264. [DOI] [PubMed] [Google Scholar]
- 61.Weissenberger J, Loeffler S, Kappeler A, et al. IL-6 is required for glioma development in a mouse model. Oncogene. 2004;23:3308–16. doi: 10.1038/sj.onc.1207455. [DOI] [PubMed] [Google Scholar]
- 62.Tchirkov A, Rolhion C, Bertrand S, Dore JF, Dubost JJ, Verrelle P. IL-6 gene amplification and expression in human glioblastomas. Br J Cancer. 2001;85:518–22. doi: 10.1054/bjoc.2001.1942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Munaut C, Noel A, Hougrand O, Foidart JM, Boniver J, Deprez M. Vascular endothelial growth factor expression correlates with matrix metalloproteinases MT1-MMP, MMP-2 and MMP-9 in human glioblastomas. Int J Cancer. 2003;106:848–55. doi: 10.1002/ijc.11313. [DOI] [PubMed] [Google Scholar]
- 64.Maher EA, Furnari FB, Bachoo RM, et al. Malignant glioma: genetics and biology of a grave matter. Genes & Dev. 2001;15:1311–33. doi: 10.1101/gad.891601. [DOI] [PubMed] [Google Scholar]
- 65.Lal A, Glazer CA, Martinson HM, et al. Mutant epidermal growth factor receptor up-regulates molecular effectors of tumor invasion. Cancer Res. 2002;62:3335–9. [PubMed] [Google Scholar]
- 66.Nishikawa R, Ji XD, Harmon RC, et al. A mutant epidermal growth factor receptor common in human glioma confers enhanced tumorigenicity. Proc Natl Acad Sci U S A. 1994;91:7727–31. doi: 10.1073/pnas.91.16.7727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Song L, Morris M, Bagui T, Lee FY, Jove R, Haura EB. Dasatinib (BMS-354825) selectively induces apoptosis in lung cancer cells dependent on epidermal growth factor receptor signaling for survival. Cancer Res. 2006;66:5542–8. doi: 10.1158/0008-5472.CAN-05-4620. [DOI] [PubMed] [Google Scholar]
- 68.Narita Y, Nagane M, Mishima K, Huang HJ, Furnari FB, Cavenee WK. Mutant epidermal growth factor receptor signaling down-regulates p27 through activation of the phosphatidylinositol 3-kinase/Akt pathway in glioblastomas. Cancer Res. 2002;62:6764–9. [PubMed] [Google Scholar]
- 69.Deo DD, Axelrad TW, Robert EG, Marcheselli V, Bazan NG, Hunt JD. Phosphorylation of STAT-3 in response to basic fibroblast growth factor occurs through a mechanism involving platelet-activating factor, JAK-2, and Src in human umbilical vein endothelial cells. Evidence for a dual kinase mechanism. J Biol Chem. 2002;277:21237–45. doi: 10.1074/jbc.M110955200. [DOI] [PubMed] [Google Scholar]
- 70.Auguste P, Gursel DB, Lemiere S, et al. Inhibition of fibroblast growth factor/fibroblast growth factor receptor activity in glioma cells impedes tumor growth by both angiogenesis-dependent and -independent mechanisms. Cancer Res. 2001;61:1717–26. [PubMed] [Google Scholar]
- 71.Turkson J, Zhang S, Palmer J, et al. Inhibition of constitutive signal transducer and activator of transcription 3 activation by novel platinum complexes with potent antitumor activity. Mol Cancer Ther. 2004;3:1533–42. [PubMed] [Google Scholar]
- 72.Jing N, Li Y, Xu X, et al. Targeting Stat3 with G-quartet oligodeoxynucleotides in human cancer cells. DNA Cell Biol. 2003;22:685–96. doi: 10.1089/104454903770946665. [DOI] [PubMed] [Google Scholar]
- 73.Leong PL, Andrews GA, Johnson DE, et al. Targeted inhibition of Stat3 with a decoy oligonucleotide abrogates head and neck cancer cell growth. Proc Natl Acad Sci U S A. 2003;100:4138–43. doi: 10.1073/pnas.0534764100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Siddiquee K, Zhang S, Guida WC, et al. Selective chemical probe inhibitor of Stat3, identified through structure-based virtual screening, induces antitumor activity. Proc Natl Acad Sci U S A. 2007;104:7391–6. doi: 10.1073/pnas.0609757104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Konnikova L, Kotecki M, Kruger MM, Cochran BH. Knockdown of STAT3 expression by RNAi induces apoptosis in astrocytoma cells. BMC Cancer. 2003;3:23–31. doi: 10.1186/1471-2407-3-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ciardiello F, Caputo R, Bianco R, et al. Antitumor effect and potentiation of cytotoxic drugs activity in human cancer cells by ZD-1839 (Iressa), an epidermal growth factor receptor-selective tyrosine kinase inhibitor. Clin Cancer Res. 2000;6:2053–63. [PubMed] [Google Scholar]
- 77.Blaskovich MA, Sun J, Cantor A, Turkson J, Jove R, Sebti SM. Discovery of JSI-124 (cucurbitacin I), a selective Janus kinase/signal transducer and activator of transcription 3 signaling pathway inhibitor with potent antitumor activity against human and murine cancer cells in mice. Cancer Res. 2003;63:1270–9. [PubMed] [Google Scholar]
- 78.Tejerina T, Ruiz E. Calcium dobesilate: pharmacology and future approaches. Gen Pharmacol. 1998;31:357–60. doi: 10.1016/s0306-3623(98)00040-8. [DOI] [PubMed] [Google Scholar]
- 79.Cuevas P, Sanchez I, Lozano RM, Gimenez-Gallego G. Dobesilate is an angiogenesis inhibitor. Eur J Med Res. 2005;10:369–72. [PubMed] [Google Scholar]
- 80.Cuevas P, Diaz-Gonzalez D, Gimenez-Gallego G, Dujovny M. Dihydroxy-2,5 benzenesulphonate (dobesilate) elicits growth arrest and apoptosis in glioma cells. Neurol Res. 2005;27:797–800. doi: 10.1179/016164105X63665. [DOI] [PubMed] [Google Scholar]
- 81.Cuevas P, Diaz-Gonzalez D, Sanchez I, Lozano RM, Gimenez-Gallego G, Dujovny M. Dobesilate inhibits the activation of signal transducer and activator of transcription 3, and the expression of cyclin D1 and bcl-XL in glioma cells. Neurol Res. 2006;28:127–30. doi: 10.1179/016164106X97982. [DOI] [PubMed] [Google Scholar]
- 82.Cuevas P, Diaz-Gonzalez D, Garcia-Martin-Cordova C, et al. Dobesilate diminishes activation of the mitogen-activated protein kinase ERK1/2 in glioma cells. J Cell Mol Med. 2006;10:225–30. doi: 10.1111/j.1582-4934.2006.tb00303.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Stommel JM, Kimmelman AC, Ying H, et al. Coactivation of receptor tyrosine kinases affects the response of tumor cells to targeted therapies. Science. 2007;318:287–90. doi: 10.1126/science.1142946. [DOI] [PubMed] [Google Scholar]
- 84.Zhang Q, Raghunath PN, Xue L, et al. Multilevel dysregulation of STAT3 activation in anaplastic lymphoma kinase-positive T/null-cell lymphoma. J Immunol. 2002;168:466–74. doi: 10.4049/jimmunol.168.1.466. [DOI] [PubMed] [Google Scholar]
- 85.Bruna A, Darken RS, Rojo F, et al. High TGFβ-Smad activity confers poor prognosis in glioma patients and promotes cell proliferation depending on the methylation of the PDGF-B gene. Cancer Cell. 2007;11:147–60. doi: 10.1016/j.ccr.2006.11.023. [DOI] [PubMed] [Google Scholar]
- 86.Chakravarti A, Zhai G, Suzuki Y, et al. The prognostic significance of phosphatidylinositol 3-kinase pathway activation in human gliomas. J Clin Oncol. 2004;22:1926–33. doi: 10.1200/JCO.2004.07.193. [DOI] [PubMed] [Google Scholar]
- 87.Choe G, Horvath S, Cloughesy TF, et al. Analysis of the phosphatidylinositol 3′-kinase signaling pathway in glioblastoma patients in vivo. Cancer Res. 2003;63:2742–6. [PubMed] [Google Scholar]
- 88.Pelloski CE, Lin E, Zhang L, et al. Prognostic associations of activated mitogen-activated protein kinase and Akt pathways in glioblastoma. Clin Cancer Res. 2006;12:3935–41. doi: 10.1158/1078-0432.CCR-05-2202. [DOI] [PubMed] [Google Scholar]
- 89.Robe PA, Bentires-Alj M, Bonif M, et al. In vitro and in vivo activity of the nuclear factor-kappaB inhibitor sulfasalazine in human glioblastomas. Clin Cancer Res. 2004;10:5595–603. doi: 10.1158/1078-0432.CCR-03-0392. [DOI] [PubMed] [Google Scholar]
- 90.Wang H, Wang H, Zhang W, Huang HJ, Liao WS, Fuller GN. Analysis of the activation status of Akt, NFkappaB, and Stat3 in human diffuse gliomas. Lab Invest. 2004;84:941–51. doi: 10.1038/labinvest.3700123. [DOI] [PubMed] [Google Scholar]
- 91.Jang HD, Yoon K, Shin YJ, Kim J, Lee SY. PIAS3 suppresses NF-kappaB-mediated transcription by interacting with the p65/RelA subunit. J Biol Chem. 2004;279:24873–80. doi: 10.1074/jbc.M313018200. [DOI] [PubMed] [Google Scholar]
- 92.Ogata Y, Osaki T, Naka T, et al. Overexpression of PIAS3 suppresses cell growth and restores the drug sensitivity of human lung cancer cells in association with PI3-K/Akt inactivation. Neoplasia. 2006;8:817–25. doi: 10.1593/neo.06409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Wible BA, Wang L, Kuryshev YA, Basu A, Haldar S, Brown AM. Increased K+ efflux and apoptosis induced by the potassium channel modulatory protein KChAP/PIAS3beta in prostate cancer cells. J Biol Chem. 2002;277:17852–62. doi: 10.1074/jbc.M201689200. [DOI] [PubMed] [Google Scholar]
- 94.He B, You L, Uematsu K, et al. SOCS-3 is frequently silenced by hypermethylation and suppresses cell growth in human lung cancer. Proc Natl Acad Sci U S A. 2003;100:14133–8. doi: 10.1073/pnas.2232790100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Weber A, Hengge UR, Bardenheuer W, et al. SOCS-3 is frequently methylated in head and neck squamous cell carcinoma and its precursor lesions and causes growth inhibition. Oncogene. 2005;24:6699–708. doi: 10.1038/sj.onc.1208818. [DOI] [PubMed] [Google Scholar]
- 96.Sutherland KD, Lindeman GJ, Choong DY, et al. Differential hypermethylation of SOCS genes in ovarian and breast carcinomas. Oncogene. 2004;23:7726–33. doi: 10.1038/sj.onc.1207787. [DOI] [PubMed] [Google Scholar]
- 97.Niwa Y, Kanda H, Shikauchi Y, et al. Methylation silencing of SOCS-3 promotes cell growth and migration by enhancing JAK/STAT and FAK signalings in human hepatocellular carcinoma. Oncogene. 2005;24:6406–17. doi: 10.1038/sj.onc.1208788. [DOI] [PubMed] [Google Scholar]
- 98.Takeuchi K, Sakai I, Narumi H, et al. Expression of SOCS3 mRNA in bone marrow cells from CML patients associated with cytogenetic response to IFN-alpha. Leuk Res. 2005;29:173–8. doi: 10.1016/j.leukres.2004.06.006. [DOI] [PubMed] [Google Scholar]
- 99.Brender C, Lovato P, Sommer VH, et al. Constitutive SOCS-3 expression protects T-cell lymphoma against growth inhibition by IFNalpha. Leukemia. 2005;19:209–13. doi: 10.1038/sj.leu.2403610. [DOI] [PubMed] [Google Scholar]
- 100.Baus D, Pfitzner E. Specific function of STAT3, SOCS1, and SOCS3 in the regulation of proliferation and survival of classical Hodgkin lymphoma cells. Int J Cancer. 2006;118:1404–13. doi: 10.1002/ijc.21539. [DOI] [PubMed] [Google Scholar]
- 101.Evans MK, Yu CR, Lohani A, et al. Expression of SOCS1 and SOCS3 genes is differentially regulated in breast cancer cells in response to proinflammatory cytokine and growth factor signals. Oncogene. 2007;26:1941–8. doi: 10.1038/sj.onc.1209993. [DOI] [PubMed] [Google Scholar]
- 102.Fojtova M, Boudny V, Kovarik A, et al. Development of IFN-γ resistance is associated with attenuation of SOCS genes induction and constitutive expression of SOCS3 in melanoma cells. Br J Cancer. 2007;97:231–7. doi: 10.1038/sj.bjc.6603849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Komyod W, Bohm M, Metze D, Heinrich PC, Behrmann I. Constitutive suppressor of cytokine signaling 3 expression confers a growth advantage to a human melanoma cell line. Mol Cancer Res. 2007;5:271–81. doi: 10.1158/1541-7786.MCR-06-0274. [DOI] [PubMed] [Google Scholar]
- 104.Raccurt M, Tam SP, Lau P, et al. Suppressor of cytokine signalling gene expression is elevated in breast carcinoma. Br J Cancer. 2003;89:524–32. doi: 10.1038/sj.bjc.6601115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Zhou H, Miki R, Eeva M, et al. Reciprocal regulation of SOCS 1 and SOCS3 enhances resistance to ionizing radiation in glioblastoma multiforme. Clin Cancer Res. 2007;13:2344–53. doi: 10.1158/1078-0432.CCR-06-2303. [DOI] [PubMed] [Google Scholar]
- 106.Cho-Vega JH, Rassidakis GZ, Amin HM, et al. Suppressor of cytokine signaling 3 expression in anaplastic large cell lymphoma. Leukemia. 2004;18:1872–8. doi: 10.1038/sj.leu.2403495. [DOI] [PubMed] [Google Scholar]
- 107.Bao S, Wu Q, McLendon RE, et al. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. 2006;444:756–60. doi: 10.1038/nature05236. [DOI] [PubMed] [Google Scholar]