Tuning the Activity of a Short Arg-Trp Antimicrobial Peptide by Lipidation of a C- or N-Terminal Lysine Side-Chain (original) (raw)
Abstract
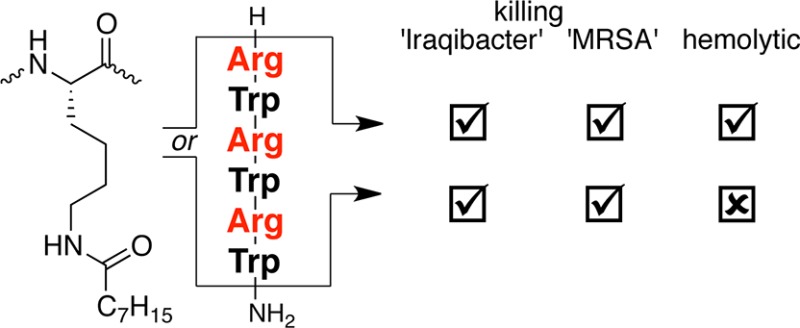
The attachment of lipids to _C_- or _N_-terminally positioned lysine side-chain amino groups increases the activity of a short synthetic (Arg-Trp)3 antimicrobial peptide significantly, making these peptides even active against pathogenic Gram-negative bacteria. Thus, a peptide with strong activity against S. aureus (1.1–2 μM) and good activity against A. baumannii and P. aeruginosa (9–18 μM) was identified. The most promising peptide causes 50% hemolysis at 285 μM and shows some selectivity against human cancer cell lines. Interestingly, the increased activity of ferrocenoylated peptides is mostly due to the lipophilicity of the organometallic fragment.
Keywords: Lipidated antimicrobial peptides, ferrocenoyl, anticancer, nonhemolytic
Next to the often-mentioned threat of resistant Gram-positive bacteria such as methicillin resistant Staphylococcus aureus (or MRSA) and vancomycin resistant Enterococcus feacium (or VRE), a threat arising from Gram-negative bacteria has been growing steadily.1 For example, it has been long recognized that late-stage chronic infections in cystic fibrosis patients are mainly caused by biofilms formed by the Gram-negative Pseudomonas aeruginosa.2 In addition, another recently emerged Gram-negative pathogen is Acinetobacter baumannii: it causes tens of thousands of deaths in the U.S. annually,3 has been mentioned to be “far worse than MRSA”,4 and was nicknamed “Iraqibacter” following the many severe A. baumannii infections brought home by U.S. soldiers that served in Iraq.5 In view of the generally higher resistance of such Gram-negative bacteria against treatments and increasing occurrence of infections by them, compounds with increased activity against this class of bacteria are urgently required.6
Interestingly, synthetic antimicrobial peptides (AMPs) hold great promise as novel antibacterial agents.7,8 Especially short sequences with relatively simple amino acid compositions are preferred: they are easy to make and can be modified according to specific requirements concerning proteolytic stability, general toxicity, circulation lifetime, or bacterial specificity. A potential downside of short sequences is their undefined or surfactant-like mode-of-action, and a lower activity when compared to, for example, known natural AMPs such as magainin, mellitin, temporin, and aurein.9 Nevertheless, the fact that these short sequences can be easily modified to tune their activity-profile makes them interesting from a therapeutic perspective. For example, synergistic approaches can be explored using synthetic methods, as was shown by combination of vancomycin with nisin(1-12),10 combination of vancomycin with metalloenzyme active site mimics,11 and dimerization of vancomycin.12
Importantly, even simple modifications can already lead to significant improvements and change the spectrum of activity, as was shown for lipidated vancomycin: the attachment of lipids to vancomycin led to a 40-fold increase in activity against vancomycin-resistant Enterococci (VRE).13 A similar effect has been shown for (short) peptides, where attachment of lipids to the peptide _N_-terminus turned otherwise nonactive peptides into nontoxic AMPs with antifungal and antibacterial activity over a broad range of pathogens.14−18 Very recently, a trivalent histidine-histidine dipeptide with a C14-lipid tail proved to be a potent nontoxic in vivo antifungal agent for the treatment of lethal lung infections in mice.19 Lastly, the attachment of an organometallic moiety to short synthetic antimicrobial peptides or antibacterial agents has been shown to modulate the antibacterial properties of those therapeutics.20−22 Next to activity-profile related considerations, it is also important to increase the circulation lifetime of a therapeutic peptide.23 Glomeruli located in the kidney are very effective in filtering compounds smaller than 8 nm from the bloodstream, resulting in rapid excretion of short peptides within minutes after administration. Albumin-binding or the formation of supramolecular structures can increase the plasma-lifetime of (lipidated) peptides.24,25 However, a major downside of lipidation is that it decreases solubility in water, which can have severe pharmacokinetic consequences, and that it can increase toxicity.
In this study, we used a short antibacterial (RW)3-sequence—that was only active against Gram-positive bacteria—and modified it with an _N_- or _C_-terminally positioned lysine residue that was lipidated on the side-chain amino-group (Chart 1). Until now, this strategy has been rarely employed: usually _N_-terminal lipidation is performed to increase activity. In doing so, we generated two groups of regioisomers of the lipidated peptides, allowing us to assess the structure–activity relationships (SARs) of both the position and length of the lipid with regard to antibacterial and hemolytic properties. Although it has been established that the attachment of lipids can increase the activity of AMPs, we wanted to determine to what extent the activity of a short (RW)3-peptide against Gram-positive bacteria could be altered and, more importantly, if its activity against Gram-negative bacteria could be enhanced.26−28 In addition, we intend to assess whether previously observed increased activities of organometallic-AMP conjugates were due to the redox-activity of the organometallic fragment or merely an effect of added lipophilicity.
Chart 1. Structures of the Two Groups of Lysine-Lipidated (RW)3-Peptides Describeda.

a R = FcCO (structure is given in the chart) or C(O)C_n_H2_n_+1 with n = 1, 3, 5, 7, 9, 11, or 13.
Synthesis of the lipidated peptides was performed using established Fmoc-based solid-phase peptide synthesis procedures (see the Supporting Information for details). The introduction of a highly acid-labile 4-methyltrityl (Mtt) protected lysine residue allowed selective modification of the side-chain amino-group. Ferrocenoyl or fatty acid derivatives with different chain lengths were then coupled using TBTU/HOBt and D_i_PEA in a mixture of 1,2-dichloroethane (DCE) and _N_-methyl-2-pyrrolidone (NMP). After cleavage of the lipidated peptides from the resin, all batches of products were found to be >90% pure by analytical HPLC. They were further purified by preparative HPLC, lyophilized, and tested for antibacterial activity (Table 1). The antibacterial activity of the prepared lysine-acylated (RW)3-peptides was tested against a representative panel of pathogenic bacteria: two strains of S. aureus (Gram-positive) and Escherichia coli, A. baumannii, and P. aeruginosa (Gram-negative); the nonpathogenic Gram-positive Bacillus subtilis was also included. As mentioned before, we were particularly interested in peptides that showed activity against P. aeruginosa and A. baumannii. For comparison, together with the fourteen lipidated peptides, the unmodified (RW)3-peptide, two nonacylated lysine-containing (RW)3-peptides, and two ferrocenoyl-labeled lysine-containing peptides were included in the activity screening.
Table 1. Minimum Inhibitory Concentrations (MIC) of (RW)3-Peptides Containing a Lipidated Lysine Side-Chain against Several Clinically Relevant Isolates of Gram-Positive and Gram-Negative Bacteria (MIC-Values Are Given in μM)a.
| | HPLC- analysis | Gram-negative bacteria | Gram-positive bacteria | | | | | | | | ---------------------------- | ---------------------- | ---------------------- | ------------------------ | ------------------------- | --------------------- | ---------------------- | ------------------------- | ------------------- | | peptideb | _t_R/min | E. coli DSM 30083 | A. baumannii DSM 30007 | P. aeruginosa DSM 50071 | S. aureus DSM 20231 | S. aureus ATCC 43300 | B. subtilis 168 DSM 402 | hemolysis 250 μg/mL | | (RW)3 | 17.3 | 21 | 85 | n.a. | 11 | 6 | 3 | <10% | | K(RW)3 | 16.7 | 18 | n.a. | n.a. | 37 | 9–18 | 2 | <10% | | N_-C2 | 16.9 | n.a. | n.a. | n.a. | n.a. | n.a. | 10 | <10% | | _N_-C4 | 17.4 | 38 | n.a. | n.a. | 38–75 | 19 | 2–5 | <10% | | _N_-C6 | 18.1 | 9 | 9–19 | 37–74 | 5–9 | 5 | 0.6–1.2 | <10% | | _N**_-C 8 | 19.0 | 5 | 9–18 | 9 | 2 | 1 | 0.6–1.1 | **>50% | | _N-C 10 | 19.9 | 5–9 | 9–18 | 9 | 2 | 1 | 2 | >50% | | _N_-C12 | 21.0 | 9–18 | 18–35 | n.a. | 4–9 | 2–4 | 9 | >50% | | _N_-C14 | 22.6 | 35 | 35 | n.a. | 9–17 | 17–35 | 9 | >50% | | (RW)3K | 16.5 | 18 | n.a. | n.a. | 37–74 | 18 | 5 | <10% | | _C_-C2 | 16.9 | n.a. | n.a. | n.a. | n.a. | n.a. | 10 | <10% | | _C_-C4 | 17.3 | 38 | n.a. | n.a. | n.a. | 38–75 | 5 | <10% | | _C_-C6 | 18.2 | 19 | 19–37 | 74 | 18–37 | 9 | 2 | <10% | | _C**_-C 8 | 19.1 | 2–5 | 5–9 | 18–37 | 2–5 | 2 | 0.6–1.1 | 10–20% | | C-C 10 | 20.2 | 2–5 | 5–9 | 9 | 1 | 1 | 0.6–1.1 | 50% | | C-C 12 | 21.4 | 4 | 9 | 18–35 | 1–2 | 1–2 | 1–2 | **>50% | | _C_-C14 | 22.4 | 17–35 | 9–17 | 70 | 2–4 | 2–4 | 4–9 | >50% | | N-FcCOc | 18.4 | 9–17 | 17 | n.a. | 4 | 2–4 | 1 | <10% | | C-FcCOc | 18.5 | 9–17 | 9–18 | n.a. | 9 | 4 | 2 | <10% |
Minimal inhibitory concentrations were determined according to CSLI guidelines (see the Supporting Information). In brief, serial dilutions of lipidated peptides were prepared in Mueller Hinton broth. The cultures were inoculated with 105 bacteria per milliliter and incubated at 37 °C for 18 h. The lowest concentration that inhibited visible growth is reported as MIC. Tests were performed twice independently.
The general trend is that lipidation increases the activity of a short active unnatural synthetic antimicrobial peptide (Table 1). Importantly, a sequence that is otherwise virtually inactive against Gram-negative pathogens becomes active upon the attachment of C6–C14 lipids. Within this window formed by the length of the lipid, the highest activities against a broad range of pathogens are found for compounds with C8 and C10. For example, introduction of a _C_-terminal lysine-residue acylated with a C8-lipid increases the activity against A. baumannii 40-fold. The lower activity of the peptides lipidated with C12 and especially C14 could be due to their poor solubility in the media used for the MIC-tests. Interestingly, attachment of the lipid at the _C_-terminal or _N_-terminal end of the peptide does not make a significant difference, although _C_-terminal lipidation is slightly favorable. In addition, the most active acylated peptides are rapid bactericidal at two times MIC-values; less than 1% survival was observed after 15 min of exposure to the peptides (see Table S2 of the Supporting Information). Control peptides (RW)3 and nonacylated peptide K(RW)3 showed 2.8% and 7% survival, respectively.
It is also interesting to note that the ferrocenoyl-derivatives—for which the lipophilicity lies in between C6 and C8 as judged from the retention times—have an activity that lies in between that of C6 and C8, except for P. aeruginosa. This indicates that for most bacteria the effect of this organometallic moiety on the activity predominantly results from its lipophilic character. Lastly, attachment of a lysine residue on either the _N_- or the _C_-terminus itself mostly reduces the activity of the (RW)3-sequence, making it less active than the parent (RW)3-sequence. Also, acetylation of the lysine side-chain residue suppresses the activity in most cases.
After this screening for antibacterial activity, we assessed the hemolytic potential of these peptides using a high concentration of lipopeptide, i.e. 250 μg/mL (Table 2). For this, a hemolytic assay was used in which leakage of hemoglobin from human red blood cells (hRBCs) as a result of exposure to the lipidated AMPs is assessed (see the Supporting Information for detailed information on the experiment). Percentages were calculated using leakage caused by 5% DMSO as the baseline and leakage caused by 1% triton X-100 as the 100%-level. Fortunately, the _C_-C8 peptide only causes 10–20% hemolysis at this high concentration of 285 μM, which is 8–16 times higher than the MIC-value against P. aeruginosa and 142 times higher than the MIC-value against the resistant Gram-positive S. aureus (type ATCC 43300). Since both _N_-C8 and _C_-C8 were considered as the most interesting candidates for further analysis, we determined their 50%-hemolysis concentration (the HC50 value): these were 200 μg/mL (114 μM) for _N_-C8 and 500 μg/mL (285 μM) for _C_-C8. Furthermore, we assessed the kinetics of hemolysis of these two peptides, which could be relevant in view of the rapid degradation and clearance rate of small peptides from the serum. To this end, hRBCs were mixed with 500 μg/mL of either _C_-C8 or _N_-C8 and, after 5, 10, 20, 30, and 60 min samples were taken, treated and analyzed (see the Supporting Information). The end-point was taken at 60 min, and this value was set at 100% hemolysis even though not all hRBCs were lysed. From this experiment, it became clear that most hemolysis occurs in the initial stages of the experiment, indicating surfactant-like properties of these peptides at the high concentrations used. Specifically, in the first 5 min 48% of all hRBCs that are lysed in 1 h were already destroyed using _C_-C8; for _N_-C8 this number is almost double of that with 88%. Clearly, the _N_-C8 peptide is more hemolytic than the _C_-C8 peptide, each of which are more hemolytic than the parent (RW)3 sequence. Apparently, the presence of a positively charged _N_-terminus next to the membrane-anchoring lipid in the _N_-series results in faster and more efficient hemolysis. This might be due to the fact that hRBCs are richly coated with sialic-rich glycoproteins and are hence negatively charged.29
Table 2. Comparison of MIC and HC50 Concentrations (in μM) of _N_-C8 and _C_-C8a.
| peptide | MIC | HC50 | HC50/MIC |
|---|---|---|---|
| _N_-C8 | 5–18 (−), 0.6–2 (+) | 114 | 6 (−), 57 (+) |
| _C_-C8 | 2–37 (−), 0.6–5 (+) | 285 | 8 (−), 57 (+) |
Finally, the effect of _N_-C8 and _C_-C8 on the cell-viability of two cancer-cell lines (MCF7 and HT29) and against healthy fibroblast cells was assessed. For this, solutions with increasing amounts of the lipidated peptides were applied after 12 h of preincubation of the cells (in duplicate). After incubation with the peptides for 48 h, the crystal violet assay30 was performed in order to determine the cell numbers. The relative cell numbers were calculated as the percentage absorption of treated cells compared to untreated cells. Concerning the cell-viability of the cancer cell-lines when exposed to either _C_-C8 or _N_-C8, both of these peptides were very active (Table 3). At only 4–5 μM of the peptide, 50% of the cells were dead, which is a strong indication that these two peptides are very toxic to malignant eukaryotic cells, although hardly hemolytic at those low concentrations. Against nonmalignant fibroblast-cells (GM5657), IC50-values of 31–33 μM, i.e. seven times higher than those against the two tested cancer-cell lines, were observed. These concentrations are in the range of the MIC-values observed against P. aeruginosa but they are 15–30 times higher than the lowest MIC-values (against S. aureus). Interestingly, whereas a large difference was observed in the hemolytic activity of the two regioisomers of the C8-lipidated peptides, in these cell-culture tests no significant difference was observed.
Table 3. Cell Viability of MCF7, HT29, and Fibroblast (GM5657) Cells in the Presence of _N_-C8 or _C_-C8 (IC50, in μM).
| peptide | MCF7 | HT29 | fibroblast |
|---|---|---|---|
| _N_-C8 | 4.6 | 4.3 | 32.5 |
| _C_-C8 | 4.7 | 4.5 | 31.3 |
In conclusion, attachment of a side-chain lipidated lysine residue on a short (RW)3-sequence substantially increases its activity against both Gram-positive and -negative bacteria. Interestingly, it appears that the increased activity of a ferrocenoyl-conjugated peptide, as observed in other organometallic AMPs, is primarily due to the added lipophilicity rather than to the redox-properties of the metal-center. The retention times of the Fc-derivatized AMPs are in between those of the C6- and C8-lipidated AMPs, a trend that is roughly followed by the MIC-values. Importantly, with activities ranging from 37 to 0.6 μM, both _N_- and _C_-terminally C8-lipidated peptides show promising activity against a broad spectrum of bacterial pathogens, including P. aeruginosa and A. baumannii. For these two peptides a significant difference in hemolytic activity was observed, of which the _C_-terminally lipidated peptide had an HC50 value of 285 μM. In addition to this, IC50-values of these two regioisomers of the C8-lipidated peptides equally showed moderate selectivity activity against cancer cells over healthy fibroblast cells; IC50-values were seven times higher for the healthy cells. Whether these peptides retain therapeutic potential in animal model systems remains to be determined. Depending on this outcome, these peptides can find applications in a therapeutic setting or will be limited to a technological setting.
Acknowledgments
We thank Annegret Knüfer for determining the IC50-values.
Glossary
Abbreviations
D_i_PEA
N,_N_′-diisopropylethylamine
HOBt
1-hydroxybenzotriazole
TBTU
_O_-(benzotriazol-1-yl)-N,N,_N_′,_N_′-tetramethyluronium tetrafluoroborate
DCE
1,2-dichloroethane
NMP
_N_-methylpyrrolidone
DMSO
dimethylsulfoxide
Fmoc
9-fluorenylmethoxycarbonyl
ESI-MS
electron spray ionization mass spectrometry
HPLC
high pressure liquid chromatography
R
arginine
W
tryptophan, K, lysine
Supporting Information Available
Detailed synthetic procedures, high-resolution ESI-MS data, HPLC chromatograms. and details on the biological assays. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
All authors have given approval to the final version of the manuscript.
Financial support is from a grant from the state of North Rhine-Westphalia (NRW) and the European Union, European Regional Development Fund “Investing in your future”, to J.E.B. and N.M.N. (“Innovative Antibiotics from North-Rhine Westfalia”).
The authors declare no competing financial interest.
Supplementary Material
References
- See, for example, the European Antimicrobial Resistance Surveillance Network at http://ecdc.europa.eu/ or its North-American counterparts ABCs (http://www.cdc.gov/abcs/index.html) and NARMS-EB (http://www.cdc.gov/narms/index.htm).
- Gibson R. L.; Burns J. L.; Ramsey B. W. Pathophysiology and management of pulmonary infections in cystic fibrosis. Am. J. Respir. Crit. Care Med. 2003, 168, 918–953. [DOI] [PubMed] [Google Scholar]
- Karageorgopoulos D. E.; Falagas M. E. Current control treatment of multidrug-resistant Acinetobacter baumannii infections. Lancet Infect. Dis. 2008, 8, 751–762. [DOI] [PubMed] [Google Scholar]
- Pollack A.Rising threat of infections unfazed by antibiotics; New York Times, February 26, 2010. [Google Scholar]
- Dijkshoorn L.; Nemec A.; Seifert H. An increasing threat in hospitals: multidrug-resistant Acinetobacter baumannii. Nat. Rev. Microbiol. 2007, 5, 939–951. [DOI] [PubMed] [Google Scholar]
- Bassetti M.; Righi E.; Esposito S.; Petrosillo N.; Nicolini L. Drug treatment for multidrug-resistant Acinetobacter baumannii infections. Future Microbiol. 2008, 3, 649–660. [DOI] [PubMed] [Google Scholar]
- Zasloff M. Antimicrobial peptides of multicellular organisms. Nature 2002, 415, 389–395. [DOI] [PubMed] [Google Scholar]
- Hancock R. E. W.; Sahl H.-G. Antimicrobial and host-defense peptides as new anti-infective therapeutic strategies. Nat. Biotechnol. 2006, 24, 1551–1557. [DOI] [PubMed] [Google Scholar]
- Antimicrobial Peptides: Discovery, Design and Novel Therapeutic Strategies; Wang G., Eds.; CABI: Wallingford, England, 2010. [Google Scholar]
- Arnusch C. J.; Bonvin A. M. J. J.; Verel A. M.; Jansen W. T. M.; Liskamp R. M. J.; De Kruijff B.; Pieters R. J.; Breukink E. The vancomycin-nisin(1-12) hybrid restores activity against vancomycin resistant enterococci. Biochemistry 2008, 47, 12661–12663. [DOI] [PubMed] [Google Scholar]
- Albada H. B.; Arnusch C. J.; Branderhorst H. M.; Verel A.-M.; Janssen W. T. M.; Breukink E.; De Kruijff B.; Pieters R. J.; Liskamp R. M. J. Potential scorpionate antibiotics: Targeted hydrolysis of lipid II containing model membranes by vancomycin-TACzyme conjugates and modulation of their antibacterial activity by Zn-ions. Bioorg. Med. Chem. Lett. 2009, 19, 3721–3724. [DOI] [PubMed] [Google Scholar]
- Griffin J. H.; Linsell M. S.; Nodwell M. B.; Chen Q.; Pace J. L.; Quast K. L.; Krause K. M.; Farringon L.; Wu T. X.; Higgins D. L.; Jenkins T. E.; Christensen B. G.; Judice J. K. Multivalent drug design. Synthesis and in vitro analysis of an array of vancomycin dimers. J. Am. Chem. Soc. 2003, 125, 6517–6531. [DOI] [PubMed] [Google Scholar]
- Kerns R.; Dong S. D.; Fukuzawa S.; Carbeck J.; Kohler J.; Silver L.; Kahne D. The role of hydrophobic substituents in the biological activity of glycopeptide antibiotics. J. Am. Chem. Soc. 2000, 122, 12608–12609. [Google Scholar]
- Makovitzki A.; Avrahami D.; Shai Y. Ultrashort antibacterial and antifungal lipopeptides. Proc. Natl. Acad. Sci. U.S.A. 2006, 103, 15997–16002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makovitzki A.; Baram J.; Shai Y. Antimicrobial lipopeptides composed of palmitoyl di- and tricationic peptides: in vitro and in vivo activities, self-assembly to nanostructures, and a plausible mode of action. Biochemistry 2008, 47, 10630–10636. [DOI] [PubMed] [Google Scholar]
- Chu-Kung A. F.; Bozzelli K. N.; Lockwood N. A.; Haseman J. R.; Mayo K. H.; Tirrell M. Promotion of peptide antimicrobial activity by fatty acid conjugation. Bioconjugate Chem. 2004, 15, 530–535. [DOI] [PubMed] [Google Scholar]
- Majerle A.; Kidric J.; Jerala R. Enhancement of antibacterial and lipopolysaccharide binding activities of a human lactoferrin peptide fragment by the addition of acyl chain. J. Antimicrob. Chemother. 2003, 51, 1159–1165. [DOI] [PubMed] [Google Scholar]
- Laverty G.; McLaughlin M.; Shaw C.; Gorman S. P.; Gilmore B. F. Antimicrobial activity of short, synthetic cationic lipopeptides. Chem. Biol. Drug. Des. 2010, 75, 563–569. [DOI] [PubMed] [Google Scholar]
- Arnusch C. J.; Albada H. B.; Van Vaardegem M.; Liskamp R. M. J.; Sahl H.-G.; Shadkchan Y.; Osherov N.; Shai Y. Trivalent ultrashort lipopeptides are potent pH dependent antifungal agents. J. Med. Chem. 2012, 55, 1296–1302. [DOI] [PubMed] [Google Scholar]
- Chantson J. T.; Falzacappa M. V. V; Crovella S.; Metzler-Nolte N. Solid-phase synthesis, characterization, and antibacterial activities of metallocene-peptide conjugates. ChemMedChem 2006, 1, 1268–1274. [DOI] [PubMed] [Google Scholar]
- Chantson J. T.; Falzacappa M. V. V; Crovella S.; Metzler-Nolte N. Antibacterial activities of ferrocenoyl- and cobaltocenium-peptide bioconjugates. J. Organomet. Chem. 2005, 690, 4564–4572. [Google Scholar]
- Patra M.; Gasser G.; Metzler-Nolte N. Small organometallic compounds as antibacterial agents. Dalton Trans. 2012, 41, 6350–6358. [DOI] [PubMed] [Google Scholar]
- Pollaro L.; Heinis C. Strategies to prolong the plasma residence time of peptide drugs. Med. Chem. Commun. 2010, 1, 319–324. [Google Scholar]
- Veronese F. M.; Pasut G. PEGylation, successful approach to drug delivery. Drug Discovery Today 2005, 10, 1451–1458. [DOI] [PubMed] [Google Scholar]
- Havelund S.; Plum A.; Ribel U.; Jonassen I.; Volund A.; Markussen J.; Kurtzhals P. The mechanism of protraction of insulin detemir, a long-acting, acylated analog of human insulin. Pharm. Res. 2004, 21, 1498–1504. [DOI] [PubMed] [Google Scholar]
- Strøm M. B.; Rekdal Ø.; Svendsen J. S. Antimicrobial activity of short arginine- and tryptophan-rich peptides. J. Peptide Sci. 2002, 8, 431–437. [DOI] [PubMed] [Google Scholar]
- Strøm M. B.; Haug B. E.; Skar M. L.; Stensen W.; Stiberg T.; Svendsen J. S. The pharmacophore of short cationic antibacterial peptides. J. Med. Chem. 2003, 46, 1567–1570. [DOI] [PubMed] [Google Scholar]
- Liu Z.; Brady A.; Young A.; Rasimick B.; Chen K.; Zhou C.; Kallenbach N. R. Length effects in antimicrobial peptides of the (RW)n series. Antimicrob. Agents Chemother. 2007, 51, 597–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jan K.-M.; Chien S. Role of surface charge in red cell interactions. J. Gen. Physiol. 1973, 61, 638–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernhardt G.; Reile H.; Birnböck H.; Spruß T.; Schönenberger H. Standardized kinetic microassay to quantify differential chemosensitivity on the basis of proliferative activity. J. Cancer Res. Clin. Oncol. 1992, 118, 35–43. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.