Single-molecule Dynamics of Enhanceosome Assembly in Embryonic Stem Cells (original) (raw)
. Author manuscript; available in PMC: 2014 Sep 13.
SUMMARY
Enhancer-binding pluripotency regulators (Sox2 and Oct4) play a seminal role in embryonic stem (ES) cell specific gene regulation. Here, we combine in vivo and in vitro single-molecule imaging, transcription factor (TF) mutagenesis and ChIP-exo mapping to determine how TFs dynamically search for and assemble on their cognate DNA target sites. We find that enhanceosome assembly is hierarchically ordered with kinetically favored Sox2 engaging the target DNA first, followed by assisted binding of Oct4. Sox2/Oct4 follow a trial-and-error sampling mechanism involving 84~97 events of 3D diffusion (3.3~3.7_s_) interspersed with brief non-specific collisions (0.75~0.9_s_) before acquiring and dwelling at specific target DNA (12.0~14.6_s_). Sox2 employs a 3D diffusion-dominated (~80 % of time) search mode facilitated by 1D sliding along open DNA to efficiently seek for targets. Our findings also reveal fundamental aspects of gene and developmental regulation by fine tuning TF dynamics and influence of the epigenome on TF search parameters.
INTRODUCTION
Precise spatiotemporal regulation of gene expression underpins the finely balanced lineage-specification and morphogenetic events during early embryonic development (Ben-Tabou de-Leon and Davidson, 2007; Levine and Tjian, 2003; Tam and Loebel, 2007). Although a powerful combination of classical biochemistry, genetics and genomic approaches (Consortium et al., 2012; Levine and Tjian, 2003) has revealed many aspects of mammalian gene regulation, the kinetic principles that govern transcription factor (TF) dynamics as it searches for specific target sites in the nucleus of living cells remained elusive. With recent advances in molecular imaging, it has become possible to track individual protein molecules in single live cells (Abrahamsson et al., 2013; Elf et al., 2007; Gebhardt et al., 2013; Mazza et al., 2012). These rapidly emerging super-resolution platforms provide a means for elucidating the search pattern and efficiency of TFs in finding and binding specific target sites.
It has been appreciated that in a fractal and compact nuclear environment, a TF must manage to search for its specific binding sites while colliding and navigating past many non-specific decoy sites (Fudenberg and Mirny, 2012; Mirny et al., 2009; Mueller et al., 2013). However, to date, the dynamic balance between specific and non-specific binding events of individual TF molecules in mammalian cells remains largely unknown limiting our understanding of TF search modalities. Likewise, the order of events of TF transactions and mechanisms that direct multiple TFs homing in on a cis-regulatory DNA element to form an enhanceosome complex have been challenging to dissect.
Here, we report a single-cell single-molecule imaging strategy that allows us to quantitatively measure in individual live cells both the specific and non-specific TF residence times on chromatin DNA and thus compute the “in vivo_” target search time. We developed our approach by tracking the DNA binding dynamics of two key pluripotency regulators, Sox2 and Oct4, in mouse embryonic stem cells as well as in a reconstituted in vitro single molecule binding assay. These single-molecule tracking results were complemented with an analysis of TF mutants and genome-wide TF binding studies to establish that the search for a specific DNA target follows a trial-and-error sampling mechanism: each TF undergoes multiple rounds of short-lived non-specific chromatin binding events (τns =0.75 ~ 0.9_s) punctuated by 3D diffusion episodes (τ_3_D = 3.3 ~ 3.7_s_) before eventually encountering a specific DNA target to which it binds more stably. Interestingly, we also observe that Sox2 slides along short stretches of DNA non-specifically searching for target sites in vitro and that Sox2 and Oct4 assemble on their in vivo enhancer sites in a hierarchically ordered fashion with Sox2 serving as the lead factor that first engages with its specific DNA target followed by assisted binding of Oct4 that subsequently stabilizes the ternary complex. We also show by inducing specific chromatin modifications that the global epigenetic state of ES cells influences the search parameters and kinetics of Sox2 and Oct4 DNA binding. Our results thus reveal important in vivo kinetic properties controlling TF dynamics and unmask new enhanceosome assembly mechanisms underlying key pluripotency transcription programs.
RESULTS
Imaging TF-DNA Dissociation Kinetics
We harnessed both epi-illumination and Bessel plane illumination (Gao et al., 2012; Planchon et al., 2011) for 2D Single Molecule Tracking (SMT). In order to selectively track DNA-bound Sox2 molecules, we imaged live ES cells expressing a fluorescently tagged Sox2 (halo-TMR labeled) using a combination of low excitation power (50 W cm−2) and long integration times (500 ms) (Figure 1A). As a result, the blurred images of fast diffusing molecules blend into the background, while less mobile Sox2 molecules that are interacting with chromatin appeared as bright individual diffraction-limited spots (Figure 1A–B and Movie S1–2). The dwell time of each “immobile” Sox2 single molecule was then directly measured as the lifetime of the fluorescence spot as demonstrated by single step of photobleaching. We found that dwell times of engaged Sox2 molecules failed to be described by a single-component decay model (Figure S1D). However, a two-component exponential decay model was in good agreement with our data (Figure S1E), with lifetimes of 0.8 s and 12.03 s for the short- and long-lived population respectively (after photobleaching correction; Figure S1A–B, Eq. S2–4). In order to test whether the two classes of relatively “immobile” particles corresponded to non-specific and specific DNA-bound Sox2 molecules, we deleted the Sox2 DNA binding domain (Sox2-TAD, Sox2 121–319aa, Figure S5A) and tracked its movement. Removal of the DNA binding domain resulted in the disappearance of the long-lived population from the dwell time histograms of the truncated protein, suggesting that long-lived immobile particles most likely correspond to Sox2 occupying its specific DNA target sequences (Figure 1D). Mutation of amino acids on the Sox2-DNA binding surface (Sox2M, M47G:F50:M51G, Figure S5A) also reduced the fraction and lifetime of the long-lived population (Figure 1E–F and Table S1). Both epi-illumination and Bessel plane illumination techniques gave convergent results, ruling out any bias induced by the imaging modality (Figure S2). To test whether some artifact might have been introduced to our non-specific residence time measurements by the 500ms acquisition time, we applied a time-lapse imaging method described by Gebhardt et al. (Gebhardt et al., 2013) to independently characterize the nonspecific chromatin binding events of both halo-Sox2 and a control protein halo-NLS. We found that halo-Sox2 and halo-NLS non-specifically interact with chromatin with residence times of 0.75 s and 0.19 s respectively (Figure S1F–G). Importantly, the halo-Sox2 nonspecific residence time (0.75 s) derived from this method agreed well with our 500ms long acquisition measurements (~0.74~0.9 s). Furthermore, two photon FCS measurements revealed that compared to ~23.5% bound halo-Sox2 molecules, only ~3% halo-NLS molecules are bound to chromatin in the live ES cells (Figure S7E). Both the shorter non-specific residence time (0.19 s) and the much smaller bound faction (~3%) of the control halo-NLS protein suggest that the shorter-lived (0.75 s) component we observed in our 2D SMT experiments (Figure 1A–C) are likely due to intrinsic nonspecific interactions of Sox2 with chromatin and cannot be accounted for by the presence of the halo-tag. Together, these observations suggest that the long-lived component (~12sec) likely reflects the residence time of Sox2 at specific DNA binding sites while the short-lived component is most likely due to non-specific Sox2 protein-DNA or protein-protein interactions.
Figure 1. Discriminating Specific and Non-specific TF-DNA Dissociation Kinetics in Live Cells by Single-molecule Imaging.

(A) Selective visualization of immobile Sox2 molecules by 2D imaging using long exposure times (low excitation power: 50 W/cm2 and long integration time: 500 ms). Fast particles blend into the background while immobile ones appear as spots. The dissociation rate (koff) is extracted from the measured residence time.
(B) Immobile Sox2 molecules imaged with the 2D imaging set-up in ES cells. Top left: Immobile single Sox2 molecules are detected as near diffraction-limited spots in the nucleus. Top right: At 500 ms, halo-tag-NLS (Nuclear Localization Signal) displays negligible stable binding but diffused fluorescence background. Bottom left: A construct lacking the DNA binding domain (Sox2-TAD) displays a great reduction in the number of immobile molecules. Bottom right: At faster frame-rates (10ms), freely diffusing halo-tag-NLS molecules can now be detected as spots. Yellow dotted circle represents the nucleus outline. Scale bar: 2 μm.
(C–D) 1-Cumulative Distribution Function (1-CDF) of Sox2 (D) and Sox2-TAD (E) residence time was respectively fitted with a two-component (long-lived and short-lived component) and a single-component exponential decay model. For wtSox2, the fitted lifetimes are _τ_1 = 12.03 ± 1.8 _s (_long-lived component) and _τ_2 = 0.8 ± 0.07 _s (_short-lived component). In the case of Sox2 DNA binding domain deletion (Sox2-TAD), τ = 0.75 ± 0.03 s.
(E) Residence lifetime of the long-lived component for Sox2 (12.03 ± 1.8 s), a Sox2 construct with mutations on Sox2-DNA binding surface (Sox2M) (9.11 ± 1.93 s) and Sox2D (8.62 ± 0.98 s) and mean residence lifetime for Sox2-TAD (0.75 ± 0.03 s).
(F) Long-lived bound fraction of all bound molecules for Sox2, Sox2M and Sox2D determined by our 2D dwell time analysis. (See Figure S2 and Eq. S1 for details.)
*: p<0.05. Error bars represent SD.
See also Figures S1 and S2, Movies S1 and S2 and Table S1.
To further verify this interpretation, we reconstituted a minimal in vitro purified TF system to study Sox2-DNA interaction kinetics with surface attached specific and non-specific DNA at single-molecule resolution by TIRF (Revyakin et al., 2012). Single-molecule trace analysis confirmed that the average residence time of Sox2 on a DNA probe containing a canonical Sox2 binding site is 16.9 s while the average residence time on a non-specific probe is 0.9_s_ (Figure 2A–B, Movie S3A–B). These numbers are remarkably consistent with our “_in vivo_” residence time measurements, confirming that our 2D imaging analysis successfully resolved specific from non-specific TF-DNA dissociation kinetics in live cells.
Figure 2. Distinct Sox2 Residence Time Distributions at Specific versus non-specific DNA sequences and Sox2 sliding on DNA measured by in vitro single molecule binding.
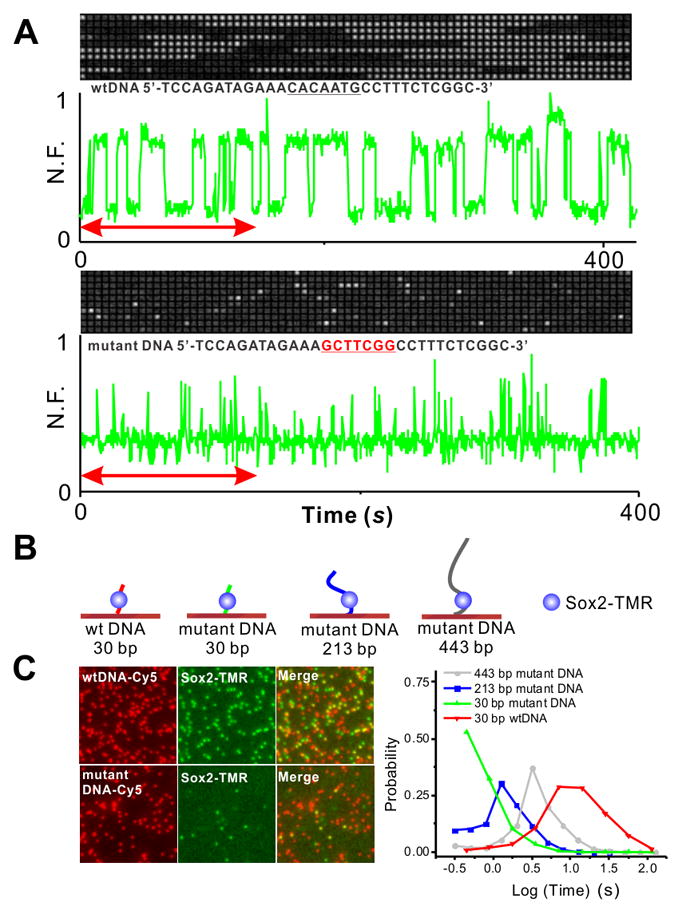
(A) In vitro single-molecule imaging to determine Sox2 specific and non-specific residence time on DNA. Single-molecule fluorescence traces of halo-TMR Sox2 molecule interacting with surface-tethered wild-type DNA (Upper panel) and mutant DNA (Bottom panel) (N.F. – Normalized Fluorescence) are presented along with the corresponding raw images (The red arrow on the graph indicates the time interval displayed on top of each plot).
(B) Schematic of Sox2 interaction with wild type DNA probe (30 bp) and different lengths mutant DNA probes (30 bp, 213 bp and 443 bp) measured by in vitro single molecule imaging.
(C) Left panel: Co-localization of wild type DNA probe (Top) or mutant DNA probe (Bottom) (cy5 channel) with TMR-halo-Sox2 molecules (TMR channel).
Right panel: histogram of TMR-halo-Sox2 residence time on 30 bp (Green), 213 bp (Blue) and 443 bp mutant DNA (Gray) and 30 bp wild-type (Red). Mean residence time of Sox2 on the 30 bp wild-type DNA is 16.9 s; that on the 30 bp, 213 bp and 443 bp mutant DNA are 0.9 s, 1.6 s and 4.5 s.
See also Figure S3, Movie S3 and Table S1.
Transcription Factor Target Search Mechanism
To dissect the association kinetics of TF binding to DNA, we turned to multi-focus microscopy with fast 3D single-molecule tracking (Abrahamsson et al., 2013) that is able to acquire simultaneous whole-nucleus imaging in live ES cells (9 focal plates, 4μm in z) at a 33Hz frame rate (Figure 3A and S4A, movie S4). This allowed us to track individual Sox2 particles undergoing rapid diffusion, DNA/chromatin binding, or a combination of these activities (Figure 3B). In order to attribute the contributions of the free and DNA-bound states to the observed trajectories, we developed a kinetic model based on a previously established 2D single molecule tracking (SMT) analysis (Mazza et al., 2012) incorporating both 3D diffusion and DNA/chromatin binding that was then fitted to our SMT measurements (Details see Supplementary Information, Eq. S5–13). Our model assumes that TFs alternate between a freely diffusing and a chromatin bound state. We set the dissociation rate from the bound state (koff) to the average value obtained from our dwell time histograms (Figure 1C). The observed association rate (k*on), bound fraction in the whole population (Ceq), and the diffusion coefficients of the free (Df) and bound species (Db) were derived through fitting (Figure 3C, Table S1). The diffusion coefficient for the bound population was in the range of 0.1–0.2 μm2/s, closely matching the diffusion coefficient of immobile H2B molecules in the nucleus, consistent with a stable DNA-bound state (Table S1, Figure S4B). Applying this kinetic model to Sox2 yielded an average duration of the freely diffusing state of 1/k*on ~ 3.7 s (Figure 4C, Table S1), which is the average time (τ_3_D) Sox2 spends diffusing in the nucleus between two association events.
Figure 3. TF-Chromatin Association Kinetics Determined by Fast 3D Single-molecule Imaging.

(A) Fast 3D tracking of TF movement by simultaneous Multi-focus microscopy (axial coverage, 4um; 33Hz). The average association rate (K_on_) is determined through fitting the displacement histogram with a 3D kinetic model (Details see Supplementary information, Eq. S5–13).
(B) Volume rendering of 3D Sox2 single-molecule image (purple) superimposed with single-molecule trajectories. Three molecules with distinct behaviors were selectively displayed on the right (from top to bottom: freely diffusing particle; particle undergoing a free/bound transition; immobile molecule). Color bar shows the corresponding frame number. Scale bar: 2 μm.
(C) 3D displacement histogram fitted by our 3D kinetic model (Eq. S11) for the indicated factor. Histogram bin was set as 30 nm. The τ_3_D is equal to 1/kon∗ of different TFs was calculated through Eq. S9–12.
See also Figure S4, Movie S4 and Table S1.
Figure 4. A Trial-and-error TF Target Search Mechanism.
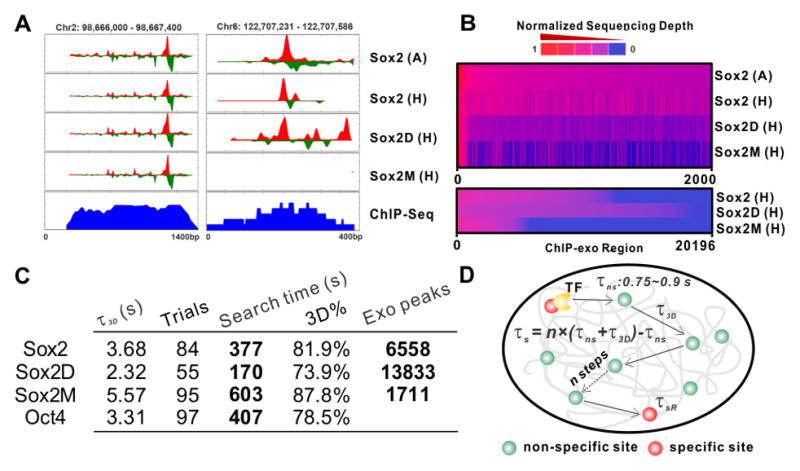
(A) Representative ChIP-exo tracks for Sox2 (Antibody), Sox2 (Halo), Sox2D (Halo) and Sox2M (Halo) at two different enhancer regions. Sox2 ChIP-seq data was previously published by Chen et. al (Chen et al., 2008). Auto-scale was applied to each track.
(B) Correlation analysis of ChIP-exo data. Upper, enrichment heat-map for all factors ranked by descending order of Sox2 (antibody) enrichment (top 2000 loci). Bottom, heat-map for Sox2 (halo), Sox2D (halo) and Sox2M (halo) showing their own enrichment in the same data set. The read counts within ±100bp regions of exo-peaks were taken into account in this analysis.
(C) Calculated duration of the 3D free diffusion state (τ_3_D), the number of trials, the target search time, the ratio that a TF spends in 3D diffusion (3D%) and the number of binding sites determined from genomic analysis (Exo-peak) for Sox2, Sox2D, Sox2M and Oct4. The enrichment cut-off for ChIP-exo peak-pairs is >12 reads for both the left and the right peak (Also see Figure S5C).
(D) The model of in vivo target search - a TF goes through on average n episodes of 3D diffusion (τ_3_D = 2.0 ~ 5.6 s, Table S1) interspersed by non-specific binding at random accessible chromatin sites (τns = 0.75 ~ 0.9s, Table S1) before reaching a specific site. τs and τsR denote the search time and the specific residence time, respectively.
See also Figures S5 and S6, and Table S1.
Using the measured ratio of specific to non-specific binding events (Figure 1D), we computed the average number of random trials before a TF molecule reaches a cognate target site (Figure 4D, Table S1, Eq. S14–17) and found that for Sox2 it is ~84 collisions. This means that after leaving a specific target, a Sox2 molecule on average samples ~ 83 non-specific sites in ES cells before reaching another cognate target site where it stably binds. Combining the information that includes the number of trials, the non-specific residence time and the τ_3_D (Eq. S18), we calculated a search time of ~377.2 s (Figure 4C–D) for Sox2 to find and bind to a specific recognition site in the chromatin of ES cells. It has previously been proposed that TF target search follows a 1D–3D facilitated diffusion model in which TFs undergo multiple rounds of 1D search (local sliding and hopping) interspersed with 3D jumps before reaching a specific site (Berg et al., 1981; Hager et al., 2009; Slutsky and Mirny, 2004). To test whether 1D sliding might also contribute to nonspecific Sox2 interactions with DNA, we performed in vitro Sox2 single-molecule binding assays with varying lengths of nonspecific DNA templates (30, 213 and 443bp). Consistent with a 1D sliding mode, we observed that Sox2 dwell times increased as a function of DNA template length (Figure 2B–C and S3B, Movie S3C–D). These in vitro binding results suggest that the in vivo non-specific chromatin binding events we observed in ES cells may also contain a 1D sliding component that could contribute to a more efficient target search by sampling short stretches of native open chromatin regions (ie. DNAase hypersensitive sites) during nonspecific collisions (Figure 2B–C).
Complementing SMT with High-resolution ChIP-exo and Sox2 Mutants
To validate our kinetic model, we next performed 2D/3D SMT experiments with Sox2 mutant proteins. As expected, mutations interfering with the protein-DNA interaction domain of Sox2 (Sox2M) significantly increased the τ_3_D (from 3.6s to 5.6_s_), the number of encounters (from 83 to 95 trials) and the search time (from 377 s to 603.2 s) (Figure, 4C, Table S1). Consistent with these observations, the Sox2M DNA bound fraction (C_eq_ = 23.5%) and long-lived bound faction (Fl = 11.3%) were also significantly decreased compared with the wild type Sox2 protein (C_eq_ = 36.9%, Fl = 15.1%) (Table S1). These data suggest that the number of Sox2M specific binding sites is substantially decreased in the cells. By contrast, deletion of the trans-activation domain (Sox2D, Sox2 1 – 120aa) accelerated the τ_3_D from ~3.7 s to ~2.3 s but decreased the number of trials from 83 to ~ 55, which together result in a much shortened search time of 170.2 s. As a result, the Sox2D DNA bound fraction (C_eq_ = 43.6%) was also significantly increased (Figure 1F, Table S1). We next confirmed that the shortened τ_3_D and search times observed for Sox2D were not merely due to a change in the DNA association rate (kon) as determined independently by surface plasmon resonance measurements (Figure S6). These results taken together suggest that the Sox2D activation domain deletion mutant binds to many more “pseudo-target” sites than WT Sox2 in the nucleus of ES cells.
As an independent and complementary test of the findings from our imaging experiments, we carried out high-resolution genome-wide mapping of TF binding sites for Sox2, Sox2M and Sox2D with the recently-developed ChIP-exo technique (Rhee and Pugh, 2011). We detected 6558 sites bound by halo-Sox2, comparable to the number of endogenous Sox2 sites (7153) detected by a Sox2 antibody. Our ChIP-exo derived halo-Sox2 binding enrichment profile also correlated well with endogenous Sox2 binding sites (Figure 4A–B), suggesting that halo-Sox2 binds to DNA in a manner similar to WT endogenous Sox2. By contrast, we found only 1711 sites bound by the DNA binding mutant Sox2M, ~ 3 fold less than the sites bound by Sox2. On the other hand, we detected 13833 binding sites for the activation domain mutant Sox2D, almost twice as many as WT Sox2-binding sites (Figure 4C). In line with this observation, a DNA sequence motif analysis revealed that, compared with Sox2, Sox2D binds with greater frequency to degenerate DNA-binding sequences (Figure S5D). Importantly, these genome-wide binding studies underscore a remarkable consistency between our single-molecule imaging results and more conventional genome-wide bulk binding assays thus confirming the robustness of the 3D SMT kinetic model. The SMT imaging analysis presented here therefore enabled us for the first time to fully characterize the dynamic aspects of specific and non-specific TF-DNA binding kinetics in live cells.
Hierarchically Ordered Enhanceosome Assembly
Sox2 and Oct4 are thought to bind to closely spaced composite DNA binding site that can serve as a nucleating element for the assembly of an enhanceosome complex and thus function coordinately as a key enhancer module regulating ES cell pluripotency (Avilion et al., 2003; Chen et al., 2008; Nichols et al., 1998; Remenyi et al., 2003). Our single-molecule imaging technique provides a direct way to dissect potential order of events during enhanceosome assembly on chromatin/DNA in the nucleus of living cells. To discern whether Sox2 and Oct4 interact with their targets in a random or ordered fashion (Hager et al., 2009), we first designed a 3T3 cell line that stably expressed halo-Sox2 protein together with an inducible expression system for Oct4 (Figure 5A). Since the 3T3 cell line does not normally express either Sox2 or Oct4, it was possible to express Sox2 and/or Oct4 without any background of competing endogenous TFs. In our inducible transgenic system, GFP was co-expressed on the same transcription unit as Oct4 by use of an Internal Ribosome Entry Site (IRES) that provided a convenient way to correlate green fluorescence intensities in each cell with levels of Oct4 expression. We then tracked Sox2 binding behavior in cells containing or lacking Oct4. With Oct4 over-expression, we only observed a modest increase in Sox2 specific residence time (from 11.6 s to 14.1 s); the Sox2 long-lived bound faction largely remained the same while its τ_3_D and search times showed slight increases (Figure 5A, movie S5). These results suggested that Oct4 mainly helps stabilize the binding of Sox2 to DNA but has little ability to assist Sox2 in its target search. The slightly increased Sox2 target search time in the presence of Oct4 suggests that non-productive Oct4 occupancy may, at times, mask target sites and hinder Sox2 binding. By contrast, when we performed the converse experiment by inducing Sox2 expression, we found substantial decreases in Oct4 τ_3_D (from 3.3 s to 2.6 s) and search times (from 366.6 s to 158.9 s), as well as significant enhancement of its long-lived bound fraction (from 10.7 % to 18.4 %) (Figure 5B, movie S6). Taken together, these results support a model in which Sox2 engages with the chromatin first and primes the target site for subsequent Oct4 binding (Figure 6E). The Sox2 assisted binding of Oct4, in turn, appears to help stabilize the Sox2-Oct4 complex at composite recognition sites. This stepwise ordered assembly at endogenous chromatin sites also suggests that Sox2 and Oct4 do not form a complex prior to binding DNA, consistent with a previous report of DNA-dependent interactions between Sox2 and Oct4 in live cells (Lam et al., 2012).
Figure 5. Sox2 Assists Oct4 Target Search.

(A–B) Sox2 and Oct4 Induction experiment in 3T3 cells. (A), Halo-Sox2 was stably expressed downstream of a CMV promoter. The expression of Oct4 and GFP (IRES) was under the control of an inducible Cumate switch. Thus, GFP is an expression indicator for Oct4. Left: images of Sox2 2D SMT experiments performed with GFP negative/positive cells without or with Oct4 induction. Right: Measured values for the long-lived bound fraction of all bound molecules (from 2D SMT analysis), the specific residence time, the_τ_3_D_, the number of trials and the target search time for Sox2. The results from the converse experiment with Sox2 induction are presented in (B). *: p<0.05, **: p<0.01. Error bars represent SD.
See also Movies S5 and S6, and Table S1.
Figure 6. Hierarchically Ordered Enhanceosome Assembly.

(A) Structural illustration of mutations (Sox2O and Oct4S) that selectively disrupt Sox2-Oct4 interaction surface. Crystal structures of Sox2 HMG domain (blue) and Oct1 POU domain (red) binding to two different enhancer DNAs (yellow) (FGF4: 1GT0 and UTF1: 1O4X; (Remenyi et al., 2003)) are presented in cartoon model by PyMOL. Mutations on Sox2 (K97E, R100E, M104E and R115E) and reciprocal mutations on Oct4 (I151Y and D159R) are highlighted by purple and green spheres respectively. Notably, these mutations interfere with Sox2-Oct4 interaction in both conformations.
(B) Sox2 and Oct4 western blot analysis to examine Sox2 K/D and Oct4 K/D efficiencies in ES cells. Tubulin served as a loading control.
(C–D) Changes in the long-lived fraction of all bound molecules, the long-lived residence time, the τ_3_D, the number of trials and the search time of Sox2 and Oct4 after the indicated perturbation experiment. Sox2O and Oct4S as described in (A); Oct4 K/D and Sox2 K/D as described in (B). Error bars represent SD.
(E) Sox2 and Oct4 assemble on DNA in an asymmetrically regulated fashion in live cells. We calculated the probability for each reaction route based on Oct4 and Sox2 DNA dissociation and association kinetic information obtained from the 3T3 cell induction experiments (See Eq. S19–26 for calculation details).
*: p<0.05, **: p<0.01.
See also Table S1.
To test if this ordered enhanceosome assembly also occurs in ES cells, we mutated amino acids on the interaction surfaces between Sox2 and Oct4 (Figure 6A and 6C) that do not directly affect the ability of Sox2 or Oct4 to bind DNA (Remenyi et al., 2003). Consistent with our induction experiments, mutations on Sox2 that disrupted its interaction with Oct4 only moderately decreased the Sox2 residence time but did not significantly affect its τ_3_D and search times in ES cells. By contrast, reciprocal mutations on Oct4 that impeded Sox2 interactions not only reduced Oct4 residence times (from 14.6 s to 12.66 s), but also substantially increased its τ_3_D (from 3.3 s to 4.6 s) and search times (from 407 s to 567.9 s) (Figure 6D).
Next, we performed lentivirus-mediated shRNA knockdown (K/D) depletion of Sox2 in ES cells that stably express halo-Oct4 and vice versa. Results from subsequent SMT experiments after loss of either Sox2 or Oct4 in ES cells confirmed our previous findings in 3T3 cells of an ordered enhanceosome assembly process (Figure 6D–E). Specifically, loss of Sox2 by K/D in ES cells greatly increased Oct4 τ_3_D (from 3.3 s to 4.7 s) and search times (from 407 s to 659 s). However, Sox2 search times and τ_3_D were only modestly altered after depletion of Oct4 by K/D. It is unlikely that the hierarchical assembly mechanism we observed here is due to some artifact of the halo-tagging of TFs. First, untagged-Sox2 decreased Oct4 search time, while untagged-Oct4 did not have the same effect on Sox2, suggesting that this asymmetric regulation is independent of the halo-tag (Figure 5A–B). Also, halo-Oct4 showed a normal pattern of activities during its target search in ES cells but, as expected, the mutant halo-Oct4S displayed a compromised search mode (Figure 6D). These results confirm that the ordered binding and cooperativity between Sox2, Oct4 and DNA contributes to the ability of Oct4 to execute a productive target search with little or no interference by the halo-tag. Based on our induction experiments in 3T3 cells, we could estimate the probabilities for each enhanceosome assembly pathway. We determined that minimally, the preferred pathway (Sox2 first) occurs ~75.3% of the time while the reverse order (Oct4 first) occurs at most 24.7% of the time (Figure 6E, Eq. S19–26).
Epigenetic Regulation of TF Target search
To explore a potential link between epigenetic regulation of chromatin and TF search dynamics, we imaged ES cells that were treated with either an HDAC inhibitor (TSA) or a DNA methylation inhibitor (5-Aza). Both treatments are expected to open chromatin globally and thus potentially influence TF search modes: 1) the number and quality of accessible non-specific sites is expected to increase, which could potentially require a greater number of random trials needed to find a cognate target and thus increase the search time; 2) the number and quality of accessible specific binding sites is likely to increase as well, which should limit the number of trials before a successful engagement and thus decrease the search time. With both drug treatments, we actually observed much shortened τ_3_D times, consistent with the notion that the total number of available DNA binding sites increased (Figure 7A–B). Importantly, we also observed significant decreases in the search time (Figure 7A–B), suggesting that an increased accessibility of specific binding sites and a shortened τ_3_D time accounts for the observed chromatin modification effects on TF search behavior.
Figure 7. Epigenetic Modulation of TF dynamics and Target Site Occupancy.

(A–B) Changes in specific residence time, τ_3_D, number of trials and search time of Sox2 and Oct4 after TSA and 5-AZA treatment in ES cells. ES cells were treated with 50nM TSA for 6 hours or 5μM 5-AZA for 12 hours before the imaging experiment. Error bars represent SD.
(C) Simulation heat-map to illustrate the relationship between TF residence time, TF concentration (binding site sampling frequency), and TF target site temporal occupancy. For each pairs of mean TF residence time (x axis) and sampling frequency (y axis), 1000 continuous binding events were simulated and then the temporal occupancy of the target site was calculated based on the total binding On-Off durations. The expected temporal occupancies of Sox2 at specific and non-specific sites derived based on data from Figure S7 were marked with the indicated black lines. The percentage of temporal occupancy is presented in ‘Jet’ color map. See “TF Dynamics Simulation” in the “EXPERIMENTAL PROCEDURES” for details of the parameter set-up. A.U. - Arbitrary Units
(D) Individual simulation tracks of distinct TF dynamics regimes (indicated in A) are presented. Specifically, track 1 represents high frequency non-specific chromatin binding. Track 5 represents the TF temporal occupancy of the target site by a TF with a relatively long TF residence time but at low concentrations in the cells.
*: p<0.05, **: p<0.01.
See also Figure S7 and Table S1.
TF Dynamics and Temporal Patterns of Target Site Occupancy
Our data reveal that TFs Sox2 and Oct4 execute a trial-and-error mechanism where they spend >98% of the search time sampling non-specific sites in the nucleus of mammalian cells before acquiring a cognate binding site. These results suggest that in an ES cell nucleus containing > 7000 potential Sox2 binding sites (Figure S5C), a Sox2 molecule would require ~6 min (377 s) to find and bind to a non-allele specific target. Thus, if there is only one cognate binding site available in the ES cell genome, it would take a Sox2 molecule ~31 days to find that site. From the perspective of DNA sites, if one target needs to be sampled by Sox2 every minute, our data indicate that ES cells must contain at least ~40,000 Sox2 molecules to carry out its function in the appropriate time scale. Simulations of TF dynamics based on our SMT data (Figure 7C–D) strongly suggest that the sampling frequency and target site occupancies of a cognate target by Sox2 and Oct4 molecules would be exquisitely sensitive to their nuclear concentrations. To better understand Sox2 and Oct4 assembly dynamics at target sites, we combined FCS measurements and Western blot analysis to quantify the endogenous Sox2 concentrations in live ES cells (Figure S7). We found that Sox2 concentrations are in the micro molar range (Sox2, ~ 0.73 μM). If we assume the ES cell nucleus approximates a 10 × 10 × 5 μm ellipsoid, one ES cell nucleus would generally have ~ 115,000 Sox2 (Eq. S30). Thus one cognate Sox2-Oct4 site is sampled by Sox2 ~ every 24 s (Eq. S31). The Sox2 specific residence times are about 12 ~ 16 s and non-specific residence time is about 0.7 ~ 0.9 s. These results suggest a 50 ~ 70% Sox2 temporal occupancy at a specific target site and ~ 4% (Sox2) temporal occupancies at a nonspecific target site in ES cells (Figure 7C, Eq. S32~33). In short, these measurements and calculations inform us of the exquisite and differential concentration dependence of key TFs in ES cells.
DISCUSSION
Mechanisms of TF Target Search
The TF target search process for binding to endogenous single copy genes has been the subject of considerable interest and theoretical discussion over the last ~40 years but direct measurements particularly in animal cells have proven elusive (reviewed in (Halford, 2009; Zakrzewska and Lavery, 2012)). By harnessing a powerful combination of recent advances in molecular imaging, the multi-faceted and cross-validated measurements of TF behavior reported here offers new insights into several long-standing TF target search questions: we first describe a method capable of discriminating specific DNA binding events at endogenous diploid allele sites from non-specific macro molecular interactions; these studies also describe several key kinetic features (3D diffusion periods, number of trials, search times, residence times etc.) associated with in vivo TF dynamics. Broadly consistent with a previously proposed facilitated diffusion model (Berg et al., 1981; Hager et al., 2009; Slutsky and Mirny, 2004), our imaging data indicate the involvement of at least two kinetically distinct steps in the specific target search process including a dominant phase involving multiple intervals of free 3D diffusion (τ_3_D = 3.3 ~ 3.7 s) and many (83–96) non-specific chromatin binding events (τns = 0.75 ~ 0.9s) prior to finding a cognate recognition site. Our in vitro single-molecule results suggest that Sox2 likely spends at least some time nonspecifically sliding along naked DNA searching for its target. If we assume that some periods of non-specific TF chromatin binding (τns) reflects the 1D search time (τ_1_D), the time partition for the 3D search is then calculated to be 72 ~ 88 %, significantly greater than 50% - the 3D search time partition that gives rise theoretically to the fastest target search (Slutsky and Mirny, 2004). It is worth noting that live-cell two-photon FCS measurements independently confirmed that > 74% Sox2 molecules in ES cells are in a diffusing state (Figure S7), also consistent with a TF target search mechanism dominated by 3D diffusion. Thus, in contrast to single-molecule studies in E.coli in which the LacI TF was found to spend more than 90% of its time nonspecifically bound to and diffusing along DNA in a 1D search (Elf et al., 2007; Hammar et al., 2012), our results record a significantly greater contribution of 3D diffusion to the TF target search process in mammalian cells. It is perhaps not entirely surprising that metazoan TFs might adopt different target search strategies/mechanisms given its distinct nuclear environment and genomic landscape relative to bacteria. Indeed, we provide evidence that the acetylation and/or methylation state of chromatin in ES cells can significantly alter the search mode by reducing 3D search time partition or/and number of trials – thus linking changes in chromatin to TF function in a manner that has no likely parallel in bacteria. Together, these results give us a first glimpse of TF target search dynamics in mammalian cells and how it’s evolutionarily adapted mechanisms may operate in a developmental context (Figure 4).
Despite the obvious differences between bacterial and mammalian TF search mechanisms, our findings suggest that the less dominant 1D sliding mode may, nevertheless, contribute to an efficient target search process. The haploid mouse genome is ~2.5 Gbp (Mouse Genome Sequencing et al., 2002) and bears ~7000 potential specific Sox2 binding sites. If we assumed that only 1~2% of the mammalian genome is accessible to TF binding due to nucleosome impedance according to hypersensitive site mapping experiments (Natarajan et al., 2012; Sabo et al., 2006), it would still require Sox2 to effectively scan and sample a DNA length of 43bp~86bp (2.5_Gbp_/7000(sites)/84(trials) × 1%~2%) during each non-specific chromatin collision to reach a non-allele specific target within ~84 trials. Our in vitro single molecule experiments revealed that Sox2 likely spends at least some time scanning along the naked DNA nonspecifically searching for its target. If we assume that Sox2 has similar sliding speeds as LacI (4×105 bp2/s) (Hammar et al., 2012) or P53 (2.6×106 bp2/s) (Tafvizi et al., 2008), during each nonspecific binding transaction (0.7 ~ 0.9 s), Sox2 would cover the expected sliding length (43bp~86bp) multiple times to ensure a specific target site recognition and acquisition. We note that multiple rounds of sliding and sampling DNA has been reported as a requirement for efficient TF target site recognition consistent with our calculations (Hammar et al., 2012).
Biological Implications of TF Function in Stem Cells
Critical lineage commitment events during animal development must be exquisitely timed and orchestrated by dynamic molecular transactions of multiple transcription factors (TFs) in the nucleus. However, to date, there have been few adequate tools to characterize the kinetic features that can discriminate specific from non-specific TF-chromatin binding events at endogenous diploid loci in living cells. Equally important has been a paucity of reliable means to determine the order of multiple TF binding events in live mammalian cells. The desirability of single-cell single-molecule imaging and tracking is particularly acute because the complex multi-step process of transcription is highly stochastic with dynamic behaviors relevant to function that are difficult to discern by conventional ensemble methods.
With the suite of new imaging modalities adopted here, we were able to dissect the dynamics of an ordered TF/enhancer assembly at endogenous loci in live mammalian cells with a minimal of perturbation. Unexpectedly, we observed that Sox2 is the lead TF that assists Oct4 to assemble on their in vivo targets in a hierarchically ordered mechanism. Oct4, in turn, appears to stabilize the Sox2/Oct4 complex on what are presumably composite cognate DNA recognition sites. We note that although Sox2 evidently performs a rather important function as a lead TF, we are not sure whether it qualifies, strictly speaking, as a “pioneer” factor since it is difficult to reconcile a 12 sec residence time with presumably highly stable chromatin interactions with condensed mitotic chromosomes that are a defining property of pioneer factors.
The dynamic behavior of individual TFs in living cells revealed by our studies establish that the sampling frequency of a cognate target by Sox2 and Oct4 molecules would be exquisitely sensitive to their nuclear concentrations in order to achieve coordinated co-binding at composite target sites to nucleate enhanceosome assembly and trigger the appropriate transcriptional programs underlying pluripotency (Figure 7). Specifically, upon ES cell differentiation the concentrations of both Sox2 and Oct4 become gradually down regulated. Consequently, the target site sampling frequency will be reduced accordingly. A critical time and TF concentration would be reached when a bound Sox2 molecule would no longer meet an Oct4 molecule within the duration of its average residence time on chromatin (~12s). The direct consequence is that a Sox2-Oct4 dependent enhanceosome could no longer be formed efficiently and the maintenance of the ES cell self-renewing pluripotent transcriptional circuitry would be disrupted. Thus, this critical tip in the balance of just a few TFs would then lead to cellular differentiation. We can also now explain how it is that merely reducing the Oct4 concentration but largely preserving Sox2 levels in ES cells is sufficient to trigger a major change in the pluripotency program given the distinct division of labor and yet co-dependence between Sox2/Oct4. These data also revealed that TFs with different target search and chromatin binding kinetics would display distinct temporal patterns of target site occupancy (Figure 7). These results thus highlight the importance of dissecting the behavior and kinetic parameters intrinsic to different types of TFs as they carry out critical time dependent activities in living cells. These essential measurements of TF search and binding modes provide new insights into the dynamic and stochastic behavior of TF-DNA transactions that form the basis for the production of mRNA outputs and cell fate determination.
A key aspect of this study is that we complemented single molecule tracking experiments with a range of genetic loss-of-function studies to probe biologically relevant regulatory aspects of TF behavior. For example, we gained new insights into how TF target search dynamics could be affected by the presence of trans-activation domains, by the specific interplay of two functionally linked TFs (ie Sox2/Oct4) and by changes in the global epigenetic state of the cell. A combination of these studies directly unmasked the molecular underpinnings that give rise to a specific target search mode. For instance, deletion of the Sox2 transcription activation domain substantially decreased the target search time by reducing the number of non-specific binding trials, suggesting that removal of a transcription activation domain might lead to reduced non-specific decoy protein-protein tethering and thus significantly truncate the target search process. This surprising finding also suggests that from the standpoint of maximal efficiency in the search process, TFs with multiple high affinity activation domains might actually be at a disadvantage for target search. Thus one might posit that combinatorial regulation may in part derive from the need for different TFs to assume distinct and complementary targeting duties: with some, like Sox2 serving as pioneer or lead factors adept at target search but perhaps limited in diversity of activation domains while other partner factors like Oct4 may be more suited for executing multi-faceted interactions with co-activators and the core promoter complex etc. that require a greater panoply of activation surfaces but at the expense of a less efficient search capacity. Our studies also provide evidence that the acetylation and/or methylation state of chromatin in ES cells can significantly alter the search mode of TFs, suggesting that epigenetic regulation may be critical not only in effecting DNA binding but also influence the TF target search pattern.
Potential Impact on Interpreting Genome-wide TF binding Studies
Our data revealed several general and fundamental aspects of TF binding to endogenous chromatin in live cells that may impact the way we interpret results from widely adopted chromatin immuno-precipitation experiments (ChIP-qPCR, ChIP-seq and ChIP-exo). Specifically, our single molecule tracking data establish that non-specific chromatin binding via either protein:protein or protein:DNA transactions is a major component of the TF target search process. Although the non-specific binding residence time of Sox2 (0.75 ~ 0.9 sec) is 15-fold shorter than its residence time at specific sites (~12 sec), when TF concentrations are relatively high as we have measured in ES cells (in the micro molar range), even non-specific sites will be sampled frequently enough to be efficiently cross-linked by commonly used agents such as formaldehyde (Figure 7 and S7). Thus, high frequency sampling of non-specific sites during the target search process could partly explain the large number of non-consensus decoy or false TF binding sites detected by ensemble assays such as those reported by ENCODE (Consortium et al., 2012; Gerstein et al., 2012). Many putative target sites identified by such genome-wide binding studies when tested more rigorously were indeed found to be non-functional (Fisher et al., 2012). The preponderance of transient non-specific interactions detected by SMT is also consistent with the proposed weak protein-protein tethering mechanisms reported by the ENCODE project (Consortium et al., 2012; Gerstein et al., 2012). Indeed, our Sox2 ChIP-exo data generated using ultra-high sequencing depth (40 million mapped reads), also detected many weak or degenerate Sox2 binding sites in ES cells (Figure S5C). These SMT experiments, thus, underscore the need to be cautious in interpreting high throughput ensemble binding assays with respect to identifying meaningful or functionally relevant binding interactions.
EXPERIMENTAL PROCEDUES
Culture Condition for Live-cell Imaging
ES cell imaging experiments were performed in the imaging medium (DMEM without phenol-red (Invitrogen), 15% FBS, 1mM glutamax, 0.1mM nonessential amino acids, 0.1mM 2-mercaptoethanol and 1000 units of LIF). We used a Tokai-hit PI live-cell chamber and GM-8000 digital gas mixer to maintain cell culturing condition (37°C, 5% CO2 and humidity) during the imaging experiment.
Stable Cell Line Generation
Stable cell lines were generated by co-transfection of ES cells or 3T3 Cells with the piggybac vector and a helper plasmid that over-expresses Piggybac transposase (Supper Piggybac Transposase, System Biosciences). 48 hours post-transfection, cells were subjected to G418 (invitrogen) selection (500 μg/ml). After a week of selection, cells were maintained in their culturing medium with a 250 μg/ml final concentration of G418.
Live-cell Single-molecule Imaging
2D single molecule experiments were conducted on a Nikon Eclipse Ti microscope equipped with a 100X Oil-immersion Objective lens (Nikon, N.A. = 1.4). 3D single molecule tracking experiments were performed at the same instrument for 2D. The multifocus optical elements are appended after the primary image plane. The details for the multifocus microscopy instrumentation were described in our previous publication (Abrahamsson et al., 2013). See supplementary Information for the procedures of single-molecule localization and tracking.
In vitro TIRF Single-molecule Imaging
Total Internal Reflection Fluorescence Microscope (TIRFM) imaging system set-up and fluorescent molecule spots co-localization analysis were essentially as described previously (Revyakin et al., 2012). See supplementary Information for experimental details and imaging analysis.
ChIP-exo
ChIP-exo library was prepared by following the published protocol with minor modifications (Rhee and Pugh). Specifically, we adapted the SoLid sequencer adaptors/primers to make the final library compatible with the illumina Tru-seq seq small-RNA system. The detailed primer information is in Table S1. See supplementary Information for the procedures of genomic data analysis.
Genomic data are available from GEO with the accession number (GSE54103).
Supplementary Material
01
02
03
04
05
06
07
08
Highlights.
- Single-cell, single-molecule imaging shows Sox2/Oct4 dynamics in live ES cells.
- Sox2 locates target via a 3D diffusion-dominated search and 1D sliding along DNA.
- Sox2/Oct4 enhanceosome forms in a hierarchical binding order.
- Temporal patterns of target site occupancy are modulated by TF dynamics.
Acknowledgments
We thank Xavier Darzacq, Carl Wu, Robert Singer, Yick W. Fong, Wulan Deng, Michael Levine and Sean B. Carroll for general discussion and proof reading the manuscript, Yick W. Fong for the Sox2 shRNA vector, Florian Mueller for FISH-QUANT software, Davide Mazza for the mathematic modeling validation, John Macklin for the assistance of FCS, C. Morkunas and S. Moorehead for assistance.
Footnotes
AUTHOR CONTRIBUTION
Z.Z. and L.L. contributed to this work equally. Z.Z. and L.L. performed in vitro single-molecule and ChIP-exo experiments respectively.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abrahamsson S, Chen J, Hajj B, Stallinga S, Katsov AY, Wisniewski J, Mizuguchi G, Soule P, Mueller F, Dugast Darzacq C, et al. Fast multicolor 3D imaging using aberration-corrected multifocus microscopy. Nature methods. 2013;10:60–63. doi: 10.1038/nmeth.2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avilion AA, Nicolis SK, Pevny LH, Perez L, Vivian N, Lovell-Badge R. Multipotent cell lineages in early mouse development depend on SOX2 function. Genes & development. 2003;17:126–140. doi: 10.1101/gad.224503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Tabou de-Leon S, Davidson EH. Gene regulation: gene control network in development. Annual review of biophysics and biomolecular structure. 2007;36:191. doi: 10.1146/annurev.biophys.35.040405.102002. [DOI] [PubMed] [Google Scholar]
- Berg OG, Winter RB, von Hippel PH. Diffusion-driven mechanisms of protein translocation on nucleic acids. 1. Models and theory. Biochemistry. 1981;20:6929–6948. doi: 10.1021/bi00527a028. [DOI] [PubMed] [Google Scholar]
- Chen X, Xu H, Yuan P, Fang F, Huss M, Vega VB, Wong E, Orlov YL, Zhang W, Jiang J, et al. Integration of external signaling pathways with the core transcriptional network in embryonic stem cells. Cell. 2008;133:1106–1117. doi: 10.1016/j.cell.2008.04.043. [DOI] [PubMed] [Google Scholar]
- Consortium EP, Bernstein BE, Birney E, Dunham I, Green ED, Gunter C, Snyder M. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elf J, Li GW, Xie XS. Probing transcription factor dynamics at the single-molecule level in a living cell. Science (New York, NY. 2007;316:1191–1194. doi: 10.1126/science.1141967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher WW, Li JJ, Hammonds AS, Brown JB, Pfeiffer BD, Weiszmann R, MacArthur S, Thomas S, Stamatoyannopoulos JA, Eisen MB, et al. DNA regions bound at low occupancy by transcription factors do not drive patterned reporter gene expression in Drosophila. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:21330–21335. doi: 10.1073/pnas.1209589110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fudenberg G, Mirny LA. Higher-order chromatin structure: bridging physics and biology. Current opinion in genetics & development. 2012;22:115–124. doi: 10.1016/j.gde.2012.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao L, Shao L, Higgins CD, Poulton JS, Peifer M, Davidson MW, Wu X, Goldstein B, Betzig E. Noninvasive imaging beyond the diffraction limit of 3D dynamics in thickly fluorescent specimens. Cell. 2012;151:1370–1385. doi: 10.1016/j.cell.2012.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gebhardt JC, Suter DM, Roy R, Zhao ZW, Chapman AR, Basu S, Maniatis T, Xie XS. Single-molecule imaging of transcription factor binding to DNA in live mammalian cells. Nature methods. 2013;10:421–426. doi: 10.1038/nmeth.2411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerstein MB, Kundaje A, Hariharan M, Landt SG, Yan KK, Cheng C, Mu XJ, Khurana E, Rozowsky J, Alexander R, et al. Architecture of the human regulatory network derived from ENCODE data. Nature. 2012;489:91–100. doi: 10.1038/nature11245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hager GL, McNally JG, Misteli T. Transcription dynamics. Molecular cell. 2009;35:741–753. doi: 10.1016/j.molcel.2009.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halford SE. An end to 40 years of mistakes in DNA-protein association kinetics? Biochemical Society transactions. 2009;37:343–348. doi: 10.1042/BST0370343. [DOI] [PubMed] [Google Scholar]
- Hammar P, Leroy P, Mahmutovic A, Marklund EG, Berg OG, Elf J. The lac repressor displays facilitated diffusion in living cells. Science (New York, NY. 2012;336:1595–1598. doi: 10.1126/science.1221648. [DOI] [PubMed] [Google Scholar]
- Lam CS, Mistri TK, Foo YH, Sudhaharan T, Gan HT, Rodda D, Lim LH, Chou C, Robson P, Wohland T, et al. DNA-dependent Oct4-Sox2 interaction and diffusion properties characteristic of the pluripotent cell state revealed by fluorescence spectroscopy. The Biochemical journal. 2012;448:21–33. doi: 10.1042/BJ20120725. [DOI] [PubMed] [Google Scholar]
- Levine M, Tjian R. Transcription regulation and animal diversity. Nature. 2003;424:147–151. doi: 10.1038/nature01763. [DOI] [PubMed] [Google Scholar]
- Mazza D, Abernathy A, Golob N, Morisaki T, McNally JG. A benchmark for chromatin binding measurements in live cells. Nucleic acids research. 2012;40:e119. doi: 10.1093/nar/gks701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirny L, Slutsky M, Wunderlich Z, Tafvizi A, Leith J, Kosmrlj A. How a protein searches for its site on DNA: the mechanism of facilitated diffusion. Journal of Physics A: Mathematical and Theoretical. 2009;42:434013. [Google Scholar]
- Waterston RH, Lindblad-Toh K, Birney E, Rogers J, Abril JF, Agarwal P, Agarwala R, Ainscough R, Alexandersson M, et al. Mouse Genome Sequencing C. Initial sequencing and comparative analysis of the mouse genome. Nature. 2002;420:520–562. doi: 10.1038/nature01262. [DOI] [PubMed] [Google Scholar]
- Mueller F, Stasevich TJ, Mazza D, McNally JG. Quantifying transcription factor kinetics: at work or at play? Critical reviews in biochemistry and molecular biology. 2013;48:492–514. doi: 10.3109/10409238.2013.833891. [DOI] [PubMed] [Google Scholar]
- Natarajan A, Yardimci GG, Sheffield NC, Crawford GE, Ohler U. Predicting cell-type-specific gene expression from regions of open chromatin. Genome research. 2012;22:1711–1722. doi: 10.1101/gr.135129.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichols J, Zevnik B, Anastassiadis K, Niwa H, Klewe-Nebenius D, Chambers I, Scholer H, Smith A. Formation of pluripotent stem cells in the mammalian embryo depends on the POU transcription factor Oct4. Cell. 1998;95:379–391. doi: 10.1016/s0092-8674(00)81769-9. [DOI] [PubMed] [Google Scholar]
- Planchon TA, Gao L, Milkie DE, Davidson MW, Galbraith JA, Galbraith CG, Betzig E. Rapid three-dimensional isotropic imaging of living cells using Bessel beam plane illumination. Nature methods. 2011;8:417–423. doi: 10.1038/nmeth.1586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remenyi A, Lins K, Nissen LJ, Reinbold R, Scholer HR, Wilmanns M. Crystal structure of a POU/HMG/DNA ternary complex suggests differential assembly of Oct4 and Sox2 on two enhancers. Genes & development. 2003;17:2048–2059. doi: 10.1101/gad.269303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Revyakin A, Zhang Z, Coleman RA, Li Y, Inouye C, Lucas JK, Park SR, Chu S, Tjian R. Transcription initiation by human RNA polymerase II visualized at single-molecule resolution. Genes & development. 2012;26:1691–1702. doi: 10.1101/gad.194936.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhee HS, Pugh BF. Comprehensive genome-wide protein-DNA interactions detected at single-nucleotide resolution. Cell. 147:1408–1419. doi: 10.1016/j.cell.2011.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhee HS, Pugh BF. Comprehensive genome-wide protein-DNA interactions detected at single-nucleotide resolution. Cell. 2011;147:1408–1419. doi: 10.1016/j.cell.2011.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabo PJ, Kuehn MS, Thurman R, Johnson BE, Johnson EM, Cao H, Yu M, Rosenzweig E, Goldy J, Haydock A, et al. Genome-scale mapping of DNase I sensitivity in vivo using tiling DNA microarrays. Nature methods. 2006;3:511–518. doi: 10.1038/nmeth890. [DOI] [PubMed] [Google Scholar]
- Slutsky M, Mirny LA. Kinetics of protein-DNA interaction: facilitated target location in sequence-dependent potential. Biophysical journal. 2004;87:4021–4035. doi: 10.1529/biophysj.104.050765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tafvizi A, Huang F, Leith JS, Fersht AR, Mirny LA, van Oijen AM. Tumor suppressor p53 slides on DNA with low friction and high stability. Biophys J. 2008;95:L01–03. doi: 10.1529/biophysj.108.134122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tam PP, Loebel DA. Gene function in mouse embryogenesis: get set for gastrulation. Nature reviews Genetics. 2007;8:368–381. doi: 10.1038/nrg2084. [DOI] [PubMed] [Google Scholar]
- Zakrzewska K, Lavery R. Towards a molecular view of transcriptional control. Current opinion in structural biology. 2012;22:160–167. doi: 10.1016/j.sbi.2012.01.004. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
01
02
03
04
05
06
07
08