Biology of the Eosinophil (original) (raw)
. Author manuscript; available in PMC: 2014 Jul 24.
Abstract
In this review, we aim to put in perspective the biology of a multifunctional leukocyte, the eosinophil, by placing it in the context of innate and adaptive immune responses. Eosinophils have a unique contribution in initiating inflammatory and adaptive responses due to their bidirectional interactions with dendritic cells and T cells, as well as their large panel of secreted cytokines and soluble mediators. The mechanisms and consequences of eosinophil responses in experimental inflammatory models are discussed.
Keywords: Eosinophil, chemokine, cytokine, pathogenesis, gastrointestinal, asthma
I. The Eosinophil: from an hematopoietic stem cell to a mature eosinophil
A. Eosinophil differentiation
Eosinophils are produced in the bone marrow from multipotent hematopoietic stem cells. Hematopoietic differentiation involves the commitment of multipotent progenitors to a given lineage, followed by the maturation of the committed cells. From these stem cells, the myeloid lineage allows the development of the myeloblast with shared properties of basophils and eosinophils, and then into a separate eosinophil lineage (Boyce et al., 1995). Each of the steps that ultimately lead to mature eosinophils is under the fine regulation of soluble mediators and transcription factors (Figure 1).
Figure 1. From the hematopoietic stem cell to the mature eosinophil.
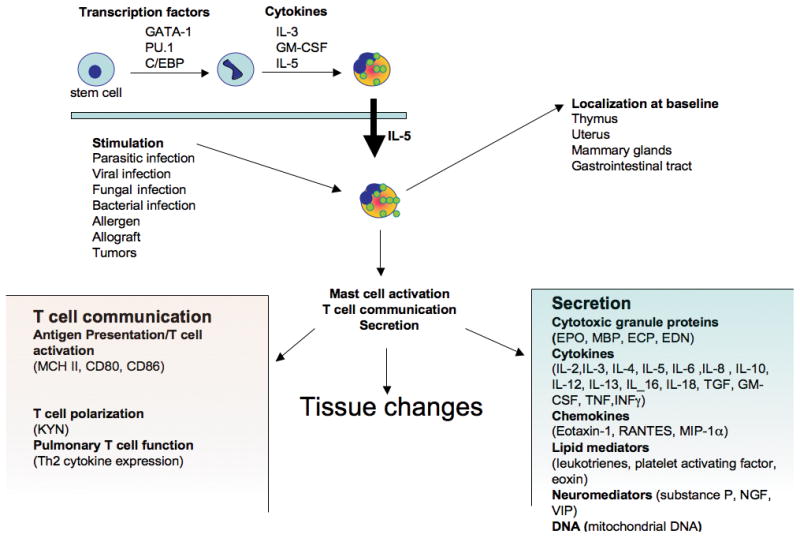
Eosinophils develop in the bone marrow. Transcription factor (such as Δdbl-GATA-1) and cytokines (such as IL-5, IL-3 and GM-CSF) are essential for their differentiation from an hematopoietic stem cell into the mature eosinophil. Once mature, IL-5 controls the eosinophil migration from the bone marrow to the blood. At baseline, eosinophils localize in the thymus, GI tract, uterus and mammary gland. Eosinophils are able to express and to secrete, at baseline or upon stimulation, a large variety of mediators (cytokines, granule proteins, lipid mediators). Eosinophils are putative antigen presenting cells and play a role in mast cell activation, T cell communication and function.
1) Transcription factors
Several transcription factors are involved in the eosinophilic lineage. Forced expression of the transcription C/EPB members (CCAAT/enhancer-binding protein family) in progenitor cells induces myeloid and eosinophil differentiation (Nerlov et al., 1998). Conversely, dominant-negative versions of C/EBP inhibits myeloid differentiation (Nerlov et al., 1998). As such, C/EBP-induced eosinophil differentiation can be separated into two distinct events, lineage commitment and maturation. Indeed, a transient activation of a conditional C/EBP form in multipotent progenitors lead to the formation of immature eosinophils, whereas sustained activation produced mature eosinophils, suggesting that C/EBP functions are required during eosinophil lineage commitment and maturation. PU.1, an ets transcription factor family member, is only expressed in hematopoietic cells. At an early time point of the differentiation, PU.1 is involved in the switching between lymphoid and myeloid lineage. PU.1 expression level determines the faith of the cell. PU.1 gene-disrupted mice are devoid of B and dendritic cells, monocytes/macrophages, and mature neutrophils. PU.1 is necessary for dictating monocyte/macrophage and dendritic cell commitment and differentiation, and for neutrophil differentiation. On the other side, high levels of PU.1 lead to an increase myeloid differentiation (McNagny and Graf, 2002; Nerlov and Graf, 1998; Nerlov et al., 1998). In most cells, PU.1 antagonizes with GATA-1, but they have synergistic activity in regulating eosinophil lineage specification and eosinophil granule protein transcription (Du et al., 2002). Of these transcription factors, GATA-1 is clearly the most important for eosinophil lineage specification. Located on the chromosome X in humans and mice, GATA-1 transcription factor was named by its ability to bind the promoter sequence composed of the bases GATA. The GATA-1 binding site is present as a palindromic sequence (double GATA site) in numerous eosinophil related genes (granule protein genes, CC-chemokine receptor 3, IL-5 receptor) and in the GATA-1 gene itself (Du et al., 2002; Yu et al., 2002; Zimmermann et al., 2000a). The targeted mutation of the double GATA binding site present in the GATA-1 gene leads to the loss of the eosinophil lineage in mice (Yu et al., 2002). The critical role for GATA-1 in eosinophil lineage was also confirmed by in vitro experiments (Hirasawa et al., 2002; Iwasaki et al., 2005). While expressing GATA-1 in other myeloid cells; mast cells, megakaryocytes, and erythroid cells of the double GATA-1 deficient mice, they do not appear to be affected by the mutation in the high affinity palindromic GATA site (Du et al., 2002).
2) Soluble mediators
Cytokines are indispensable for hematopoietic cell development differentiation and maturation. Located on chromosome 5 in position q31, IL-3, IL-5, and GM-CSF are particularly important in regulating eosinophil development (Lopez et al., 1986; Lopez et al., 1988; Rothenberg et al., 1988; Takatsu et al., 1994). In addition to a close proximity on the chromosome and a relative homology of sequence, IL-3, IL-5 and GM-CSF also share the common beta chain in their receptor in addition to the specific alpha chains (Vadas et al., 1994). IL-3 and GM-CSF also induce the differentiation of other myeloid cells such as the mast cell, but IL-3, GM-CSF and IL-5 synergize toward the differentiation of eosinophils. Indeed, of these three cytokines, IL-5 is the most specific to the eosinophil lineage. In 1995, using a high systemic level of IL-5, after intravenous injection, Collins et al have shown that mice developed blood eosinophilia and a depletion of bone marrow eosinophils, suggesting that IL-5 stimulates the release of eosinophils from the bone marrow into the peripheral circulation (Collins et al., 1995). But IL-5 is also responsible for selective differentiation of eosinophils and this has been clearly demonstrated in genetically modified animals (Sanderson, 1992). Mice overexpressing IL-5 under the promoter of CD2, see a profound eosinophilia in the blood and spleen but also in the bone marrow (Dent et al., 1990; Lee et al., 1997b; Mishra et al., 2002b; Tominaga et al., 1991). In contrast, IL-5 deficient animals show a marked reduction in eosinophil levels in the blood, or in the tissues in allergic models (Foster et al., 1996; Kopf et al., 1996; Mishra and Rothenberg, 2003).
B. The mature eosinophil: a complex granulocyte
In 1879, Paul Ehrlich reported the avidity of a subtype of blood leukocytes for the acidic stain eosin and thus named these cell types “eosinophil”. The basic components when stained will then be identified in the eosinophil granules as major basic protein (MBP), eosinophil cationic protein (ECP), eosinophil peroxidase (EPO), and eosinophil-derived neurotoxin (EDN) (Hamann et al., 1991).
Eosinophil granules contain a crystalloid core composed of MBP-1 (and MBP-2), and a matrix composed of ECP, EDN, and EPO (Gleich and Adolphson, 1986). These granules are capable of inducing tissue damage and dysfunction (Gleich and Adolphson, 1986) since MBP, EPO, and ECP are toxic to a variety of tissues, including heart, brain and bronchial epithelium (Frigas et al., 1980; Gleich et al., 1979; Tai et al., 1982; Venge et al., 1980).
1) ECP
ECP was cloned in 1989 by Rosenberg et al. ECP is a small, basic protein found in the matrix of the eosinophil-specific granule that has cytotoxic, helminthotoxic, and ribonuclease activity. On molecular sizing, ECP displays marked heterogeneity, probably as a result of differential glycosylation, with a molecular weight ranging between 16-21.4 kD. Two isoforms, ECP-1 and ECP-2, have been identified using heparin sepharose chromatography (Gleich and Adolphson, 1986). The cDNA sequence shows 89% sequence identity with that reported for the related granule protein, eosinophil-derived neurotoxin (EDN). The amino acid sequence is 66% homologous to EDN and 31% homologous to human pancreatic ribonuclease including conservation of the essential structural cysteine and cataytic lysine and histidine residues. ECP does have ribonuclease activity but is 100 times less potent than EDN (Slifman et al., 1986). ECP has been shown to possess anti-viral activity and causes voltage-insensitive, ion-selective toxic pores in the membranes of target cells, possibly facilitating the entry of other cytotoxic molecules (Gleich and Adolphson, 1986; Rosenberg and Domachowske, 2001; Slifman et al., 1986; Young et al., 1986). ECP also has a number of additional non-cytotoxic activities including suppression of T cell proliferative responses, immunoglobulin synthesis by B cells, mast cell degranulation stimulation of airway mucus secretion and glycosaminoglycan production by human fibroblasts (Venge et al., 1999).
2) MBP
MBP is expressed as two different homologs (MBP-1 and MBP-2) derived from two separate genes. MBP-1 is a small protein that consists of 117 amino acids, with a molecular weight of 13.8 kD, and a high isoelectric point (>11) which cannot be measured accurately due to its extremely basic nature (Hamann et al., 1991). Mature eosinophils loose the ability to transcribe mRNA encoding MBP-1, indicating that all of the MBP-1 stored in crystalloid granules is synthesized during early eosinophil development prior to maturation (Popken-Harris et al., 1998; Voehringer et al., 2007). MBP2 is exclusively expressed by eosinophils, and may be a more specific marker for elevated eosinophils in patients with eosinophilia than MBP1 (Plager et al., 2006).
The toxicity of MBP to helminthic worms has supported the role of eosinophils in host defense (Ackerman et al., 1985; Butterworth, 1984; Gleich, 1986; Gleich and Adolphson, 1986; O'Donnell et al., 1983). MBP has also been shown to be cytotoxic to airways and may be at least partly responsible for tissue damage associated with eosinophil infiltration in bronchial mucosa in asthma (Frigas et al., 1980; Furuta et al., 2005; Hisamatsu et al., 1990). The toxic effect of MBP is thought to result from increased membrane permeability through surface charge interactions leading to perturbation of the cell surface lipid bilayer (Wasmoen et al., 1988). MBP2 is two-fold less positively charged than MBP1, and this difference may explain MBP2's similar, but less potent, in vitro biological activities. While conservation of MBP2's amino acid sequence (63% identity with MBP1) suggests a common functions with MBP1, MBP2's substantially reduced charge and the existence of the similar murine MBP2 argue for additional, unique functions for MBP2.
3) EPO
EPO, which has peroxidase activity, is localized in the matrix of the granule. It is composed of two subunits, a heavy chain of 50 to 57 kD and a light chain of 11 to 15 kDa. EPO has 68% sequence identity to the neutrophil myeloperoxidase (MPO). Enzymatic reaction of EPO, but not MPO, is resistant to inhibition by potassium cyanide (Hamann et al., 1991; Ten et al., 1989). EPO constitutes ∼25% of the total protein mass of specific granules. EPO has been shown to catalyze the oxidation of halides, pseudohalides, and nitric oxide to form highly reactive oxygen species (hypohalous acids), reactive nitrogen metabolites (nitric dioxide) and perioxynitrate-like oxidants. These electrophil species oxidize nucleophil targets on proteins, promoting oxidative stress, and subsequent cell death by apoptosis and necrosis (Agosti et al., 1987; MacPherson et al., 2001; Wu et al., 1999).
4) EDN
Eosinophil-derived neurotoxin (EDN) is an eosinophil granule-derived secretory protein with ribonuclease and antiviral activity. EDN has also been shown to induce the migration and maturation of dendritic cells (DCs). Yang et al (Yang et al., 2008) recently reported that EDN is an endogenous ligand of Toll-like receptor (TLR)2 and can activate myeloid DCs by triggering the TLR2 – myeloid differentiation factor 88 (Myd88) signaling pathway. In the same study, the authors have also shown that EDN enhanced antigen-specific T helper (Th)2-biased immune responses (IL-5, IL-6, IL-10, and IL-13, and higher levels of IgG1 than IgG2a). EDN thus has the propensity to alert the adaptive immune system for preferential enhancement of antigen-specific Th2 immune responses.
While these molecules share similarity in function, they demonstrate differences in their mode of action on helminthic worms. ECP is 8 to 10 times more potent than MBP. Purified ECP produced complete fragmentation and disruption of schistosomula, whereas MBP produced a distinctive ballooning and detachment of the tegumental membrane. In contrast, EDN is only marginally toxic at high concentrations and caused crinkling of the tegumental membrane (Ackerman et al., 1985).
In a recent study, it has been shown that eosinophils, but not other cell types including neutrophils, contain nitrotyrosine-positive proteins in specific granules. Nitration of tyrosine residues has been observed during various acute and chronic inflammatory diseases. This recent study demonstrates that the human eosinophil toxins, EPO, MBP, EDN, ECP, and the respective murine toxins, are post-translationally modified by nitration the tyrosine residues during cell maturation. This mechanism depends on the presence of EPO and targets specific single nitration sites at Tyr-349 in EPO and Tyr-33 in both ECP and EDN. The study also suggests that the nitrated tyrosine residues in ECP, EDN and EPO are surface exposed and occur in mature eosinophils independently of inflammation (Ulrich et al., 2008).
C. Eosinophil secretion/degranulation
Most secretory cells hold a molecular system allowing docking and fusion of vesicles to the membrane. As such, regulated exocytosis occurs by the formation of a docking complex composed of soluble N-ethylmaleimide-sensitive factor attachment protein receptors (SNAREs) located on the vesicle (v-SNAREs) and the target membrane (t-SNAREs). SNAREs are classified into two categories based upon the presence of a conserved amino acid (arginine (R) or glutamine (Q)). Human eosinophils have been shown to express the Q-SNAREs SNAP-23 and syntaxin 4, which are predominantly localized to the plasma membrane (Logan et al., 2002) and the R-SNARE VAMP-2, which is localized to cytoplasmic secretory vesicles. A recent study has shown the IkB kinase 2 phosphorylates SNAP-23 (in an NFKB-independent manner) regulates mast cell degranulation (Suzuki and Verma, 2008). Whether the same mechanism occurs in eosinophils is not known, and regulation of degranulation may follow other pathways. Interestingly, a recent study has shown that purified eosinophil granules express extracellular domains of the receptors for IFN-γ and CCR3 and can respond upon stimulation and increase ECP release (Neves et al., 2007).
D. Eosinophil DNA trap
Yousefi et al have recently demonstrated that eosinophils are able to generate extracellular traps. The phenomenon previously described in neutrophils, presented genomics DNA traps having microbial activity, certainly due to the presence of histones. Interestingly, eosinophils rapidly release mitochondrial DNA in response to exposure to bacteria, C5a or CCR3 ligands. In contrast to neutrophils, eosinophils do not undergo cell death upon release of their DNA; in addition, this process requires free radical production via NADP oxidase. The traps contain the granule protein ECP and MBP, and display antimicrobial activity (Yousefi et al., 2008). This indicates that eosinophils may have an essential role in innate immunity against bacteria, using a unique mechanism.
E. Cytokine production
Eosinophils have the propensity to synthesize numerous cytokines and growth factors that have implicated eosinophils in numerous homeostatic processes and inflammatory conditions. While usually produced in small amounts in the resting eosinophils, some cytokines are largely induced in inflammatory conditions and triggering of eosinophils by engagement of receptors for cytokines, immunoglobulins, and complement can lead to the secretion of a large variety of pro-inflammatory cytokines (IL-2, IL-4, IL-5, IL-10, IL-12, IL-13, IL-16, IL-18 and TGF-α/β), chemokines (RANTES and eotaxin-1) and lipid mediators (platelet activating factor and leukotriene C4).
II. A Role for Eosinophils at Baseline
Some organs are rich in eosinophils, such as the gastrointestinal (GI) tract, spleen, lymph nodes, thymus, mammary glands and uterus. Their presence in normal condition suggests a role for eosinophils in some homeostatic processes.
A. Eosinophils and reproduction
The uterus is home to a large number of eosinophils mainly localized to the endometrial stroma and at the endometrial-myometrial junction (Sferruzzi-Perri et al., 2003). While regulated by IL-5, their presence in the subepithelial stroma is not affected by IL-5 deficiency. (Robertson et al., 2000). The infiltration in the uterus is correlated with the expression of eotaxin-1, RANTES and MIP-1α whose expression is modified by steroid hormones. (Gouon-Evans and Pollard, 2001; Robertson et al., 1998; Zhang et al., 2000). Indeed, eotaxin-1 deficient mice have a deficiency of eosinophils in the uterus, and a delay in estrus onset (Gouon-Evans et al., 2002). These suggest a role for eosinophils in uterus maturation.
While the role of eosinophils during implantation and pregnancy has yet to be proven, it is interesting to note that eosinophil MBP is ectopically expressed in the uterus by placental X and giant cells (Maddox et al., 1984) during pregnancy, and its production peaks 2-3 weeks before parturition but this is not directly related to eosinophils (Wagner et al., 1994).
In the mammary gland, increased expression of eotaxin-1 coincides with eosinophil infiltration into the head of the terminal end bud (Gouon-Evans et al., 2000). Using eotaxin-1 deficient mice, the presence of eosinophils in the mammary gland has been associated with the terminal end bud formation and the branching complexity of the ductal tree (Gouon-Evans et al., 2000). Eosinophil participation in mammary gland development might be due to the eosinophil secretion of TGF -β (Gouon-Evans et al., 2000).
B. Thymic eosinophils
In a compelling study, Throsby et al have analyzed murine eosinophils in the thymus. Indeed, thymic eosinophils are preferentially recruited during the neonatal period. In mice, the absolute numbers increased 10-fold between 7–14 days to reach parity with dendritic cells before diminishing (Throsby et al., 2000). Eosinophils primarily localize to the cortico-medullary region of the thymus and reach basal levels by 28 days of age. Subsequently, an increase in thymic eosinophil levels at 16 weeks of age corresponds to the commencement of thymic involution. Notably, eosinophils at this stage localize to the medullary region.
Previous studies of eosinophil cytokine expression suggest that different combinations of cytokines may be linked to activation or disease states (Throsby et al., 2000). Thymic eosinophils expressed TGF-β and IL-16 mRNA consistent with their wide distribution among leukocytes. Detectable mRNA levels of the pro-inflammatory cytokines, IL-1α, IL-6, and TNF-α are present in activated eosinophils (Throsby et al., 2000); the expression of eotaxin, IL-2, IL-3, IL-10, IL-12, IFN-γ GM-CSF, and IL-5 are undetectable. GM-CSF and IL-5 have been reported to act as autocrine survival and recruitment factors for activated eosinophils in inflammatory foci (Throsby et al., 2000). However, thymic eosinophils expressed mRNA for the closely related Th2 cytokines IL-4 and IL-13. Both are linked to eosinophil involvement in certain pathologies and are reported to aid recruitment, activation, and survival.
Eosinophils can act as antigen presenting cells (APC) and express costimulatory molecules under activating conditions. Throsby et al. have shown that thymic eosinophils are CD11b/CD11c double-positive and express class II molecules and intermediate levels of class I molecules (Throsby et al., 2000). Low surface expression of the costimulatory molecules, CD86 (B7.2) and CD30L (CD153), suggest that thymic eosinophils may be able to present antigens.
Matthews et al. demonstrated that the recruitment of eosinophils into the thymus is regulated by eotaxin-1, which is constitutively expressed in the thymus (Matthews et al., 1998). The recruitment of eosinophils and their anatomical localization within discrete compartments of the thymus coincides with negative selection of double positive thymocytes (Throsby et al., 2000). Thymic eosinophils are increased in models of acute negative selection. In addition, eosinophils are associated with clusters of apoptotic bodies suggesting eosinophil-mediated MHC-I-restricted thymocyte deletion. Of note, eosinophils express costimulatory molecules that are involved in clonal deletions such as CD30 ligand (CD153), and CD66 (Throsby et al., 2000) and may promote developing-thymocyte apoptosis since thymic eosinophils have a high level of free radicals due to high levels of NADPH oxidase activity (Throsby et al., 2000).
III. Eosinophils and Immune Regulation
A. Antigen presentation / T cells proliferation
Early studies on the role of eosinophils in antigen presentation and T cell activation have raised controversy. Eosinophils can effectively present soluble antigens to CD4+ T cells, promoting T cell proliferation and polarization (Shi et al., 2000; van Rijt et al., 2003). But the ability of eosinophils to present antigen seems closely linked to the extraction methods. The use, in the lysis buffer, of amonium chloride, an inhibitor of lysosome acidification (needed for antigen presentation), negatively correlates with eosinophil antigen presentation activity (Shi et al., 2000; van Rijt et al., 2003), likely explaining the discrepancy in the results between studies. Eosinophils are capable of processing and presenting a variety of microbial, viral (human rhinovirus-16) (Handzel et al., 1998), and parasitic antigens, as well as superantigens (Staphylococcus enterotoxins A, B and E) (Mawhorter et al., 1994) and allergen (MacKenzie et al., 2001) to promote T cell proliferation (Shi, 2004).
Eosinophils secrete a panel of cytokines capable of promoting T cell proliferation, and activation of Th1 or Th2 polarization (IL-2, IL-4, IL-6, IL-12, IL-10) (Kita, 1996; Lacy and Moqbel, 2000; MacKenzie et al., 2001; Shi et al., 2000). Furthermore, murine eosinophils promote IL-4, IL-5 and IL-13 secretion by CD4+ T cells (MacKenzie et al., 2001). The eosinophil mediates T cell proliferative and cytokine secretion responses in a CD80, CD86 and CTLA-4-dependent manner (Bashir et al., 2004). Additionally, eosinophils are involved in T cell polarization via indoleamine 2,3-dioxygenase or IDO. IDO is an enzyme involved in oxidative metabolism of tryptophan by converting tryptophan to kynurenines that can regulate Th1 and Th2 imbalance by promoting Th1 cell apoptosis (Odemuyiwa et al., 2004).
Fluorescent labeling studies have demonstrated that eosinophils traffic into the draining lymph nodes and localize to the T cell rich paracortical regions (Hogan et al., 2001; Korsgren et al., 1997; MacKenzie et al., 2001; Mishra et al., 2000; Shi et al., 2000). It has been proposed that eosinophils can only promote proliferation of effector T cells, but not naïve T cells (van Rijt et al., 2003). Eosinophils may thus traffic to draining lymph nodes in order to recruit activated effector T cells and promote proliferation of effector T cells.
B. Mast cell regulation
Eosinophils also have the propensity to regulate mast cell functions, notably through the release of granule protein and cytokines. Incubation of rat peritoneal mast cells with native MBP, EPO and ECP (but not EDN,) results in concentration-dependent histamine release (Zheutlin et al., 1984). Human umbilical cord blood derived mast cells can be activated by MBP to release histamine, PGD-2, GM-CSF, TNFα, and IL-8 (Piliponsky et al., 2002). A cross talk exists between mast cells and eosinophils, which is characterized by the fact that mast cells are also able to activate eosinophils; for example, the mast cell protease chymase promotes production of eosinophil derived stem cell factor; interestingly, this is a critical mast cell growth factor. Finally, the production by eosinophils of nerve growth factor (NGF) (Solomon et al., 1998), a cytokine involved in mast cell survival and activation (Bullock and Johnson, 1996; Horigome et al., 1994) is induce in an autocrine manner following activation by EPO (Solomon et al., 1998).
IV. Eosinophil Trafficking
The trafficking of eosinophils involves three interacting components: 1) cytokines that upregulated chemokines, 2) chemokines that activate eosinophils, and 3) adhesion molecules and other molecules (Figure 2).
Figure 2. Eosinophil trafficking.
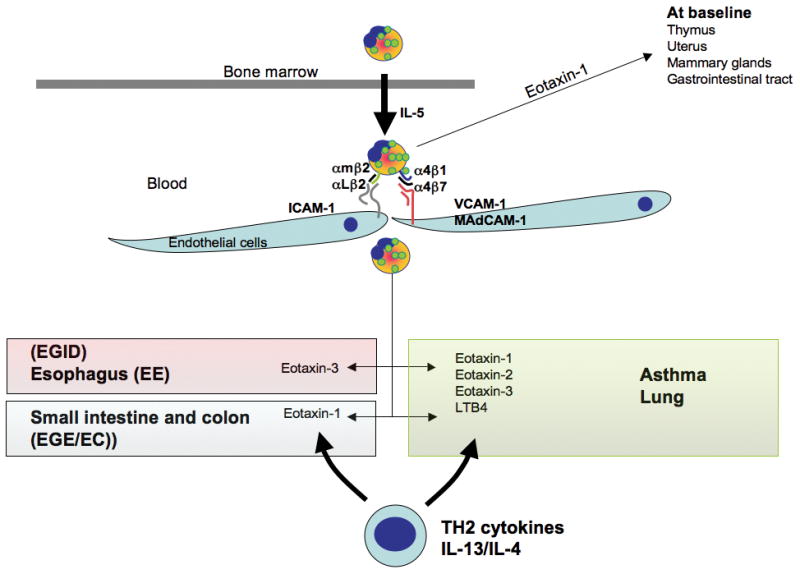
Once mature in the bone marrow, IL-5 controls the migration of eosinophils into the blood. At baseline, eotaxin-1 drives eosinophils in the thymus, uterus, mammary gland and gastrointestinal tract. Eosinophils express adhesion molecules (integrins) that allow attachment to the endothelial surface (VCAM-1, MadCAM-1, ICAM-1). Tissue chemokine expression allows the formation of a gradient chemotactic that guide the eosinophils in the tissue. In Th2 diseases, Th2 cytokines increase chemokine expression. In the asthmatic lung, eotaxin-1, 2 and -3 are increased and/or involved in chemotaxis of eosinophils, as well as other molecules such as LTB4. In the gastrointestinal tract eotaxin-3 is a key player in the eosinophilia observed in eosinophilic esophagitis while eotaxin-1 has been shown to be involved in the lower gastrointestinal eosinophilic diseases.
A. The cytokines
In inflammatory conditions, a large number of cytokines have been shown to be involved in eosinophil trafficking (most notably the Th2 cell products IL-4, IL-5, and IL-13) (Horie et al., 1997; Moser et al., 1992; Sher et al., 1990).
Using IL-5 injection, IL-5 neutralization, IL-5 transgenic mice and IL-5 deficient mice, it has been shown that IL-5 is an essential signal for the expansion and mobilization of eosinophils from the bone marrow into the lung following allergen exposure (Collins et al., 1995; Foster et al., 1996; Hogan et al., 1997). However, antigen-induced tissue eosinophilia can occur independent of IL-5, as demonstrated by residual tissue eosinophils in trials using anti-IL-5 in patients with asthma (Flood-Page et al., 2003c), and IL-5-deficient mice (Foster et al., 1996; Hogan et al., 1997).
The implications of IL-4 and IL-13 in eosinophil trafficking are mainly indirect, due to their propensity to increase chemokine expression and more particularly eotaxins. Indeed, the IL4/13 induced eotaxins by a STAT6-dependent pathway, provides an integrated mechanism to explain the eosinophilia associated with Th2 responses (Zimmermann et al., 2003).; however, recent results with IL-13Ralpha1 deficient mice, have dissociated eosinophilia from IL-13 signaling in the lung (Munitz et al., 2008).
B. The chemokines
Eotaxins, MCPs and RANTES are the main chemokines involved in eosinophil trafficking. Three eotaxins have been identified in the human genome: Eotaxin-1, 2, and 3 (Jose et al., 1994; Rankin et al., 2000; Rothenberg et al., 1995; Zimmermann et al., 2003). Eotaxin-2 and eotaxin-3 are only distantly related to eotaxin-1 since they are only ∼30% identical in sequence and are located in a different chromosomal position (Shinkai et al., 1999; Zimmermann et al., 2000b). While not sharing sequence homology, their 3-dimensional structure allows the three chemokines to bind to the same receptor: CC chemokine receptor 3 (CCR3). CCR3 is a seven-transmembrane spanning, G-protein coupled receptor primarily expressed on eosinophils (Daugherty et al., 1996; Murphy, 1994; Ponath et al., 1996). Of further interest, CCR3 has been shown to deliver a powerful negative signal in eosinophils, depending on the ligand engaged. For example, pretreatment with the chemokine Mig inhibits eosinophil responses by a CCR3 and Rac2 dependent mechanism (Fulkerson et al., 2005).
Genetic manipulation of eotaxin-1 expression and its receptor CCR3 has helped the understanding of their involvement in eosinophil infiltration in the GI tract at baseline, and in the Th2 inflammatory models such as asthma or eosinophilic esophagitis (Blanchard et al., 2006b). In an asthma model, using CCR3 deficient animals, the involvement of CCR3 in eosinophil accumulation in the lung has been shown to be modest or more marked depending on the sensitization route used (systemic or epicutaneous, respectively) (Humbles et al., 2002; Ma et al., 2002) or the origin of the mice (Pope et al., 2005).
Studies suggest that tissue and cell specificity of the expression of eotaxin-1, -2 and -3, in addition to a different kinetic expression and affinity for CCR3, influences the course of asthma pathogenesis (Zimmermann et al., 2003). Indeed, using eotaxin-1 and eotaxin-2 single and double gene-deficient mice or neutralizing antibodies, both chemokines, eotaxin-1 and 2 have been shown to have non-overlapping roles in regulating the temporal and regional distribution of eosinophils in an allergic inflammatory site in asthma models (Gonzalo et al., 1998; Pope et al., 2005; Rothenberg et al., 1997).
In humans, experimental induction of cutaneous and pulmonary late phase responses have demonstrated that the eotaxin chemokines are produced by both tissue resident cells (e.g. respiratory epithelial cells and skin fibroblasts) and allergen-induced infiltrative cells (e.g. macrophages and eosinophils). Finally, the time course of eotaxin expression in the lung is organized in time. Eotaxin-1 is induced early (6 hrs) and correlates with early eosinophil recruitment; in contrast, eotaxin-2 and 3 correlate with eosinophil accumulation at 24 hours (Zimmermann et al., 2003). Additionally, antibodies against RANTES, MCP-3, MCP-4, and eotaxin-1 are able to inhibit the chemotactic activity of the bronchoalveolar lavage fluid (Zimmermann et al., 2003). Finally, single nucleotide polymorphisms (SNPs) in eotaxin-1, 2 and 3 genes have been associated with atopy, IgE levels, eosinophilia, improved lung function (e.g. FEV1) and eosinophilic esophagitis (EE), further supporting an important role for eotaxins in human allergic diseases (Blanchard et al., 2006b; Chae et al., 2005; Nakamura et al., 2001).
C. Adhesion molecules
The involvement of adhesion molecules has been demonstrated mainly in inflammatory models and/or at baseline in the GI tract. Eosinophils express numerous adhesion molecules; some are highly expressed (Bochner and Schleimer, 1994).
1) The CD18 family of molecules or lymphocyte function antigen (LFA)-1 and Mac-1
Highly expressed by eosinophils, these molecules interact with endothelial cells via intercellular adhesion molecule (ICAM)-1. Indeed, despite the availability of alternate adhesion pathways in ICAM-1 deficient mice, the absence of ICAM-1 prevented eosinophils from entering the airways, although this reduction is due, in part, to the important role of ICAM-1 in ligand mediating T-cell proliferation in response to antigen.
2) Integrin α4β7
The α4β7 integrin interacts with the mucosal addressin cell adhesion molecule-1 (MAdCAM-1). MadCAM-1 is expressed by the vascular endothelium, more particularly in the intestinal tract. β7 gene targeted mice display a delay and reduced magnitude in the development of intestinal eosinophilia following Trichinella spiralis infection (Artis et al., 2000) and when the intestinal eotaxin-1 transgene is expressed. However, no changes in the baseline level of small intestine eosinophils is seen in β7 deficient mice (Mishra et al., 2002b).
3) The very late antigen (VLA)-4 molecules (β1-integrins)
VLA-4 interacts with endothelial cells via vascular cell adhesion molecule (VCAM)-1 and fibronectin. Anti-β1 treated mice and VLA-4-deficient mice have demonstrated the critical participation of these integrin molecules in regulating eosinophil trafficking to the allergic lung (Gonzalo et al., 1996; Nakajima et al., 1994; Pretolani et al., 1994).
4) Periostin
Finally, in asthma and EE models, it has recently been shown that the extracellular matrix protein periostin, an IL-13 induced gene that is highly overexpressed in EE patients compared to control biopsy samples, correlates with eosinophil numbers in the biopsies. Interestingly, in experimental asthma and EE, periostin-deficient mice have decreased eosinophil recruitment to the lung and the esophagus. A direct role of periostin on eosinophil adhesion was shown in vitro using spleen eosinophils from CD2-IL-5 transgenic mice. This study suggests that periostin may facilitate eosinophil infiltration in the tissues (Blanchard et al., 2008).
D. Other molecules involved in eosinophil trafficking
The arachidonic acid metabolites have been implicated in eosinophil trafficking. In particular, leukotriene (LT) B4, the cysteinyl-leukotrienes (LTC4, LTCD4, and LTE4), and prostaglandin (PG) D2 are thought to participate in eosinophilia. Indeed, cysteinyl leukotriene-type-1-receptor antagonists have been shown to reduce blood and lung eosinophilia. Additionally, mice with the targeted deletion of the LTB4 receptor have markedly reduced allergen-induced lung eosinophilia (Tager et al., 2000). The high affinity PGD2 type 2 receptor or (chemoattractant receptor Th2 cells (CRTH2)) has been shown to mediate Th2 cell and eosinophil/basophil recruitment (Hirai et al., 2001). Eosinophils have also been shown to express high levels of the histamine receptor 4 (H4) that mediates eosinophil chemoattraction and activation in vitro (O'Reilly et al., 2002). Additionally the induction of 15-lipoxygenase-1 (15-LO-1), an enzyme involved in the arachidonic pathway, has been observed in the airways of subjects with asthma. LOX15 is a Th2 induced gene and has been reported in several other Th2 mediated diseases such as eosinophilic esophagitis (Blanchard et al., 2006b). In a recent study, it has been shown that 12/15-LO knockout mice are protected from the development of mucosal allergic sensitization and airway inflammation but not against a systemic model involving IP sensitization. This suggested the presence of a lung-restricted protective role for 12/15-LO deficiency that potentially accounts for activation of mucosal B cells and increased production of secretory IgA (Hajek et al., 2008). Eoxins are new proinflammatory arachidonic acid metabolites produced via the 15-lipoxygenase pathway in human eosinophils and mast cells. These compounds were uncovered after incubation of eosinophils with exogenous arachidonic acid. Because eosinophils are such an abundant source of these metabolites and to avoid confusion with leukotrienes, these new compounds were named eoxin (EX)-C4, -D4, and -E4. Interestingly, cord blood-derived mast cells and surgically removed nasal polyps from allergic subjects produce EXC4. Eosinophils produce EXC4 after challenge with the proinflammatory agents LTC4, prostaglandin D2, and IL-5, demonstrating that EXC4 can be synthesized from the endogenous reservoir of arachidonic acid. Eoxins can increase permeability of endothelial cell monolayer in vitro, a hallmark of inflammation. Interestingly, in this study, the authors also demonstrate that eoxins are 100 times more potent than histamine and almost as potent as LTC4 and LTD4 (Feltenmark et al., 2008).
Recent attention has been given to chitin, a polymer that provides structural rigidity to fungi, crustaceans, helminths and insects. When given intranasally to mice, chitin induces the accumulation in tissue of eosinophils. This effect is reduced when the injected chitin was pre-treated with the IL-4- and IL-13-inducible mammalian chitinase, or if the chitin was injected into mice that overexpressed AMCase. Indeed chitin mediates the production of leukotriene B4, which is required in this model for optimal eosinophil recruitment (Reese et al., 2007).
E. Negative regulation of eosinophil trafficking
Leukocyte negative signaling is an important process involved in homeostatic, inflammatory, and repair responses, yet these processes have not yet been examined in eosinophils. A recent study has shown that the paired immunoglobulin-like receptor B (PIR-B), an inhibitory receptor of the Ig superfamily, is highly expressed by eosinophils. Notably, PIR-B deficient mice have increased gastrointestinal eosinophils and evidence that PIR-B directly negatively regulates eotaxin-dependent eosinophil chemotaxis in vivo and in vitro was demonstrated (Munitz et al., 2008).
V. Role of Eosinophils In Disease
Eosinophils are multifunctional leukocytes implicated in the pathogenesis of numerous inflammatory processes (Figure 3).
Figure 3. Eosinophil function.
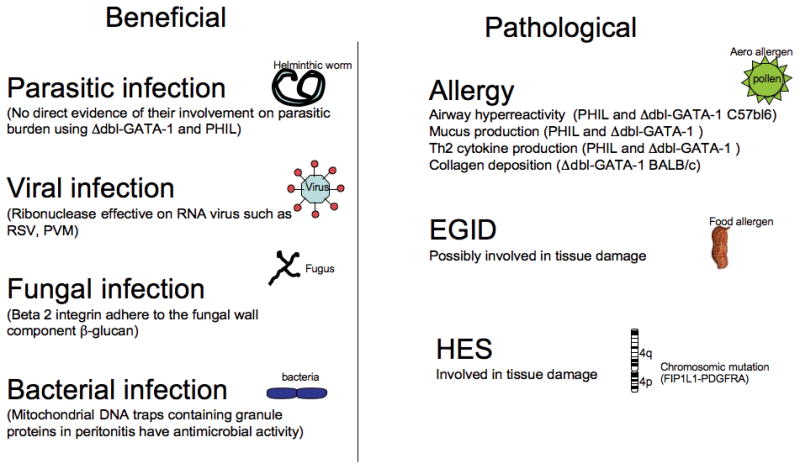
Eosinophils are believed to have a beneficial role in helminthic infection while no direct evidence was provided in the eosinophil depleted mice. Ribonuclease in granules display anti viral property on RNA virus such as RSV. Eosinophils are able to bind to fungal wall via their β2 integrin chain. Finally, DNA traps possess antibacterial activities. Eosinophils are involved in the pathological features of several diseases. In asthma, using eosinophil depleted mice (Δdbl-GATA-1 and PHIL) eosinophils have been shown involved in AHR, mucus production, TH2 cytokine production and collagen deposition. The role of eosinophils in EGID and HES might lead to tissue damage but needs to be better studied.
A. Infections
1) Helminth
Eosinophil function has primarily been associated with its contribution in host defense against parasitic infection. Several studies using helminth infection models have evaluated the propensity of eosinophils to 1) mediate antibody (or complement) dependent cellular toxicity against helminths in vitro (Butterworth, 1977), 2) to aggregate, 3) to degranulate in the local vicinity of damaged parasites in vivo during helminthic infections, and 4) to be required in experimental parasite infected mice that have been depleted of eosinophils by IL-5 neutralization and/or gene targeting (Behm and Ovington, 2000). Humans and rodents do not share many common natural helminthic hosts making the studies difficult to interpret. A role for IL-5 in protective immunity has been suggested following infection with Strongyloides venezuelensis, Strongyloides ratti, Heligmosomoides polygyrus, and Nippostrongyloides brasiliensis (Behm and Ovington, 2000; Korenaga et al., 1991). The contribution of other IL-5 receptor bearing cells such as B cell, and basophils (Bischoff et al., 1990; Erickson et al., 2001; Hakonarson et al., 1999; Sanderson, 1992), has not been ruled out in this model. The analysis of CCR3 and eotaxin-1-deficient mice have suggested a role for eosinophils in controlling the Brugia malayi microfilariae infection and in the encystment of larvae in Trichinella spiralis (Gurish et al., 2002; Simons et al., 2005); but the ultimate evidence for a role of eosinophils in host defense against parasites has not been provided yet. Swartz et al., explored the role of eosinophils in host defense against helminthic parasites in Schistosoma mansoni infection model in the two eosinophil lineage ablation mice lines (ΔdblGATA and PHIL). They found that eosinophil ablation had no effect on worm burden or on egg deposition, indicating that eosinophil ablation has no impact on traditional measures of disease in the S. mansoni infection model in mice. However, the authors concluded: “eosinophils may have unexplored immunomodulatory contributions to this disease process” (Swartz et al., 2006).
2) Viral infection
Eosinophil granule proteins are known for their ribonuclease activity (such as human ECP and EDN, and at least 11 eosinophil associated ribonucleases (EAR) orthologs in mice) and have been shown to degrade single stranded RNA containing viruses (Rosenberg and Domachowske, 2001). Interestingly, it has recently been shown that viruses (parainfluenza virus, respiratory syncytial virus, or rhinovirus) induce the release of EPO by eosinophils when coincubated in the presence of antigen-presenting cells and T cells (Davoine et al., 2008). Eosinophils may also have a protective role in other infections, especially against RNA viruses such as respiratory syncytial virus (RSV) and the related natural rodent pathogen, pneumonia virus of mice (PVM), in vivo (Adamko et al., 1999; Rosenberg and Domachowske, 2001). Despite divergence of the coding regions, (Rosenberg and Domachowske, 2001), the conserved ribonuclease activity of these molecules across species, suggests a strong evolutionary pressure to preserve this critical enzymatic activity. Paradoxally, in vitro study has shown that eosinophils may be an important reservoir for the HIV-1 virus in vivo (Freedman et al., 1991).
3) Fungal infection
Recent investigation has focused on the role of eosinophils in fungi infections. Indeed, eosinophils release their cytotoxic granule proteins into the extracellular milieu and onto the surface of fungal organisms and kill fungi in a contact-dependent manner. Yoon et al has recently shown that eosinophils use their versatile β2-integrin molecule, CD11b, to adhere to a major cell wall component, beta-glucan, but eosinophils do not express other common fungal receptors, such as dectin-1 and lactosylceramide. The I-domain of CD11b is distinctively involved in eosinophil interaction with beta-glucan. Interestingly, eosinophils do not react with chitin, another fungal cell wall component (Yoon et al., 2008).
4) Bacterial infection
As previously discussed, eosinophils rapidly release mitochondrial DNA in response to exposure to bacteria, C5a or CCR3 ligands. The traps contain the granule protein ECP and MBP, and display antimicrobial activity (Yousefi et al., 2008). In the extracellular space, the mitochondrial DNA and the granule proteins form extracellular structures that bind and kill bacteria both in vitro and under inflammatory conditions in vivo. After cecal ligation and puncture, IL5-transgenic but not wild-type mice show intestinal eosinophil infiltration and extracellular DNA deposition in association with protection against microbial sepsis. This data suggests a previously undescribed mechanism of eosinophil-mediated innate immune responses that might be crucial for maintaining the intestinal barrier function after inflammation-associated epithelial cell damage, preventing the host from uncontrolled invasion of bacteria (Yousefi et al., 2008).
B. Role of eosinophils in asthma
Granule proteins such as MBP, have been found in bronchoalveolar lavage fluid from patients with asthma in sufficient concentrations to induce cytotoxicity of a variety of host tissue including respiratory epithelial cells in vitro (Rothenberg, 1998). As previously discussed, MBP has been indirectly involved in airway hyperreactivity (AHR) due to the ability to directly increases smooth muscle reactivity (Jacoby et al., 1993). In addition to its effect on tissue, MBP can trigger the degranulation of mast cells and basophils, that is thought to also be involved in disease pathogenesis (Rothenberg, 1998). Additionally, eosinophils generate large amounts of the cysteinyl leukotrienes (Bandeira-Melo et al., 2002) that may lead to increased vascular permeability, mucus secretion, and are potent smooth muscle constrictors. Indeed, inhibitors of cysteinyl leukotrienes are effective therapeutic agents for the treatment of allergic airway disease.
However, during the past decade, the exact involvement of eosinophils in asthma pathogenesis has been very controversial due to the gap between rodent models, murine strains and contradictory observation in human disease.
- While observed in humans, eosinophil degranulation is not always consistent in murine models (Denzler et al., 2000; Shinkai et al., 2002).
- Elevated levels of blood and/or lung eosinophils are not constitutively associated with lung changes in studies with transgenic mice over-expressing IL-5 (in T cells, lung epithelial cells, or enterocytes) (Dent et al., 1990; Lee et al., 1997a; Lee et al., 1997b; Mishra et al., 2002b; Tominaga et al., 1991).
- Neutralization of IL-5 or IL-5 deficient mice, have reduced lung eosinophilia in allergen challenged lungs, (Foster et al., 1996; Foster et al., 2001; Hamelmann and Gelfand, 2001; Hogan et al., 1998; Mattes et al., 2002) but this reduction is not total (Corry et al., 1996; Foster et al., 1996; Hamelmann et al., 1997) and does not always correlate with lung function (AHR). For example, antigen-induced airway hyperreactivity occurs in allergic IL-5-deficient BALB/c mice but not in IL-5-deficient mice of the C57BL/6 strain (Foster et al., 1996; Foster et al., 1997).
- A modest effect is seen in human asthma studies using anti-IL-5 antibodies. Patients with mild to moderate asthma were shown to have decreased circulating and sputum eosinophil levels (Leckie et al., 2000); however, no clinical benefit (e.g. improvement in FEV1) was demonstrated. Indeed, clinical studies have shown that airway hyperreactivity correlates with mast cell localization near pulmonary nerves, whereas pulmonary eosinophilia relates more strongly with chronic cough (Brightling et al., 2002).
The role of eosinophils in asthma has also been investigated by targeting the receptor CCR3 and its eotaxin ligands. Using these strategies, the depletion of murine eosinophils in the lung have suggested an important role for eosinophils in the development of asthma associated airway hyperreactivity (Justice et al., 2003).
However, all these studies, using neutralizing antibody or deficient mice, do not account for the possible action of these molecules on other cell types. Indeed, two different lines of eosinophil-deficient mice were developed almost simultaneously, using two different approaches.
- The PHIL mice. Lee, et al., targeted eosinophils using the EPO promoter to drive expression of diphtheria toxin A chain (Lee et al., 2004b). The eosinophil-deficient character of these mice (called PHIL mice) was assessed by examination of peripheral blood and by immunohistochemistry of tissues with such as bone marrow, uterus, small intestine, and thymus using antibodies specific for eosinophil granule proteins.
- The Δdbl-GATA mice. In comparison, Yu et al developed mice harboring a deletion of a high affinity GATA binding site in the GATA-1 promoter (Δdbl-GATA) which led to the specific ablation of the eosinophil lineage (Humbles et al., 2004). RT-PCR analysis of gene expression in the bone marrow of the Δdbl-GATA mice revealed no expression of EPO, but expression of MBP was only partially reduced and CCR3 expression remained unchanged. Eosinophil deficiency in these mice was assessed by morphological observation of cells from the blood, bone marrow and spleen.
Indeed, utilizing both eosinophil-deficient mice, eosinophils were shown to have an integral role in experimental allergic asthma. Their specific contribution toward allergen-induced airway hyperresponsiveness and mucus cell metaplasia was however different. It is possible that Δdbl-GATA mice have residual eosinophils or unappreciated hematological abnormalities, or that alternatively, diptheria toxin treatment of PHIL mice may induce toxic effects on non-eosinophil cells. It should be noted that Δdbl-GATA mice had impaired development of lung remodeling in a chronic model of asthma, consistent with the results of anti-human IL-5 in patients with asthma (Foster et al., 2002; Kips et al., 2003). In addition, in a recent study, using transgenic murine models, Walsh et al. reported that in contrast to results obtained on a BALB/c background, eosinophil-deficient C57BL/6 Δdbl-GATA mice have reduced airway hyperresponsiveness, and cytokine production of IL-4, -5, and -13 in OVA-induced allergic airway inflammation. This was caused by reduced T cell recruitment into the lung, as these mouse lungs had reduced expression of CCL7/MCP-3, CC11/eotaxin-1, and CCL24/eotaxin-2. These studies indicate that on the C57BL/6 background, eosinophils are integral to the development of airway allergic responses by modulating chemokine and/or cytokine production in the lung, leading to T cell recruitment (Walsh et al., 2008). Finally, recent attention has been drawn to the contribution of eosinophils in regulating T cell responses in the asthmatic lung. The current paradigm surrounding allergen-mediated Th2 immune responses in the lung suggests an almost hegemonic role for T cells. Lee et al. proposed an alternative hypothesis implicating eosinophils in the regulation of pulmonary T cell responses. This was supported by OVA-sensitized/challenged mice devoid of eosinophils (the transgenic line PHIL) that have reduced airway levels of Th2 cytokines that correlated with a reduced ability to recruit effector T cells to the lung. Indeed, they have shown that adoptive transfer of Th2-polarized OVA-specific transgenic T cells (OT-II) alone into OVA-challenged PHIL recipient mice failed to restore Th2 cytokines, airway histopathologies, and, the recruitment of pulmonary effector T cells (Jacobsen et al., 2008). Using _Aspergillus fumigatus_-induced allergic airway inflammation, Fulkerson et al. have shown that mice deficient in CCR3, mice deficient in both eotaxin-1 and eotaxin-2 and Δ dbl-GATA have eosinophilic infiltration abolished by 94%, 98%, and 99% respectively. Importantly, Th2 lymphocyte cytokine production was impaired in the lungs of eosinophil- and CCR3-deficient mice as well as in allergen-induced mucus production (Fulkerson et al., 2006). All together these studies present multiple lines of independent evidence that eosinophils have a central role in chronic allergic airway disease.
C. Atopic dermatitis
In spite of the progress regarding the description of immunological phenomena associated with atopic dermatitis (AD), the pathogenesis of this disease still remains unclear. The presence of eosinophils in the inflammatory infiltrate of AD has long been established. Eosinophil numbers as well as eosinophil granule protein levels in peripheral blood are elevated in most AD patients and appear to correlate with disease activity. Moreover, eosinophil granule proteins, which possess cytotoxic activity, are deposited in the skin lesions. Interestingly, Davis et al. have shown abundant MBP positive staining in the skin of AD patients even in the absence of eosinophils (Davis et al., 2003). These observations indicate a role for eosinophils in the pathogenesis of AD. Furthermore, AD is associated with increased production of T helper 2 cytokines including IL-5 and IL-4. In AD, IL-5 would specifically act on eosinophils, resulting in accelerated eosinophilopoiesis, chemotaxis, cell activation, and delayed apoptosis and IL-4 would be responsible for the Th2 response and eosinophil specific chemokine production. Therefore, IL-5 is an interesting target for experimental therapy in this inflammatory disorder of the skin. Such studies might result in new insights into the pathogenic role of eosinophils in AD.
D. Gastrointestinal disorders
While present in multiple tissues, only GI eosinophils are associated with a marked eosinophil degranulation (Kato et al., 1998). In healthy patients or normal mice, eosinophils are present in the lamina propria throughout the GI tract from the stomach to the colon (DeBrosse et al., 2006; Kato et al., 1998). However, eosinophils are not found in Peyers patches, or intra-epithelial locations (Mishra et al., 2000; Rothenberg, 2004; Rothenberg et al., 2001a; Rothenberg et al., 2001b). Murine models have demonstrated that eosinophil infiltration in the gastrointestinal tract is not dependent upon the colonic flora or the endotoxin load of the gut as assessed by the high eosinophil level observed in prenatal mice (Mishra et al., 1999). Indeed, germ-free mice have normal levels of GI eosinophils. The accumulation of eosinophils in the GI tract is a common feature of numerous disorders such as drug reactions, helminth infections, gastroesophageal reflux disease, hypereosinophilic syndromes, eosinophilic gastroenteritis, allergic colitis and inflammatory bowel disease (Rothenberg, 2004). A subset of these diseases, are referred to as primary eosinophilic gastrointestinal disorders (EGID), including eosinophilic esophagitis (EE), eosinophilic gastritis (EG), and eosinophilic gastroenteritis (EGE). EGID usually occurs independent of peripheral blood eosinophilia, indicating the significance of gastrointestinal-specific mechanisms for regulating eosinophil levels. Interestingly, the intestine of eotaxin-1 deficient mice is almost completely devoid of eosinophils (Matthews et al., 1998; Mishra et al., 1999); and similar results were observed in CCR3 deficient mice, which show a decreased eosinophil level at baseline, in the jejunum. The residual presence of eosinophils in the GI tract of CCR3 deficient mice and eotaxin-1 deficient mice suggest a modest involvement of other chemotactic factors for eosinophils in the jejunum (Gurish et al., 2002; Humbles et al., 2002; Matthews et al., 1998). Under baseline conditions, the receptor PIR-B, expressed by eosinophils, provides an inhibitory signal that limits eosinophil accumulation into the gastrointestinal tract, including the esophagus (Munitz et al., 2008).
While absent in the normal esophagus, eosinophils markedly accumulate in the esophagus of eosinophilic esophagitis patients. A minimum of 15 eosinophils per high power field is now used as pathological criteria for EE (Gonsalves et al., 2006; Lim et al., 2004; Potter et al., 2004). Murine models have demonstrated that IL-5 maintains the systemic eosinophil levels needed for esophageal eosinophilia accumulation (Mishra et al., 2002a; Mishra et al., 1999). Substantial evidence is accumulating that human EE is also associated with a Th2 type immune response and local or systemic Th2 cytokine overproduction; IL-5 mRNA expression is induced in the biopsies of EE patients compared to healthy controls ((Straumann et al., 2001; Straumann et al., 2005) and unpublished data).
Eosinophil accumulation has been shown to be CCL11/eotaxin-1 and CCR3 dependent using the respective gene targeted mice (Blanchard et al., 2006b; Mishra and Rothenberg, 2003). However, eosinophils are still infiltrating the esophagus of CD2-IL-5tg/CCL11KO mice, suggesting that other factors are involved (Mishra et al., 1999).
In human EE, eotaxin-3 expression strongly correlates with eosinophil numbers (Blanchard et al., 2006b). Other factors such as chemokines, extracellular matrix component (periostin) or adhesion molecules may facilitate the entry of eosinophils in esophageal tissue. Eotaxin-3 is a Th2 induced molecule and interestingly, IL-13 has recently been shown to induce 20% of the EE transcriptome, and in particular, to induce eotaxin-3 expression in primary esophageal epithelial cells (Blanchard et al., 2007). Interestingly, the EE transcriptome of the diseased biopsies revealed that the accumulation of esophageal eosinophils is not associated by an increase in eosinophil specific transcripts (Blanchard et al., 2006b). As discussed earlier, Locksley's group demonstrated that eosinophil granule protein mRNAs were detectable in the early development of eosinophils but not once the eosinophils infiltrate into the tissues (Voehringer et al., 2007). Therefore, although not actively transcribed in the esophagus, granule proteins, such as MBP, EPO and EDN are present in the esophageal eosinophils, and MBP deposition has been detected by immunohistochemistry, in EE patient esophageal biopsies. As such, granule proteins may influence disease via their cytotoxic activity.
Indeed, most of the published models so far have shown an association between lung and esophageal eosinophilia; but EE and atopic dermatitis also share common features, including squamous epithelial cell hyperplasia, eosinophil infiltration, eosinophil degranulation, suggesting that common pathogenic mechanisms may be taking place.
While in human atopic dermatitis, eotaxin-3 is also increased, two murine models have shown that skin sensitization primes for EE (Akei et al., 2006; Akei et al., 2005). Epicutaneous exposure to the allergens OVA or Aspergillus fumigatus induces AD-like skin inflammation but eosinophils do not migrate into the esophagus despite a strong systemic Th2 response, chronic cutaneous antigen exposure, accelerated bone marrow eosinophilopoiesis and circulating eosinophilia. However, when epicutaneously sensitized mice are subsequently exposed only once to intranasal antigen, esophageal eosinophilia (and lung inflammation) is powerfully induced (Akei et al., 2006; Akei et al., 2005). Collectively, these experimental systems demonstrate an intimate connection between the development of eosinophilic inflammation in the respiratory tract, the skin and esophagus.
There is a paucity of studies on the molecular pathogenesis of EG, EGE and EC, mainly due to the lack of murine models. In mice, the overexpression of eotaxin-1 by epithelial cells is sufficient to induce intestinal eosinophilia in eotaxin-deficient mice suggesting a possible role for eotaxin-1 in small bowel eosinophilia (Mishra et al., 2002a). Additionally, RANTES expression has been shown to correlate with eosinophilia in food allergy model in mice (OVA) (Lee et al., 2004a) and, like eotaxin-1, RANTES mRNA is highly expressed in the jejunum of mice (Lee et al., 2004a). RANTES is expressed at baseline in the human GI tract and may contribute to hematopoietic cell recruitment in healthy and in EGID patients (Beyer et al., 2002). RANTES is increased in the colon of atopic dermatitis patients (Yamada et al., 1996) and in a rat colitis model (Ajuebor et al., 2001). No studies on RANTES deficient mice have determined the ultimate role of this cytokine in eosinophil recruitment at baseline and in EGID.
Using an experimental gastrointestinal allergy model an essential role for eotaxin-1 in regulating eosinophil-associated gastrointestinal pathology (Forbes et al., 2004), as well as the development of eosinophilia in DSS-induced colitis has been demonstrated (Hogan et al., 2000). Indeed the use of eosinophil deficient mice will uncover the role of eosinophils in these gastrointestinal models.
E. Hyper eosinophilic diseases
Idiopathic hypereosinophilic syndrome (HES) and chronic eosinophilic leukemia (CEL) are related hematological malignancies characterized by sustained, unexplained hypereosinophilia (>1,500 eosinophils/microL). The term CEL is used when there is evidence that the disease is of clonal origin. A subset of patients with HES have a 800 kb interstitial deletion on chromosome 4 (4q12) that results in the fusion of an unknown gene Fip1-like1 (FIP1L1) with the platelet derived growth factor receptor-alpha (PDGFRA) (Cools et al., 2003; Cools et al., 2004). Dysregulated tyrosine kinase activity by the FIP1L1-PDGFRA fusion gene has been identified as a cause of clonal hypereosinophilic syndrome (HES), called FIP1L1-PDGFRA-positive chronic eosinophilic leukemia (CEL) in humans. However, transplantation of FIP1L1-PDGFRA-transduced hematopoietic stem cells/progenitors (HSC/Ps) into mice results in a chronic myelogenous leukemia-like disease, which does not resemble HES. Because a subgroup of patients with HES show T-cell-dependent IL-5 overexpression, the expression of the FIP1L1-PDGFRA fusion gene in the presence of transgenic T-cell IL-5 overexpression in mice induces HES-like disease was studied. Mice that received a transplant of CD2-IL-5-transgenic FIP1L1-PDGFRA positive HSC/Ps (IL-5Tg-F/P) developed intense leukocytosis, strikingly high eosinophilia, and eosinophilic infiltration of nonhematopoietic as well as hematopoietic tissues, a phenotype resembling human HES. The disease phenotype was transferable to secondary transplant recipients, suggesting involvement of a short-term repopulating stem cell or an early myeloid progenitor. Induction of significant eosinophilia is in this model specific for FIP1L1-PDGFRA since expression of another fusion oncogene, p210-BCR/ABL, in the presence of IL-5 overexpression is characterized by a significantly lower eosinophilia than IL-5Tg-F/P recipients. These results suggest that FIP1L1-PDGFRA fusion gene is not sufficient to induce a HES/CEL-like disease but requires a second event associated with IL-5 overexpression (Yamada et al., 2006).
VI. Anti-Eosinophil Therapeutics
A. Therapeutics available
Several therapeutics help in the control of systemic and tissue eosinophilia (Rothenberg and Hogan, 2005).
- Glucocorticoids are the most common agents for reducing eosinophilia (Rothenberg, 1998). They seems to act on the transcription of a number of genes for inflammatory mediators including the genes for IL-3, IL-4, IL-5, GM-CSF, and various chemokines including the eotaxins. Glucocorticoids have also been shown to destabilize the mRNA of eosinophil active cytokines; thus, reducing the half-life of cytokines such as eotaxins (Stellato et al., 1999). In addition, glucocorticoids inhibit the cytokine-dependent survival of eosinophils (Schleimer and Bochner, 1994). Systemic or topical (inhaled or intranasal) glucocorticoid treatment causes a rapid reduction in eosinophils. Systemic and topical glucocorticoids are indeed widely used in controlling eosinophil infiltration in EE, when the diet modification is too restricting (Aceves et al., 2005; Blanchard et al., 2006a; Konikoff et al., 2006; Rothenberg et al., 2001c). Unfortunately long-term use of glucocorticoids is usually accompanied by side effects. Indeed glucocorticoids are not effective for everyone since few patients are glucocorticoid-resistant and maintain eosinophilia despite high doses (Barnes and Adcock, 1995). The mechanism of glucocorticoid resistance is unclear, but a reduced level of glucocorticoid receptors, polymorphism and alterations in transcription factor activator protein-1 (AP-1) appear to be at least partially responsible in some of them (Barnes and Adcock, 1995). Glucocorticoid-resistant patients thus require other therapy such as myelosuppressive drugs (hydroxyurea, vincristine) or alpha-interferon (Rothenberg, 1998).
- Alpha-interferon can be especially helpful because it inhibits eosinophil degranulation and effector function (Aldebert et al., 1996). Notably, patients with myeloproliferative variants of hypereosinophilic syndrome can often go into remission with alpha-interferon therapy.
- Cyclosporine A has also been used because it blocks the transcription of numerous eosinophil-active cytokines (e.g. IL-5, GM-CSF) (Meng et al., 1997; Rolfe et al., 1997).
- Lidocaine is another drug that has been shown to shorten eosinophil survival, and while its effects mimic those of glucocorticoids they are non-cytotoxic (Bankers-Fulbright et al., 1998). Indeed, a clinical trial has shown that nebulized lidocaine is effective in subjects with asthma (Hunt et al., 2004).
- The arachidonic acid products are also targeted and drugs that interfere with eosinophil chemotactic signals include leukotriene antagonists and inhibitors. For example, 5-lipoxygenase inhibition, blocks the rate-limiting step in leukotriene synthesis and inhibits the generation of the eosinophil chemoattractant, LTB4, and the cysteinyl leukotrienes (Kane et al., 1996). The inhibition of cysteinyl leukotriene receptor, using antagonists blocks the increased vascular permeability mediated by leukocyte-derived leukotrienes (Gaddy et al., 1992).
- Some of the third generation antihistamines inhibit the vacuolization (Snyman et al., 1992) and accumulation (Redier et al., 1992) of eosinophils after challenge and directly inhibit eosinophils in vitro (Rand et al., 1988; Snyman et al., 1992).
- Cromoglycate and nedocromil inhibit the effector function of eosinophils such as antibody-dependent cellular cytotoxicity (Rand et al., 1988).
- Imatinib therapy. The etiology of the primary disease often specifies the best therapeutic strategy. Patients with hypereosinophilic syndrome with FIP1L1-PDGFRA+ disease are now treated with Imatinib mesylate or STI 571 as first line therapy (Gleich et al., 2002). This anti-tyrosine-kinase was the first of its categories to be prescribed in patients and has been shown active on non- FIP1L1-PDGFRA+ cancer patients. Indeed, a variety of other activated tyrosine kinases have been associated with hypereosinophilic syndromes including PDGFR-beta, Janus kinase-2, and fibroblast growth factor receptor-1 (Klion, 2005).
- Alemtuzumab is a monoclonal anti-CD52 antibody that depleted CD52+ cells including lymphocytes; it is used in the treatment of chronic lymphocytic leukemia (CLL) and T-cell lymphoma. As CD52+ cells, it was hypothesized that eosinophils may also be depleted by this therapy. Indeed, recent clinical trials have shown that Alemtuzumab can lower eosinophils and induced disease remission in patients with refractory idiopathic HES with abnormal T cells (CD3-CD4+) (Pitini et al., 2004; Sefcick et al., 2004). This targeted therapy hold great promise for the treatment of certain HES patients who are resistant to other therapies.
B. Therapeutics in development
The identification of molecules that specifically regulate eosinophil function and/or production offers new therapeutic strategies in the pipeline. Agents that interrupt eosinophil adhesion to the endothelium through the interaction of CD18/ICAM-1 (Wegner et al., 1990) or VLA-4 /VCAM-1 may be useful (Kuijpers et al., 1993; Weg et al., 1993). Inhibitors of the IL-13/eotaxin/CCR3 axis including small molecule inhibitors of CCR3 and a human anti-human eotaxin-1 and IL-13 antibody are being developed (Blanchard et al., 2005; Zimmermann et al., 2003) and looks promising for lowering tissue eosinophil levels. A recently identified eosinophil surface molecule Siglec-8 may offer a therapeutic opportunity (Nutku et al., 2003). Siglec-8 is a member of the sialic acid binding lectin family and contains ITIMs (immunoreceptor tyrosine-based inhibitory motifs) that can induce efficient eosinophil apoptosis when crosslinked. Finally, CD48 is an activation molecule on eosinophils, its neutralization has been shown to decrease eosinophil infiltration in the lung in vivo (Munitz et al., 2006). Siglec-8, as well as CCR-3 and the chemoattractant receptor-homologous molecule expressed on Th2 lymphocytes (CRTH2) are co-expressed by other cells involved in Th2 responses including Th2 cells, mast cells, and basophils and may thus target several other aspects of allergic disorders than just eosinophils (Rothenberg and Hogan, 2005).
The critical role of humanized anti-IL-5 in decreasing eosinophil load in humans has been demonstrated by several clinical trials with humanized anti-IL-5 antibody (Egan et al., 1995; Mauser et al., 1995). This drug dramatically lowers eosinophil levels in the blood, decreases eosinophil activation and to a lesser extent eosinophil levels in the inflamed lung and esophagus (Flood-Page et al., 2003a; Flood-Page et al., 2003b; Kips et al., 2003; Leckie et al., 2000; Stein et al., 2008). In a recent study, 85 HES patients (whose condition remained stable on steroid monotherapy) were randomized to placebo or 750 mg of mepolizumab every four weeks for 36 weeks (Rothenberg et al., 2008). Indeed, all but one patient in the mepolizumab group (95%) had eosinophil counts below 600 for at least eight weeks, versus 45% of the placebo group. Additionally, the mean prednisone dose increased to 6.2 mg/day in the mepolizumab group, compared with 21.8 mg/day in the placebo group confirming that anti-IL-5 therapies such as mepolizumab have potential as clinical therapies for hypereosinophilic syndrome.
In a recent study, Nair et al. studied, in a randomized placebo-controlled trial, the prednisone-sparing effect of mepolizumab on eosinophilic bronchitis with or without asthma. They found that patients who received mepolizumab were able to reduce their prednisone dose by 90% of their maximum possible compared to 55% in the placebo arm (p<0.05). Mepolizumab treatment was accompanied by a significant decrease in sputum and blood eosinophils and improvements in asthma control, FEV1 and asthma quality of life that were maintained for 8 weeks after the last infusion, suggesting that mepolizumab is an effective prednisone-sparing therapy in patients with eosinophilic bronchitis with or without asthma (Nair et al., 2008)
Acknowledgments
The Authors wish to thank the whole eosinophil field that built the concepts presented. Andrea lippelman, Katherine Henderson and LaWanda Bryant for administrative assistance.
This work was supported by in part by the Thrasher Research Fund NR-0014 (C.B.), the PHS Grant P30 DK0789392 (C.B.), the NIH AI079874-01 (C.B.) AI070235, AI45898, and DK076893 (M.E.R.), the Food Allergy and Anaphylaxis Network (M.E.R.), Campaign Urging Research for Eosinophil Disorders (CURED), the Buckeye Foundation (M.E.R.) and The Food Allergy Project (M.E.R).
References
- Aceves SS, Dohil R, Newbury RO, Bastian JF. J Allergy Clin Immunol. 2005;116:705. doi: 10.1016/j.jaci.2005.05.011. [DOI] [PubMed] [Google Scholar]
- Ackerman SJ, Gleich GJ, Loegering DA, Richardson BA, Butterworth AE. Am J Trop Med Hyg. 1985;34:735. doi: 10.4269/ajtmh.1985.34.735. [DOI] [PubMed] [Google Scholar]
- Adamko DJ, Yost BL, Gleich GJ, Fryer AD, Jacoby DB. J Exp Med. 1999;190:1465. doi: 10.1084/jem.190.10.1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agosti JM, Altman LC, Ayars GH, Loegering DA, Gleich GJ, Klebanoff SJ. J Allergy Clin Immunol. 1987;79:496. doi: 10.1016/0091-6749(87)90368-x. [DOI] [PubMed] [Google Scholar]
- Ajuebor MN, Hogaboam CM, Kunkel SL, Proudfoot AE, Wallace JL. J Immunol. 2001;166:552. doi: 10.4049/jimmunol.166.1.552. [DOI] [PubMed] [Google Scholar]
- Akei HS, Brandt EB, Mishra A, Strait RT, Finkelman FD, Warrier MR, Hershey GK, Blanchard C, Rothenberg ME. J Allergy Clin Immunol. 2006;118:62. doi: 10.1016/j.jaci.2006.04.046. [DOI] [PubMed] [Google Scholar]
- Akei HS, Mishra A, Blanchard C, Rothenberg ME. Gastroenterology. 2005;129:985. doi: 10.1053/j.gastro.2005.06.027. [DOI] [PubMed] [Google Scholar]
- Aldebert D, Lamkhioued B, Desaint C, Gounni AS, Goldman M, Capron A, Prin L, Capron M. Blood. 1996;87:2354. [PubMed] [Google Scholar]
- Artis D, Humphreys NE, Potten CS, Wagner N, Muller W, McDermott JR, Grencis RK, Else KJ. European Journal of Immunology. 2000;30:1656. doi: 10.1002/1521-4141(200006)30:6<1656::AID-IMMU1656>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- Bandeira-Melo C, Woods LJ, Phoofolo M, Weller PF. J Exp Med. 2002;196:841. doi: 10.1084/jem.20020516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bankers-Fulbright JL, Kephart GM, Loegering DA, Bradford AL, Okada S, Kita H, Gleich GJ. J Immunol. 1998;160:5546. [PubMed] [Google Scholar]
- Barnes PJ, Adcock IM. QJM. 1995;88:455. [PubMed] [Google Scholar]
- Bashir ME, Louie S, Shi HN, Nagler-Anderson C. J Immunol. 2004;172:6978. doi: 10.4049/jimmunol.172.11.6978. [DOI] [PubMed] [Google Scholar]
- Behm CA, Ovington KS. Parasitol Today. 2000;16:202. doi: 10.1016/s0169-4758(99)01620-8. [DOI] [PubMed] [Google Scholar]
- Beyer K, Castro R, Birnbaum A, Benkov K, Pittman N, Sampson HA. J Allergy Clin Immunol. 2002;109:707. doi: 10.1067/mai.2002.122503. [DOI] [PubMed] [Google Scholar]
- Bischoff SC, Brunner T, De WA, Dahinden CA. J Exp Med. 1990;172:1577. doi: 10.1084/jem.172.6.1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanchard C, Mingler MK, MacBride M, Putnam PE, Collins MH, Chang G, Stringer K, Abonia JP, Molkentin JD, Rothenberg ME. Mucosal Immunology. 2008;1:289. doi: 10.1038/mi.2008.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanchard C, Mingler MK, Vicario M, Abonia JP, Wu YY, Lu TX, Collins MH, Putnam PE, Wells SI, Rothenberg ME. J Allergy Clin Immunol. 2007;120:1292. doi: 10.1016/j.jaci.2007.10.024. [DOI] [PubMed] [Google Scholar]
- Blanchard C, Mishra A, Saito-Akei H, Monk P, Anderson I, Rothenberg ME. Clin Exp Allergy. 2005;35:1096. doi: 10.1111/j.1365-2222.2005.02299.x. [DOI] [PubMed] [Google Scholar]
- Blanchard C, Wang N, Rothenberg ME. J Allergy Clin Immunol. 2006a;118:1054. doi: 10.1016/j.jaci.2006.07.038. [DOI] [PubMed] [Google Scholar]
- Blanchard C, Wang N, Stringer KF, Mishra A, Fulkerson PC, Abonia JP, Jameson SC, Kirby C, Konikoff MR, Collins MH, Cohen MB, Akers R, Hogan SP, Assa'ad AH, Putnam PE, Aronow BJ, Rothenberg ME. J Clin Invest. 2006b;116:536. doi: 10.1172/JCI26679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bochner BS, Schleimer RP. Journal of Allergy & Clinical Immunology. 1994;94:427. doi: 10.1016/0091-6749(94)90195-3. [DOI] [PubMed] [Google Scholar]
- Boyce JA, Friend D, Matsumoto R, Austen KF, Owen WF. Journal of Experimental Medicine. 1995;182:49. doi: 10.1084/jem.182.1.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brightling CE, Bradding P, Symon FA, Holgate ST, Wardlaw AJ, Pavord ID. N Eng J Med. 2002;346:1699. doi: 10.1056/NEJMoa012705. [DOI] [PubMed] [Google Scholar]
- Bullock ED, Johnson EM., Jr J Biol Chem. 1996;271:27500. doi: 10.1074/jbc.271.44.27500. [DOI] [PubMed] [Google Scholar]
- Butterworth AE. Current Topics in Microbiology & Immunology. 1977;77:127. doi: 10.1007/978-3-642-66740-4_5. [DOI] [PubMed] [Google Scholar]
- Butterworth AE. Adv Parasitol. 1984;23:143. doi: 10.1016/s0065-308x(08)60287-0. [DOI] [PubMed] [Google Scholar]
- Chae SC, Park YR, Shim SC, Lee IK, Chung HT. Hum Immunol. 2005;66:314. doi: 10.1016/j.humimm.2005.01.002. [DOI] [PubMed] [Google Scholar]
- Collins PD, Marleau S, Griffiths-Johnson DA, Jose PJ, Williams TJ. J Exp Med. 1995;182:1169. doi: 10.1084/jem.182.4.1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cools J, DeAngelo DJ, Gotlib J, Stover EH, Legare RD, Cortes J, Kutok J, Clark J, Galinsky I, Griffin JD, Cross NC, Tefferi A, Malone J, Alam R, Schrier SL, Schmid J, Rose M, Vandenberghe P, Verhoef G, Boogaerts M, Wlodarska I, Kantarjian H, Marynen P, Coutre SE, Stone R, Gilliland DG. N Engl J Med. 2003;348:1201. doi: 10.1056/NEJMoa025217. [DOI] [PubMed] [Google Scholar]
- Cools J, Stover EH, Wlodarska I, Marynen P, Gilliland DG. Curr Opin Hematol. 2004;11:51. doi: 10.1097/00062752-200401000-00008. [DOI] [PubMed] [Google Scholar]
- Corry D, Folkesson H, Warnock M, Erle D, Matthay M, Wiener-Kronish J, Locksley R. J Exp Med. 1996;183:109. doi: 10.1084/jem.183.1.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daugherty BL, Siciliano SJ, Demartino JA, Malkowitz L, Sirotina A, Springer MS. Journal of Experimental Medicine. 1996;183:2349. doi: 10.1084/jem.183.5.2349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis MD, Plager DA, George TJ, Weiss EA, Gleich GJ, Leiferman KM. J Allergy Clin Immunol. 2003;112:988. doi: 10.1016/j.jaci.2003.08.028. [DOI] [PubMed] [Google Scholar]
- Davoine F, Cao M, Wu Y, Ajamian F, Ilarraza R, Kokaji AI, Moqbel R, Adamko DJ. J Allergy Clin Immunol. 2008;122:69. doi: 10.1016/j.jaci.2008.03.028. [DOI] [PubMed] [Google Scholar]
- DeBrosse CW, Case JW, Putnam PE, Collins MH, Rothenberg ME. Pediatr Dev Pathol. 2006;9:210. doi: 10.2350/11-05-0130.1. [DOI] [PubMed] [Google Scholar]
- Dent LA, Strath M, Mellor AL, Sanderson CJ. J Exp Med. 1990;172:1425. doi: 10.1084/jem.172.5.1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denzler KL, Farmer SC, Crosby JR, Borchers MT, Cieslewicz G, Larson KA, Cormier-Regard S, Lee NA, Lee JJ. J Immunol. 2000;165:5509. doi: 10.4049/jimmunol.165.10.5509. [DOI] [PubMed] [Google Scholar]
- Du J, Stankiewicz MJ, Liu Y, Xi Q, Schmitz JE, Lekstrom-Himes JA, Ackerman SJ. J Biol Chem. 2002;277:43481. doi: 10.1074/jbc.M204777200. [DOI] [PubMed] [Google Scholar]
- Egan RW, Athwahl D, Chou CC, Emtage S, Jehn CH, Kung TT, Mauser PJ, Murgolo NJ, Bodmer MW. International Archives of Allergy & Immunology. 1995;107:321. doi: 10.1159/000237014. [DOI] [PubMed] [Google Scholar]
- Erickson LD, Foy TM, Waldschmidt TJ. J Immunol. 2001;166:1531. doi: 10.4049/jimmunol.166.3.1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feltenmark S, Gautam N, Brunnstrom A, Griffiths W, Backman L, Edenius C, Lindbom L, Bjorkholm M, Claesson HE. Proc Natl Acad Sci U S A. 2008;105:680. doi: 10.1073/pnas.0710127105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flood-Page P, Menzies-Gow A, Phipps S, Ying S, Wangoo A, Ludwig MS, Barnes N, Robinson D, Kay AB. J Clin Invest. 2003a;112:1029. doi: 10.1172/JCI17974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flood-Page P, Phipps S, Menzies-Gow A, Ong YE, Kay AB. J Allergy Clin Immunol. 2003b;111:S261. doi: 10.1067/mai.2003.1382. [DOI] [PubMed] [Google Scholar]
- Flood-Page PT, Menzies-Gow AN, Kay AB, Robinson DS. Am J Respir Crit Care Med. 2003c;167:199. doi: 10.1164/rccm.200208-789OC. [DOI] [PubMed] [Google Scholar]
- Forbes E, Murase T, Yang M, Matthaei KI, Lee JJ, Lee NA, Foster PS, Hogan SP. J Immunol. 2004;172:5664. doi: 10.4049/jimmunol.172.9.5664. [DOI] [PubMed] [Google Scholar]
- Foster P, Hogan S, Ramsay A, Matthaei K, Young I. J Exp Med. 1996;183:195. doi: 10.1084/jem.183.1.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster P, Mould A, Yang M, Mackenzie J, Mattes J, Hogan S, Mahalingam S, M ANJ, Rothenberg M, Young I, Matthaei K, Webb D. Immunological Reviews. 2001;179:173. doi: 10.1034/j.1600-065x.2001.790117.x. [DOI] [PubMed] [Google Scholar]
- Foster PS, Hogan SP, Matthaei KI, Young IG. Mem Inst Oswaldo Cruz. 1997;92 Suppl 2:55. doi: 10.1590/s0074-02761997000800009. [DOI] [PubMed] [Google Scholar]
- Foster PS, Hogan SP, Yang M, Mattes J, Young IG, Matthaei KI, Kumar RK, Mahalingam S, Webb DC. Trends Mol Med. 2002;8:162. doi: 10.1016/s1471-4914(02)02302-x. [DOI] [PubMed] [Google Scholar]
- Freedman AR, Gibson FM, Fleming SC, Spry CJ, Griffin GE. J Exp Med. 1991;174:1661. doi: 10.1084/jem.174.6.1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frigas E, Loegering DA, Gleich GJ. Lab Invest. 1980;42:35. [PubMed] [Google Scholar]
- Fulkerson PC, Fischetti CA, McBride ML, Hassman LM, Hogan SP, Rothenberg ME. Proc Natl Acad Sci U S A. 2006;103:16418. doi: 10.1073/pnas.0607863103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulkerson PC, Zhu H, Williams DA, Zimmermann N, Rothenberg ME. Blood. 2005;106:436. doi: 10.1182/blood-2005-02-0489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuta GT, Nieuwenhuis EE, Karhausen J, Gleich G, Blumberg RS, Lee JJ, Ackerman SJ. Am J Physiol Gastrointest Liver Physiol. 2005;289:G890. doi: 10.1152/ajpgi.00015.2005. [DOI] [PubMed] [Google Scholar]
- Gaddy JN, Margolskee DJ, Bush RK, Williams VC, Busse WW. American Review of Respiratory Disease. 1992;146:358. doi: 10.1164/ajrccm/146.2.358. [DOI] [PubMed] [Google Scholar]
- Gleich GJ. Ann Inst Pasteur Immunol. 1986;137D:136. [PubMed] [Google Scholar]
- Gleich GJ, Adolphson CR. Adv Immunol. 1986;39:177. doi: 10.1016/s0065-2776(08)60351-x. [DOI] [PubMed] [Google Scholar]
- Gleich GJ, Frigas E, Loegering DA, Wassom DL, Steinmuller D. J Immunol. 1979;123:2925. [PubMed] [Google Scholar]
- Gleich GJ, Leiferman KM, Pardanani A, Tefferi A, Butterfield JH. Lancet. 2002;359:1577. doi: 10.1016/S0140-6736(02)08505-7. [DOI] [PubMed] [Google Scholar]
- Gonsalves N, Policarpio-Nicolas M, Zhang Q, Rao MS, Hirano I. Gastrointest Endosc. 2006;64:313. doi: 10.1016/j.gie.2006.04.037. [DOI] [PubMed] [Google Scholar]
- Gonzalo JA, Lloyd CM, Kremer L, Finger E, Martinez AC, Siegelman MH, Cybulsky M, Gutierrez-Ramos JC. J Clin Invest. 1996;98:2332. doi: 10.1172/JCI119045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalo JA, Lloyd CM, Wen D, Albar JP, Wells TN, Proudfoot A, Martinez AC, Dorf M, Bjerke T, Coyle AJ, Gutierrez-Ramos JC. J Exp Med. 1998;188:157. doi: 10.1084/jem.188.1.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouon-Evans V, Lin EY, Pollard JW. Breast Cancer Res. 2002;4:155. doi: 10.1186/bcr441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouon-Evans V, Pollard JW. Endocrinology. 2001;142:4515. doi: 10.1210/endo.142.10.8459. [DOI] [PubMed] [Google Scholar]
- Gouon-Evans V, Rothenberg ME, Pollard JW. Development. 2000;127:2269. doi: 10.1242/dev.127.11.2269. [DOI] [PubMed] [Google Scholar]
- Gurish MF, Humbles A, Tao H, Finkelstein S, Boyce JA, Gerard C, Friend DS, Austen KF. J Immunol. 2002;168:5730. doi: 10.4049/jimmunol.168.11.5730. [DOI] [PubMed] [Google Scholar]
- Hajek AR, Lindley AR, Favoreto S, Jr, Carter R, Schleimer RP, Kuperman DA. J Allergy Clin Immunol. 2008 doi: 10.1016/j.jaci.2008.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hakonarson H, Maskeri N, Carter C, Chuang S, Grunstein MM. J Clin Invest. 1999;104:657. doi: 10.1172/JCI7137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamann KJ, Barker RL, Ten RM, Gleich GJ. Int Arch Allergy Appl Immunol. 1991;94:202. doi: 10.1159/000235362. [DOI] [PubMed] [Google Scholar]
- Hamelmann E, Gelfand EW. Immunol Rev. 2001;179:182. doi: 10.1034/j.1600-065x.2001.790118.x. [DOI] [PubMed] [Google Scholar]
- Hamelmann E, Oshiba A, Loader J, Larsen GL, Gleich G, Lee J, Gelfand EW. Am J Respir Crit Care Med. 1997;155:819. doi: 10.1164/ajrccm.155.3.9117011. [DOI] [PubMed] [Google Scholar]
- Handzel ZT, Busse WW, Sedgwick JB, Vrtis R, Lee WM, Kelly EA, Gern JE. J Immunol. 1998;160:1279. [PubMed] [Google Scholar]
- Hirai H, Tanaka K, Yoshie O, Ogawa K, Kenmotsu K, Takamori Y, Ichimasa M, Sugamura K, Nakamura M, Takano S, Nagata K. J Exp Med. 2001;193:255. doi: 10.1084/jem.193.2.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirasawa R, Shimizu R, Takahashi S, Osawa M, Takayanagi S, Kato Y, Onodera M, Minegishi N, Yamamoto M, Fukao K, Taniguchi H, Nakauchi H, Iwama A. J Exp Med. 2002;195:1379. doi: 10.1084/jem.20020170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hisamatsu K, Ganbo T, Nakazawa T, Murakami Y, Gleich GJ, Makiyama K, Koyama H. J Allergy Clin Immunol. 1990;86:52. doi: 10.1016/s0091-6749(05)80123-x. [DOI] [PubMed] [Google Scholar]
- Hogan SP, Koskinen A, Foster PS. Immunol Cell Biol. 1997;75:284. doi: 10.1038/icb.1997.43. [DOI] [PubMed] [Google Scholar]
- Hogan SP, Koskinen A, Matthaei KI, Young IG, Foster PS. Am J Respir Crit Care Med. 1998;157:210. doi: 10.1164/ajrccm.157.6.mar-1. [DOI] [PubMed] [Google Scholar]
- Hogan SP, Mishra A, Brandt EB, Foster PS, Rothenberg ME. Proc Natl Acad Sci U S A. 2000;97:6681. doi: 10.1073/pnas.97.12.6681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogan SP, Mishra A, Brandt EB, Royalty MP, Pope SM, Zimmermann N, Foster PS, Rothenberg ME. Nat Immunol. 2001;2:353. doi: 10.1038/86365. [DOI] [PubMed] [Google Scholar]
- Horie S, Okubo Y, Hossain M, Sato E, Nomura H, Koyama S, Suzuki J, Isobe M, Sekiguchi M. Intern Med. 1997;36:179. doi: 10.2169/internalmedicine.36.179. [DOI] [PubMed] [Google Scholar]
- Horigome K, Bullock ED, Johnson EM., Jr J Biol Chem. 1994;269:2695. [PubMed] [Google Scholar]
- Humbles AA, Lloyd CM, McMillan SJ, Friend DS, Xanthou G, McKenna EE, Ghiran S, Gerard NP, Yu C, Orkin SH, Gerard C. Science. 2004;305:1776. doi: 10.1126/science.1100283. [DOI] [PubMed] [Google Scholar]
- Humbles AA, Lu B, Friend DS, Okinaga S, Lora J, Al-Garawi A, Martin TR, Gerard NP, Gerard C. Proc Natl Acad Sci U S A. 2002;99:1479. doi: 10.1073/pnas.261462598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt LW, Frigas E, Butterfield JH, Kita H, Blomgren J, Dunnette SL, Offord KP, Gleich GJ. J Allergy Clin Immunol. 2004;113:853. doi: 10.1016/j.jaci.2004.02.039. [DOI] [PubMed] [Google Scholar]
- Iwasaki H, Mizuno S, Mayfield R, Shigematsu H, Arinobu Y, Seed B, Gurish MF, Takatsu K, Akashi K. J Exp Med. 2005;201:1891. doi: 10.1084/jem.20050548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobsen EA, Ochkur SI, Pero RS, Taranova AG, Protheroe CA, Colbert DC, Lee NA, Lee JJ. J Exp Med. 2008;205:699. doi: 10.1084/jem.20071840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacoby DB, Gleich GJ, Fryer AD. J Clin Invest. 1993;91:1314. doi: 10.1172/JCI116331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jose PJ, Griffiths-Johnson DA, Collins PD, Walsh DT, Moqbel R, Totty NF, Truong O, Hsuan JJ, Williams TJ. J Exp Med. 1994;179:881. doi: 10.1084/jem.179.3.881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Justice JP, Borchers MT, Crosby JR, Hines EM, Shen HH, Ochkur SI, McGarry MP, Lee NA, Lee JJ. Am J Physiol Lung Cell Mol Physiol. 2003;284:L169. doi: 10.1152/ajplung.00260.2002. [DOI] [PubMed] [Google Scholar]
- Kane GC, Pollice M, Kim CJ, Cohn J, Dworski RT, Murray JJ, Sheller JR, Fish JE, Peters SP. Journal of Allergy & Clinical Immunology. 1996;97:646. doi: 10.1016/s0091-6749(96)70310-x. [DOI] [PubMed] [Google Scholar]
- Kato M, Kephart GM, Talley NJ, Wagner JM, Sarr MG, Bonno M, McGovern TW, Gleich GJ. Anat Rec. 1998;252:418. doi: 10.1002/(SICI)1097-0185(199811)252:3<418::AID-AR10>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- Kips JC, O'Connor BJ, Langley SJ, Woodcock AA, Kerstjens HA, Postma DS, Danzig M, Cuss F, Pauwels RA. American Journal of Respiratory and Critical Care Medicine. 2003;167:1655. doi: 10.1164/rccm.200206-525OC. [DOI] [PubMed] [Google Scholar]
- Kita H. J Allergy Clin Immunol. 1996;97:889. doi: 10.1016/s0091-6749(96)80061-3. [DOI] [PubMed] [Google Scholar]
- Klion AD. Hematology Am Soc Hematol Educ Program. 2005:209. doi: 10.1182/asheducation-2005.1.209. [DOI] [PubMed] [Google Scholar]
- Konikoff MR, Noel RJ, Blanchard C, Kirby C, Jameson SC, Buckmeier BK, Akers R, Cohen MB, Collins MH, Assa'ad AH, Aceves SS, Putnam PE, Rothenberg ME. Gastroenterology. 2006;131:1381. doi: 10.1053/j.gastro.2006.08.033. [DOI] [PubMed] [Google Scholar]
- Kopf M, Brombacher F, Hodgkin PD, Ramsay AJ, Milbourne EA, Dai WJ, Ovington KS, Behm CA, Kohler G, Young IG, Matthaei KI. Immunity. 1996;4:15. doi: 10.1016/s1074-7613(00)80294-0. [DOI] [PubMed] [Google Scholar]
- Korenaga M, Hitoshi Y, Yamaguchi N, Sato Y, Takatsu K, Tada I. Immunology. 1991;72:502. [PMC free article] [PubMed] [Google Scholar]
- Korsgren M, Erjefalt JS, Korsgren O, Sundler F, Persson CGA. Journal of Experimental Medicine. 1997;185:885. doi: 10.1084/jem.185.5.885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuijpers TW, Mul EP, Blom M, Kovach NL, Gaeta FC, Tollefson V, Elices MJ, Harlan JM. J Exp Med. 1993;178:279. doi: 10.1084/jem.178.1.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacy P, Moqbel R. Chem Immunol. 2000;76:134. doi: 10.1159/000058782. [DOI] [PubMed] [Google Scholar]
- Leckie MJ, ten Brinke A, Khan J, Diamant Z, O'Connor BJ, Walls CM, Mathur AK, Cowley HC, Chung KF, Djukanovic R, Hansel TT, Holgate ST, Sterk PJ, Barnes PJ. Lancet. 2000;356:2144. doi: 10.1016/s0140-6736(00)03496-6. [DOI] [PubMed] [Google Scholar]
- Lee JB, Matsumoto T, Shin YO, Yang HM, Min YK, Timothy O, Bae SJ, Quan FS. Immunol Invest. 2004a;33:27. doi: 10.1081/imm-120027682. [DOI] [PubMed] [Google Scholar]
- Lee JJ, Dimina D, Macias MP, Ochkur SI, McGarry MP, O'Neill KR, Protheroe C, Pero R, Nguyen T, Cormier SA, Lenkiewicz E, Colbert D, Rinaldi L, Ackerman SJ, Irvin CG, Lee NA. Science. 2004b;305:1773. doi: 10.1126/science.1099472. [DOI] [PubMed] [Google Scholar]
- Lee JJ, McGarry MP, Farmer SC, Denzler KL, Larson KA, Carrigan PE, Brenneise IE, Horton MA, Haczku A, Gelfand EW, Leikauf GD, Lee NA. J Exp Med. 1997a;185:2143. doi: 10.1084/jem.185.12.2143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee NA, McGarry MP, Larson KA, Horton MA, Kristensen AB, Lee JJ. J Immunol. 1997b;158:1332. [PubMed] [Google Scholar]
- Lim JR, Gupta SK, Croffie JM, Pfefferkorn MD, Molleston JP, Corkins MR, Davis MM, Faught PP, Steiner SJ, Fitzgerald JF. Gastrointest Endosc. 2004;59:835. doi: 10.1016/s0016-5107(04)00364-5. [DOI] [PubMed] [Google Scholar]
- Logan MR, Lacy P, Bablitz B, Moqbel R. J Allergy Clin Immunol. 2002;109:299. doi: 10.1067/mai.2002.121453. [DOI] [PubMed] [Google Scholar]
- Lopez AF, Begley CG, Williamson DJ, Warren DJ, Vadas MA, Sanderson CJ. Journal of Experimental Medicine. 1986;163:1085. doi: 10.1084/jem.163.5.1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez AF, Sanderson CJ, Gamble JR, Campbell HD, Young IG, Vadas MA. Source (Bibliographic Citation): J Exp Med. 1988;167:219. doi: 10.1084/jem.167.1.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma W, Bryce PJ, Humbles AA, Laouini D, Yalcindag A, Alenius H, Friend DS, Oettgen HC, Gerard C, Geha RS. J Clin Invest. 2002;109:621. doi: 10.1172/JCI14097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacKenzie JR, Mattes J, Dent LA, Foster PS. J Immunol. 2001;167:3146. doi: 10.4049/jimmunol.167.6.3146. [DOI] [PubMed] [Google Scholar]
- MacPherson JC, Comhair SA, Erzurum SC, Klein DF, Lipscomb MF, Kavuru MS, Samoszuk MK, Hazen SL. J Immunol. 2001;166:5763. doi: 10.4049/jimmunol.166.9.5763. [DOI] [PubMed] [Google Scholar]
- Maddox DE, Kephart GM, Coulam CB, Butterfield JH, Benirschke K, Gleich GJ. J Exp Med. 1984;160:29. doi: 10.1084/jem.160.1.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattes J, Yang M, Mahalingam S, Kuehr J, Webb DC, Simson L, Hogan SP, Koskinen A, McKenzie AN, Dent LA, Rothenberg ME, Matthaei KI, Young IG, Foster PS. J Exp Med. 2002;195:1433. doi: 10.1084/jem.20020009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews AN, Friend DS, Zimmermann N, Sarafi MN, Luster AD, Pearlman E, Wert SE, Rothenberg ME. Proc Natl Acad Sci U S A. 1998;95:6273. doi: 10.1073/pnas.95.11.6273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mauser PJ, Pitman AM, Fernandez X, Foran SK, Adams Gr, Kreutner W, Egan RW, Chapman RW. American Journal of Respiratory & Critical Care Medicine. 1995;152:467. doi: 10.1164/ajrccm.152.2.7633694. [DOI] [PubMed] [Google Scholar]
- Mawhorter SD, Kazura JW, Boom WH. Immunology. 1994;81:584. [PMC free article] [PubMed] [Google Scholar]
- McNagny K, Graf T. J Exp Med. 2002;195:F43. doi: 10.1084/jem.20020636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng Q, Ying S, Corrigan CJ, Wakelin M, Assoufi B, Moqbel R, Kay AB. Allergy. 1997;52:1095. doi: 10.1111/j.1398-9995.1997.tb00181.x. [DOI] [PubMed] [Google Scholar]
- Mishra A, Hogan SP, Brandt EB, Rothenberg ME. Blood. 2000;96:1538. [PubMed] [Google Scholar]
- Mishra A, Hogan SP, Brandt EB, Rothenberg ME. J Immunol. 2002a;168:2464. doi: 10.4049/jimmunol.168.5.2464. [DOI] [PubMed] [Google Scholar]
- Mishra A, Hogan SP, Brandt EB, Wagner N, Crossman MW, Foster PS, Rothenberg ME. J Biol Chem. 2002b;277:4406. doi: 10.1074/jbc.M110424200. [DOI] [PubMed] [Google Scholar]
- Mishra A, Hogan SP, Lee JJ, Foster PS, Rothenberg ME. J Clin Invest. 1999;103:1719. doi: 10.1172/JCI6560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra A, Rothenberg ME. Gastroenterology. 2003;125:1419. doi: 10.1016/j.gastro.2003.07.007. [DOI] [PubMed] [Google Scholar]
- Moser R, Fehr J, Bruijnzeel PL. Journal of Immunology. 1992;149:1432. [PubMed] [Google Scholar]
- Munitz A, Bachelet I, Eliashar R, Khodoun M, Finkelman FD, Rothenberg ME, Levi-Schaffer F. J Immunol. 2006;177:77. doi: 10.4049/jimmunol.177.1.77. [DOI] [PubMed] [Google Scholar]
- Munitz A, McBride ML, Bernstein JS, Rothenberg ME. Blood. 2008;111:5694. doi: 10.1182/blood-2007-12-126748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy PM. Annu Rev Immunol. 1994;12:593. doi: 10.1146/annurev.iy.12.040194.003113. [DOI] [PubMed] [Google Scholar]
- Nakajima H, Sano H, Nishimura T, Yoshida S, Iwamoto I. Journal of Experimental Medicine. 1994;179:1145. doi: 10.1084/jem.179.4.1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura H, Luster AD, Nakamura T, In KH, Sonna LA, Deykin A, Israel E, Drazen JM, Lilly CM. J Allergy Clin Immunol. 2001;108:946. doi: 10.1067/mai.2001.120135. [DOI] [PubMed] [Google Scholar]
- Nerlov C, Graf T. Genes Dev. 1998;12:2403. doi: 10.1101/gad.12.15.2403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nerlov C, McNagny KM, Doderlein G, Kowenz-Leutz E, Graf T. Genes Dev. 1998;12:2413. doi: 10.1101/gad.12.15.2413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nutku E, Aizawa H, Hudson SA, Bochner BS. Blood. 2003;101:5014. doi: 10.1182/blood-2002-10-3058. [DOI] [PubMed] [Google Scholar]
- O'Donnell MC, Ackerman SJ, Gleich GJ, Thomas LL. J Exp Med. 1983;157:1981. doi: 10.1084/jem.157.6.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Reilly M, Alpert R, Jenkinson S, Gladue RP, Foo S, Trim S, Peter B, Trevethick M, Fidock M. J Recept Signal Transduct Res. 2002;22:431. doi: 10.1081/rrs-120014612. [DOI] [PubMed] [Google Scholar]
- Odemuyiwa SO, Ghahary A, Li Y, Puttagunta L, Lee JE, Muscat-Marcu S, Ghahary A, Moqbel R. J Immunol. 2004;173:5909. doi: 10.4049/jimmunol.173.10.5909. [DOI] [PubMed] [Google Scholar]
- Piliponsky A, Gleich G, Bar I, Levi-Schaffer F. Mol Immunol. 2002;38:1369. doi: 10.1016/s0161-5890(02)00090-1. [DOI] [PubMed] [Google Scholar]
- Plager DA, Loegering DA, Checkel JL, Tang J, Kephart GM, Caffes PL, Adolphson CR, Ohnuki LE, Gleich GJ. 2006;177:7340. doi: 10.4049/jimmunol.177.10.7340. [DOI] [PubMed] [Google Scholar]
- Ponath PD, Qin S, Post TW, Wang J, Wu L, Gerard NP, Newman W, Gerard C, Mackay CR. J Exp Med. 1996;183:2437. doi: 10.1084/jem.183.6.2437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pope SM, Zimmermann N, Stringer KF, Karow ML, Rothenberg ME. J Immunol. 2005;175:5341. doi: 10.4049/jimmunol.175.8.5341. [DOI] [PubMed] [Google Scholar]
- Popken-Harris P, Checkel J, Loegering D, Madden B, Springett M, Kephart G, Gleich GJ. Blood. 1998;92:623. [PubMed] [Google Scholar]
- Potter JW, Saeian K, Staff D, Massey BT, Komorowski RA, Shaker R, Hogan WJ. Gastrointest Endosc. 2004;59:355. doi: 10.1016/s0016-5107(03)02713-5. [DOI] [PubMed] [Google Scholar]
- Pretolani M, Ruffie C, Lapa e Silva JR, Joseph D, Lobb RR, Vargaftig BB. Journal of Experimental Medicine. 1994;180:795. doi: 10.1084/jem.180.3.795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rand TH, Lopez AF, Gamble JR, Vadas MA. International Archives of Allergy & Applied Immunology. 1988;87:151. doi: 10.1159/000234665. [DOI] [PubMed] [Google Scholar]
- Rankin SM, Conroy DM, Williams TJ. Mol Med Today. 2000;6:20. doi: 10.1016/s1357-4310(99)01635-4. [DOI] [PubMed] [Google Scholar]
- Redier H, Chanez P, De VC, Rifai N, Clauzel AM, Michel FB, Godard P. Journal of Allergy & Clinical Immunology. 1992;90:215. doi: 10.1016/0091-6749(92)90074-c. [DOI] [PubMed] [Google Scholar]
- Reese TA, Liang HE, Tager AM, Luster AD, Van Rooijen N, Voehringer D, Locksley RM. Nature. 2007;447:92. doi: 10.1038/nature05746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson SA, Allanson M, Mau VJ. Trophoblast Research. 1998;11:101. [Google Scholar]
- Robertson SA, Mau VJ, Young IG, Matthaei KI. J Reprod Fertil. 2000;120:423. doi: 10.1530/jrf.0.1200423. [DOI] [PubMed] [Google Scholar]
- Rolfe FG, Valentine JE, Sewell WA. Am J Respir Cell Mol Biol. 1997;17:243. doi: 10.1165/ajrcmb.17.2.2819. [DOI] [PubMed] [Google Scholar]
- Rosenberg HF, Domachowske JB. J Leukoc Biol. 2001;70:691. [PubMed] [Google Scholar]
- Rothenberg ME. N Engl J Med. 1998;338:1592. doi: 10.1056/NEJM199805283382206. [DOI] [PubMed] [Google Scholar]
- Rothenberg ME. J Allergy Clin Immunol. 2004;113:11. doi: 10.1016/j.jaci.2003.10.047. [DOI] [PubMed] [Google Scholar]
- Rothenberg ME, Hogan SP. Annu Rev Immunol. 2005 doi: 10.1146/annurev.immunol.24.021605.090720. [DOI] [PubMed] [Google Scholar]
- Rothenberg ME, Klion AD, Roufosse FE, Kahn JE, Weller PF, Simon HU, Schwartz LB, Rosenwasser LJ, Ring J, Griffin EF, Haig AE, Frewer PI, Parkin JM, Gleich GJ. N Engl J Med. 2008;358:1215. doi: 10.1056/NEJMoa070812. [DOI] [PubMed] [Google Scholar]
- Rothenberg ME, Luster AD, Leder P. Proc Natl Acad Sci U S A. 1995;92:8960. doi: 10.1073/pnas.92.19.8960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothenberg ME, MacLean JA, Pearlman E, Luster AD, Leder P. J Exp Med. 1997;185:785. doi: 10.1084/jem.185.4.785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothenberg ME, Mishra A, Brandt EB, Hogan SP. Immunol Rev. 2001a;179:139. doi: 10.1034/j.1600-065x.2001.790114.x. [DOI] [PubMed] [Google Scholar]
- Rothenberg ME, Mishra A, Brandt EB, Hogan SP. Adv Immunol. 2001b;78:291. doi: 10.1016/s0065-2776(01)78007-8. [DOI] [PubMed] [Google Scholar]
- Rothenberg ME, Mishra A, Collins MH, Putnam PE. J Allergy Clin Immunol. 2001c;108:891. doi: 10.1067/mai.2001.120095. [DOI] [PubMed] [Google Scholar]
- Rothenberg ME, Pomerantz JL, Owen WF, Jr, Avraham S, Soberman RJ, Austen KF, Stevens RL. J Biol Chem. 1988;263:13901. [PubMed] [Google Scholar]
- Sanderson CJ. Blood. 1992;79:3101. [PubMed] [Google Scholar]
- Schleimer RP, Bochner BS. Journal of Allergy and Clinical Immunology. 1994;94:1202. doi: 10.1016/0091-6749(94)90333-6. [DOI] [PubMed] [Google Scholar]
- Sferruzzi-Perri AN, Robertson SA, Dent LA. Biol Reprod. 2003;69:224. doi: 10.1095/biolreprod.102.010611. [DOI] [PubMed] [Google Scholar]
- Sher A, Coffman RL, Hieny S, Scott P, Cheever AW. Proc Natl Acad Sci USA. 1990;87:61. doi: 10.1073/pnas.87.1.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi H. J Leuko Biol. 2004;76:520. doi: 10.1189/jlb.0404228. [DOI] [PubMed] [Google Scholar]
- Shi HZ, Humbles A, Gerard C, Jin Z, Weller PF. J Clin Invest. 2000;105:945. doi: 10.1172/JCI8945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinkai A, Yoshisue H, Koike M, Shoji E, Nakagawa S, Saito A, Takeda T, Imabeppu S, Kato Y, Hanai N, Anazawa H, Kuga T, Nishi T. J Immunol. 1999;163:1602. [PubMed] [Google Scholar]
- Shinkai K, Mohrs M, Locksley RM. Nature. 2002;420:825. doi: 10.1038/nature01202. [DOI] [PubMed] [Google Scholar]
- Simons JE, Rothenberg ME, Lawrence RA. Eur J Immunol. 2005;35:189. doi: 10.1002/eji.200425541. [DOI] [PubMed] [Google Scholar]
- Slifman NR, Loegering DA, McKean DJ, Gleich GJ. J Immunol. 1986;137:2913. [PubMed] [Google Scholar]
- Snyman JR, Sommers DK, Gregorowski MD, Boraine H. European Journal of Clinical Pharmacology. 1992;42:359. doi: 10.1007/BF00280118. [DOI] [PubMed] [Google Scholar]
- Solomon A, Aloe L, Pe'er J, Frucht-Pery J, Bonini S, Levi-Schaffer F. J Allergy Clin Immunol. 1998;102:454. doi: 10.1016/s0091-6749(98)70135-6. [DOI] [PubMed] [Google Scholar]
- Stein ML, Villanueva JM, Buckmeier BK, Yamada Y, Filipovich AH, Assa'ad AH, Rothenberg ME. J Allergy Clin Immunol. 2008;121:1473. doi: 10.1016/j.jaci.2008.02.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stellato C, Matsukura S, Fal A, White J, Beck LA, Proud D, Schleimer RP. J Immunol. 1999;163:5624. [PubMed] [Google Scholar]
- Straumann A, Bauer M, Fischer B, Blaser K, Simon HU. J Allergy Clin Immunol. 2001;108:954. doi: 10.1067/mai.2001.119917. [DOI] [PubMed] [Google Scholar]
- Straumann A, Kristl J, Conus S, Vassina E, Spichtin HP, Beglinger C, Simon HU. Inflamm Bowel Dis. 2005;11:720. doi: 10.1097/01.mib.0000172557.39767.53. [DOI] [PubMed] [Google Scholar]
- Suzuki K, Verma IM. Cell. 2008;134:485. doi: 10.1016/j.cell.2008.05.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swartz JM, Dyer KD, Cheever AW, Ramalingam T, Pesnicak L, Domachowske JB, Lee JJ, Lee NA, Foster PS, Wynn TA, Rosenberg HF. Blood. 2006;108:2420. doi: 10.1182/blood-2006-04-015933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tager AM, Dufour JH, Goodarzi K, Bercury SD, von Andrian UH, Luster AD. J Exp Med. 2000;192:439. doi: 10.1084/jem.192.3.439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tai PC, hayes DJ, Clark JB, Spry CJF. Biochem J. 1982;204:75. doi: 10.1042/bj2040075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takatsu K, Takaki S, Hitoshi Y. Advances in Immunology. 1994;57:45. doi: 10.1016/s0065-2776(08)60673-2. [DOI] [PubMed] [Google Scholar]
- Ten RM, Pease LR, McKean DJ, Bell MP, Gleich GJ. J Exp Med. 1989;169:1757. doi: 10.1084/jem.169.5.1757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Throsby M, Herbelin A, Pleau JM, Dardenne M. J Immunol. 2000;165:1965. doi: 10.4049/jimmunol.165.4.1965. [DOI] [PubMed] [Google Scholar]
- Tominaga A, Takaki S, Koyama N, Katoh S, Matsumoto R, Migata M, Hitoshi Y, Hosoya Y, Yamauchi S, Kanai Y, Miyazaki JI, Usuku G, Yamamura KI, Takatsu K. Journal of Experimental Medicine. 1991;173:429. doi: 10.1084/jem.173.2.429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulrich M, Petre A, Youhnovski N, Promm F, Schirle M, Schumm M, Pero RS, Doyle A, Checkel J, Kita H, Thiyagarajan N, Acharya KR, Schmid-Grendelmeier P, Simon HU, Schwarz H, Tsutsui M, Shimokawa H, Bellon G, Lee JJ, Przybylski M, Doring G. J Biol Chem. 2008 doi: 10.1074/jbc.M801196200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vadas M, Lopez A, Gamble J, Khew GY, Smith W, Bernard C, Cockerill G, Cockerill P, Shannon F, Sun Q, et al. J Allergy Clin Immunol. 1994 doi: 10.1016/0091-6749(94)90344-1. [DOI] [PubMed] [Google Scholar]
- van Rijt LS, Vos N, Hijdra D, De Vries VC, Hoogsteden HC, Lambrecht BN. J Immunol. 2003;171:3372. doi: 10.4049/jimmunol.171.7.3372. [DOI] [PubMed] [Google Scholar]
- Venge P, Bystrom J, Carlson M, Hakansson L, Karawacjzyk M, Peterson C, Seveus L, Trulson A. Clin Exp Allergy. 1999;29:1172. doi: 10.1046/j.1365-2222.1999.00542.x. [DOI] [PubMed] [Google Scholar]
- Venge P, Dahl R, Hallgren R, Olsson I. In: The eosinophil in health and disease. Mahmoud AAF, Austin KF, editors. New York: Grune and Stratton; 1980. p. 1131. [Google Scholar]
- Voehringer D, van Rooijen N, Locksley RM. J Leukoc Biol. 2007 doi: 10.1189/jlb.1106686. [DOI] [PubMed] [Google Scholar]
- Wagner JM, Hustin J, Bonno M, Kephart GM, Gurian KV, Gleich GJ. Placenta. 1994;15:625. doi: 10.1016/s0143-4004(05)80409-6. [DOI] [PubMed] [Google Scholar]
- Walsh ER, Sahu N, Kearley J, Benjamin E, Kang BH, Humbles A, August A. J Exp Med. 2008;205:1285. doi: 10.1084/jem.20071836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wasmoen TL, Bell MP, Loegering DA, Gleich GJ, Prendergast FG, McKean DJ. J Biol Chem. 1988;263:12559. [PubMed] [Google Scholar]
- Weg VB, Williams TJ, Lobb RR, Nourshargh S. Journal of Experimental Medicine. 1993;177:561. doi: 10.1084/jem.177.2.561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wegner CD, Gundel RH, Reilly P, Haynes N, Letts LG, Rothlein R. Science. 1990;247:456. doi: 10.1126/science.1967851. [DOI] [PubMed] [Google Scholar]
- Wu W, Chen Y, Hazen SL. J Biol Chem. 1999;274:25933. doi: 10.1074/jbc.274.36.25933. [DOI] [PubMed] [Google Scholar]
- Yamada H, Izutani R, Chihara J, Yudate T, Matsukura M, Tezuka T. Int Arch Allergy Immunol. 1996;111 Suppl 1:19. doi: 10.1159/000237408. [DOI] [PubMed] [Google Scholar]
- Yamada Y, Rothenberg ME, Lee AW, Saito Akei H, Brandt EB, Williams DA, Cancelas JA. Blood. 2006 doi: 10.1182/blood-2005-08-3153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang D, Chen Q, Su SB, Zhang P, Kurosaka K, Caspi RR, Michalek SM, Rosenberg HF, Zhang N, Oppenheim JJ. J Exp Med. 2008;205:79. doi: 10.1084/jem.20062027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon J, Ponikau JU, Lawrence CB, Kita H. J Immunol. 2008;181:2907. doi: 10.4049/jimmunol.181.4.2907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young JD, Peterson CG, Venge P, Cohn ZA. Nature. 1986;321:613. doi: 10.1038/321613a0. [DOI] [PubMed] [Google Scholar]
- Yousefi S, Gold JA, Andina N, Lee JJ, Kelly AM, Kozlowski E, Schmid I, Straumann A, Reichenbach J, Gleich GJ, Simon HU. Nat Med. 2008 doi: 10.1038/nm.1855. [DOI] [PubMed] [Google Scholar]
- Yu C, Cantor AB, Yang H, Browne C, Wells RA, Fujiwara Y, Orkin SH. J Exp Med. 2002;195:1387. doi: 10.1084/jem.20020656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Lathbury LJ, Salamonsen LA. Biol Reprod. 2000;62:404. doi: 10.1095/biolreprod62.2.404. [DOI] [PubMed] [Google Scholar]
- Zheutlin LM, Ackerman SJ, Gleich GJ, Thomas LL. J Immunol. 1984;133:2180. [PubMed] [Google Scholar]
- Zimmermann N, Daugherty BL, Kavanaugh JL, El-Awar FY, Moulton EA, Rothenberg ME. Blood. 2000a;96:2346. [PubMed] [Google Scholar]
- Zimmermann N, Hershey GK, Foster PS, Rothenberg ME. J Allergy Clin Immunol. 2003;111:227. doi: 10.1067/mai.2003.139. [DOI] [PubMed] [Google Scholar]
- Zimmermann N, Hogan SP, Mishra A, Brandt EB, Bodette TR, Pope SM, Finkelman FD, Rothenberg ME. J Immunol. 2000b;165:5839. doi: 10.4049/jimmunol.165.10.5839. [DOI] [PubMed] [Google Scholar]