Oxidative stress, mitochondrial damage and neurodegenerative diseases (original) (raw)
Abstract
Oxidative stress and mitochondrial damage have been implicated in the pathogenesis of several neurodegenerative diseases, including Alzheimer's disease, Parkinson's disease and amyotrophic lateral sclerosis. Oxidative stress is characterized by the overproduction of reactive oxygen species, which can induce mitochondrial DNA mutations, damage the mitochondrial respiratory chain, alter membrane permeability, and influence Ca2+ homeostasis and mitochondrial defense systems. All these changes are implicated in the development of these neurodegenerative diseases, mediating or amplifying neuronal dysfunction and triggering neurodegeneration. This paper summarizes the contribution of oxidative stress and mitochondrial damage to the onset of neurodegenerative eases and discusses strategies to modify mitochondrial dysfunction that may be attractive therapeutic interventions for the treatment of various neurodegenerative diseases.
Keywords: neural regeneration, neurodegenerative diseases, Alzheimer's disease, Parkinson's disease, amyotrophic lateral sclerosis, oxidative stress, reactive oxygen species, mitochondrial damage, respiratory chain, grants-supported paper, neuroregeneration
Research Highlights
- (1)
Growing evidence has highlighted that oxidative stress and mitochondrial dysfunction may trigger neurodegenerative diseases. - (2)
The contribution of oxidative stress-caused mitochondrial damage in Alzheimer's disease, Parkinson's disease and amyotrophic lateral sclerosis is summarized and analyzed, in a broad attempt to provide evidence for the potential treatment of neurodegenerative diseases.
INTRODUCTION
Neurodegenerative diseases, including Alzheimer's disease (AD), Parkinson's disease (PD) and amyotrophic lateral sclerosis (ALS)[1], are a heterogeneous group of orders characterized by gradually sive, selective loss of anatomically or ologically related neuronal systems[2]. though the precise pathophysiological chanisms underlying neurodegenerative disorders remain unclear, oxidative stress and mitochondrial involvement may be jor triggering factors in these diseases[3,4]. This review aims to summarize the evidence for the involvement of oxidative stress and mitochondrial damage in AD, PD and ALS.
OXIDATIVE STRESS AND MITOCHONDRIAL DAMAGE
Oxidative stress is characterized by an overproduction of reactive oxygen species (ROS), which can lead to mitochondrial damage in the following ways.
Oxidative damage and ROS
The balance of ROS and antioxidants under normal physiological conditions is disrupted with the overproduction of free radicals. These excess free radicals can lead to DNA damage, degradation of protein and lipids, neurodegenerative diseases including AD, PD and ALS (Figure 1), and can cause protein deposition.
Figure 1.
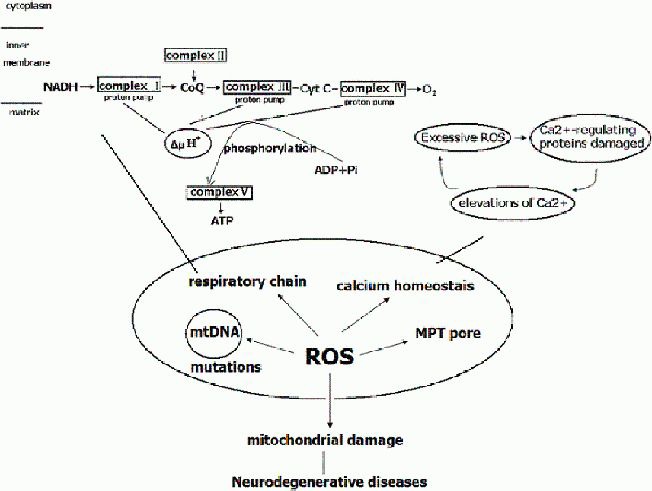
Reactive oxygen species (ROS) and mitochondrial damage.
The mitochondrial respiratory chain complexes consist of five complexes: complexes I, II, III, IV and V, which catalyze the phosphorylation of adenosine diphosphate (ADP) to adenosine triphosphate (ATP). Complex I: nicotinamide adenine dinucleotide (NADH)-coenzyme Q; Complex II: succinate dehydrogenase-coenzyme Q; Complex III: coenzyme Q-cytochrome c reductase; Complex IV: cytochrome c oxidase; Complex V: ATP synthase. More than 80 proteins comprise these complexes, 13 of them encoded by mitochondrial DNA. Complex I, II, III, and IV constitute the electron-transport chain. The electrons, borne on NAD+, are transferred to NADH and then to coenzyme Q, and the electrons are transferred to complex II and to coenzyme Q. From coenzyme Q, the electrons are passed to complex III, then to cytochrome c, then to complex IV and finally to 1/2 O2 to give H2O. ATP is generated by the influx of these protons back into the mitochondrial matrix through ATP synthase.
The highly ROS can damage NADH dehydrogenase, cytochrome c oxidase, and ATP synthase, resulting in shutdown of mitochondrial energy production. Under normal physiological conditions, Ca2+ fluxes are properly controlled across the plasma membrane and between intracellular compartments. Excessive ROS generation can directly damage Ca2+-regulating proteins (in the plasma membrane, such as ligand- and voltage-gated Ca2+ channels, endoplasmic reticulum Ca2+-ATP synthases, and mitochondria electron-transport chain proteins), resulting in elevations of Ca2+, which disturb Ca2+ homeostasis. Overproduction of ROS can lead to mitochondrial damage, including mutations in mitochondrial DNA, damage to the mitochondrial respiratory chain and mitochondrial membrane permeability, and disruption to Ca2+ homeostasis. Therefore, mitochondrial damage plays an important role in the pathogenesis of neurodegenerative diseases.
CoQ: Coenzyme Q; Cyt c: cytochrome c; mtDNA: mitochondrial DNA; MPT: mitochondrial permeability transition.
ROS and mitochondrial damage
ROS and mitochondrial DNA mutations
Mitochondria are the only organelles in cells, besides the nucleus, that contain their own DNA (called mitochondrial DNA) and their own machinery for synthesizing RNA and proteins[5]. Mitochondrial DNA makes up approximately 1% of total cellular DNA and is thought to be particularly susceptible to ROS attack associated with oxidative stress[6]. The persistence of mitochondrial DNA damage ultimately leads to mutations in the mitochondrial genome[6] and gives rise to further mitochondrial dysfunction, which induces and aggravates the diseases (Figure 1).
ROS induces damage to the mitochondrial respiratory chain
The mitochondrial respiratory chain, located in the inner mitochondrial membrane[5], is one of the major structural and functional parts of mitochondria. It consists of five complexes, namely complexes I, II, III, IV, V[7], which catalyze the phosphorylation of adenosine diphosphate to adenosine triphosphate, working as an integrated system composed of a total of five protein complexes[8]: reduced nicotinamide adenine dinucleotide dehydrogenase-ubiquinone reductase (coenzyme Q) (complex I, approximately 46 subunits), succinate dehydrogenase (FADH2)-coenzyme Q (complex II, 4 subunits), ubiquinone-cytochrome c reductase (complex III, 11 subunits), cytochrome c oxidase (complex IV, 13 subunits), and adenosine triphosphate synthase (complex V, 16 subunits)[5]. More than 80 proteins comprise these complexes, 13 of them encoded by mitochondrial DNA[7], and all of them components of the oxidative phosphorylation system[8]. Complexes I, II, III, and IV constitute the respiratory electron-transport chain[4] (Figure 1). The electron-transport chain oxidizes hydrogen derived from the oxidation of organic acids such as pyruvate and fatty acids with atomic oxygen to generate water. The electrons, borne on nicotinamide adenine dinucleotide, are transferred to respiratory complex I (nicotinamide adenine dinucleotide dehydrogenase) and coenzyme Q, and from succinate in the tricarboxylic acid cycle to complex II (succinate dehydrogenase) and coenzyme Q. From coenzyme Q, the electrons are passed to complex III, cytochrome c, complex IV (cytochrome c oxidase), and finally to 1/2 O2 to give H2O[9]. The electron-transport chain also requires two small electron carriers, ubiquinone and cytochrome c. Adenosine triphosphate synthesis entails two coordinated processes. First, electrons (actually hydrogen ions derived from nicotinamide adenine dinucleotide and reduced flavin adenine dinucleotide in intermediary metabolism) are transported along the complexes to molecular oxygen, thereby producing water[4]. At the same time, protons are pumped across the mitochondrial inner membrane (i.e., from the matrix to the intermembrane space) by complexes I, III, and IV[10]. Adenosine triphosphate is generated by the influx of these protons back into the mitochondrial matrix through complex V (adenosine triphosphate synthase)[11]. Under normal physiological conditions, 1–5% of the oxygen is converted to ROS[12], thus most estimates suggest that the majority of intracellular ROS production is derived from mitochondria. The production of mitochondrial superoxide radicals occurs primarily at two discrete points in the electron-transport chain, namely at complex I (nicotinamide adenine dinucleotide dehydro genase) and complex III (ubiquinone-cytochrome c reductase)[13]. Under normal metabolic conditions, complex III is the main site of ROS production[14].
Thereby, free radical attack occurs directly at complexes in the mitochondrial respiratory chain. Complexes I and III are also thought to be major sites for the production of superoxide and other reactive oxygen species[6]. The highly reactive hydroxyl radical can damage macromolecules within mitochondria, including lipids, proteins and DNA[15], and unrepaired mitochondrial DNA damage leading to defective complex I and/or III function can result in increased electron reduction of O2 to form superoxide[16,17,18]. The increased flux of superoxide resulting from these types of mitochondrial DNA lesions could subsequently contribute to metabolic oxidative stress, genomic instability, and cellular injury[6]. Mitochondrial DNA represents a critical target for such oxidative damage. Once damaged, mitochondrial DNA can amplify oxidative stress by decreased expression of critical proteins important for electron transport, leading to a vicious cycle of ROS and organelle dysregulation that eventually triggers apoptosis[15]. In addition, the mitochondrial respiratory chain is particularly sensitive to both NO− and ONOO− mediated damage[4]. Mitochondrial proteins can be modified by nitration. Protein oxidation and nitration result in altered function of many metabolic enzymes in the mitochondrial electron-transport chain, including nicotinamide adenine dinucleotide dehydrogenase, cytochrome c oxidase, and adenosine triphosphate synthase[19]. Chronic ROS exposure can result in oxidative damage to mitochondrial and cellular proteins, lipids, and nucleic acids, and acute ROS exposure can inactivate the iron-sulfur (Fe-S) centers of electron-transport chain complexes I, II, and III, and tricarboxylic acid cycle aconitase, resulting in shut-down of mitochondrial energy production[8].
ROS leads to changes in mitochondrial membrane permeability and structure
Mitochondria are important organelles in eukaryotic cells[7]. Mitochondria have an inner and an outer membrane separated by an intermembrane space. The outer membrane is more permeable than the inner membrane because of the presence of porin proteins[20]. The inner membrane is the site of oxidative phosphorylation and electron transport, which is involved in adenosine triphosphate production[21]. Under normal physiological conditions, the inner membrane is only permeable to neutral small molecules. Therefore, it is difficult for exogenous antioxidants to reach mitochondria. However, the inner mitochondrial membrane, near the site of ROS production, is prone to lipid peroxidation[21]. Peroxidation of mitochondrial phospholipids can increase proton permeability of the inner mitochondrial membrane[22], alter the fluidity and other biophysical properties of mitochondrial membranes and impair biochemical functions of various transporters and respiratory enzymes in the inner and outer membranes. In addition, the iron-sulfur centers of the respiratory enzymes (e.g., succinate dehydrogenase and nicotinamide adenine dinucleotide dehydrogenase) are prone to oxidative modification and the electron-transport function of mitochondria may be impaired by ROS under oxidative stress[12]. ROS may promote mitochondrial permeability transition by causing oxidation of thiol groups on the adenine nucleotide translocator, which is believed to form part of the mitochondrial permeability transition pore[23]. The mitochondrial permeability transition pore is considered to be a channel with a large conductance provided by proteins residing in both the inner and outer mitochondrial membrane, which is activated by mitochondrial Ca2+ over-loading and other factors including oxidative stress[24]. Its activation increases inner mitochondrial membrane permeability to solutes with a molecular mass of up to 1.5 kDa[25].
ROS disturbed Ca2+ homeostasis
Ca2+ is a major player in the intracellular signaling system that translates extracellular stimuli into the regulation of a bewildering number of phenomena such as muscle contraction, neurotransmitter release and other secretion processes, cell proliferation, gene expression and cell death[26]. Ca2+ is also important for the activation of the mitochondrial enzymes pyruvate dehydrogenase, 2-oxoglutarate dehydrogenase, and NAD-linked isocitrate dehydrogenase[27]. The influx of Ca2+ through voltage-dependent and ligand-gated channels in the plasma membrane is a critical signal for the release of neurotransmitters from presynaptic terminals and for responses of the postsynaptic neuron[28]. Ca2+ is removed from the cytoplasm by the plasma membrane Na+/Ca2+ exchanger, plasma membrane and endoplasmic reticulum Ca2+-adenosine triphosphate synthases, and Ca2+-binding proteins such as calbindin and parvalbumin[19]. By buffering intracellular Ca2+ loads, Ca2+-binding proteins such as calbindin may serve as endogenous anti-excitotoxic proteins[29]. Ca2+ can also be transported into and released from mitochondria and play important roles in the regulation of neuronal Ca2+ homeostasis. In fact, genetic and pharmacological manipulations that enhance mitochondrial Ca2+ sequestration can protect neurons against excitotoxicity[30]. The concentration of Ca2+ in the cytosol is tightly regulated in all cells[31]: under resting conditions, Ca2+ concentrations are typically in the range of 75–200 nmol/L, and transiently increase to 1–10 μmol/L in response to membrane depolarization and the opening of voltage-dependent Ca2+ and N-methyl-D-aspartic acid receptor channels. Mitochondria are also capable of sequestering Ca2+ and then releasing these ions into the cytosol[31].
However, excessive ROS generation can directly damage proteins and nucleic acids, cause oxidative stress, and further alter mitochondrial Ca2+ homeostasis, particularly affecting the oxidation of specific thiol groups in proteins[32]. Peroxynitrite can also avidly react with thiols to form nitrosothiols, affecting the function of proteins[33]. In addition, peroxynitrite can affect the energy status of a cell by inactivating key mitochondrial enzymes and triggering calcium release from the mitochondria[5]. Elevation in Ca2+ levels also causes a change in mitochondrial potential and leads to the production of superoxide ion radicals[34], resulting in a vicious cycle. When mitochondria become overloaded with Ca2+, they undergo mitochondrial permeability transition, resulting in osmotic swelling and rupture of the outer mitochondrial membrane[34]. The sustained elevations in intracellular Ca2+ concentrations can cause neuronal degeneration and cell death[35]. In addition, ROS produced in the mitochondria promote Ca2+ uptake and increase membrane permeability, which results in the release of cytochrome c and initiates apoptosis[35]. Hydroxyl radicals can directly attack DNA bases, and if the damage is extensive, a cell death pathway is activated. Perturbed neuronal Ca2+ homeostasis is a consequence of those mutations that contribute to the degeneration of neurons[35].
ROS undermines the mitochondrial defense system
Mitochondria are normally protected from oxidative damage by a multilayer network of mitochondrial anti-oxidant systems[36], which consist of superoxide dismutase, catalase, glutathione peroxidase and glutathione reductase together with a number of low molecular weight antioxidants such as α-tocopherol and ubiquinol[12]. These molecules are particularly effective in scavenging lipid peroxyl radicals and preventing free radical chain reactions of lipid peroxidation[37]. However, these antioxidant systems are not perfect. Hydrogen peroxide produced by superoxide dismutase is relatively unreactive; it can form highly reactive hydroxyl radicals in the presence of ferrous ion via Fenton chemistry. These hydroxyl radicals can initiate lipid peroxidation cascades in membranes[36,37]. Furthermore, the products of sugar, protein, and lipid oxidation can cause secondary damage to proteins, which may lose catalytic function and undergo selective degradation[38]. Because no defense system is completely efficient, the entire array of available endogenous antioxidant enzymes cannot fully neutralize the ROS being emitted from mitochondria. Cumulative oxidative injuries to mitochondria, triggered by endogenous metabolic processes and/or exogenous oxidative influences, cause mitochondria to progressively become less efficient. As mitochondria progressively lose their functional integrity, ever-greater proportions of oxygen molecules reaching them are converted to ROS[38]. As mentioned above, ROS undermine the mitochondrial defense system.
MITOCHONDRIAL DAMAGE AND NEURODEGENERATIVE DISEASES
Mitochondrial structure and function
Structurally, mitochondria have four compartments: the outer membrane, the inner membrane, the intermembrane space, and the matrix (the region inside the inner membrane)[37]. The porous outer membrane encompasses the whole organelle and contains many important enzymes and receptors[39], and is freely permeable to small molecules and ions. The convoluted and invaginated inner membrane contains the enzymes of oxidative phosphorylation: cofactor coenzyme Q (ubiquinone Q), F0F1-adenosine triphosphate synthase, and some carrier proteins[40]. In addition, cardiolipin is an important component of the inner mitochondrial membrane, which is impermeable to most small molecules and ions, including H+[40]. Between the inner and outer membrane is the intermembrane space, with contains specialized proteins[40]. In the matrix, bordered by the inner membrane, there are many enzymes for different metabolic pathways, including the citric acid cycle, fatty acid oxidation and urea cycle, mitochondrial DNA synthesis, and peptidases and chaperones[4]. Mitochondria perform numerous tasks, with the most crucial probably being the generation of energy as adenosine triphosphate by means of the electron-transport chain and the oxidative phosphorylation system (respiratory chain)[41]. In addition to adenosine triphosphate synthesis, mitochondria can accumulate Ca2+. Changes in mitochondrial Ca2+ can regulate tricarboxylic acid cycle enzymes[42]. Through the adenosine triphosphate/adenosine diphosphate pool, mitochondria can influence glycolysis, the activity of Ca2+ and Na+-K+-adenosine triphosphate synthases at the plasma membrane and consequently the activity of Na+-coupled plasma membrane transporters[43]. Each mammalian mitochondrion contains 2–10 copies of mitochondrial DNA, resulting in 1 000–100 000 copies in each human cell. Individual mitochondrial DNA molecules replicate at random, making one or more copies at a time while maintaining a relatively constant total number of mitochondrial DNA molecules within the cell. If there are two or more different types of mitochondrial DNA molecules within a cell, by chance, any one type of molecule may replicate more frequently than another type, resulting in a change in the level of heteroplasmy within the cell (intracellular drift)[44]. The number of organelles varies among cells, depending in large part on the metabolic requirements of that cell. Thus, skin fibroblasts contain a few hundred mitochondria, whereas neurons may contain thousands, and cardiomyocytes tens of thousands[41]. In short, mitochondria are the seat of a number of important cellular functions, including essential pathways of intermediate metabolism, amino acid biosynthesis, fatty acid oxidation, steroid metabolism, and apoptosis[45].
Mitochondrial damage and the development of neurodegenerative diseases
Neurodegenerative diseases are characterized by gradually progressive selective loss of anatomically or physiologically related neuronal systems[1,46,47]. Despite different clinical symptoms and pathology in these diseases, increasing evidence suggests that mitochondrial damage plays an important role in the pathogenesis of these diseases[46].
Mitochondrial damage and AD
AD is the most common neurodegenerative disorder worldwide. Mitochondrial degeneration and oxidative damage are involved in the pathogenesis of AD[48,49]. Oxidative stress from mitochondrial dysfunction occurs early in AD. Postmortem analyses have revealed that overall levels of oxidative damage to proteins, lipids, and DNA are elevated in AD brains[50]. Oxidative changes to proteins such as β-amyloid in AD may result in protein misfolding and aggregate formation[51]. In addition, β-amyloid is targeted to mitochondria, where it has been shown to bind β-amyloid binding alcohol dehydrogenase and inhibit cytochrome c oxidase[52]. Direct associations between ROS production and amyloid plaques have also been demonstrated in transgenic mice and in human brain tissue from AD sufferers[53,54]. Decreased activity of the α-ketoglutarate dehydrogenase enzyme complex in brains from AD patients has been observed[54]. The α-ketoglutarate dehydrogenase enzyme complex is a crucial mitochondrial enzyme complex that mediates oxidative metabolism. Dumont et al [53] found that mitochondrial dihydrolipoyl succinyltransferase enzyme deficiency increased amyloid plaque burden, β-amyloid oligomers and nitrotyrosine levels in female Tg19959 mice. The ketoglutarate dehydrogenase enzyme complex can participate in oxidative stress and ROS production. Dihydrolipoyl succinyltransferase is one of the key subunits specific to α-ketoglutarate dehydrogenase enzyme complex activity. In late-onset AD, agerelated free radicals, which are generated in mitochondria, are carried to the cytoplasm where they activate β-secretase and facilitate the cleavage of amyloid precursor protein molecules[54,55]. The cleaved amyloid precursor protein molecule further generates free radicals, leading to the disruption of the electron-transport chain and enzyme activities, the inhibition of adenosine triphosphate, and the subsequent oxidation of both nuclear and mitochondrial DNA proteins[56]. Indeed, Alzheimer's brains harbor somatic mitochondrial DNA mutations that suppress mitochondrial transcription and replication[51]. Finally, the damage caused by mitochondria ultimately leads to neuronal damage, neurodegeneration, and cognitive decline in AD patients[56,57] (Figure 2).
Figure 2.
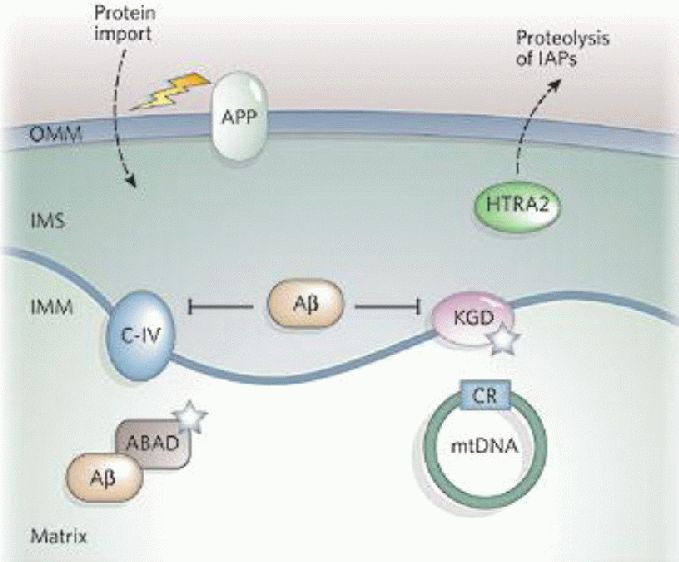
The role of mitochondria in Alzheimer's disease[1]. Mitochondrial reactive oxygen species (ROS) generation can increase β-amyloid protein (Aβ) levels, and Aβ can interact with mitochondria and further cause mitochondrial dysfunction. Aβ inhibits mitochondrial respiratory chain complex IV (C-IV) and α-ketoglutarate dehydrogenase (KGD), and binds Aβ-binding alcohol dehydrogenase (ABAD). Both KGD and ABAD produce ROS (white stars).
The amyloid precursor protein (APP) may be targeted to the outer mitochondrial membrane (OMM) and interfere with protein import. Mitochondria have also been reported to contain active γ-secretase complexes, which are involved in cleaving the APP to form Aβ and contain presenilin 1, which increases the proteolytic activity of high temperature requirement protein A2 (HTRA2) towards the inhibitor of apoptosis proteins (IAPs). Alzheimer's disease patients have on average more somatic mutations in the mitochondrial DNA (mtDNA) control region than control subjects.
IMM: Inner mitochondrial membrane; IMS: intermembrane space.
Interestingly, a broad range of pathological disorders and molecular mechanisms in AD have been linked to oxidative stress, such as c-Abl signaling, DNA damage, and redox regulation of protein function[58,59,60,61,62,63]. Aberrant c-Abl activation is linked to many neuronal disorders and c-Abl colocalized with granulovacuolar degeneration in brains of AD patients[64,65]. Aberrant S-nitrosylation of dynamin related protein 1 occurs in the presence of high levels of NO. S-nitrosylation causes a dramatic increase in mitochondrial fission and consequent bioenergetic compromise. S-nitrosylation-dynamin related protein 1 levels are significantly increased in postmortem sporadic AD patients when compared with controls[66,67,68].
Mitochondrial damage and PD
PD is the second most common neurodegenerative disorder[69], affecting approximately 2% of the population over the age of 60 years and 4% of those over the age of 80 years[70]. The pathological hallmark of the disease is the accumulation of fibrous protein deposits in neuronal cytoplasm (Lewy bodies) and nerve fibers (Lewy neurites) in the brain[71].
Growing evidence indicates that defects in mitochondria (such as mutations or polymorphisms in mitochondrial DNA, and defects in complex I of the mitochondrial electron chain transport) are implicated in PD[72,73,74,75,76] (Figure 3).
Figure 3.
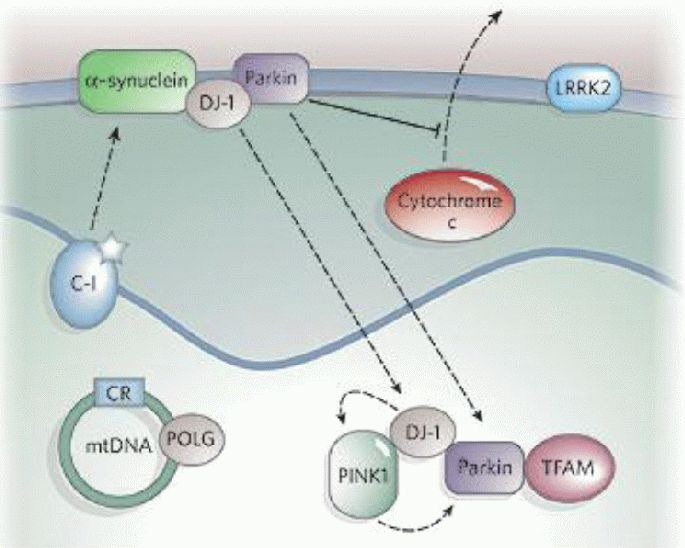
The role of mitochondria in Parkinson's disease[1]. Complex I (C-I) activity is decreased in Parkinson's disease, and inhibition of C-I by mitochondrial permeability transition pore or rotenone causes Parkinsonism. Mutations in mitochondrial DNA (mtDNA)-encoded C-I subunits and mtDNA polymerase γ (POLG) also cause Parkinsonism. Many genes associated with Parkinson's disease (such as α-synuclein, parkin, DJ-1, PINK1, and LRRK2) also implicate mitochondria in disease pathogenesis. α-synuclein overexpression can impair mitochondrial function and enhance the toxicity of the mitochondrial permeability transition pore.
Parkin associates with the outer mitochondrial membrane and protects against cytochrome c release. It may also associate with mitochondrial-transcription-factor A (TFAM). Physical associations have been reported between DJ-1 and α-synuclein, DJ-1 and PINK1, and DJ-1 and parkin, and there is genetic evidence that DJ-1, PINK1 and parkin are functionally linked in the same pathway. About 10% of the kinase LRRK2 is localized to mitochondria, and Parkinson's disease-related mutations augment its kinase activity. A mutation in high temperature requirement protein A2 was found in 1% of sporadic Parkinson's disease patients. CR: Control region.
So far, mutations or polymorphisms in mitochondrial DNA and at least nine named nuclear genes have been identified as causing PD or affecting PD risk: α-synuclein, parkin, ubiquitin C-terminal hydrolase-L1, DJ-1, mitochondrial phosphatase and tensin homologue-induced kinase 1, leucine-rich-repeat kinase 2, nuclear receptor NURR1, HTRA2 and tau[77,78,79,80,81,82,83,84,85,86,87,88,89,90,91,92]. Of the nuclear genes, α-synuclein, parkin, DJ-1, phosphatase and tensin homologue-induced kinase 1, leucine-rich-repeat kinase 2 and HTRA2 directly or indirectly involve mitochondria[1].
α-Synuclein is a major component of Lewy bodies[1], and three missense mutations (A53T, A30P and E46K) in the gene encoding α-synuclein are linked to dominantly inherited PD[93]. Researchers discovered secretion of α-synuclein is elevated in response to proteasomal and mitochondrial dysfunction, resulting in cellular defects in PD[94,95].
Mutations in parkin account for the majority of early-onset familial PD cases[96]. Loss-of-function mutations in the gene encoding parkin cause mitochondrial dysfunction[97] and the recessively inherited form of PD[98]. Phosphatase and tensin homologue-induced kinase 1 and parkin are functionally linked, as their expression induces mitochondrial fission[99,100]. Phosphatase and tensin homologue-induced kinase 1 is localized to the mitochondria. Loss-of-function of phosphatase and tensin homologue-induced kinase 1 leads to decreased mitochondrial protection against oxidative stress, causing enhanced mitochondrial dysfunction[101]. Parkin is recruited to dysfunctional mitochondria to promote their autophagic degradation and rescues degeneration in phosphatase and tensin homologue-induced kinase 1-null flies[102].
Cacciatore et al [103] selected rotenone, a specific mitochondrial complex I inhibitor, to produce Parkinsonism like symptoms in rats and induce mitochondrial dysfunction and ultrastructural damage. Impaired mitochondrial function leads to an excessive production of ROS. Mitochondrial complex I alterations are a major source of ROS generation in patients with PD. Dysfunctional mitochondria are the primary intracellular source of ROS in PD models[104]. Mitochondrial dysfunction, especially selective loss of complex I activity and oxidative metabolism, are critical components of most current theories of nigrostriatal degeneration in PD[105,106].
Mitochondrial damage and ALS
ALS is a progressive neurological disease that is associated with degeneration of motor neurons, causing muscle atrophy and, ultimately, paralysis. ALS is predominantly a sporadic disorder, but approximately 10% of cases are familial[107]. Abundant evidence indicates that mitochondrial degeneration has been directly or indirectly implicated in the pathogenesis of ALS[108,109,110,111,112,113,114] (Figure 4).
Figure 4.
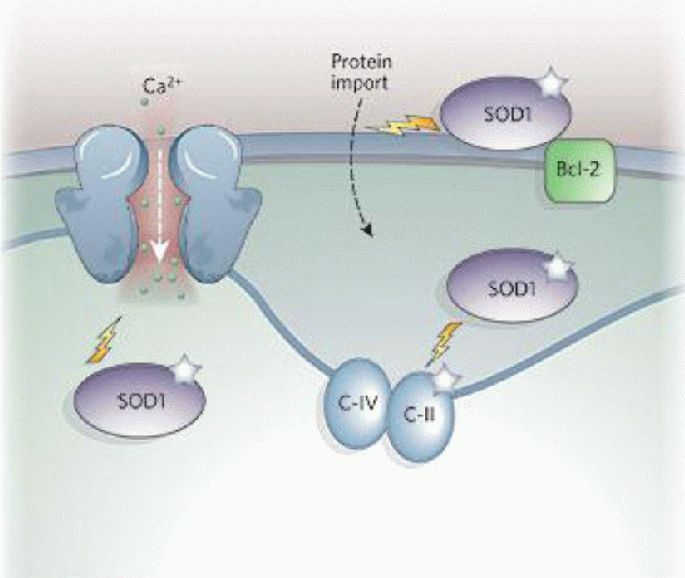
The role of mitochondria in amyotrophic lateral sclerosis[1].
Overexpression of mutant superoxide dismutase 1 (SOD1) in amyotrophic lateral sclerosis impairs electron transport chain activities and decreases mitochondrial calcium-loading capacity. SOD1 has been localized to the outer mitochondrial membrane, intermembrane space and matrix, and targeting of mutant SOD1 to mitochondria causes cytochrome c release and apoptosis. Mutant SOD1 promotes aberrant mitochondrial reactive oxygen species production and forms aggregates that may clog the outer mitochondrial membrane protein importation machinery or bind and sequester the antiapoptotic protein Bcl-2.
C-II: Complex II; C-IV: complex IV.
Approximately 20% of familial ALS cases are due to mutations in the gene encoding superoxide dismutase 1[108]. The expression of mutant superoxide dismutase 1 is not only associated with mitochondrial morphological changes, but also with mitochondrial dysfunction[93]. Postmortem tissue, as well as biopsy samples of a variety of tissues, show mitochondrial ab-normalities in both sporadic and familial ALS patients[109]. Aggregated and swollen mitochondria have been observed in postmortem sporadic ALS[109]. Massive mitochondrial vacuolation was observed in transgenic mice expressing the superoxide dismutase 1 mutants G93A or G37R in their motor neurons, which appear to derive from degenerating mitochondria[110,111]. Morphological abnormalities in the form of swollen and vacuolated mitochondria of motor neurons were recognized early and have been described extensively in mutant superoxide dismutase 1 transgenic mice[110,111]. Swollen and fragmented mitochondria were observed in N2A and NSC34 cells[112].
Presynaptic terminals on the somata of normal-appearing Betz cells showed a wide range of changes including increased mitochondria, and the dark type of degenerative change in synapses such as dense conglomerates of dark mitochondria with dense cristae and densely aggregated presynaptic vesicles[113]. These alterations were significantly more common in ALS patients than in controls, suggesting that a substantial synaptic change occurs in Betz cells even in the early stage of ALS[113]. Menzies et al [115] and Sasaki et al [116] have also reported mitochondrial morphology abnormalities in skeletal muscle, liver, spinal motor neurons and cortical upper motor neuron regions of ALS patients using electron microscopy.
Mitochondrial alterations may represent an early event triggering the onset of ALS, rather than simply a by product of cell degeneration[107]. In the mutant superoxide dismutase 1 mouse model, mitochondrial abnormalities appear before symptoms of paralysis and motor neuron degeneration. At the time of disease onset, mitochondrial respiration and adenosine triphosphate synthesis are defective in the brain and spinal cord of G93A mutant superoxide dismutase 1 transgenic mice[114]. Some ALS patients with defects in mitochondrial oxidative phosphorylation in skeletal muscle have a novel superoxide dismutase 1 mutation[117]. Concomitantly, the release of mitochondrial Ca2+ as well as pro-apoptotic factors (e.g., apoptosis-inducing factor, cytochrome c) into the cytosol triggers signaling cascades leading to apoptosis that seem to cause neuronal decline in neurodegenerative diseases[44]. Observations on the function of mitochondria have produced evidence consistent with these morphological observations[102].
Mitochondrial damage and other neurodegenerative diseases
Mitochondrial damage plays an important role in the pathogenesis of other neurodegenerative diseases besides AD, PD and ALS. Helguera et al [118] reported that mitochondrial dysfunction and oxidative stress are common features of Down syndrome. Siddiqui et al [119] suggested that mitochondrial DNA is a major target of mutant Huntington-associated oxidative stress, and that mitochondrial damage is implicated in Huntington's disease. The activity of complex II in the electron transport chain is decreased in Huntington's disease brains[1]. Friedreich ataxia is a common form of ataxia caused by decreased expression of the mitochondrial protein frataxin[120,121]. Oxidative damage of mitochondria is thought to play a key role in the pathogenesis of the disease[120,122].
CONCLUSIONS AND PROSPECTS
Accumulating data suggest that oxidative stress and mitochondrial damage are involved in the pathogenesis of neurodegenerative diseases, and antioxidant administration to modify mitochondrial dysfunction may be useful in the prevention and treatment of neurodegenerative diseases[123,124,125,126]. Mitochondria are a major source of intracellular ROS and are particularly vulnerable to oxidative stress. Mitochondrial dysfunction is a prominent feature of neurodegenerative diseases[37], such as PD, AD, and ALS[51]. As mentioned above, ROS can induce mitochondrial DNA mutations, and damage the mitochondrial respiratory chain, membrane permeability, Ca2+ homeostasis and mitochondrial defense systems; all these aspects are implicated in the development of neurodegenerative diseases, which mediate or amplifying neuronal dysfunction during the course of neurodegeneration. Therefore, strategies to modify mitochondrial dysfunction may be an attractive therapeutic intervention for the treatment of various neurodegenerative diseases.
Footnotes
Funding: This work was supported by the National Natural Science Foundation of China, No. 81274005; Medical Science Research, Health Department of Hebei Province, No. 20110173, 20090588; Hebei Education Department Science Foundation, No. 2007302.
Conflict of Interest: None declared.
(Reviewed by Diwakarla S, Norman C, Feng HL, Zhang GR)
(Edited by Wang LM, Yang Y, Li CH, Song LP, Liu WJ, Zhao M)
REFERENCES
- [1].Lin MT, Beal MF. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature. 2006;443(7113):787–795. doi: 10.1038/nature05292. [DOI] [PubMed] [Google Scholar]
- [2].Martin LJ. Mitochondrial and cell death mechanisms in neurodegenerative diseases. Pharmaceuticals (Basel) 2010;3(4):839–915. doi: 10.3390/ph3040839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Crunkhorn S. Neurodegenerative disorders: Restoring the balance. Nat Rev Drug Discov. 2011;10(8):576. doi: 10.1038/nrd3521. [DOI] [PubMed] [Google Scholar]
- [4].Sas K, Robotka H, Toldi J, et al. Mitochondria, metabolic disturbances, oxidative stress and the kynurenine system, with focus on neurodegenerative disorders. J Neurol Sci. 2007;257(1-2):221–239. doi: 10.1016/j.jns.2007.01.033. [DOI] [PubMed] [Google Scholar]
- [5].Douarre C, Sourbier C, Dalla Rosa I, et al. Mitochondrial topoisomerase I is critical for mitochondrial integrity and cellular energy metabolism. PLoS One. 2012;7(7):e41094. doi: 10.1371/journal.pone.0041094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Hollensworth SB, Shen C, Sim JE, et al. Glial cell type-specific responses to menadione-induced oxidative stress. Free Radic Biol Med. 2000;28(8):1161–1174. doi: 10.1016/s0891-5849(00)00214-8. [DOI] [PubMed] [Google Scholar]
- [7].Chen JQ, Yager JD, Russo J. Regulation of mitochondrial respiratory chain structure and function by estrogens/ estrogen receptors and potential physiological/ pathophysiological implications. Biochim Biophys Acta. 2005;1746(1):1–17. doi: 10.1016/j.bbamcr.2005.08.001. [DOI] [PubMed] [Google Scholar]
- [8].Ghezzi D, Zeviani M. Assembly factors of human mitochondrial respiratory chain complexes: physiology and pathophysiology. Adv Exp Med Biol. 2012;748:65–106. doi: 10.1007/978-1-4614-3573-0_4. [DOI] [PubMed] [Google Scholar]
- [9].Shoubridge EA, Wai T. Mitochondrial DNA and the mammalian oocyte. Curr Top Dev Biol. 2007;77:87–111. doi: 10.1016/S0070-2153(06)77004-1. [DOI] [PubMed] [Google Scholar]
- [10].Porter RK, Brand MD. Mitochondrial proton conductance and H+/O ratio are independent of electron transport rate in isolated hepatocytes. Biochem J. 1995;310(Pt 2):379–382. doi: 10.1042/bj3100379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Zhang Y, Wang M, Li H, et al. Accumulation of nuclear and mitochondrial DNA damage in the frontal cortex cells of patients with HIV-associated neurocognitive disorders. Brain Res. 2012;1458:1–11. doi: 10.1016/j.brainres.2012.04.001. [DOI] [PubMed] [Google Scholar]
- [12].Wei YH, Lu CY, Wei CY, et al. Oxidative stress in human aging and mitochondrial disease? consequences of defective mitochondrial respiration and impaired antioxidant enzyme system. Chin J Physiol. 2001;44(1):1–11. [PubMed] [Google Scholar]
- [13].Selivanov VA, Votyakova TV, Pivtoraiko VN, et al. Reactive oxygen species production by forward and reverse electron fluxes in the mitochondrial respiratory chain. PLoS Comput Biol. 2011;7(3):e1001115. doi: 10.1371/journal.pcbi.1001115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Finkel T, Holbrook NJ. Oxidants, oxidative stress and the biology of ageing. Nature. 2000;408(6809):239–247. doi: 10.1038/35041687. [DOI] [PubMed] [Google Scholar]
- [15].Van Houten B, Woshner V, Santos JH. Role of mitochondrial DNA in toxic responses to oxidative stress. DNA Repair (Amst) 2006;5(2):145–152. doi: 10.1016/j.dnarep.2005.03.002. [DOI] [PubMed] [Google Scholar]
- [16].Voets AM, Huigsloot M, Lindsey PJ, et al. Transcriptional changes in OXPHOS complex I deficiency are related to anti-oxidant pathways and could explain the disturbed calcium homeostasis. Biochim Biophys Acta. 2012;1822(7):1161–1168. doi: 10.1016/j.bbadis.2011.10.009. [DOI] [PubMed] [Google Scholar]
- [17].Castro Mdel R, Suarez E, Kraiselburd E, et al. Aging increases mitochondrial DNA damage and oxidative stress in liver of rhesus monkeys. Exp Gerontol. 2012;47(1):29–37. doi: 10.1016/j.exger.2011.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Alexeyev MF. Is there more to aging than mitochondrial DNA and reactive oxygen species? FEBS J. 2009;276(20):5768–5787. doi: 10.1111/j.1742-4658.2009.07269.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Andreazza AC, Shao L, Wang JF, et al. Mitochondrial complex I activity and oxidative damage to mitochondrial proteins in the prefrontal cortex of patients with bipolar disorder. Arch Gen Psychiatry. 2010;67(4):360–368. doi: 10.1001/archgenpsychiatry.2010.22. [DOI] [PubMed] [Google Scholar]
- [20].Vajrala V, Claycomb JR, Sanabria H, et al. Effects of oscillatory electric fields on internal membranes: an analytical model. Biophys J. 2008;94(6):2043–2052. doi: 10.1529/biophysj.107.114611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Kirkinezos IG, Bacman SR, Hernandez D, et al. Cytochrome c association with the inner mitochondrial membrane is impaired in the CNS of G93A-SOD1 mice. J Neurosci. 2005;25(1):164–172. doi: 10.1523/JNEUROSCI.3829-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Stewart VC, Heales SJ. Nitric oxide-induced mitochondrial dysfunction implications for neurodegeneration. Free Radic Biol Med. 2003;34(3):287–303. doi: 10.1016/s0891-5849(02)01327-8. [DOI] [PubMed] [Google Scholar]
- [23].Valko M, Leibfritz D, Moncol J, et al. Free radicals and antioxidants in normal physiological functions and human disease. Int J Biochem Cell Biol. 2007;39(1):44–84. doi: 10.1016/j.biocel.2006.07.001. [DOI] [PubMed] [Google Scholar]
- [24].Sultana R, Poon HF, Cai J, et al. Identification of nitrated proteins in Alzheimer's disease brain using a redox proteomics approach. Neurobiol Dis. 2006;22(1):76–87. doi: 10.1016/j.nbd.2005.10.004. [DOI] [PubMed] [Google Scholar]
- [25].Beal MF. Energetics in the pathogenesis of neurodegenerative diseases. Trends Neurosci. 2000;23(7):298–304. doi: 10.1016/s0166-2236(00)01584-8. [DOI] [PubMed] [Google Scholar]
- [26].Fu JD, Yang HT. Developmental regulation of intracellular calcium homeostasis in early cardiac myocytes. Sheng Li Xue Bao. 2006;58(2):95–103. [PubMed] [Google Scholar]
- [27].Martín M, Macías M, Escames G, et al. Melatonin-induced increased activity of the respiratory chain complexes I and IV can prevent mitochondrial damage induced by ruthenium red in vivo. J Pineal Res. 2000;28(4):242–248. doi: 10.1034/j.1600-079x.2000.280407.x. [DOI] [PubMed] [Google Scholar]
- [28].Chang Y, Huang SK, Wang SJ. Coenzyme Q10 inhibits the release of glutamate in rat cerebrocortical nerve terminals by suppression of voltage-dependent calcium influx and mitogen-activated protein kinase signaling pathway. J Agric Food Chem. 2012;60(48):11909–11918. doi: 10.1021/jf302875k. [DOI] [PubMed] [Google Scholar]
- [29].Martel MA, Soriano FX, Baxter P, et al. Inhibiting pro-death NMDA receptor signaling dependent on the NR2 PDZ ligand may not affect synaptic function or synaptic NMDA receptor signaling to gene expression. Channels (Austin) 2009;3(1):12–15. doi: 10.4161/chan.3.1.7864. [DOI] [PubMed] [Google Scholar]
- [30].Mattson MP. Calcium and neurodegeneration. Aging Cell. 2007;6(3):337–350. doi: 10.1111/j.1474-9726.2007.00275.x. [DOI] [PubMed] [Google Scholar]
- [31].Mattson MP, Magnus T. Ageing and neuronal vulnerability. Nat Rev Neurosci. 2006;7(4):278–294. doi: 10.1038/nrn1886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Toescu EC. Normal brain ageing: models and mechanisms. Philos Trans R Soc Lond B Biol Sci. 2005;360(1464):2347–2354. doi: 10.1098/rstb.2005.1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].van der Vliet A, Hoen PA, Wong PS, et al. Formation of S-nitrosothiols via direct nucleophilic nitrosation of thiols by peroxynitrite with elimination of hydrogen peroxide. J Biol Chem. 1998;273(46):30255–30262. doi: 10.1074/jbc.273.46.30255. [DOI] [PubMed] [Google Scholar]
- [34].Joshi G, Sultana R, Perluigi M, et al. In vivo protection of synaptosomes from oxidative stress mediated by Fe2+/H2O2 or 2, 2-azobis-(2-amidinopropane) dihydrochloride by the glutathione mimetic tricyclodecan-9-yl-xanthogenate. Free Radic Biol Med. 2005;38(8):1023–1031. doi: 10.1016/j.freeradbiomed.2004.12.027. [DOI] [PubMed] [Google Scholar]
- [35].Ross WN. Understanding calcium waves and sparks in central neurons. Nat Rev Neurosci. 2012;13(3):157–168. doi: 10.1038/nrn3168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Martin LJ. The mitochondrial permeability transition pore: A molecular target for amyotrophic lateral sclerosis therapy. Bio-chim Biophys Acta. 2010;1802(1):186–197. doi: 10.1016/j.bbadis.2009.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Szeto HH. Mitochondria-targeted peptide antioxidants: novel neuroprotective agents. AAPS J. 2006;8(3):E521–531. doi: 10.1208/aapsj080362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Brand MD, Affourtit C, Esteves TC, et al. Mitochondrial superoxide: production, biological effects, and activation of uncoupling proteins. Free Radic Biol Med. 2004;37(6):755–767. doi: 10.1016/j.freeradbiomed.2004.05.034. [DOI] [PubMed] [Google Scholar]
- [39].Zeviani M, Simonati A, Bindoff LA. Ataxia in mitochondrial disorders. Handb Clin Neurol. 2012;103:359–372. doi: 10.1016/B978-0-444-51892-7.00022-X. [DOI] [PubMed] [Google Scholar]
- [40].Haas RH. The evidence basis for coenzyme Q therapy in oxidative phosphorylation disease. Mitochondrion. 2007;7(Suppl):S136–145. doi: 10.1016/j.mito.2007.03.008. [DOI] [PubMed] [Google Scholar]
- [41].Wilson DF, Harrison DK, Vinogradov SA. Oxygen, pH, and mitochondrial oxidative phosphorylation. J Appl Physiol. 2012;113(12):1838–1845. doi: 10.1152/japplphysiol.01160.2012. [DOI] [PubMed] [Google Scholar]
- [42].Hansford RG. Physiological role of mitochondrial Ca2+ transport. J Bioenerg Biomembr. 1994;26(5):495–508. doi: 10.1007/BF00762734. [DOI] [PubMed] [Google Scholar]
- [43].Nicholls DG, Budd SL. Mitochondria and neuronal survival. Physiol Rev. 2000;80(1):315–360. doi: 10.1152/physrev.2000.80.1.315. [DOI] [PubMed] [Google Scholar]
- [44].Chinnery PF, Elliott HR, Hudson G, et al. Epigenetics, epidemiology and mitochondrial DNA diseases. Int J Epidemiol. 2012;41(1):177–187. doi: 10.1093/ije/dyr232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Schon EA, Manfredi G. Neuronal degeneration and mitochondrial dysfunction. J Clin Invest. 2003;111(3):303–312. doi: 10.1172/JCI17741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Kasote DM, Hegde MV, Katyare SS. Mitochondrial dysfunction in psychiatric and neurological diseases: Cause(s), consequence(s), and implications of antioxidant therapy. Biofactors. doi: 10.1002/biof.1093. in press. [DOI] [PubMed] [Google Scholar]
- [47].Wojda U, Salinska E, Kuznicki J. Calcium ions in neuronal degeneration. IUBMB Life. 2008;60(9):575–590. doi: 10.1002/iub.91. [DOI] [PubMed] [Google Scholar]
- [48].Ferreiro E, Baldeiras I, Ferreira IL, et al. Mitochondrial-and endoplasmic reticulum-associated oxidative stress in Alzheimer's disease: from pathogenesis to biomarkers. Int J Cell Biol 2012. 2012 doi: 10.1155/2012/735206. 735206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Anandatheerthavarada HK, Biswas G, Robin MA, et al. Mitochondrial targeting and a novel transmembrane arrest of Alzheimer's amyloid precursor protein impairs mitochondrial function in neuronal cells. J Cell Biol. 2003;161(1):41–54. doi: 10.1083/jcb.200207030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Giasson BI, Ischiropoulos H, Lee VM, et al. The relationship between oxidative/nitrative stress and pathological inclusions in Alzheimer's and Parkinson's diseases. Free Radic Biol Med. 2002;32(12):1264–1275. doi: 10.1016/s0891-5849(02)00804-3. [DOI] [PubMed] [Google Scholar]
- [51].Andersen JK. Oxidative stress in neurodegeneration: cause or consequence? Nat Med. 2004;10(Suppl):S18–25. doi: 10.1038/nrn1434. [DOI] [PubMed] [Google Scholar]
- [52].Lustbader JW, Cirilli M, Lin C, et al. ABAD directly links Abeta to mitochondrial toxicity in Alzheimer's disease. Science. 2004;304(5669):448–452. doi: 10.1126/science.1091230. [DOI] [PubMed] [Google Scholar]
- [53].Dumont M, Ho DJ, Calingasan NY, et al. Mitochondrial dihydrolipoyl succinyltransferase deficiency accelerates amyloid pathology and memory deficit in a transgenic mouse model of amyloid deposition. Free Radic Biol Med. 2009;47(7):1019–1027. doi: 10.1016/j.freeradbiomed.2009.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Shi Q, Xu H, Yu H, et al. Inactivation and reactivation of the mitochondrial α-ketoglutarate dehydrogenase complex. J Biol Chem. 2011;286(20):17640–17648. doi: 10.1074/jbc.M110.203018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Coskun PE, Beal MF, Wallace DC. Alzheimer's brains harbor somatic mtDNA control-region mutations that suppress mitochondrial transcription and replication. Proc Natl Acad Sci U S A. 2004;101(29):10726–10731. doi: 10.1073/pnas.0403649101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Reddy PH. Mitochondrial oxidative damage in aging and Alzheimer's disease: implications for mitochondrially targeted antioxidant therapeutics. J Biomed Biotechnol. 2006;2006(3):31372. doi: 10.1155/JBB/2006/31372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Tranah GJ, Nalls MA, Katzman SM, et al. Mitochondrial DNA sequence variation associated with dementia and cognitive function in the elderly. J Alzheimers Dis. 2012;32(2):357–372. doi: 10.3233/JAD-2012-120466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Estrada LD, Zanlungo SM, Alvarez AR. C-Abl tyrosine kinase signaling: a new player in AD tau pathology. Curr Alzheimer Res. 2011;8(6):643–651. doi: 10.2174/156720511796717249. [DOI] [PubMed] [Google Scholar]
- [59].Cancino GI, Perez de Arce K, Castro PU, et al. c-Abl tyrosine kinase modulates tau pathology and Cdk5 phosphorylation in AD transgenic mice. Neurobiol Aging. 2011;32(7):1249–1261. doi: 10.1016/j.neurobiolaging.2009.07.007. [DOI] [PubMed] [Google Scholar]
- [60].Jing Z, Caltagarone J, Bowser R. Altered subcellular distribution of c-Abl in Alzheimer's disease. J Alzheimers Dis. 2009;17(2):409–422. doi: 10.3233/JAD-2009-1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Zivković L, Spremo-Potparević B, Siedlak SL, et al. DNA damage in Alzheimer disease lymphocytes and its relation to premature centromere division. Neurodegener Dis. doi: 10.1159/000346114. in press. [DOI] [PubMed] [Google Scholar]
- [62].López-Erauskin J, Galino J, Bianchi P, et al. Oxidative stress modulates mitochondrial failure and cyclophilin D function in X-linked adrenoleukodystrophy. Brain. 2012;135(Pt 12):3584–3598. doi: 10.1093/brain/aws292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Akhtar MW, Sunico CR, Nakamura T, et al. Redox regulation of protein function via cysteine s-nitrosylation and its relevance to neurodegenerative diseases. Int J Cell Biol 2012. 2012 doi: 10.1155/2012/463756. 463756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Bemporad F, Chiti F. Protein misfolded oligomers: experimental approaches, mechanism of formation, and structure-toxicity relationships. Chem Biol. 2012;19(3):315–327. doi: 10.1016/j.chembiol.2012.02.003. [DOI] [PubMed] [Google Scholar]
- [65].Gonfloni S, Maiani E, Di Bartolomeo C, et al. Oxidative Stress, DNA damage, and c-Abl signaling: at the crossroad in neurodegenerative diseases? Int J Cell Biol 2012. 2012 doi: 10.1155/2012/683097. 683097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Cho DH, Nakamura T, Fang J, et al. S-nitrosylation of Drp1 mediates beta-amyloid-related mitochondrial fission and neuronal injury. Science. 2009;324(5923):102–105. doi: 10.1126/science.1171091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Qu J, Nakamura T, Holland EA, et al. S-nitrosylation of Cdk5: potential implications in amyloid-β-related neurotoxicity in Alzheimer disease. Prion. 2012;6(4):364–370. doi: 10.4161/pri.21250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Nakamura T, Cho DH, Lipton SA. Redox regulation of protein misfolding, mitochondrial dysfunction, synaptic damage, and cell death in neurodegenerative diseases. Exp Neurol. 2012;238(1):12–21. doi: 10.1016/j.expneurol.2012.06.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Feeney CJ, Frantseva MV, Carlen PL, et al. Vulnerability of glial cells to hydrogen peroxide in cultured hippocampal slices. Brain Res. 2008;1198:1–15. doi: 10.1016/j.brainres.2007.12.049. [DOI] [PubMed] [Google Scholar]
- [70].Giasson BI, Lee VM. A new link between pesticides and Parkinson's disease. Nat Neurosci. 2000;3(12):1227–1228. doi: 10.1038/81737. [DOI] [PubMed] [Google Scholar]
- [71].Lindqvist D, Kaufman E, Brundin L, et al. Non-motor symptoms in patients with Parkinson's disease -correlations with inflammatory cytokines in serum. PLoS One. 2012;7(10):e47387. doi: 10.1371/journal.pone.0047387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Abeliovich A. Parkinson's disease: Mitochondrial damage control. Nature. 2010;463(7282):744–745. doi: 10.1038/463744a. [DOI] [PubMed] [Google Scholar]
- [73].Matsui H, Gavinio R, Asano T, et al. PINK1 and Parkin complementarily protect dopaminergic neurons in vertebrates. Hum Mol Genet. 2013;22(12):2423–2434. doi: 10.1093/hmg/ddt095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Momb J, Lewandowski JP, Bryant JD, et al. Deletion of Mthfd1l causes embryonic lethality and neural tube and craniofacial defects in mice. Proc Natl Acad Sci U S A. 2013;110(2):549–554. doi: 10.1073/pnas.1211199110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Alleyne T, Mohan N, Adogwa A. Elevated ferric, calcium and magnesium ions in the brain induce protein aggregation in brain mitochondria. West Indian Med J. 2012;61(2):122–127. [PubMed] [Google Scholar]
- [76].Chaturvedi RK, Beal MF. Mitochondria targeted therapeutic approaches in Parkinson's and Huntington's diseases. Mol Cell Neurosci. 2013;55:101–114. doi: 10.1016/j.mcn.2012.11.011. [DOI] [PubMed] [Google Scholar]
- [77].Puschmann A. Monogenic Parkinson's disease and Parkinsonism: Clinical phenotypes and frequencies of known mutations. Parkinsonism Relat Disord. 2013;19(4):407–415. doi: 10.1016/j.parkreldis.2013.01.020. [DOI] [PubMed] [Google Scholar]
- [78].Plaitakis A, Zaganas I, Spanaki C. Deregulation of glutamate dehydrogenase in human neurologic disorders. J Neurosci Res. doi: 10.1002/jnr.23176. in press. [DOI] [PubMed] [Google Scholar]
- [79].Ciccone S, Maiani E, Bellusci G, et al. Parkinson's Disease: a complex interplay of mitochondrial DNA alterations and oxidative stress. Int J Mol Sci. 2013;14(2):2388–2409. doi: 10.3390/ijms14022388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Duplan E, Giaime E, Viotti J, et al. ER-stress-associated functional link between Parkin and DJ-1 via a transcriptional cascade involving the tumor suppressor p53 and the spliced X-box binding protein XBP-1. J Cell Sci. 2013;126(Pt 9):2124–2133. doi: 10.1242/jcs.127340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Song S, Jang S, Park J, et al. Characterization of PINK1 (PTEN-induced Putative Kinase 1) mutations associated with Parkinson disease in mammalian cells and Drosophila. J Biol Chem. 2013;288(8):5660–5672. doi: 10.1074/jbc.M112.430801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Lenzi P, Marongiu R, Falleni A, et al. A subcellular analysis of genetic modulation of PINK1 on mitochondrial alterations, autophagy and cell death. Arch Ital Biol. 2012;150(2-3):194–217. doi: 10.4449/aib.v150i2/3.1417. [DOI] [PubMed] [Google Scholar]
- [83].Cartier AE, Ubhi K, Spencer B, et al. Differential effects of UCHL1 modulation on alpha-synuclein in PD-like models of alpha-synucleinopathy. PLoS One. 2012;7(4):e34713. doi: 10.1371/journal.pone.0034713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Shimshek DR, Schweizer T, Schmid P, et al. Excess α-synuclein worsens disease in mice lacking ubiquitin carboxy-terminal hydrolase L1. Sci Rep. 2012;2:262. doi: 10.1038/srep00262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Hartfield EM, Fernandes HJ, Vowles J, et al. Cellular re-programming: a new approach to modelling Parkinson's disease. Biochem Soc Trans. 2012;40(5):1152–1157. doi: 10.1042/BST20120159. [DOI] [PubMed] [Google Scholar]
- [86].Orenstein SJ, Kuo SH, Tasset I, et al. Interplay of LRRK2 with chaperone-mediated autophagy. Nat Neurosci. 2013;16(4):394–406. doi: 10.1038/nn.3350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Federoff HJ. Nur (R1) turing a notion on the etiopathogenesis of Parkinson's disease. Neurotox Res. 2009;16(3):261–270. doi: 10.1007/s12640-009-9056-7. [DOI] [PubMed] [Google Scholar]
- [88].Lin X, Parisiadou L, Sgobio C, et al. Conditional expression of Parkinson's disease-related mutant α-synuclein in the midbrain dopaminergic neurons causes progressive neurodegeneration and degradation of transcription factor nuclear receptor related 1. J Neurosci. 2012;32(27):9248–9264. doi: 10.1523/JNEUROSCI.1731-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Lin CH, Chen ML, Chen GS, et al. Novel variant Pro143Ala in HTRA2 contributes to Parkinson's disease by inducing hyperphosphorylation of HTRA2 protein in mitochondria. Hum Genet. 2011;130(6):817–827. doi: 10.1007/s00439-011-1041-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Aasly JO, Shi M, Sossi V, et al. Cerebrospinal fluid amyloid β and tau in LRRK2 mutation carriers. Neurology. 2012;78(1):55–61. doi: 10.1212/WNL.0b013e31823ed101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Vandrovcova J, Anaya F, Kay V, et al. Disentangling the role of the tau gene locus in sporadic tauopathies. Curr Alzheimer Res. 2010;7(8):726–734. doi: 10.2174/156720510793611619. [DOI] [PubMed] [Google Scholar]
- [92].Coskun P, Wyrembak J, Schriner SE, et al. A mitochondrial etiology of Alzheimer and Parkinson disease. Biochim Biophys Acta. 2012;1820(5):553–564. doi: 10.1016/j.bbagen.2011.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Obeso JA, Rodriguez-Oroz MC, Goetz CG, et al. Missing pieces in the Parkinson's disease puzzle. Nat Med. 2010;16(6):653–661. doi: 10.1038/nm.2165. [DOI] [PubMed] [Google Scholar]
- [94].Orzi F, Casolla B, Rocchi R, et al. Prion-like mechanisms in epileptogenesis. Neurol Sci. doi: 10.1007/s10072-012-1148-0. in press. [DOI] [PubMed] [Google Scholar]
- [95].Vila M, Ramonet D, Perier C. Mitochondrial alterations in Parkinson's disease: new clues. J Neurochem. 2008;107(2):317–328. doi: 10.1111/j.1471-4159.2008.05604.x. [DOI] [PubMed] [Google Scholar]
- [96].Horowitz MP, Greenamyre JT. Gene-environment interactions in Parkinson's disease: the importance of animal modeling. Clin Pharmacol Ther. 2010;88(4):467–474. doi: 10.1038/clpt.2010.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Narendra D, Walker JE, Youle R. Mitochondrial quality control mediated by PINK1 and Parkin: links to Parkin-sonism. Cold Spring Harb Perspect Biol. 2012;4(11):pii:a011338. doi: 10.1101/cshperspect.a011338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Lee HJ, Patel S, Lee SJ. Intravesicular localization and exocytosis of alpha-synuclein and its aggregates. J Neurosci. 2005;25(25):6016–6024. doi: 10.1523/JNEUROSCI.0692-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Lutz AK, Exner N, Fett ME, et al. Loss of parkin or PINK1 function increases Drp1-dependent mitochondrial fragmentation. J Biol Chem. 2009;284(34):22938–22951. doi: 10.1074/jbc.M109.035774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Narendra D, Tanaka A, Suen DF, et al. Parkin-induced mitophagy in the pathogenesis of Parkinson disease. Autophagy. 2009;5(5):706–708. doi: 10.4161/auto.5.5.8505. [DOI] [PubMed] [Google Scholar]
- [101].Varçin M, Bentea E, Michotte Y, et al. Oxidative stress in genetic mouse models of Parkinson's disease. Oxid Med Cell Longev. 2012;2012:624925. doi: 10.1155/2012/624925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Manfredi G, Xu Z. Mitochondrial dysfunction and its role in motor neuron degeneration in ALS. Mitochondrion. 2005;5(2):77–87. doi: 10.1016/j.mito.2005.01.002. [DOI] [PubMed] [Google Scholar]
- [103].Cacciatore I, Baldassarre L, Fornasari E, et al. Recent advances in the treatment of neurodegenerative diseases based on GSH delivery systems. Oxid Med Cell Longev 2012. 2012 doi: 10.1155/2012/240146. 240146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Kumar H, Lim HW, More SV, et al. The role of free radicals in the aging brain and Parkinson's disease: convergence and parallelism. Int J Mol Sci. 2012;13(8):10478–10504. doi: 10.3390/ijms130810478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Lin TK, Cheng CH, Chen SD, et al. Mitochondrial dysfunction and oxidative stress promote apoptotic cell death in the striatum via cytochrome c/caspase-3 signaling cascade following chronic rotenone intoxication in rats. Int J Mol Sci. 2012;13(7):8722–8739. doi: 10.3390/ijms13078722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Schapira AH. Mitochondria in the aetiology and pathogenesis of Parkinson's disease. Lancet Neurol. 2008;7(1):97–109. doi: 10.1016/S1474-4422(07)70327-7. [DOI] [PubMed] [Google Scholar]
- [107].Rosen DR. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;364(6435):362. doi: 10.1038/364362c0. [DOI] [PubMed] [Google Scholar]
- [108].Ferri A, Cozzolino M, Crosio C, et al. Familial ALS- superoxide dismutases associate with mitochondria and shift their redox potentials. Proc Natl Acad Sci U S A. 2006;103(37):13860–13865. doi: 10.1073/pnas.0605814103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Sasaki S, Iwata M. Mitochondrial alterations in the spinal cord of patients with sporadic amyotrophic lateral sclerosis. J Neuropathol Exp Neurol. 2007;66(1):10–16. doi: 10.1097/nen.0b013e31802c396b. [DOI] [PubMed] [Google Scholar]
- [110].Sasaki S, Warita H, Murakami T, et al. Ultrastructural study of aggregates in the spinal cord of transgenic mice with a G93A mutant SOD1 gene. Acta Neuropathol. 2005;109(3):247–255. doi: 10.1007/s00401-004-0939-7. [DOI] [PubMed] [Google Scholar]
- [111].Israelson A, Arbel N, Da Cruz S, et al. Misfolded mutant SOD1 directly inhibits VDAC1 conductance in a mouse model of inherited ALS. Neuron. 2010;67(4):575–587. doi: 10.1016/j.neuron.2010.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Magrané J, Manfredi G. Mitochondrial function, morphology, and axonal transport in amyotrophic lateral sclerosis. Antioxid Redox Signal. 2009;11(7):1615–1626. doi: 10.1089/ars.2009.2604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Kawamata H, Manfredi G. Mitochondrial dysfunction and intracellular calcium dysregulation in ALS. Mech Ageing Dev. 2010;131(7-8):517–526. doi: 10.1016/j.mad.2010.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Corti S, Donadoni C, Ronchi D, et al. Amyotrophic lateral sclerosis linked to a novel SOD1 mutation with muscle mitochondrial dysfunction. J Neurol Sci. 2009;276(1-2):170–174. doi: 10.1016/j.jns.2008.09.030. [DOI] [PubMed] [Google Scholar]
- [115].Menzies FM, Ince PG, Shaw PJ. Mitochondrial involvement in amyotrophic lateral sclerosis. Neurochem Int. 2002;40(6):543–551. doi: 10.1016/s0197-0186(01)00125-5. [DOI] [PubMed] [Google Scholar]
- [116].Sasaki S, Horie Y, Iwata M. Mitochondrial alterations in dorsal root ganglion cells in sporadic amyotrophic lateral sclerosis. Acta Neuropathol. 2007;114(6):633–639. doi: 10.1007/s00401-007-0299-1. [DOI] [PubMed] [Google Scholar]
- [117].Reddy PH, Beal MF. Are mitochondria critical in the pathogenesis of Alzheimer's disease? Brain Res Brain Res Rev. 2005;49(3):618–632. doi: 10.1016/j.brainresrev.2005.03.004. [DOI] [PubMed] [Google Scholar]
- [118].Helguera P, Seiglie J, Rodriguez J, et al. Adaptive down-regulation of mitochondrial function in down syndrome. Cell Metab. 2013;17(1):132–140. doi: 10.1016/j.cmet.2012.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Siddiqui A, Rivera-Sánchez S, Castro Mdel R, et al. Mitochondrial DNA damage is associated with reduced mitochondrial bioenergetics in Huntington's disease. Free Radic Biol Med. 2012;53(7):1478–1488. doi: 10.1016/j.freeradbiomed.2012.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Marobbio CM, Pisano I, Porcelli V, et al. Rapamycin reduces oxidative stress in frataxin-deficient yeast cells. Mitochondrion. 2012;12(1):156–161. doi: 10.1016/j.mito.2011.07.001. [DOI] [PubMed] [Google Scholar]
- [121].Xia H, Cao Y, Dai X, et al. Novel frataxin isoforms may contribute to the pathological mechanism of friedreich ataxia. PLoS One. 2012;7(10):e47847. doi: 10.1371/journal.pone.0047847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Santos R, Lefevre S, Sliwa D, et al. Friedreich ataxia: molecular mechanisms, redox considerations, and therapeutic opportunities. Antioxid Redox Signal. 2010;13(5):651–690. doi: 10.1089/ars.2009.3015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Cooper JM, Korlipara LV, Hart PE, et al. Coenzyme Q10 and vitamin E deficiency in Friedreich's ataxia: predictor of efficacy of vitamin E and coenzyme Q10 therapy. Eur J Neurol. 2008;15(12):1371–1379. doi: 10.1111/j.1468-1331.2008.02318.x. [DOI] [PubMed] [Google Scholar]
- [124].Murphy MP. Targeting lipophilic cations to mitochondria. Biochim Biophys Acta. 2008;1777(7-8):1028–1031. doi: 10.1016/j.bbabio.2008.03.029. [DOI] [PubMed] [Google Scholar]
- [125].Moreira PI, Zhu X, Wang X, et al. Mitochondria: a therapeutic target in neurodegeneration. Biochim Biophys Acta. 2010;1802(1):212–220. doi: 10.1016/j.bbadis.2009.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Herrero MT, Pagonabarraga J, Linazasoro G. Neuroprotective role of dopamine agonists: evidence from animal models and clinical studies. Neurologist. 2011;17(6 Suppl 1):S54–66. doi: 10.1097/NRL.0b013e31823968fc. [DOI] [PubMed] [Google Scholar]