Maresin 1 biosynthesis during platelet–neutrophil interactions is organ-protective (original) (raw)
Significance
Neutrophil accumulation is fundamental to acute inflammation. In response to tissue injury, circulating neutrophil–platelet aggregates (N-PAs) form for secondary capture. Counterregulation of acute inflammatory processes by specialized proresolving mediators is essential to mitigate collateral injury to healthy bystander tissue. Here, we identified a biosynthetic route in human platelets for the proresolving mediator maresin 1 (MaR1) that is amplified during platelet–neutrophil interactions. In a self-resolving murine model of acute lung injury, N-PAs rapidly formed and a MaR1 counterregulatory circuit was engaged to restrain N-PAs and acute inflammation and restore homeostasis of the injured lung.
Keywords: maresin, platelet, inflammation, lung, resolution
Abstract
Unregulated acute inflammation can lead to collateral tissue injury in vital organs, such as the lung during the acute respiratory distress syndrome. In response to tissue injury, circulating platelet–neutrophil aggregates form to augment neutrophil tissue entry. These early cellular events in acute inflammation are pivotal to timely resolution by mechanisms that remain to be elucidated. Here, we identified a previously undescribed biosynthetic route during human platelet–neutrophil interactions for the proresolving mediator maresin 1 (MaR1; 7R,14S-dihydroxy-docosa-4Z,8E,10E,12Z,16Z,19Z-hexaenoic acid). Docosahexaenoic acid was converted by platelet 12-lipoxygenase to 13S,14S-epoxy-maresin, which was further transformed by neutrophils to MaR1. In a murine model of acute respiratory distress syndrome, lipid mediator metabololipidomics uncovered MaR1 generation in vivo in a temporally regulated manner. Early MaR1 production was dependent on platelet–neutrophil interactions, and intravascular MaR1 was organ-protective, leading to decreased lung neutrophils, edema, tissue hypoxia, and prophlogistic mediators. Together, these findings identify a transcellular route for intravascular maresin 1 biosynthesis via platelet–neutrophil interactions that regulates the extent of lung inflammation.
One critical function of acute inflammatory responses is delivery of leukocytes to sites of tissue injury to protect the host and restore homeostasis (1, 2). Activated vascular endothelial cells recruit leukocytes by direct interaction (primary capture) or via platelets (secondary capture) that engage circulating leukocytes rapidly after tissue injury (3, 4). If excessive, leukocyte recruitment and activation can injure healthy host bystander tissues and cause organ damage (2, 5)—a pathobiology common to several diseases, including the acute respiratory distress syndrome (ARDS), a devastating condition of acute lung inflammation with excess morbidity and mortality and without available medical therapy (6). Platelet–neutrophil interactions are integral to the early inflammatory response in ARDS (7, 8).
In health, acute inflammatory responses are terminated by specialized proresolving mediators (SPMs) that restrain the inflammatory response and signal for resolution (2). Early cellular and biochemical events in acute inflammation are pivotal to timely resolution (9, 10). A new family of SPMs of macrophage origin was recently uncovered in murine peritonitis and human macrophages and coined maresins (macrophage mediators in resolving inflammation) (11). Maresins are biosynthesized via initial lipoxygenation and molecular oxygen insertion at the carbon 14 position of docosahexaenoic acid (DHA) followed by epoxidation and oxygen incorporation at the carbon 7 position from a molecule of H2O (11). The complete stereochemistry of maresin 1 (MaR1; 7R,14S-dihydroxy-docosa-4Z,8E,10E,12Z,16Z,19Z-hexaenoic acid) (12) and its biosynthesis via a 13S,14S-epoxy-maresin intermediate (13_S_,14_S_-epoxy-docosa-4Z,7Z,9E,11E,16Z,19Z-hexaenoic acid) (13) were recently established. In addition to macrophages, human and murine platelets contain arachidonate 12-lipoxygenase that may be capable of generating the 13S,14S-epoxy-maresin intermediate and participating in MaR1 production at sites of vascular inflammation. Here, we provide evidence for a MaR1 biosynthetic route during platelet–neutrophil interactions that is operative in vivo in a murine model of ARDS to restrain inflammation and restore homeostasis of the injured lung.
Results
To determine if platelets can participate in vascular MaR1 biosynthesis, platelets were isolated from fresh human whole blood, then incubated with d5-DHA in the presence or absence of autologous peripheral blood neutrophils, and activated with either platelet-activating factor (PAF) or the platelet-specific agonist thrombin. MaR1 biosynthesis was assessed by lipid mediator (LM) metabololipidomics (SI Methods). Small amounts of MaR1 were identified in platelet–d5-DHA incubations by matching MS/MS fragmentation patterns and retention times (Fig. 1 A and B). Coincubation of platelets with neutrophils in the presence of PAF markedly increased MaR1 generation (Fig. 1_A_) in a neutrophil-dependent manner (Fig. 1_C_); platelet activation with PAF (Fig. 1_A_) or thrombin (Fig. 1_C_) or increased platelet number (Fig. 1_C_) did not lead to a significant increase in MaR1 production in the absence of neutrophil activation. These findings support a previously unidentified MaR1 biosynthetic route in platelets that was significantly increased via transcellular biosynthesis during platelet–neutrophil interactions.
Fig. 1.
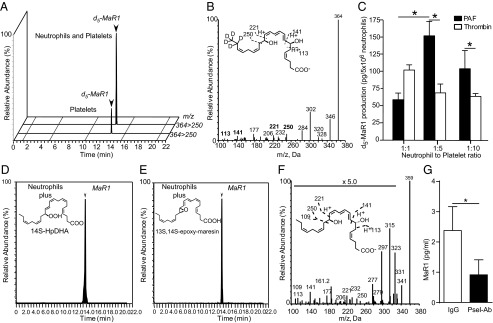
MaR1 is produced by N-PAs via transcellular biosynthesis. (A–C) MaR1 production by isolated human peripheral blood platelets incubated with d5-DHA before incubation with or without neutrophils in the presence of PAF or thrombin. Levels of d5-MaR1 were assessed by liquid chromatography (LC)–MS/MS metabololipidomics. (A) Representative multiple reaction monitoring (MRM) chromatograms (m/z = 364–250) and (B) MS/MS spectrum from PAF-treated samples used for the identification of d5-MaR1. (C) d5-MaR1 levels in incubations with different neutrophil-to-platelet ratios. Results for A and B are representative of n = 3 healthy donor blood isolations. Results for C are mean ± SEM for n = 3 healthy donor blood isolations. *P < 0.05. (D–F) Human peripheral blood neutrophils were isolated by density centrifugation. Cells were suspended at 1 × 108 in PBS with 10 mg/mL BSA, then incubated with (D) 10 µM 14HpDHA or (E) 10 µM 13S,14S-eMaR and 0.1 mg of serum-treated zymosan (15 min, 37 °C, pH 7.45). (F) Representative MS/MS spectrum used for the identification of MaR1 (7R,14S-dihydroxydocosa-4Z,8E,10E,12Z,16Z,19Z-hexaenoic acid). (G) Levels of MaR1 in fresh human whole blood pretreated with anti-human P-selectin or isotype control antibody after treatment with PAF (500 nM). Values represent the mean ± SEM for n = 7 healthy volunteers. *P < 0.05.
To test this biosynthetic role for neutrophil conversion, the human recombinant 12-lipoxygenase product 14S-hydro(peroxy)-docosahexaenoic acid (14S-HpDHA) (Fig. 1_D_) and synthetic 13S,14S-epoxy-maresin (Fig. 1_E_) were added to activated neutrophils, which converted these human platelet 12-lipoxygenase–derived intermediates to MaR1 (n = 3 for each intermediate). To determine the impact of direct platelet–neutrophil interactions on MaR1 biosynthesis, fresh human blood was incubated with an anti–P-selectin antibody before cell activation with PAF. Relative to an isotype control antibody, blockade of P-selectin decreased neutrophil–platelet aggregate (N-PA) formation (Fig. S1_A_) and concomitantly decreased MaR1 production (Fig. 1_G_). Taken together, these results provide evidence for transcellular biosynthesis of MaR1 during platelet neutrophil interactions, including as N-PAs. MaR1 was identified by retention time (Fig. 1 D and E) and diagnostic ions in MS/MS (Fig. 1_F_ and inset ions). Each gave m/z 359 = M-H, m/z 341 = M-H-H2O, m/z 315 = M-H-CO2, m/z 297 = M-H-H2O-CO2, m/z 279 = M-H-2H2O-CO2, m/z 177 = 221-CO2, m/z 141 = 177-2H2O, and m/z 123 = 141-H2O, matching published values (11–13).
Because platelet–neutrophil interactions are linked to the development of ARDS, we next determined if this new route for MaR1 production was operative in vivo by collecting murine lung tissues at timed intervals after selective injury to the left lung with intratracheal HCl (SI Methods). In this murine model of gastric acid aspiration, a leading cause of ARDS (14), LM metabololipidomic profiling of lung tissue revealed MaR1, as well as the nonenzymatic hydrolysis maresin pathway products from the labile 13,14-epoxy-maresin (11–13), namely 7_S_,14_S_-dihydroxydocosa-4_Z_,8_E_,10_E_,12_E_,16_Z_,19_Z_-hexaenoic acid (7-epi-Δ12-trans-MaR1) and 7_R_,14_S_-dihydroxydocosa-4_Z_,8_E_,10_E_,12_E_,16_Z_,19_Z_-hexaenoic acid (Δ12-trans-MaR1) (Fig. 2 A and B). MaR1 and its nonenzymatic hydrolysis product isomers were identified by their retention times (Fig. 2_A_) and diagnostic ions in MS/MS. After intratracheal HCl, lung MaR1 was significantly increased within 2 h and peaked at 24 h, returning to baseline levels by 72 h (Fig. 2_B_). Relative to other SPMs, lung MaR1 levels had a distinct temporal regulation that was notable for an earlier maximum (Table S1).
Fig. 2.

MaR1 is produced during acid-induced ALI in a temporally regulated manner. Lipid mediator metabololipidomics was performed to identify lung MaR1 and its biosynthetic pathway markers 7-epi-Δ12-trans-MaR1 and Δ12-trans-MaR1. (A) MRM ion chromatograms, where Q1 is the parent ion and Q3 a characteristic daughter ion (m/z = 359–221), present in lung homogenates obtained 24 h after intratracheal HCl. (B) Levels of MaR1 in lung homogenates from injured mice were determined at baseline, 2, 24, 48, and 72 h after intratracheal HCl administration. (C) Levels of MaR1 and nonenzymatic hydrolysis products 2 h after HCl in neutrophil-depleted mice (anti-Ly6G) and mice pretreated with an isotype control antibody. (D) Levels of MaR1 2 h after HCl in mice administered anti-mouse P-selectin or isotype control antibody. (E) Levels of MaR1 and nonenzymatic hydrolysis products 24 h after HCl in neutrophil-depleted mice (anti-Ly6G) and mice pretreated with an isotype control antibody. Values represent the mean ± SEM for n ≥ 4 mice. *P < 0.05.
The relative contribution of platelet–neutrophil interactions to MaR1 generation in vivo was first investigated by neutrophil depletion with an anti-Ly6G (lymphocyte antigen 6G) antibody before lung injury. In neutrophil-depleted mice (Fig. S2_A_), MaR1 levels in lungs obtained 2 h after intratracheal HCl were significantly decreased, with concomitant increases in MaR1 nonenzymatic hydrolysis products (Fig. 2_C_). We next used a P-selectin antibody to prevent in vivo N-PA formation. When administered immediately before intratracheal HCl, the P-selectin antibody decreased lung MaR1 (Fig. 2_D_) and N-PA in blood (Fig. S2_B_) 2 h after injury. Together, these results were consistent with a pivotal role for neutrophil transformation of platelet-derived 14S-HpDHA and 13_S_,14_S_-epoxy-maresin–producing MaR1 via neutrophil–platelet interactions that was diminished by neutrophil depletion and N-PA blockade, leading to nonenzymatic hydrolysis of 13_S_,14_S_-epoxy-maresin intermediate to 7-epi-Δ12-trans-MaR1 and Δ12-trans-MaR1 rather than enzymatic conversion to MaR1 (Fig. 2_A_).
Because lung MaR1 remained elevated 24 h after intratracheal HCl (Fig. 2_B_), we next assessed the contributions of neutrophils to MaR1 biosynthesis at this later time point. In contrast to the earlier time point, neutrophil depletion had no impact on lung MaR1 levels 24 h after intratracheal HCl (Fig. 2_E_ and Fig. S2_C_), consistent with a neutrophil-independent MaR1 biosynthetic pathway.
Lung inflammation after intratracheal HCl was monitored by total leukocyte, neutrophil, and macrophage counts in bronchoalveolar lavage fluid (BALF) (SI Methods). Lung neutrophils peaked 24 h after HCl (Fig. S3_A_), so the impact of early vascular MaR1 on the progression of lung injury and inflammation was next determined via administration of MaR1 (10 ng i.v., ∼0.5 µg/kg) 1 h after HCl. This dose of MaR1 gave tissue levels of 8.8 ± 1.4 pg per lung (mean ± SEM, n = 3), which was ∼threefold higher than endogenous levels (Fig. S3_B_). When assessed after 24 h, injury to the left lung was notable for tissue leukocyte infiltration and alveolar and interstitial edema that were in sharp contrast to the uninjured right lung (Fig. 3_A_). MaR1 markedly reduced lung injury with decreased tissue edema and cellular infiltration (Fig. 3_A_) and diminished leakage permeability changes (Fig. 3_B_). MaR1 improved lung tissue hypoxia (Fig. 3_C_) and lung mechanics (Fig. S3_C_). MaR1 also selectively regulated BALF and plasma levels of proinflammatory mediators linked to ARDS pathogenesis (Fig. 3_D_ and Fig. S3_D_), including levels of mediators important for neutrophil activation (TNF-α, GM-CSF) (4) and endothelial barrier function (cysteinyl leukotrienes, CysLTs) (15). These findings indicated that following self-limited tissue injury, MaR1 activated the endogenous host resolution program to restore lung tissue homeostasis.
Fig. 3.

MaR1 reduces lung inflammation and restores pulmonary capillary endothelial permeability. (A) Hematoxylin and eosin stain of right and left lungs from mice treated with MaR1 or vehicle. Results are representative from n = 3. (Scale bar, 50 µm.) (B) Endothelial barrier integrity determined by measurement of Evans blue dye in perfused whole lung in mice exposed to MaR1 or vehicle. (C) Tissue hypoxia determined by pimonidazole (Hypoxyprobe) immunostaining in injured left lung sections obtained from mice treated with MaR1 or vehicle. Pimonidazole staining appears brown. (D) Prophlogistic mediator levels in BALF and plasma (CysLTs) were analyzed by cytokine bead array or ELISA (CysLTs). In all experiments, tissues and samples were harvested 24 h after intratracheal HCl, and MaR1 was administered i.v 1 h after HCl. All values represent the mean ± SEM for n ≥ 5 mice from two independent experiments. *P < 0.05.
MaR1 administration gave dose-dependent reduction in BALF neutrophil counts (Fig. 4_A_) without significant changes in total leukocyte and macrophage numbers (Fig. S4_A_). To determine mechanisms for MaR1 regulation of neutrophil trafficking to the lungs, single cell suspensions from left (injured) and right (uninjured) lungs were examined by flow cytometry using a double-labeling immunofluorescence protocol to discriminate between interstitial and intravascular neutrophils (SI Methods) (16). Intratracheal HCl led to increased interstitial neutrophils in the injured left lung relative to the right lung (Fig. 4_B_). MaR1 significantly decreased lung interstitial neutrophils (Fig. 4_B_) without further decrease in peripheral blood neutrophil counts (Fig. S4_B_), suggesting regulation at the point of neutrophil entry into the lung interstitium.
Fig. 4.
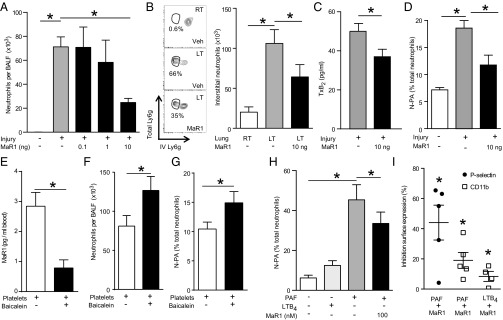
Intravascular MaR1 reduces neutrophil recruitment by decreasing N-PAs. (A) BALF cell counts after administration of MaR1 or vehicle. (B) Lung interstitial neutrophils (circled in flow cytometry dot plots) determined by differential immunostaining of intravascular and whole lung neutrophils. (C) Plasma TxB2 levels 2 h after intratracheal HCl from mice given MaR1 or vehicle determined by ELISA. (D) N-PAs determined by flow cytometry of murine whole blood 24 h after intratracheal HCl administration and treatment with MaR1 or vehicle. (E) Whole blood MaR1 levels, (F) airway neutrophil counts, and (G) whole blood N-PA 24 h after intratracheal HCl in mice transfused with platelets (500 × 106 platelets, intravenously) pretreated with baicalein or vehicle. Values represent the mean ± SEM for n ≥ 5 mice from two independent experiments. *P < 0.05. (H) Human N-PA in whole blood after incubation with PAF or LTB4 and pretreatment with MaR1 or vehicle determined by flow cytometry. (I) Platelet and neutrophil activation after incubation with PAF or LTB4 and coincubation with MaR1 or vehicle quantified by flow cytometry analysis of P-selectin and CD11b surface expression, respectively. Values represent the mean ± SEM for n ≥ 5 blood donors. *P < 0.05 by paired t test.
The regulation of lung neutrophil capture by MaR1 was linked to decreased platelet activation. MaR1 administration 1 h after HCl decreased plasma thromboxane (TxB2) (Fig. 4_C_) and circulating N-PA (Fig. 4_D_ and Fig. S4_C_). To assess roles for platelets in intravascular MaR1 production and lung neutrophil accumulation, freshly isolated platelets were exposed to baicalein, a lipoxygenase inhibitor, or vehicle before transfusion (500 × 106 platelets intravenously) immediately after intratracheal HCl. Twenty-four hours later, intravascular (whole blood) MaR1 levels were decreased in mice receiving baicalein-treated platelet transfusions (Fig. 4_E_). In addition, lung MaR1 levels significantly decreased (49.1 ± 14.8%, n = 4), whereas BALF neutrophil counts (Fig. 4_F_) and circulating N-PA levels (Fig. 4_G_) increased in these mice after injury. These results indicate that inhibition of intravascular MaR1 via blockade of platelet 12-lipoxygenase increased inflammation following acute lung injury (ALI).
To investigate the translational potential of MaR1, fresh human whole blood was collected from healthy volunteers and incubated with MaR1 or vehicle before the addition of inflammatory mediators. Activation of both neutrophils and platelets with PAF markedly increased N-PA, in contrast to leukotriene B4 (LTB4), which activates only neutrophils and did not increase N-PA (Fig. 4_H_). Despite potent N-PA formation by PAF (500 nM), MaR1 decreased N-PA (Fig. 4_E_). PAF increased platelet P-selectin surface expression, and both PAF and LTB4 increased neutrophil CD11b expression—proadhesive actions that were decreased by MaR1 (Fig. 4_I_). Taken together, these findings indicate that MaR1 reduced both murine and human neutrophil and platelet activation to decrease N-PA formation.
Collectively, these experiments uncovered a temporal regulation of MaR1 biosynthesis involving intravascular neutrophil and platelet interactions that predominated in the early phases of acute inflammation (Fig. 5_A_), whereas macrophages contribute the majority of MaR1 biosynthesis at later time points (Fig. 5_B_).
Fig. 5.

Proposed (platelet–neutrophil) and established (macrophage) biosynthetic routes for MaR1. (A) During neutrophil–platelet interactions, platelet 12-lipoxyenase converts DHA to 14_S_-hydroperoxy-DHA and 13_S_,14_S_-epoxy-maresin, which is then converted by an epoxide hydrolase in neutrophils to MaR1. (B) Macrophages express both 12-lipoxygenase and the hydrolase required to conduct MaR1 biosynthesis.
Discussion
Results presented herein establish MaR1 biosynthesis by platelets and its impact in ALI. During cell–cell interactions with platelets, neutrophils amplified MaR1 generation via conversion of the platelet 12-lipoxygenase–derived intermediates 14S-HpDHA and 13S,14S-epoxy-maresin to MaR1. The new epoxide hydrolase responsible for MaR1 biosynthesis present in human leukocytes may also be present in human platelets, as these cells produce low amounts of MaR1 in the absence of leukocytes. The amounts of MaR1 identified (∼15 pg in the left lung with an estimated residual volume of 80 µL) (17) approximated a concentration of 0.5 nM, which is within the bioactive range of this immunoresolvent in regulating leukocyte and epithelial cells (11–13, 18). Of note, MaR1 is an autacoid that is rapidly formed and rapidly metabolized; therefore, static tissue quantitation at a given time point may underrepresent biosynthesis, particularly in the lung, where enzymes involved in further metabolism of proresolving mediators are highly expressed (19). Transcellular biosynthesis of lipoxins, a family of proresolving mediators derived from arachidonic acid, occurs during neutrophil–platelet interactions using a distinct biosynthetic pathway that requires different enzymes (neutrophil 5-lipoxygenase) and a sequence of events to produce LTA4 that is converted to lipoxins via the human 12-lipoxygenase (20).
Heterotypic neutrophil–platelet interactions play important roles in early inflammatory responses (21). Platelets bound to endothelial cells can interact with neutrophils as they slowly transit through the pulmonary capillaries and facilitate neutrophil–endothelial interactions at sites of inflammation through TxA2-dependent endothelial activation, leading to secondary capture of neutrophils for their recruitment to the lung (8). Endovascular N-PA can also directly injure the pulmonary capillary endothelium and contribute to lung injury (6). Accordingly, regulation of these cellular events represents a key target for controlling tissue inflammation and minimizing injury.
In response to intratracheal HCl, endogenous MaR1 biosynthesis was regulated in a temporal manner with intravascular sources (e.g., N-PA), providing the majority of MaR1 early after airway injury. Although platelet activation and N-PA are linked to ARDS pathogenesis (6), MaR1 biosynthesis by these cells suggests counterregulatory actions for platelets as well that were uncovered in this self-limited model of ALI. MaR1 increased early within the response to acid-induced injury compared with other LMs (Table S1), placing this proresolving mediator in a unique position. In contrast to antiinflammatory agents, this mediator does not appear to be immunosuppressive (12) and regulates hallmarks of resolution that are characteristics of SPMs (22), such as efferocytosis of apoptotic neutrophils (13) and shifting macrophage phenotype to proresolving (13), as well as tissue regeneration (12). Treatment of mice with this proresolving mediator at amounts produced in situ in this model gave statistically significant protection from acid-mediated lung injury exemplified by decreasing plasma TxB2, P-selectin expression, as well as neutrophil activation (i.e., CD11b surface expression) and influx into lung interstitium, whereas disruption of this pathway led to increased inflammatory responses. Taken together, these results provide evidence for an endogenous host-protective response to ALI centered on N-PA and MaR1. Of note, 15-epi-LXA4, AT-RvD1, and RvE1, each produced from separate biosynthetic precursors (arachidonic acid, DHA, and eicosapentaenoic acid, respectively), are each potent in the protective actions engaged by ALI (16, 23–25). Additional experiments are required to determine their relative therapeutic potential for humans.
In addition to MaR1, the 12-lipoxygenase–derived 13S,14S-epoxy-maresin was produced by platelets, and this epoxide is bioactive, blocking 12-lipoxygenase conversion of arachidonic acid to 12-hydro-peroxy-eicosatetraenoic acid (12-HpETE) (13) and inhibiting the formation of proinflammatory mediators from 12-HpETE. In addition, 13S,14S-epoxy-maresin inhibits LTA4 hydrolase, leading to decreased production of the neutrophil chemoattractant and secretagogue LTB4 (13). Because LTB4 is a neutrophil product that is linked to neutrophil activation in ARDS (26), the MaR1 pathway can use this additional mechanism to regulate neutrophil recruitment and activation to protect the lung from bystander tissue injury from these “first responders.” Along these lines, MaR1 reduced levels of several cytokines with known roles in recruitment and activation of neutrophils in ALI, such as GM-CSF and TNFα, which are reported to play important roles in acid-induced lung injury (27, 28). Indeed, MaR1’s antineutrophil actions in the lung, including P-selectin expression, were recently reported in an LPS model of lung inflammation (29). In addition to its actions on platelets and neutrophils, MaR1 also regulates macrophages and airway epithelial cells. These cell types are the lung’s principal sources of the BALF cytokines that were decreased here by MaR1. MaR1 is a potent regulator of macrophage function (11, 12) and reduces proinflammatory cytokine release from bronchial epithelial cells exposed to organic dust extract (18). In response to lung injury, MaR1 also decreased CysLT levels and endothelial barrier permeability, suggesting an additional role for MaR1 in promoting the restoration of endothelial barrier function (15). In each of these, cell-type-specific actions and MaR1 structure–activity relationship (11–13) suggest highly stereoselective actions.
In this self-limited model of ARDS, MaR1 biosynthesis was temporally regulated. Early after initiation of injury, N-PAs were the principal source of MaR1 (Fig. 5_A_), and in sharp contrast nonneutrophil lung cells carried out MaR1 biosynthesis at a later time point. The late generation of MaR1 was likely derived from macrophages, as this class of cells was the source of MaR1 in its original identification (Fig. 5_B_) (11). It is also possible that macrophages played a minor role in early MaR1 production, as low levels of MaR1 were identified 2 h after injury in neutrophil-depleted mice. Multipronged generation of MaR1 emphasizes its pivotal antiinflammatory and proresolving roles in lung catabasis and highlights the temporally coordinated activation of distinct counterregulatory circuits. As illustrated in Fig. 5, the macrophage-mediated MaR1 biosynthetic pathway and the platelet–neutrophil pathway for MaR1 have similar components, enhancing the versatility of MaR1 formation and actions.
In summary, our findings have shed light on endogenous counterregulation during self-limited tissue injury and uncovered temporal biosynthesis and regulation of the potent SPM MaR1. The kinetics of MaR1 formation paralleled the initial course of ALI, also raising its potential as a marker of acute inflammatory responses characterized by intraluminal platelet activation. MaR1’s antiinflammatory, proresolving, and tissue regenerative actions in response to tissue injury were organ-protective (11–13), suggesting potential new therapeutic approaches that emphasize proresolving mediators, SPM mechanisms, and the MaR1 pathway elucidated herein.
Methods
Maresin 1 was prepared by total organic synthesis as reported in ref. 13, and just prior to use was validated by LC-MS/MS and physical criteria reported earlier (11, 13). SI Methods details LM metabololipidomics, human blood cell isolation and incubations, P-selectin inhibition, neutrophil depletion in mice, platelet transfusion, flow cytometry and i.v. neutrophil staining, histology, measurement of lung mechanics, assessment of pulmonary capillary permeability, and determination of mediator levels.
Supplementary Material
Supplementary File
Acknowledgments
We thank Guangli Zhu and Bonna Ith for technical assistance. This research was supported in part by National Institutes of Health Grants T32-HL07633 (to R.E.A. and B.D.L.), R01-HL068669 (to B.D.L.), and P01-GM095467 (to B.D.L., N.A.P., and C.N.S.).
Footnotes
Conflict of interest statement: C.N.S. is an inventor on patents (resolvins) assigned to Brigham and Women's Hospital (BWH) and licensed to Resolvyx Pharmaceuticals. C.N.S. was the scientific founder of Resolvyx Pharmaceuticals and owns founder stock in the company. C.N.S.’s interests were reviewed and are managed by the Brigham and Women's Hospital and Partners HealthCare in accordance with their conflict of interest policies. B.D.L. is an inventor on patents (resolvins) assigned to BWH and licensed to Resolvyx Pharmaceuticals. B.D.L.’s interests were reviewed and are managed by the Brigham and Women’s Hospital and Partners HealthCare in accordance with their conflict of interest policies.
This article is a PNAS Direct Submission. D.W.G. is a guest editor invited by the Editorial Board.
References
- 1.Morris T, et al. Dichotomy in duration and severity of acute inflammatory responses in humans arising from differentially expressed proresolution pathways. Proc Natl Acad Sci USA. 2010;107(19):8842–8847. doi: 10.1073/pnas.1000373107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Levy BD, Serhan CN. Resolution of acute inflammation in the lung. Annu Rev Physiol. 2014;76:467–492. doi: 10.1146/annurev-physiol-021113-170408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lehr HA, Frei B, Arfors KE. Vitamin C prevents cigarette smoke-induced leukocyte aggregation and adhesion to endothelium in vivo. Proc Natl Acad Sci USA. 1994;91(16):7688–7692. doi: 10.1073/pnas.91.16.7688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ley K, Laudanna C, Cybulsky MI, Nourshargh S. Getting to the site of inflammation: The leukocyte adhesion cascade updated. Nat Rev Immunol. 2007;7(9):678–689. doi: 10.1038/nri2156. [DOI] [PubMed] [Google Scholar]
- 5.Tabas I, Glass CK. Anti-inflammatory therapy in chronic disease: Challenges and opportunities. Science. 2013;339(6116):166–172. doi: 10.1126/science.1230720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Matthay MA, Ware LB, Zimmerman GA. The acute respiratory distress syndrome. J Clin Invest. 2012;122(8):2731–2740. doi: 10.1172/JCI60331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Looney MR, et al. Platelet depletion and aspirin treatment protect mice in a two-event model of transfusion-related acute lung injury. J Clin Invest. 2009;119(11):3450–3461. doi: 10.1172/JCI38432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zarbock A, Singbartl K, Ley K. Complete reversal of acid-induced acute lung injury by blocking of platelet-neutrophil aggregation. J Clin Invest. 2006;116(12):3211–3219. doi: 10.1172/JCI29499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fukunaga K, Kohli P, Bonnans C, Fredenburgh LE, Levy BD. Cyclooxygenase 2 plays a pivotal role in the resolution of acute lung injury. J Immunol. 2005;174(8):5033–5039. doi: 10.4049/jimmunol.174.8.5033. [DOI] [PubMed] [Google Scholar]
- 10.Levy BD, Clish CB, Schmidt B, Gronert K, Serhan CN. Lipid mediator class switching during acute inflammation: Signals in resolution. Nat Immunol. 2001;2(7):612–619. doi: 10.1038/89759. [DOI] [PubMed] [Google Scholar]
- 11.Serhan CN, et al. Maresins: Novel macrophage mediators with potent antiinflammatory and proresolving actions. J Exp Med. 2009;206(1):15–23. doi: 10.1084/jem.20081880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Serhan CN, et al. Macrophage proresolving mediator maresin 1 stimulates tissue regeneration and controls pain. FASEB J. 2012;26(4):1755–1765. doi: 10.1096/fj.11-201442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dalli J, et al. The novel 13S,14S-epoxy-maresin is converted by human macrophages to maresin 1 (MaR1), inhibits leukotriene A4 hydrolase (LTA4H), and shifts macrophage phenotype. FASEB J. 2013;27(7):2573–2583. doi: 10.1096/fj.13-227728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Marik PE. Aspiration pneumonitis and aspiration pneumonia. N Engl J Med. 2001;344(9):665–671. doi: 10.1056/NEJM200103013440908. [DOI] [PubMed] [Google Scholar]
- 15.Hilberath JN, et al. Resolution of Toll-like receptor 4-mediated acute lung injury is linked to eicosanoids and suppressor of cytokine signaling 3. FASEB J. 2011;25(6):1827–1835. doi: 10.1096/fj.10-169896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Eickmeier O, et al. Aspirin-triggered resolvin D1 reduces mucosal inflammation and promotes resolution in a murine model of acute lung injury. Mucosal Immunol. 2013;6(2):256–266. doi: 10.1038/mi.2012.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mitzner W, Brown R, Lee W. In vivo measurement of lung volumes in mice. Physiol Genomics. 2001;4(3):215–221. doi: 10.1152/physiolgenomics.2001.4.3.215. [DOI] [PubMed] [Google Scholar]
- 18.Nordgren TM, et al. Maresin-1 reduces the pro-inflammatory response of bronchial epithelial cells to organic dust. Respir Res. 2013;14(1):51. doi: 10.1186/1465-9921-14-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Clish CB, Levy BD, Chiang N, Tai HH, Serhan CN. Oxidoreductases in lipoxin A4 metabolic inactivation: A novel role for 15-onoprostaglandin 13-reductase/leukotriene B4 12-hydroxydehydrogenase in inflammation. J Biol Chem. 2000;275(33):25372–25380. doi: 10.1074/jbc.M002863200. [DOI] [PubMed] [Google Scholar]
- 20.Serhan CN, Sheppard KA. Lipoxin formation during human neutrophil-platelet interactions. Evidence for the transformation of leukotriene A4 by platelet 12-lipoxygenase in vitro. J Clin Invest. 1990;85(3):772–780. doi: 10.1172/JCI114503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Weyrich AS, Zimmerman GA. Platelets in lung biology. Annu Rev Physiol. 2013;75:569–591. doi: 10.1146/annurev-physiol-030212-183752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Serhan CN. Pro-resolving lipid mediators are leads for resolution physiology. Nature. 2014;510(7503):92–101. doi: 10.1038/nature13479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.El Kebir D, et al. 15-epi-lipoxin A4 inhibits myeloperoxidase signaling and enhances resolution of acute lung injury. Am J Respir Crit Care Med. 2009;180(4):311–319. doi: 10.1164/rccm.200810-1601OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Seki H, et al. The anti-inflammatory and proresolving mediator resolvin E1 protects mice from bacterial pneumonia and acute lung injury. J Immunol. 2010;184(2):836–843. doi: 10.4049/jimmunol.0901809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ortiz-Muñoz G, et al. Aspirin-triggered 15-epi-lipoxin A4 regulates neutrophil-platelet aggregation and attenuates acute lung injury in mice. Blood. 2014 doi: 10.1182/blood-2014-03-562876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mantovani A, Cassatella MA, Costantini C, Jaillon S. Neutrophils in the activation and regulation of innate and adaptive immunity. Nat Rev Immunol. 2011;11(8):519–531. doi: 10.1038/nri3024. [DOI] [PubMed] [Google Scholar]
- 27.Wiedermann FJ, et al. Alveolar granulocyte colony-stimulating factor and alpha-chemokines in relation to serum levels, pulmonary neutrophilia, and severity of lung injury in ARDS. Chest. 2004;125(1):212–219. doi: 10.1378/chest.125.1.212. [DOI] [PubMed] [Google Scholar]
- 28.Patel BV, Wilson MR, O’Dea KP, Takata M. TNF-induced death signaling triggers alveolar epithelial dysfunction in acute lung injury. J Immunol. 2013;190(8):4274–4282. doi: 10.4049/jimmunol.1202437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gong J, et al. Maresin 1 mitigates lipopolysaccharide-induced acute lung injury in mice. Br J Pharmacol. 2014;171(14):3539–3550. doi: 10.1111/bph.12714. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary File