The many faces of α-synuclein: from structure and toxicity to therapeutic target (original) (raw)
. Author manuscript; available in PMC: 2015 Jan 15.
Published in final edited form as: Nat Rev Neurosci. 2013 Jan;14(1):38–48. doi: 10.1038/nrn3406
Abstract
Disorders characterized by α-synuclein (α-syn) accumulation, Lewy body formation and parkinsonism (and in some cases dementia) are collectively known as Lewy body diseases. The molecular mechanism(s) through which α-syn abnormally accumulates and contributes to neurodegeneration in these disorders remains unknown. Here, we provide an overview of current knowledge and prevailing hypotheses regarding the conformational, oligomerization and aggregation states of α-syn and their role in regulating α-syn function in health and disease. Understanding the nature of the various α-syn structures, how they are formed, and their relative contributions to α-syn-mediated toxicity may inform future studies aiming to develop therapeutic prevention and intervention.
Introduction
Lewy body diseases (LBDs) are a heterogeneous group of disorders that include Parkinson disease (PD), PD with dementia (PDD) and dementia with Lewy bodies (DLB).1 Together, these diseases affect over 5 million worldwide. LBDs are mainly characterized pathologically by the progressive accumulation of the presynaptic protein α-synuclein (α-syn), and hence they are often referred to as synucleinopathies, and by the degeneration of neocortical, limbic and nigral-striato circuitries2, 3. A pathogenic role for α-syn in these disorders is supported by various genetic data. Multiplications of 4 and various mutations (A53T, A30P, and E46K)5-7 in the gene encoding α-syn (SNCA) result in dominant familial parkinsonism. Moreover, certain polymorphisms in SNCA are major risk factors for sporadic PD8.
An increasing body of evidence from animal models as well as data from genetic, biochemical and biophysical studies support the hypothesis that the processes of α-syn oligomerization9, 10 and fibril growth11, 12 have central roles in the pathogenesis of PD and other synucleinopathies13. In addition, it is possible that the α-syn monomers might also have a role in synucleonopathies by their displacement from their physiological location, resulting in a loss of cellular function or by disrupting the activity of other molecular or signaling pathways14. Nevertheless, the molecular mechanisms by which α-syn aggregation contributes to neurodegeneration, the nature of the toxic forms of α-syn and the cellular pathways that are affected by α-syn remain unknown. Addressing these knowledge gaps is crucial for understanding the molecular basis of synucleinopathies, developing tools to diagnose and monitor disease progression, and assessing the effectiveness of preventive and therapeutic strategies. Here, we provide an overview of the current state of knowledge regarding the role of α-syn aggregation states in regulating α-syn function under physiological and pathophysiological conditions. We also discuss the implications of these findings for therapeutic interventions.
Physiological function of α-synuclein
Although the normal function(s) of α-syn remains unknown, its localization at pre-synaptic terminals (FIG 1A, B)15-17, its association with the distal reserve pool of synaptic vesicles18-20 and the deficiencies in synaptic transmissions observed in response to knock down or overexpression of α-syn suggest that α-syn has a role in the regulation of neurotransmitter release, synaptic function and plasticity (FIG. 1C).
Figure 1. Functional properties of α-synuclein.

a, b | Wide field and magnified images of cultured cortical neurons from a postnatal day 1 wild-type mouse showing a neuronal dendrite (as revealed by MAP2 immunostaining; red) opposed to α-synuclein (α-syn)-positive presynaptic densities (green), indicating that α-syn is located in the presynaptic terminals. c | The schematic depicts the various roles of α-syn at the pre-synaptic terminal in the regulation of vesicle trafficking and vesicle refilling (α-syn; blue), as well as the interaction with membrane-associated t-SNARE or the vesicle-associated v-SNARE proteins and neurotransmitter release. Accumulation of α-syn induces an impairment of neurotransmitter release, vesicle recycling and trafficking between synaptic buttons and influence t-SNARE complex assembly stability (α-syn; red), whereas its depletion induces an impairment of vesicle trafficking between the reserve pool and the ready releasable pool and a deficiency in vesicle refilling and neurotransmitter uptake.
Snca knock-out mice exhibit an impairment in hippocampal synaptic responses to prolonged trains of high-frequency stimulation that deplete the docked and reserve pool of synaptic vesicles, as well as impairments in replenishment of docked pools from the reserve pool, indicating that α-syn may control the refilling and the trafficking of synaptic vesicles from the reserve pool to the site of synaptic vesicle release21, 22. Moreover, depletion of α-syn, through use of antisense oligonucleotides, induces a decrease in the availability of reserve synaptic vesicle pool in primary cultured hippocampal neurons23.
Transgenic mice overexpressing human α-syn exhibit impairment in synaptic vesicle exocytosis and a reduction in neurotransmitter release24-26. Similar effects have been observed after α-syn overexpression in genetic rodent models of PD27, 28 and in the PC12 stable cell-line29. At the ultrastructural level, overexpression of α-syn induces a decrease in readily releasable vesicles27 and affects the recycling of synaptic vesicles following endocytosis, inducing a reduction in the size of the synaptic vesicle recycling pool26. Moreover, excess α-syn induces a reduction in dopamine reuptake in the dopaminergic terminals28 and inhibits intersynaptic trafficking of vesicles, leading to a smaller reserve pool of vesicles30.
The possible role of α-syn in regulating synaptic homeostasis is not exclusively related to its direct interaction with synaptic vesicles. α-Syn interacts with synaptic proteins controlling vesicle exocytosis, such as phospholipase D231 and the family of Rab small GTPases32. Recent studies reported that α-syn can act as a chaperone, and controls the degradation and affects the assembly, maintenance and distribution of the pre-synaptic SNARE protein complex (FIG. 1C), which is directly implicated in the release of neurotransmitters, including dopamine33. Together, these observations indicate that α-syn has an important role in the trafficking of synaptic vesicles and in the regulation of vesicle exocytosis, and may contribute to more subtle regulatory phenomena by controlling synaptic homeostasis-associated proteins.
The fact that the individual synuclein knock-outs (α-, β-, and γ-syn) are viable suggeststhat synucleins are not essential components of the neurotransmitter release machinery but may contribute to the long-term regulation and maintenance of the nerve terminal function34. The neuroprotective effects of α-syn against progressive neurodegeneration in cysteine-string protein-α-deficient mice35 strongly suggest that its functional properties become more prominent or essential under conditions of stress. The neuroprotective function of α-syn appears to be mediated by its ability to bind to membranes and vesicles, since the A30P α-syn mutant, which is deficient in membrane binding failed to show protection in CSP α-knockout mice.
Structure of α-synuclein
Since a protein's sequence and structure are linked to its function, a concerted effort has been made to characterize the sequence and structural determinants that govern the cellular properties of α-syn and its aberrant behaviour in PD and other synucleinopathies. α-Synuclein is a 14 kDa protein (140 amino acids; pKa of 4.7)36 that is characterized by an amphipathic lysine-rich amino terminus, which plays a critical role in modulating its interactions with membranes, and a disordered, acidic carboxy-terminal tail that has been implicated in regulating its nuclear localization and interactions with metals, small molecules and proteins (FIG. 2A)37, 38. The central region of α-syn was first purified from amyloid plaques in patients with Alzheimer's disease (AD)36, 39, and contains a highly hydrophobic motif that comprises amino acid residues 65–90, known as the non-amyloid-β component of AD amyloid plaques (NAC; FIG 2A)36, 40, 41. The NAC region is indispensable for α-syn aggregation; the deletion of large segments within this motif greatly diminishes α-syn oligomerization and fibrilogenesis in vitro42, 43 and in a cell-based assay44.
Figure 2. Biochemical structure of α-synuclein and its pathological distribution in Parkinson's disease and a mouse model of Lewy body disease.
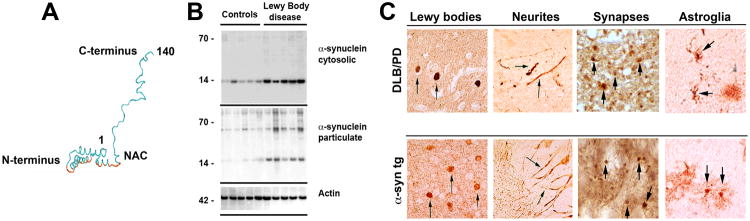
a | Computer-generated model of α-synuclein (α-syn) representing the N-terminal α helices, non-amyloid-β component of Alzheimer's disease amyloid plaques (NAC; depicted in red), and unstructured C-terminal regions. b | Western blot identifying α-syn in brain homogenates from control and Lewy body disease cases that were divided into cytosolic and particulate fractions. α-Syn migrated to 57–60 kDa as well as to 14 kDa in the particulate but not cytosolic fraction due to the different conformational states of the protein. c, d | α-Syn is present in Lewy bodies, neurites, synapses and astroglia in dementia with Lewy bodies (DLB) and Parkinson's disease (PD) and in PDGFβ-human _α_-syn wild type transgenic mice, as indicated by arrows.
Studies by several groups using different biophysical methods (for example, nuclear magnetic resonance (NMR), light scattering and circular dichroism (CD)) consistently showed that α-syn purified from Escherichia coli under native or denaturing conditions exists predominantly as stable unfolded monomers38, 45. When α-syn was extracted from patients diagnosed with LBD and age-matched controls and evaluated by non-denaturing gels or size exclusion chromatography (SEC) columns, α-syn monomers migrated as 57–60 kDa proteins, but under denaturing conditions they ran at 14 kDa (FIG. 2B)42, 44. Initially, the apparent size of α-syn in native gels and SEC led many researchers to propose that this protein exists as a stable oligomer, but subsequent detailed biophysical studies demonstrated that the larger than expected size is mainly due to the fact that monomeric α-syn adopts an unfolded, extended conformation45, 46, which results in a larger than expected hydrodynamic radius (BOX 1). Together, these studies support the notion that the native and physiological form of α-syn is the monomer, but do not rule the possibility that the protein could form functional oligomers upon interaction with other proteins or biological membranes.
Box 1. Native state of α-synuclein.
The wide distribution and abundant expression of α-synuclein (α-syn) throughout the brain have been difficult to reconcile with its unfolded structure, which is expected to make the protein more susceptible to proteolysis and degradation. Thus, it was proposed that the oligomerization of α-syn or its association with other proteins might represent mechanisms by which the protein may acquire different structures and functions in vivo. Two recent studies reported that native α-syn exists as a folded tetramer126, 127. The first study showed that α-syn purified from mammalian cell lines or red blood cells exists as a stable α-helical tetramer with an apparent size of 58–60 kDa, as was discerned by size exclusion chromatography (SEC), native PAGE (clear and blue-native PAGE) and sedimentation equilibrium studies126. The second study used NMR, chemical crosslinking and SEC, and indicated that α-syn produced in Escherichia coli exists as a ‘dynamic tetramer’ that is rich in α-helical structure127. Both studies suggested that the tetrameric helical form of α-syn is resistant to aggregation and fibril formation.
The above findings have not been verified independently. Indeed, studies from our own laboratories and three other independent groups showed that α-syn produced in mammalian cell lines or red blood cells or isolated from the brain tissue of mice, rats, humans or α-syn transgenic animals exhibited identical mobility in native and denaturing gels to the unfolded monomeric α-syn produced in E. coli46. NMR data of α-syn in intact cells also ruled out the presence of stable or highly populated α-syn oligomers and confirmed the intrinsically disordered nature of the protein in E. coli regardless of its purification method128. N-terminal acetylation, which was observed in native α-syn isolated from cell lines and brain tissues, did not result in significant changes in the protein's structure, oligomeric state, membrane-binding properties or subcellular localization. These findings are in agreement with most studies on the oligomeric state of native α-syn, which have consistently shown that α-syn behaves as an unfolded monomer9, 45, 46. The migration of α-syn with an apparent molecular weight slightly above 66 kDa in native gels and SEC is probably the result of its tendency to adopt extended conformations, and not because it exists in an oligomeric form (for example, as a tetramer), as the addition of denaturants or boiling of α-syn samples from various sources did not change α-syn migration128. Although Bartels et al.126 reported that chemical crosslinking of native α-syn primarily stabilized α-syn tetramers, studies by several groups, including the work by Wang et al.127, showed that chemical or photochemical crosslinking of α-syn in mammalian cells or purified α-syn from E. coli results in the formation of a ladder of α-syn species ranging from monomers to hexamers, with dimers being the major species129, consistent with the pattern expected for nonspecific crosslinking. Interestingly, SDS-resistant dimers seem to be the most commonly detected form of α-syn oligomers in denaturing gels under conditions of oxidative stress49, 130 or in the presence of specific metals and small molecules, such as polyphenols122. These dimers are stable in the presence of dissociative conditions such as reducing agents, chemical denaturants and boiling, suggesting that they are covalently crosslinked89, 122.
The development of novel tools or methodologies that allow direct detection and monitoring of changes in α-syn structure and oligomeric states in living cells and/or in vivo is required for assessing changes in the distribution of α-syn conformations and oligomeric states in vivo and the role of these changes in regulating α-syn normal function and toxicity. This knowledge is essential for the successful design of novel strategies for the diagnosis and treatment of Parkinson's disease and related synucleinopathies.
The apparently multifunctional properties of α-syn may lie in its conformational flexibility, which may allow the protein to adopt different conformations upon interacting with biological membranes of different compositions, other proteins or protein complexes47, 48. It is well established that α-syn adopts an α-helical conformation upon binding to synthetic or biological membranes in vitro38. However, very little is known about the conformational state(s) of α-syn in the different compartments of the living cell, and there is a lack of direct evidence for a functional role of native α-syn oligomers in biological membranes. It is conceivable that native α-syn exists in equilibrium between different conformational and/or oligomeric states. Several factors, including oxidative stress49, post-translational modifications50, 51, proteolysis52, 53, as well as the concentration of fatty acids54-56, phospholipids and metal ions11, 49 were shown to induce and/or modulate α-syn structure and oligomerization in vitro, which may influence this equilibrium between the monomer and oligomer state in vivo. Together, these studies suggest that α-syn exists predominantly as a monomer, but do not rule out the possibility that it can form a stable multimer and/or adopt different structures under specific stress-induced conditions or upon interaction with other proteins, specific ligands, lipids and/or biological membranes. Understanding the molecular determinants that regulate the conformational and oligomeric states of native α-syn and the structural basis of its toxicity is crucial for our ability to generate and test different mechanistic models and hypotheses on α-syn function in health and disease and has significant implications for the development of therapeutic strategies based on targeting α-syn, as discussed in the following section.
α-Synuclein and disease
Paths to increased expression and accumulation of α-synuclein
PD, DLB and other LBDs show accumulation and redistribution of α-syn in various brains regions and cellular populations. These changes in the nature and localization of α-syn may have pathogenic roles in these disorders (FIG 2C) and can reproduced in α-syn transgenic animal models (FIG 2D).1, 3, 57-59
The levels of α-syn in the CNS depend on the balance between the rates of α-syn synthesis, aggregation and clearance (FIG. 3A,B)60. An imbalance between these mechanisms, caused by dysfunction of one or more of these pathways, can result in abnormal levels of α-syn that might favour the formation and/or accumulation of oligomeric and fibrillar species, which may be toxic. Indeed, in some familial forms of parkinsonism, multiplication of SNCA results in increased accumulation α-syn because of increased protein expression4, whereas in others, SNCA mutations enhance the propensity of α-syn to aggregate (FIG. 3A)9. A genome-wide association study (GWAS) showed that individuals with certain variations in the SNCA gene had a higher risk of PD.61 One such polymorphism is known as Rep1, which occurs in the promoter region of SNCA and might increase the susceptibility to PD by increasing the expression of α-syn62. Clearance of α-syn monomers and aggregates occurs via direct proteolysis (for example, by neurosin or matrix metalloprotease 9 (MMP9))63, binding to molecular chaperones (for example, heat shock proteins (HSPs))64, the proteasome65-67, and autophagy (involving the activity of the lysosome) (FIG. 3A)60, 68-70. In sporadic forms of PD and DLB, failure of the autophagy pathways to eliminate oligomers might enhance α-syn-mediated toxicity (FIG. 3A)69. Chaperone-mediated autophagy68 has been shown to be disrupted by oligomeric forms of wild-type and disease-associated mutant α-syn. In PD and DLB, the levels of key autophagy molecules such as ATG7, a ubiquitin-like modifier-activating enzyme, and mTOR, a serine–threonine-protein kinase, are dysregulated71.
Figure 3. Cellular events controlling intracellular α-synuclein levels and possible therapeutic strategies to combat α-synuclein accumulation and transmission.
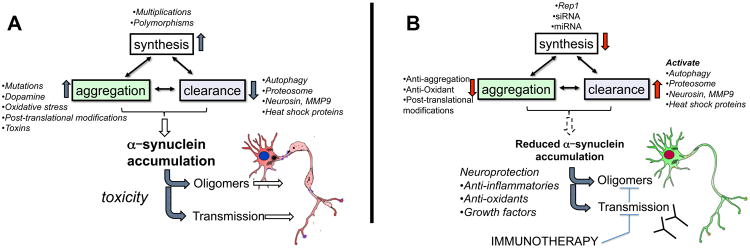
Intracellular α-synuclein (α-syn) levels are tightly regulated by the balance between the rates of α-syn synthesis, clearance and aggregation. a | Abnormalities affecting α-syn synthesis, including SNCA multiplication and polymorphisms, may increase intracellular α-syn levels and induce its accumulation. Accumulation may also be caused by a failure to degrade α-syn. Clearance deficits might arise from failure of the ubiquitin – proteasome system, chaperone-mediated autophagy dysfunction (induced by Parkinson disease-linked mutations) or dysfunction of proteases (neurosin or matrix metalloprotease 9). Finally, certain SNCA mutations, post-translational modifications, oxidative stress, toxins and interaction with oxidized dopamine increase the propensity of α-syn to aggregate and accumulate. b | Targeted mechanisms to reduce α-syn accumulation include decreasing protein synthesis by using Rep1, siRNA or miRNA. Accumulation may also be decreased by activating mechanisms or proteins involved in clearance, such as autophagy, the proteasome, neurosin, MMP9 and heat shock proteins. Additionally, aggregation of α-syn can be decreased using anti-aggregating, antioxidant, or post-translational modification approaches. Finally, immunotherapy may be used to block transmission and oligomer formation.
Evidence for different conformers of α-synuclein in disease
There are a number of different α-syn conformers that have been associated with the pathogenesis of LBDs, including oligomers, protofibrils, and fibrils. The fibrillar forms of α-syn are detected mostly in Lewy bodies72, 73 which are localized in the neuronal cell body. These intracellular structures are the neuropathological hallmark of PD and DLB (FIG. 2B) and are thought to reflect an attempt by the neurons to isolate and/or convert toxic α-syn oligomers to fibrils, which a stable, less dynamic structures, and exhibit reduced toxicity. Aggregates of α-syn can also be observed in astroglial cells from cases of LBD74. The distribution of α-syn pathology in human diseases can be recapitulated in α-syn transgenic mice (FIG. 2C)75.
Strong indirect evidence supports the existence of various oligomeric α-syn species in vivo under pathophysiological conditions. SDS-resistant dimers as well as low and high molecular weight oligomeric forms of α-syn have been detected in diseased human brains10, 76, 77 and in brains of transgenic animal models of synucleinopathies10, 18, 78, expressing wild-type or PD-associated mutant variants of α-syn79 (FIG. 2A). In contrast to fibrillar α-syn, oligomeric aggregates are most likely located in axons (FIG. 1B) and presynaptic terminals where they might damage synapses and dendrites25, 26, 28, 80-82
In vitro, several oligomeric species of different morphologies, including spherical, chain-like, and annular oligomers, have been observed prior to α-syn fibril formation (FIG. 4)83. The various oligomeric species seem to exist in equilibrium with monomeric α-syn and undergo a very slow conversion to fibrils in the absence of a high molar ratio of monomers to other species of α-syn (FIG. 4). However, the relationships between the various α-syn oligomeric species and mechanisms of the inter-conversion between these different oligomers remain poorly understood, although some studies suggest that formation of ring-like oligomers is off-pathway to amyloid formation. Additionally, both in vitro and animal model studies show that three PD-linked SNCA mutations (A30P, E46K and A53T) accelerate α-syn oligomerization, but only two of these (E46K and A53T) enhance fibrillization in vitro and in vivo9, 42, 84. Although the A30P mutation has been shown to result in enhanced α-syn fibrillization in vivo, as illustrated by an autopsy case of a patient with this mutation that had extensive Lewy body pathology61, in vitro it exhibits reduced fibrillization compared to the wild type protein and other mutants. lesser Biophysical and SEC studies suggest that α-syn SDS-resistant oligomers from post-mortem brains (human and from transgenic animals) can be in general divided into small (∼2–5 mers), medium (∼5–15 mers), and large (∼15–150 mers) oligomers85, 86. Spherical oligomers 2 to 6 nm in diameter may comprise the toxic forms of α-syn since they promote neuronal degeneration and abnormal calcium currents in cultured primary cortical neurons86.
Figure 4. Mechanisms of α-synuclein aggregation and propagation.

Unfolded monomers of α-synuclein (α-syn) interact to form two types of dimers: anti-parallel dimers, which do not propagate, and parallel dimmers, which do propagate. A dynamic equilibrium is established between unfolded monomers and both forms of dimers. Interestingly, this process can take place either in the cytoplasm or in association with the cellular membrane. Propagating α-syn dimers can grow by the addition of unfolded monomers and generate ring-like oligomers and oligomers. Ring-like α-syn oligomers interact with the cytoplasmic membrane and form trans-membrane pores, inducing abnormal intracellular calcium influx. Cytoplasmic α-syn oligomers grow by the addition of soluble monomers, forming small amyloid fibrils and then longer fibrils. The accumulation of these amyloid fibrils leads to the formation of intracellular inclusions called Lewy bodies. During α-syn fibrillogenesis and aggregation, the intermediate species (oligomers and amyloid fibrils) are highly toxic, affecting mitochondrial function, endoplasmic reticulum (ER) – Golgi trafficking, protein degradation and/or synaptic transmission. These intracellular effects are thought to induce neurodegeneration. Interestingly, α-syn oligomers and fibrils, as well as the monomers, can be transferred between cells and induce disease spreading to other brain regions. Spreading mechanisms are multiple and can occur via endocytosis, direct penetration, transynaptic transmission, or via membrane receptors.
It is important to stress that in almost all of the in vivo studies described in the literature, the detection of oligomeric forms of α-syn was solely based on indirect evidence and the use of native and/or denaturing gel electrophoresis techniques46. Thus, to what extent the oligomers formed in vitro share similar characteristics and size to those formed in vivo or isolated from human brains or brains of transgenic animal models of synucleinopathies remains unknown. Future studies would greatly benefit from tools that allow direct monitoring of protein oligomerization in living cells.
Toxicity of α-synuclein oligomers and fibrils
Given the diversity and heterogeneity of the oligomers that form under different conditions and induce α-syn aggregation and toxicity, it is likely that various α-syn oligomeric species have a role in mediating α-syn toxicity in neurons and, potentially, in glial cells.
Conversion of α-syn to a toxic oligomeric form(s) might be influenced by interactions with lipids or small molecules and post-transcriptional modifications, including phosphorylation87 of α-syn at S129 and S8788, oxidative stress,49, 89 and truncation11. Under in vitro conditions, increasing protein concentrations, long incubations at 37°C, addition of specific metal ions (Fe2+, Cu2+ and Zn2+)49, 90, nitration89 or application of various ligands, such as dopamine91, can induce α-syn self-assembly (FIG. 3A) and formation of soluble aggregates that are rich in β-sheets and are of variable sizes. These range in stability and morphology, and some go on to form amyloid fibrils (FIG. 4).
Data collected in vitro and in vivo supports the hypothesis that prefibrillar oligomers may represent the toxic species of α-syn 84, 92. In vitro studies showed that annular protofibrils alter membrane permeability resulting in an increased influx of calcium from the extracellular to intracellular space, leading to cell death 86. α-Syn oligomers might also cause toxicity by damaging mitochondria,93 triggering lysosomal leakage,94 or disrupting microtubules95. Moreover, a recent study showed that α-syn oligomers interfere with the axonal transport of synaptic proteins such as synapsin-1, resulting in dysfunctional synapses and eventual neurodegeneration25. It is likely that α-syn toxicity occurs via different mechanisms and involves different intermediates on the pathway to fibril formation. However, recent evidence suggests that the process involving the conversion of oligomers to fibrils contributes to α-syn toxicity and the progression of neurodegeneration. Mutations that promote fibrillization (such as S129A96, 97) also enhance α-syn-induced toxicity and behavioral deficits in genetic rat models of PD, whereas mutations that block α-syn oligomerization and fibrillogenesis (S87E) induce significantly less α-syn aggregates, fibre pathology and dopaminergic loss in rat midbrains88. Artificial mutant variants of α-syn (for example, E57K α-syn) that enhance the formation of α-syn oligomers but not fibrils were shown to be highly toxic to dopaminergic neurons 98 in different animal models of synucleinopathies. In addition, Taschenberger and colleagues showed that mutations that enhance α-syn oligomerization (that is, A56P and the triple mutant A30P/A56P/A76P) exhibit higher toxicity, but sustained progressive loss of dopaminergic neurons was strongly dependent on the ability of the α-syn variant to from fibrils in vivo12. These results are consistent with recent findings in the amyloid-β field99, 100, and demonstrate that the process of fibril formation, rather than the fibrils themselves, has an important role in α-syn toxicity and progressive neurodegeneration in PD and related disorders. Thus, it is possible that toxicity might be related to the process of aggregation rather than just to the final outcome (that is oligomers or fibrils).
Although the majority of the studies mentioned here suggest that α-syn oligomerization and/or fibrillogenesis has a central role in α-syn toxicity, it is important to stress that most of the evidence demonstrating comparatively reduced toxicity for monomeric α-syn is based on use recombinant proteins in extracellular toxicity assays or by showing that higher levels of α-synoligomers using SDS-PAGE gels correlated with the progression of neurodegeneration in transgenic animals and other experimental models
Of note, none of the available data excludes the following two possibilities. The first of these is that α-syn oligomerization within specific cellular compartments may alter the distribution of functional forms of monomeric α-syn or result in monomer sequestration into non-functional oligomeric forms, thus resulting in partial loss of α-syn function101. The second possibility is that the native or misfolded forms of the monomeric protein may also contribute to α-syn toxicity and PD pathogenesis via aggregation-independent mechanisms, including abarrent interactions with membranes, proteins and small molecules, retention in specific cellular compartments, and disruption of specific cellular processes. Unlike stable oligomeric forms of α-syn, which can be easily distinguished from monomers and fibrils, the existing experimental tools do not allow detection and characterization of different α-syn monomeric conformers, and thus preclude studies aimed at investigating the role the α-syn monomer in health and disease.
Propagation and transmission of α-synuclein in the pathogenesis of PD
Under physiological conditions, α-syn has been traditionally considered to be an exclusively intracellular synaptic protein that associates with vesicles15. However, recent evidence suggests that under pathological circumstances, toxic α-syn oligomers could be eliminated from neurons via unusual secretory mechanisms (FIG. 4)102-104. Failure of the intracellular clearance pathways such as autophagy might contribute to the pathological release of α-syn105. Pathways leading to the release of toxic α-syn oligomers include exocytosis in clear vesicles105, exosomal release103, 106, and penetration79, 107 from the donor cell membrane (FIG 4). Interestingly, these extracellular α-syn aggregates can then transfer from neuron to neuron or from neuron to glial cell105 where they can nucleate further intracellular aggregation and/or trigger neuro-inflammation and exacerbate the neurodegenerative process69, 108. Supporting the potential relevance of this process in the pathogenesis of α-synucleinopathies, previous studies have shown accumulation of α-syn in fetal grafted neurons in patients with PD109, 110, as well as in grafted neuronal precursor cells in the hippocampus69 and basal ganglia111 in mouse models. Interestingly, α-syn has also been shown to ectopically accumulate in oligodendroglial cells in multiple system atrophy (another synucleinopathy)112 and in astroglial cells in PD113, 114. Moreover, the ascending distribution of the Lewy body pathology in LBD, as described by Braak113, has been interpreted to support the dissemination of α-syn from subcortical to cortical brain regions. However, further in vivo experimental verification of this is needed. A definition for the terms of propagation, dissemination and infectivity is provided in (BOX 2).
Box 2. Key terminology in the field of prion-like diseases.
The term transmission means the conveyance of non-prion proteins, whereas the terms propagation, spreading, transfer and dissemination are used interchangeably to indicate the dispersal of protein over a larger area, as well as from cell to cell. Use of the word seeding is synonymous with the addition of preformed aggregates, which when added during aggregate growth eliminates the lag phase that is associated with the formation of soluble aggregation competent oligomers, nuclei, and is the theoretical basis for prion infectivity, or the conveyance of prion proteins from animal to animal.
The mechanisms through which extracellular α-syn oligomers transfer to other cells includes endocytosis115, direct penetration116, transynaptic dissemination103, and membrane-receptor mediated access105. Once inside the acceptor cell, α-syn could seed further intracellular aggregation or the protein could be targeted for degradation. Although the exact process of intracellular oligomer and fibril propagation remains unknown, evidence from in vitro biophysical studies have consistently shown that fibrillization of α-syn follows a nucleated polymerization mechanism92. This mechanism is characterized by a nucleation phase that initially involves the formation of assembly-competent oligomers (nuclei), which is followed by cooperative oligomer growth and fibril formation by monomer addition (FIG. 4)117. This process can be seeded and accelerated by the addition of preformed fibrils and is thought to serve as the underling mechanism for the spreading of α-syn pathology in the brain. This phenomenon has been observed in a cell-based assay where the introduction of recombinant α-syn fibrils results in the seeding and the recruitment of endogenous α-syn and the formation of Lewy body-like inclusions44. A recent in vivo study has shown that inoculation of α-syn transgenic mice with homogenates containing α-syn protofibrils and fibrils results in considerable enhancement of the α-syn pathology and propagation118. These observations have lead to the provocative hypothesis that extracellular α-syn seeds might behave in a prion-like fashion. Further experimental evidence is needed to confirm this idea.
Perspectives on therapeutic targets
Given the potential toxicity of various α-syn aggregates, therapeutic approaches for synucleonopathies might involve reducing the levels or accumulation of intracellular and extracellular α-syn. One way to reduce the accumulation of intracellular α-syn may be to reduce the expression of this protein, by silencing SNCA with antisense microRNA, or by repressing the SNCA promoter (FIG. 3B). Another approach may be to increase the clearance of α-syn by activating autophagy or the proteasome; increasing proteolytic breakdown of α-syn with cathepsin D, neurosin or MMP9; or by promoting the binding of α-syn to chaperone-like molecules such as β-syn and HSPs (FIG. 3B). The third approach would be to reduce the posttranslational modifications such as oxidation, nitration, phosphorylation, and C-terminal cleavage (FIG. 3B). Two other possibilities may be to reduce aggregation or stabilize that native state of α-syn (FIG. 3B).
Resolving the question of whether α-syn exists as a monomer or as an oligomer, under physiological and pathophysiological conditions, is important not only in defining the function of this molecule but also in terms of developing therapeutic approaches for synucleonopathies. For example, if native α-syn exists as a multimer, then one therapeutic strategy for PD and DLB could involve preventing this complex from breaking apart. However, if toxic α-syn oligomers are derived from single conformers then viable therapeutic approaches might include stabilization of monomers, stabilization of non-propagating α-syn dimers, inhibiting α-syn oligomerization, and prevention of interactions between monomers and protofibrils that result in fibril growth. Since in vitro studies of multiple amyloidogenic proteins119 have shown that fibril growth and disassembly occur via monomer addition to protofibrils and disassociation, respectively, α-syn toxicity would not only be dependent on the levels of oligomers, but also on the concentration of the monomers. The main issue relating to developing small molecules to stabilize monomeric α-syn or prevent the formation of toxic forms of α-syn is that the monomer lacks a well-defined structure or hydrophobic pockets. Additionally, the identity and three-dimensional structure of toxic oligomeric species and their mode of action remains the subject of active investigation and debate.
If oligomeric α-syn species are the primary cause of toxicity in synucleinopathies, then inhibition of α-synoligomerization or enhancement of α-syn fibrillization should attenuate α-syn toxicity and prevent neurodegeneration. Conversely, small molecules and/or mutations that stabilize toxic α-syn oligomers should enhance neurodegeneration. Interestingly, the large majority of molecules that inhibit α-syn fibrilization exert their effects by stabilizing high molecular weight oligomeric intermediates on the amyloid pathway120 and protect, rather than enhance, α-syn toxicity121. This protective effect might be related to the anti-oxidant and anti-inflammatory properties of some of these compounds such as polyphenols and curcuminoids122. Alternatively, it is possible that the binding of these molecules to oligomeric species of α-syn blocks interactions between α-syn and key mediators of α-syn-associated toxicity. Interestingly, this seems to be the case for most Aβ inhibitors as well123, where the majority of the compounds shown to protect against Aβ toxicity do not stabilize monomeric Aβ and promote the accumulation of oligomers and/or non-fibrillar aggregates of various sizes and morphologies. It is possible that these molecules induce remodelling of the toxic oligomers and/or promote the formation of non-toxic aggregate or pathways. A recent study by Ayrolles-Torro_et al._124 showed that small molecule inhibitors that stabilize oligomeric forms of the prion protein attenuate prion infectivity in vivo, again suggesting a direct link between the process of oligomer growth, fibrillization and prion infectivity.
The toxic extracellular α-syn aggregates can be reduced by targeting them with specific antibodies, passive and active immunotherapy, increasing clearance, blocking putative receptors for extracellular α-syn in acceptor cells, reducing α-syn endocytosis and exocytosis, or targeting exosomes. Moreover, given the pro-inflammatory effects of α-syn, reducing microglial or astroglial activation with non-steroidal anti-inflammatory drugs may also be fruitful area of research125. In addition, the considerable biophysical and structural data available for the single conformation of α-syn can be used in targeting the α-syn monomers by decreasing synthesis, increasing clearance, or stabilizing the monomer remains the most viable therapeutic strategy for PD, DLB and related disorders. Ultimately, developing a successful therapy for synucleonopathies will require multidisciplinary research combined with multimodal therapeutics.
Conclusions
Since the discovery of α-syn in the mammalian brain, research has focused on better understanding the contributions of this natively unfolded protein to vesicular synaptic function under normal conditions. The discovery of disease-linked mutations and multiplications in α-syn that cause familial PD and accelerate α-synoligomerization in vitro has resulted in greater emphasis on elucidating the role of α-syn oligomers and fibrils as well as prion-like α-syn propagation in the pathogenesis of LBD. Therapeutic interventions directed at reducing α-syn synthesis, toxicity and aggregation or at increasing α-syn clearance are currently been investigated in preclinical models and hold promise for developing future treatments for α-synucleonopathies.
Online summary.
- Current studies of structural and functional role of membrane bound α-synuclein shows participation in the vesicular synaptic transport pathways.
- Increased expression or accumulation of α-synuclein due to genetic duplication, mutations, or failure in clearance may play a role in Parkinson disease and related disorders.
- Different conformers of α-synuclein including oligomers, protofibrils, and fibrils play a role in the toxicity of α-synuclein.
- Recent studies suggest that the propagation and transmission of α-synuclein participates in the pathogenesis of PD.
- Reducing α-synuclein expression, aggregation, or propagation, or increasing clearance represent viable therapeutical targets for Parkinson's disease and related disorders.
Acknowledgments
Grant support: National Institute of Health through the following grants: AG5131, AG18440, AG022074, and NS044233 (EM); the Swiss National Science Foundation grant #31003A_120653, Swiss Federal Institute of Technology, Lausanne, ERC starting grant, and a Merck Serono grant (HAL).
Glossary
Lewy Bodies
Intraneuronal globular inclusions composed primarily of α-syn fibrils characteristic of Parkinson's disease, but are also found in other neurodegenerative diseases such as Dementia with Lewy Bodies.
Fibrils
Mature fibrils are characterized by: (i) cross-β-sheet X-ray fiber diffraction pattern; (ii) β-sheet rich CD and FTIR spectra; (iii) binding to Congo red and Thioflavin-T/S and (iv) characteristic filamentous morphology (8–12 nm in diameter, > 1 μm long) by AFM and TEM imaging. α-Syn can form fibrils of diverse morphologies depending on the solution conditions.
Oilgomers
The term ‘oligomers’ encompasses a wide range of species, ranging from low-molecular-weight (LMW) species (including dimers, trimers and tetramers) to high-molecular-weight (HMW) oligomeric species (such as spherical, chain-like and annular structures or longer structures. Different types of oligomeric species have been described and are typically distinguished by their composition, stability and molecular size distribution.
Nucleus
A thermodynamically unstable oligomeric species that is capable of nucleating further growth into amyloid fibrils.
Native state
The three dimensional structure of the protein in its normal physiological milieu in the absence of any denaturing agents or conditions.
Seed
A relatively stable entity (such as fibrils and fragmented fibrils), which, when introduced into a solution containing monomeric subunits, serve as effective nuclei and accelerate fibril formation (by eliminating the lag phase associated with nuclei formation) in a nucleation polymerization process.
Biographies
Hilal A. Lashuel: Dr. Lashuel obtained his Ph.D. from Texas AM University in 2000 and did his post-doctoral training in the laboratory of Peter T. Lansbury at Harvard Medical School. Dr. Lashuelis currently an associate professor in the Brain Mind Institute at EFPL. His research interests focus on the molecular determinants and structural basis of protein aggregation and toxicity in neurodegenerative diseases.
Eliezer Masliah: Dr. Eliezer Masliah received his Doctorate of Medicine from the National Autonomous University of Mexico in 1983. He is now Director of the Laboratory of Experimental Neuropathology in the Neurosciences Department at UCSD. He interested in understanding the molecular and cellular mechanisms of neurodegeneration in Alzheimer's and Parkinson's diseases, as well as in other neurodegenerative disorders.
Cassia R. Overk: Dr. Cassia Overk earned her doctorate at the University of Illinois at Chicago in 2007. Following a Post-Doctoral Research Fellowship at Rush University Medical Center, she is now a Project Scientist in Dr. Masliah's lab at the University of California San Diego (UCSD). Her research is focused on evaluating small molecules from natural product, as well as synthetic chemical libraries as modulators of neurodegenerative diseases.
Abid Oueslati: Dr. Abid Oueslati is a postdoctoral Fellow at the Ecole Polytechnique Federale de Lausanne (EPFL), Switzerland. He received his Ph.D. from the University of the Mediterranean, Marseilles, France. He then joined the EPFL where he is conducting research focused on elucidating the molecular basis of Parkinson's disease pathogenesis and the identification of new therapeutic targets for synucleinopathies.
References
- 1.McKeith IG, et al. Consensus guidelines for the clinical and pathologic diagnosis of dementia with Lewy bodies (DLB): report of the consortium on DLB international workshop. Neurology. 1996;47:1113–1124. doi: 10.1212/wnl.47.5.1113. [DOI] [PubMed] [Google Scholar]
- 2.Braak H, Braak E. Pathoanatomy of Parkinson's disease. J Neurol. 2000;247 Suppl 2:II3–II10. doi: 10.1007/PL00007758. Detailed description of the pathoanatomy that occurs in Parkinson's disease. [DOI] [PubMed] [Google Scholar]
- 3.Vekrellis K, Xilouri M, Emmanouilidou E, Rideout HJ, Stefanis L. Pathological roles of alpha-synuclein in neurological disorders. Lancet Neurol. 2011;10:1015–1025. doi: 10.1016/S1474-4422(11)70213-7. [DOI] [PubMed] [Google Scholar]
- 4.Chartier-Harlin MC, et al. Alpha-synuclein locus duplication as a cause of familial Parkinson's disease. Lancet. 2004;364:1167–1169. doi: 10.1016/S0140-6736(04)17103-1. [DOI] [PubMed] [Google Scholar]
- 5.Kruger R, et al. Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson's disease. Nat Genet. 1998;18:106–108. doi: 10.1038/ng0298-106. [DOI] [PubMed] [Google Scholar]
- 6.Polymeropoulos MH, et al. Mutation in the alpha-synuclein gene identified in families with Parkinson's disease. Science. 1997;276:2045–2047. doi: 10.1126/science.276.5321.2045. [DOI] [PubMed] [Google Scholar]
- 7.Zarranz JJ, et al. The new mutation, E46K, of alpha-synuclein causes Parkinson and Lewy body dementia. Ann Neurol. 2004;55:164–173. doi: 10.1002/ana.10795. [DOI] [PubMed] [Google Scholar]
- 8.Simon-Sanchez J, et al. Genome-wide association study reveals genetic risk underlying Parkinson's disease. Nat Genet. 2009;41:1308–1312. doi: 10.1038/ng.487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Conway KA, Harper JD, Lansbury PT. Accelerated in vitro fibril formation by a mutant alpha-synuclein linked to early-onset Parkinson disease. Nat Med. 1998;4:1318–1320. doi: 10.1038/3311. [DOI] [PubMed] [Google Scholar]
- 10.Tsigelny IF, et al. Mechanisms of hybrid oligomer formation in the pathogenesis of combined Alzheimer's and Parkinson's diseases. PloS one. 2008;3:e3135. doi: 10.1371/journal.pone.0003135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Oueslati A, Fournier M, Lashuel HA. Role of post-translational modifications in modulating the structure, function and toxicity of alpha-synuclein: Implications for Parkinson's disease pathogenesis and therapies. Prog Brain Res. 2010;183:115–145. doi: 10.1016/S0079-6123(10)83007-9. [DOI] [PubMed] [Google Scholar]
- 12.Taschenberger G, et al. Aggregation of alpha- synuclein promotes progressive in vivo neurotoxicity in adult rat dopaminergic neurons. Acta Neuropathol. 2011;123:671–683. doi: 10.1007/s00401-011-0926-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Galvin JE, Lee VM, Trojanowski JQ. Synucleinopathies: clinical and pathological implications. Arch Neurol. 2001;58:186–190. doi: 10.1001/archneur.58.2.186. [DOI] [PubMed] [Google Scholar]
- 14.Wang S, et al. alpha-Synuclein disrupts stress signaling by inhibiting polo-like kinase Cdc5/Plk2. Proc Natl Acad Sci U S A. 2012;109:16119–16124. doi: 10.1073/pnas.1206286109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Iwai A, et al. The precursor protein of non-A beta component of Alzheimer's disease amyloid is a presynaptic protein of the central nervous system. Neuron. 1995;14:467–475. doi: 10.1016/0896-6273(95)90302-x. Identification of NAC as a presynaptic protein. [DOI] [PubMed] [Google Scholar]
- 16.Jakes R, Spillantini MG, Goedert M. Identification of two distinct synucleins from human brain. FEBS letters. 1994;345:27–32. doi: 10.1016/0014-5793(94)00395-5. [DOI] [PubMed] [Google Scholar]
- 17.Withers GS, George JM, Banker GA, Clayton DF. Delayed localization of synelfin (synuclein, NACP) to presynaptic terminals in cultured rat hippocampal neurons. Brain research Developmental brain research. 1997;99:87–94. doi: 10.1016/s0165-3806(96)00210-6. [DOI] [PubMed] [Google Scholar]
- 18.Kahle PJ, et al. Subcellular localization of wild-type and Parkinson's disease-associated mutant alpha -synuclein in human and transgenic mouse brain. J Neurosci. 2000;20:6365–6373. doi: 10.1523/JNEUROSCI.20-17-06365.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee SJ, Jeon H, Kandror KV. Alpha-synuclein is localized in a subpopulation of rat brain synaptic vesicles. Acta Neurobiol Exp (Wars) 2008;68:509–515. doi: 10.55782/ane-2008-1717. [DOI] [PubMed] [Google Scholar]
- 20.Zhang L, et al. Semi-quantitative analysis of alpha-synuclein in subcellular pools of rat brain neurons: an immunogold electron microscopic study using a C-terminal specific monoclonal antibody. Brain Res. 2008;1244:40–52. doi: 10.1016/j.brainres.2008.08.067. [DOI] [PubMed] [Google Scholar]
- 21.Cabin DE, et al. Synaptic vesicle depletion correlates with attenuated synaptic responses to prolonged repetitive stimulation in mice lacking alpha-synuclein. J Neurosci. 2002;22:8797–8807. doi: 10.1523/JNEUROSCI.22-20-08797.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Abeliovich A, et al. Mice lacking alpha-synuclein display functional deficits in the nigrostriatal dopamine system. Neuron. 2000;25:239–252. doi: 10.1016/s0896-6273(00)80886-7. [DOI] [PubMed] [Google Scholar]
- 23.Murphy DD, Rueter SM, Trojanowski JQ, Lee VM. Synucleins are developmentally expressed, and alpha-synuclein regulates the size of the presynaptic vesicular pool in primary hippocampal neurons. J Neurosci. 2000;20:3214–3220. doi: 10.1523/JNEUROSCI.20-09-03214.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yavich L, Tanila H, Vepsalainen S, Jakala P. Role of alpha-synuclein in presynaptic dopamine recruitment. J Neurosci. 2004;24:11165–11170. doi: 10.1523/JNEUROSCI.2559-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Scott DA, et al. A pathologic cascade leading to synaptic dysfunction in alpha-synuclein-induced neurodegeneration. J Neurosci. 2010;30:8083–8095. doi: 10.1523/JNEUROSCI.1091-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nemani VM, et al. Increased expression of alpha-synuclein reduces neurotransmitter release by inhibiting synaptic vesicle reclustering after endocytosis. Neuron. 2010;65:66–79. doi: 10.1016/j.neuron.2009.12.023. α-Synuclein overexpression inhibited neurotransmitter release by reducing the size of the synaptic vesicle recycling pool. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gaugler MN, et al. Nigrostriatal overabundance of alpha-synuclein leads to decreased vesicle density and deficits in dopamine release that correlate with reduced motor activity. Acta Neuropathol. 2012;123:653–669. doi: 10.1007/s00401-012-0963-y. [DOI] [PubMed] [Google Scholar]
- 28.Lundblad M, Decressac M, Mattsson B, Bjorklund A. Impaired neurotransmission caused by overexpression of alpha-synuclein in nigral dopamine neurons. Proc Natl Acad Sci U S A. 2012;109:3213–3219. doi: 10.1073/pnas.1200575109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Larsen KE, et al. Alpha-synuclein overexpression in PC12 and chromaffin cells impairs catecholamine release by interfering with a late step in exocytosis. J Neurosci. 2006;26:11915–11922. doi: 10.1523/JNEUROSCI.3821-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Scott D, Roy S. alpha-Synuclein Inhibits Intersynaptic Vesicle Mobility and Maintains Recycling-Pool Homeostasis. J Neurosci. 2012;32:10129–10135. doi: 10.1523/JNEUROSCI.0535-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Payton JE, Perrin RJ, Woods WS, George JM. Structural determinants of PLD2 inhibition by alpha-synuclein. J Mol Biol. 2004;337:1001–1009. doi: 10.1016/j.jmb.2004.02.014. [DOI] [PubMed] [Google Scholar]
- 32.Dalfo E, Ferrer I. Alpha-synuclein binding to rab3a in multiple system atrophy. Neurosci Lett. 2005;380:170–175. doi: 10.1016/j.neulet.2005.01.034. [DOI] [PubMed] [Google Scholar]
- 33.Burre J, et al. Alpha-synuclein promotes SNARE-complex assembly in vivo and in vitro. Science. 2010;329:1663–1667. doi: 10.1126/science.1195227. Identification of α-synuclein as a non-classical chaperone which bound and promoted SNARE-complex assembly. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chandra S, et al. Double-knockout mice for alpha- and beta-synucleins: effect on synaptic functions. Proc Natl Acad Sci U S A. 2004;101:14966–14971. doi: 10.1073/pnas.0406283101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chandra S, Gallardo G, Fernandez-Chacon R, Schluter OM, Sudhof TC. Alpha-synuclein cooperates with CSPalpha in preventing neurodegeneration. Cell. 2005;123:383–396. doi: 10.1016/j.cell.2005.09.028. [DOI] [PubMed] [Google Scholar]
- 36.Ueda K, et al. Molecular cloning of cDNA encoding an unrecognized component of amyloid in Alzheimer disease. Proc Natl Acad Sci U S A. 1993;90:11282–11286. doi: 10.1073/pnas.90.23.11282. First report of the NAC being 140 amino acid protein. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ulmer TS, Bax A, Cole NB, Nussbaum RL. Structure and dynamics of micelle-bound human alpha-synuclein. J Biol Chem. 2005;280:9595–9603. doi: 10.1074/jbc.M411805200. [DOI] [PubMed] [Google Scholar]
- 38.Eliezer D, Kutluay E, Bussell R, Jr, Browne G. Conformational properties of alpha-synuclein in its free and lipid-associated states. J Mol Biol. 2001;307:1061–1073. doi: 10.1006/jmbi.2001.4538. [DOI] [PubMed] [Google Scholar]
- 39.Masliah E, Iwai A, Mallory M, Ueda K, Saitoh T. Altered presynaptic protein NACP is associated with plaque formation and neurodegeneration in Alzheimer's disease. Am J Pathol. 1996;148:201–210. [PMC free article] [PubMed] [Google Scholar]
- 40.Spillantini MG, et al. Alpha-synuclein in Lewy bodies. Nature. 1997;388:839–840. doi: 10.1038/42166. [DOI] [PubMed] [Google Scholar]
- 41.Hashimoto M, Takenouchi T, Mallory M, Masliah E, Takeda A. The role of NAC in amyloidogenesis in Alzheimer's disease. Am J Pathol. 2000;156:734–736. doi: 10.1016/s0002-9440(10)64777-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.El-Agnaf OM, Jakes R, Curran MD, Wallace A. Effects of the mutations Ala30 to Pro and Ala53 to Thr on the physical and morphological properties of alpha-synuclein protein implicated in Parkinson's disease. FEBS Lett. 1998;440:67–70. doi: 10.1016/s0014-5793(98)01419-7. [DOI] [PubMed] [Google Scholar]
- 43.Giasson BI, Murray IV, Trojanowski JQ, Lee VM. A hydrophobic stretch of 12 amino acid residues in the middle of alpha-synuclein is essential for filament assembly. J Biol Chem. 2001;276:2380–2386. doi: 10.1074/jbc.M008919200. The middle hydrophobic domain of α-synuclein is necessary and sufficient for fibrillization. [DOI] [PubMed] [Google Scholar]
- 44.Luk KC, et al. Exogenous alpha-synuclein fibrils seed the formation of Lewy body-like intracellular inclusions in cultured cells. Proc Natl Acad Sci U S A. 2009;106:20051–20056. doi: 10.1073/pnas.0908005106. α-Synuclein seeds can recruit endogenous soluble α-synuclein protein to form pathological species. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Weinreb PH, Zhen W, Poon AW, Conway KA, Lansbury PT., Jr NACP, a protein implicated in Alzheimer's disease and learning, is natively unfolded. Biochemistry. 1996;35:13709–13715. doi: 10.1021/bi961799n. [DOI] [PubMed] [Google Scholar]
- 46.Fauvet B, et al. Alpha-synuclein in the central nervous system and from erythrocytes, mammalian cells and E. coli exists predominantly as a disordered monomer. J Biol Chem. 2012 doi: 10.1074/jbc.M111.318949. This reassessed the oligomeric state of a-syn and demonstrated that native a-syn exists predominantly as an unfolded monomer and not a tetramer. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ramakrishnan M, Jensen PH, Marsh D. Association of alpha-synuclein and mutants with lipid membranes: spin-label ESR and polarized IR. Biochemistry. 2006;45:3386–3395. doi: 10.1021/bi052344d. [DOI] [PubMed] [Google Scholar]
- 48.Ullman O, Fisher CK, Stultz CM. Explaining the structural plasticity of alpha-synuclein. J Am Chem Soc. 2011;133:19536–19546. doi: 10.1021/ja208657z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hashimoto M, et al. Oxidative stress induces amyloid-like aggregate formation of NACP/alpha-synuclein in vitro. Neuroreport. 1999;10:717–721. doi: 10.1097/00001756-199903170-00011. [DOI] [PubMed] [Google Scholar]
- 50.Andringa G, et al. Tissue transglutaminase catalyzes the formation of alpha-synuclein crosslinks in Parkinson's disease. FASEB J. 2004;18:932–934. doi: 10.1096/fj.03-0829fje. [DOI] [PubMed] [Google Scholar]
- 51.Paleologou KE, et al. Phosphorylation at S87 is enhanced in synucleinopathies, inhibits alpha-synuclein oligomerization, and influences synuclein-membrane interactions. J Neurosci. 2010;30:3184–3198. doi: 10.1523/JNEUROSCI.5922-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Li W, et al. Aggregation promoting C-terminal truncation of alpha-synuclein is a normal cellular process and is enhanced by the familial Parkinson's disease-linked mutations. Proc Natl Acad Sci USA. 2005;102:2162–2167. doi: 10.1073/pnas.0406976102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dufty BM, et al. Calpain-cleavage of alpha-synuclein: connecting proteolytic processing to disease-linked aggregation. Am J Pathol. 2007;170:1725–1738. doi: 10.2353/ajpath.2007.061232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Perrin RJ, Woods WS, Clayton DF, George JM. Exposure to long chain polyunsaturated fatty acids triggers rapid multimerization of synucleins. J Biol Chem. 2001;276:41958–41962. doi: 10.1074/jbc.M105022200. [DOI] [PubMed] [Google Scholar]
- 55.Sharon R, et al. The formation of highly soluble oligomers of alpha-synuclein is regulated by fatty acids and enhanced in Parkinson's disease. Neuron. 2003;37:583–595. doi: 10.1016/s0896-6273(03)00024-2. [DOI] [PubMed] [Google Scholar]
- 56.Karube H, et al. N-terminal region of alpha-synuclein is essential for the fatty acid-induced oligomerization of the molecules. FEBS Lett. 2008;582:3693–3700. doi: 10.1016/j.febslet.2008.10.001. [DOI] [PubMed] [Google Scholar]
- 57.Takeda A, et al. Abnormal distribution of the non-Abeta component of Alzheimer's disease amyloid precursor/alpha-synuclein in Lewy body disease as revealed by proteinase K and formic acid pretreatment. Lab Invest. 1998;78:1169–1177. [PubMed] [Google Scholar]
- 58.Suh YH, Checler F. Amyloid precursor protein, presenilins, and alpha-synuclein: molecular pathogenesis and pharmacological applications in Alzheimer's disease. Pharmacol Rev. 2002;54:469–525. doi: 10.1124/pr.54.3.469. [DOI] [PubMed] [Google Scholar]
- 59.Iwai A, et al. The synaptic protein NACP is abnormally expressed during the progression of Alzheimer's disease. Brain Res. 1996;720:230–234. doi: 10.1016/0006-8993(96)00014-5. [DOI] [PubMed] [Google Scholar]
- 60.Kragh CL, Ubhi K, Wyss-Corey T, Masliah E. Autophagy in dementias. Brain Pathol. 2012;22:99–109. doi: 10.1111/j.1750-3639.2011.00545.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Seidel K, et al. First appraisal of brain pathology owing to A30P mutant alpha-synuclein. Annals of neurology. 2010;67:684–689. doi: 10.1002/ana.21966. [DOI] [PubMed] [Google Scholar]
- 62.Maraganore DM, et al. Collaborative analysis of alpha-synuclein gene promoter variability and Parkinson disease. JAMA : the journal of the American Medical Association. 2006;296:661–670. doi: 10.1001/jama.296.6.661. [DOI] [PubMed] [Google Scholar]
- 63.Iwata A, et al. Alpha-synuclein degradation by serine protease neurosin: implication for pathogenesis of synucleinopathies. Hum Mol Genet. 2003;12:2625–2635. doi: 10.1093/hmg/ddg283. [DOI] [PubMed] [Google Scholar]
- 64.Klucken J, Shin Y, Masliah E, Hyman BT, McLean PJ. Hsp70 Reduces alpha-Synuclein Aggregation and Toxicity. J Biol Chem. 2004;279:25497–25502. doi: 10.1074/jbc.M400255200. [DOI] [PubMed] [Google Scholar]
- 65.McNaught KS, et al. Impairment of the ubiquitin-proteasome system causes dopaminergic cell death and inclusion body formation in ventral mesencephalic cultures. Journal of neurochemistry. 2002;81:301–306. doi: 10.1046/j.1471-4159.2002.00821.x. [DOI] [PubMed] [Google Scholar]
- 66.McNaught KS, et al. Proteasome inhibition causes nigral degeneration with inclusion bodies in rats. Neuroreport. 2002;13:1437–1441. doi: 10.1097/00001756-200208070-00018. [DOI] [PubMed] [Google Scholar]
- 67.Webb JL, Ravikumar B, Atkins J, Skepper JN, Rubinsztein DC. Alpha-Synuclein is degraded by both autophagy and the proteasome. J Biol Chem. 2003;278:25009–59013. doi: 10.1074/jbc.M300227200. [DOI] [PubMed] [Google Scholar]
- 68.Cuervo AM, Stefanis L, Fredenburg R, Lansbury PT, Sulzer D. Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science. 2004;305:1292–1295. doi: 10.1126/science.1101738. Identification of lysosomes for degradation of wild type α-synuclein by the chaperone-mediated autophagy pathway. [DOI] [PubMed] [Google Scholar]
- 69.Desplats P, et al. Inclusion formation and neuronal cell death through neuron-to-neuron transmission of alpha-synuclein. Proc Natl Acad Sci U S A. 2009;106:13010–13015. doi: 10.1073/pnas.0903691106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Spencer B, et al. Beclin 1 gene transfer activates autophagy and ameliorates the neurodegenerative pathology in alpha-synuclein models of Parkinson's and Lewy body diseases. J Neurosci. 2009;29:13578–13588. doi: 10.1523/JNEUROSCI.4390-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Crews L, et al. Selective molecular alterations in the autophagy pathway in patients with Lewy body disease and in models of alpha-synucleinopathy. PloS one. 2010;5:e9313. doi: 10.1371/journal.pone.0009313. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 72.Kosaka K. Diffuse Lewy body disease in Japan. J Neurol. 1990;237:197–204. doi: 10.1007/BF00314594. [DOI] [PubMed] [Google Scholar]
- 73.Dickson DW, et al. Diffuse Lewy body disease: light and electron microscopic immunocytochemistry of senile plaques. Acta Neuropathol. 1989;78:572–584. doi: 10.1007/BF00691284. [DOI] [PubMed] [Google Scholar]
- 74.Braak H, Sastre M, Del Tredici K. Development of alpha-synuclein immunoreactive astrocytes in the forebrain parallels stages of intraneuronal pathology in sporadic Parkinson's disease. Acta Neuropathol. 2007;114:231–241. doi: 10.1007/s00401-007-0244-3. Association of α-synuclein with reactive astrocytes in clinically diagnosed Parkinson's disease cases. [DOI] [PubMed] [Google Scholar]
- 75.Lee HJ, et al. Direct transfer of alpha-synuclein from neuron to astroglia causes inflammatory responses in synucleinopathies. J Biol Chem. 2010;285:9262–9272. doi: 10.1074/jbc.M109.081125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kahle PJ, et al. Selective insolubility of alpha-synuclein in human Lewy body diseases is recapitulated in a transgenic mouse model. Am J Pathol. 2001;159:2215–2225. doi: 10.1016/s0002-9440(10)63072-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Baba M, et al. Aggregation of alpha-synuclein in Lewy bodies of sporadic Parkinson's disease and dementia with Lewy bodies. Am J Pathol. 1998;152:879–884. [PMC free article] [PubMed] [Google Scholar]
- 78.Lee MK, et al. Human alpha-synuclein-harboring familial Parkinson's disease-linked Ala-53 --> Thr mutation causes neurodegenerative disease with alpha-synuclein aggregation in transgenic mice. Proc Natl Acad Sci U S A. 2002;99:8968–8973. doi: 10.1073/pnas.132197599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Tsigelny IF, et al. Role of alpha-synuclein penetration into the membrane in the mechanisms of oligomer pore formation. FEBS J. 2012 doi: 10.1111/j.1742-4658.2012.08489.x. Computational modeling of α-synuclein forming ring-like structures that pentrate the membrane. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Bellucci A, Navarria L, Zaltieri M, Missale C, Spano P. Alpha-synuclein synaptic pathology and its implications in the development of novel therapeutic approaches to cure Parkinson's disease. Brain Res. 2012;1432:95–113. doi: 10.1016/j.brainres.2011.11.031. [DOI] [PubMed] [Google Scholar]
- 81.Schulz-Schaeffer WJ. The synaptic pathology of alpha-synuclein aggregation in dementia with Lewy bodies, Parkinson's disease and Parkinson's disease dementia. Acta Neuropathol. 2010;120:131–143. doi: 10.1007/s00401-010-0711-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Garcia-Reitbock P, et al. SNARE protein redistribution and synaptic failure in a transgenic mouse model of Parkinson's disease. Brain : a journal of neurology. 2010;133:2032–2044. doi: 10.1093/brain/awq132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Horvath I, et al. Mechanisms of Protein Oligomerization: Inhibitor of Functional Amyloids Templates alpha-Synuclein Fibrillation. J Am Chem Soc. 2012;134:3439–3444. doi: 10.1021/ja209829m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Conway KA, et al. Accelerated oligomerization by Parkinson's disease linked alpha-synuclein mutants. Ann N Y Acad Sci. 2000;920:42–45. doi: 10.1111/j.1749-6632.2000.tb06903.x. [DOI] [PubMed] [Google Scholar]
- 85.Cremades N, et al. Direct Observation of the Interconversion of Normal and Toxic Forms of alpha-Synuclein. Cell. 2012;149:1048–1059. doi: 10.1016/j.cell.2012.03.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Danzer KM, et al. Different species of alpha-synuclein oligomers induce calcium influx and seeding. J Neurosci. 2007;27:9220–9232. doi: 10.1523/JNEUROSCI.2617-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Iwatsubo T. Pathological biochemistry of alpha-synucleinopathy. Neuropathology : official journal of the Japanese Society of Neuropathology. 2007;27:474–478. doi: 10.1111/j.1440-1789.2007.00785.x. [DOI] [PubMed] [Google Scholar]
- 88.Oueslati A, Paleologou KE, Schneider BL, Aebischer P, Lashuel HA. Mimicking phosphorylation at serine 87 inhibits the aggregation of human alpha-synuclein and protects against its toxicity in a rat model of Parkinson's disease. J Neurosci. 2012;32:1536–1544. doi: 10.1523/JNEUROSCI.3784-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Souza JM, Giasson BI, Chen Q, Lee VM, Ischiropoulos H. Dityrosine cross-linking promotes formation of stable alpha -synuclein polymers. Implication of nitrative and oxidative stress in the pathogenesis of neurodegenerative synucleinopathies. J Biol Chem. 2000;275:18344–18349. doi: 10.1074/jbc.M000206200. [DOI] [PubMed] [Google Scholar]
- 90.Uversky VN, Li J, Fink AL. Metal-triggered structural transformations, aggregation, and fibrillation of human alpha-synuclein - A possible molecular link between Parkinson's disease and heavy metal exposure. J Biol Chem. 2001;276:44284–44296. doi: 10.1074/jbc.M105343200. [DOI] [PubMed] [Google Scholar]
- 91.Conway KA, Rochet JC, Bieganski RM, Lansbury PT., Jr Kinetic stabilization of the alpha-synuclein protofibril by a dopamine-alpha-synuclein adduct. Science. 2001;294:1346–1349. doi: 10.1126/science.1063522. [DOI] [PubMed] [Google Scholar]
- 92.Conway KA, et al. Acceleration of oligomerization, not fibrillization, is a shared property of both alpha-synuclein mutations linked to early-onset Parkinson's disease: implications for pathogenesis and therapy. Proc Nat Acad Sci. 2000;97:571–576. doi: 10.1073/pnas.97.2.571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Hsu LJ, et al. alpha-synuclein promotes mitochondrial deficit and oxidative stress. Am J Pathol. 2000;157:401–410. doi: 10.1016/s0002-9440(10)64553-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Hashimoto M, et al. The Role of alpha-synuclein assembly and metabolism in the pathogenesis of Lewy body disease. J Mol Neurosci. 2004;24:343–352. doi: 10.1385/JMN:24:3:343. [DOI] [PubMed] [Google Scholar]
- 95.Alim MA, et al. Demonstration of a role for alpha-synuclein as a functional microtubule-associated protein. J Alzheimers Dis. 2004;6:435–442. doi: 10.3233/jad-2004-6412. [DOI] [PubMed] [Google Scholar]
- 96.Gorbatyuk OS, et al. The phosphorylation state of Ser-129 in human alpha-synuclein determines neurodegeneration in a rat model of Parkinson disease. Proc Natl Acad Sci U S A. 2008;105:763–768. doi: 10.1073/pnas.0711053105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Azeredo da Silveira S, et al. Phosphorylation does not prompt, nor prevent, the formation of alpha-synuclein toxic species in a rat model of Parkinson's disease. Hum Mol Genet. 2009;18:872–887. doi: 10.1093/hmg/ddn417. [DOI] [PubMed] [Google Scholar]
- 98.Winner B, et al. In vivo demonstration that alpha-synuclein oligomers are toxic. Proc Natl Acad Sci U S A. 2011;108:4194–4199. doi: 10.1073/pnas.1100976108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Jan A, et al. Abeta42 neurotoxicity is mediated by ongoing nucleated polymerization process rather than by discrete Abeta42 species. J Biol Chem. 2011;286:8585–8596. doi: 10.1074/jbc.M110.172411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Wogulis M, et al. Nucleation-dependent polymerization is an essential component of amyloid-mediated neuronal cell death. J Neurosci. 2005;25:1071–1080. doi: 10.1523/JNEUROSCI.2381-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Colla E, et al. Accumulation of toxic alpha-synuclein oligomer within endoplasmic reticulum occurs in alpha-synucleinopathy in vivo. J Neurosci. 2012;32:3301–3305. doi: 10.1523/JNEUROSCI.5368-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Jang A, et al. Non-classical exocytosis of alpha-synuclein is sensitive to folding states and promoted under stress conditions. J Neurochem. 2010;113:1263–1274. doi: 10.1111/j.1471-4159.2010.06695.x. [DOI] [PubMed] [Google Scholar]
- 103.Danzer KM, et al. Heat-shock protein 70 modulates toxic extracellular alpha-synuclein oligomers and rescues trans-synaptic toxicity. FASEB J. 2011;25:326–336. doi: 10.1096/fj.10-164624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Alvarez-Erviti L, et al. Lysosomal dysfunction increases exosome-mediated alpha-synuclein release and transmission. Neurobiology of disease. 2011;42:360–367. doi: 10.1016/j.nbd.2011.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Lee HJ, et al. Assembly-dependent endocytosis and clearance of extracellular alpha-synuclein. Int J Biochem Cell Biol. 2008;40:1835–1849. doi: 10.1016/j.biocel.2008.01.017. [DOI] [PubMed] [Google Scholar]
- 106.Emmanouilidou E, et al. Cell-produced alpha-synuclein is secreted in a calcium-dependent manner by exosomes and impacts neuronal survival. J Neurosci. 2010;30:6838–6851. doi: 10.1523/JNEUROSCI.5699-09.2010. First report that α-synuclein is secreted by externalized vesicles in a calcium-dependent manner. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Jao CC, Hegde BG, Chen J, Haworth IS, Langen R. Structure of membrane-bound alpha-synuclein from site-directed spin labeling and computational refinement. Proc Natl Acad Sci U S A. 2008;105:19666–19671. doi: 10.1073/pnas.0807826105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Volpicelli-Daley LA, et al. Exogenous alpha-synuclein fibrils induce Lewy body pathology leading to synaptic dysfunction and neuron death. Neuron. 2011;72:57–71. doi: 10.1016/j.neuron.2011.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Crews L, et al. Alpha-synuclein alters Notch-1 expression and neurogenesis in mouse embryonic stem cells and in the hippocampus of transgenic mice. J Neurosci. 2008;28:4250–4260. doi: 10.1523/JNEUROSCI.0066-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Letarov A, Manival X, Desplats C, Krisch HM. gpwac of the T4-type bacteriophages: structure, function, and evolution of a segmented coiled-coil protein that controls viral infectivity. Journal of bacteriology. 2005;187:1055–1066. doi: 10.1128/JB.187.3.1055-1066.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Ekberg H, et al. The specific monocarboxylate transporter-1 (MCT-1) inhibitor, AR-C117977, induces donor-specific suppression, reducing acute and chronic allograft rejection in the rat. Transplantation. 2007;84:1191–1199. doi: 10.1097/01.tp.0000287541.53389.be. [DOI] [PubMed] [Google Scholar]
- 112.Karlsson J, Petersen A, Gido G, Wieloch T, Brundin P. Combining neuroprotective treatment of embryonic nigral donor tissue with mild hypothermia of the graft recipient. Cell Transplant. 2005;14:301–309. doi: 10.3727/000000005783983089. [DOI] [PubMed] [Google Scholar]
- 113.Karlsson J, Emgard M, Gido G, Wieloch T, Brundin P. Increased survival of embryonic nigral neurons when grafted to hypothermic rats. Neuroreport. 2000;11:1665–1668. doi: 10.1097/00001756-200006050-00014. [DOI] [PubMed] [Google Scholar]
- 114.Frodl EM, Duan WM, Sauer H, Kupsch A, Brundin P. Human embryonic dopamine neurons xenografted to the rat: effects of cryopreservation and varying regional source of donor cells on transplant survival, morphology and function. Brain Res. 1994;647:286–298. doi: 10.1016/0006-8993(94)91328-5. [DOI] [PubMed] [Google Scholar]
- 115.Kordower JH, Freeman TB, Olanow CW. Neuropathology of fetal nigral grafts in patients with Parkinson's disease. Movement disorders : official journal of the Movement Disorder Society. 1998;13 Suppl 1:88–95. [PubMed] [Google Scholar]
- 116.Tang B, et al. Forkhead box protein p1 is a transcriptional repressor of immune signaling in the CNS: implications for transcriptional dysregulation in Huntington disease. Hum Mol Genet. 2012;21:3097–3111. doi: 10.1093/hmg/dds132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Nonaka T, Watanabe ST, Iwatsubo T, Hasegawa M. Seeded aggregation and toxicity of alpha-synuclein and tau: cellular models of neurodegenerative diseases. J Biol Chem. 2010;285:34885–34898. doi: 10.1074/jbc.M110.148460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Luk KC, et al. Intracerebral inoculation of pathological alpha-synuclein initiates a rapidly progressive neurodegenerative alpha-synucleinopathy in mice. The Journal of experimental medicine. 2012;209:975–986. doi: 10.1084/jem.20112457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Ban T, et al. Direct observation of Abeta amyloid fibril growth and inhibition. J Mol Biol. 2004;344:757–767. doi: 10.1016/j.jmb.2004.09.078. [DOI] [PubMed] [Google Scholar]
- 120.Lashuel HA, et al. New class of inhibitors of amyloid-beta fibril formation. Implications for the mechanism of pathogenesis in Alzheimer's disease. J Biol Chem. 2002;277:42881–42890. doi: 10.1074/jbc.M206593200. [DOI] [PubMed] [Google Scholar]
- 121.Di Giovanni S, et al. Entacapone and tolcapone, two catechol O-methyltransferase inhibitors, block fibril formation of alpha-synuclein and beta-amyloid and protect against amyloid-induced toxicity. J Biol Chem. 2010;285:14941–14954. doi: 10.1074/jbc.M109.080390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Masuda M, et al. Small molecule inhibitors of alpha-synuclein filament assembly. Biochemistry. 2006;45:6085–6094. doi: 10.1021/bi0600749. [DOI] [PubMed] [Google Scholar]
- 123.Porat Y, Abramowitz A, Gazit E. Inhibition of amyloid fibril formation by polyphenols: structural similarity and aromatic interactions as a common inhibition mechanism. Chem Biol Drug Des. 2006;67:27–37. doi: 10.1111/j.1747-0285.2005.00318.x. [DOI] [PubMed] [Google Scholar]
- 124.Ayrolles-Torro A, et al. Oligomeric-induced activity by thienyl pyrimidine compounds traps prion infectivity. J Neurosci. 2011;31:14882–14892. doi: 10.1523/JNEUROSCI.0547-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Hirohata M, Ono K, Morinaga A, Yamada M. Non-steroidal anti-inflammatory drugs have potent anti-fibrillogenic and fibril-destabilizing effects for alpha-synuclein fibrils in vitro. Neuropharmacology. 2008;54:620–627. doi: 10.1016/j.neuropharm.2007.11.010. [DOI] [PubMed] [Google Scholar]
- 126.Bartels T, Choi JG, Selkoe DJ. alpha-Synuclein occurs physiologically as a helically folded tetramer that resists aggregation. Nature. 2011;477:107–110. doi: 10.1038/nature10324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Wang W, et al. A soluble alpha-synuclein construct forms a dynamic tetramer. Proc Natl Acad Sci U S A. 2011;108:17797–17802. doi: 10.1073/pnas.1113260108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Fauvet B, et al. Characterization of semisynthetic and naturally Nalpha-acetylated alpha-synuclein in vitro and in intact cells: implications for aggregation and cellular properties of alpha-synuclein. The Journal of biological chemistry. 2012;287:28243–28262. doi: 10.1074/jbc.M112.383711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Shaikh S, Nicholson LF. Advanced glycation end products induce in vitro cross-linking of alpha-synuclein and accelerate the process of intracellular inclusion body formation. J Neurosci Res. 2008;86:2071–2082. doi: 10.1002/jnr.21644. [DOI] [PubMed] [Google Scholar]
- 130.Hashimoto M, Takeda A, Hsu LJ, Takenouchi T, Masliah E. Role of cytochrome c as a stimulator of alpha-synuclein aggregation in Lewy body disease. J Biol Chem. 1999;274:28849–28852. doi: 10.1074/jbc.274.41.28849. [DOI] [PubMed] [Google Scholar]