The role of miRNAs in the regulation of inflammatory processes during hepatofibrogenesis (original) (raw)
Abstract
Liver cirrhosis represents the end stage of most chronic inflammatory liver diseases and is a major global health burden. Despite the enormous relevance of cirrhotic disease, pharmacological strategies for prevention or treatment of hepatic fibrosis are still limited, underlining the need to establish a better understanding of the molecular mechanisms underlying the pathogenesis of hepatic cirrhosis. Recently, miRNAs have emerged as a new class of RNAs that do not withhold the information to encode for proteins but regulate whole gene expression networks during different physiological and pathological processes. Various authors demonstrated that miRNA species are functionally involved in the regulation of chronic liver damage and development of liver cirrhosis in inflamed livers. Moreover, circulating miRNA patterns were suggested to serve as blood-based biomarkers indicating liver injury and progression to hepatic cirrhosis and cancer. Here we summarize current findings on a potential role of miRNAs in the cascade leading from liver inflammation to liver fibrosis and finally hepatocellular carcinoma. We compare data from animal models with findings on miRNAs dysregulated in human patients and finally highlight a potential use of miRNAs as biomarkers for liver injury, fibrosis and cancer.
Keywords: miRNAs, liver, fibrosis, inflammation, biomarker
Liver fibrosis and inflammation
Chronic liver inflammation is a major health burden worldwide, potentially leading to liver fibrosis, liver cirrhosis, hepatocellular carcinoma and finally death (1). From a pathophysiological point of view, liver fibrosis represents the result of a wound-healing response to chronic or repeated liver injury (2). Liver injury is associated with liver cell death and consecutive regeneration of parenchymal cells to replace necrotic or apoptotic cells. If the hepatic injury persists, liver regeneration may fail, and hepatocytes are replaced by extracellular matrix (ECM) (2). While in early stages, fibrotic material is only located around portal tracts (in case of viral hepatitis) or pericentral (in case of alcohol-induced liver disease), in later stages, bridging collagen bands occurs (2). Hepatic stellate cells (HSCs) represent the main source of ECM-production in the injured liver (3). Quiescent HSCs are located in space of Disse and are the major storage sites of vitamin A (4). During liver injury, HSCs become activated and acquire contractile, pro-inflammatory, and fibrogenic properties (5). Such activated HSCs secrete tremendous amounts of ECM and in advanced stages of liver fibrosis, the liver may contain up to six times more ECM than normal (2).
Chronic liver injury and liver fibrosis are associated with local and systemic inflammatory response, highlighting that hepatic cell types other than HSCs are also involved in fibrogenesis. Hepatocytes that become injured, release reactive oxygen species (ROS) and inflammatory mediators and thereby induce recruitment of inflammatory cells to the liver. Inflammatory cells, either lymphocytes or polymorphonuclear cells, activate HSCs to secrete collagen (6). Activated HSCs themselves secrete inflammatory chemokines, express cell adhesion molecules, and modulate the activation of lymphocytes and Kupffer cells that play a major role in liver inflammation by releasing proinflammatory mediators (7). Therefore, fibrosis is mediated by a vicious circle in which inflammatory and fibrogenic cells are activated and stimulate each other (2).
From a clinical point of view, liver fibrosis represents a common consequence of chronic liver diseases. At present, hepatic fibrosis is still not sufficiently treatable and often progress to liver cirrhosis representing the major risk factor for the development of hepatocellular carcinoma (8). Despite recent advances, e.g., in the treatment of viral hepatitis, pharmacological strategies in treatment or prevention of liver cirrhosis are still lacking and prognosis of patients with liver cirrhosis remains poor. Thus, a better understanding of the mechanisms underlying liver inflammation, hepatofibrogenesis and the development of liver cirrhosis are urgently needed. Here we review the available literature on the role of microRNAs (miRNA, miR) in these processes. Such knowledge might help to develop novel, innovative strategies for the prevention and therapy of acute and chronic liver diseases.
MicroRNAs
MicroRNAs represent a new class of small RNAs ~18-25 nucleotides in length, that do not withhold the information to encode for proteins but have been shown to be involved in the regulation of gene expression on a post-transcriptional level (9). miRNAs were first described in C. elegans and are highly conserved among animals, humans, plants and viruses (10). miRNAs bind via partial complementary sequences to 3’ untranslated region (UTR) of the target mRNAs and thereby inhibit gene expression (11). Due to this partial complementarity, a single miRNA can regulate multiple mRNAs simultaneously and on the other hand, multiple miRNAs can regulate the expression of a single mRNA (12), highlighting that miRNAs are highly suitable for fine-tuning of whole networks of genes. In line, in-silico prediction algorithms revealed that up to 60% of protein coding genes are regulated by miRNAs, underscoring the tremendous impact of miRNAs for the regulation of gene expression (13,14).
miRNAs have a cell- and tissue- specific expression. They control a broad spectrum of biological processes including cell proliferation, differentiation, development, metabolism, apoptosis, secretion and viral infection (15-17). Deregulation in their expression levels has been linked to human diseases such as cancer, diabetes, nervous system disorders, cardiovascular diseases, asthma and autoimmune diseases (18). The biogenesis of miRNA starts with the transcription of miRNA-encoding genes by RNA polymerase II to ~3-4 kilobases long primary transcripts (pri-miRNAs) (19). Pri-miRNAs are cleaved into ~60-70 nt long stem-loop structured precursor miRNAs (pre-miRNAs) by the nuclear RNase III endonuclease Drosha and are exported to the cytoplasm by Exportin-5. In the cytoplasm, pre-miRNA cleaves into mature miRNA/miRNA* duplex by attaching to RNase III Dicer, TAR RNA binding protein (TRBP) and Argonaute-2 (Ago-2). The miRNA* strand is degraded whereas the mature miRNA strand called guide strand is loaded into RNA-induced silencing complex (RISC) and binds to its target gene. Once a miRNA binds to its target gene, either of two mechanisms of action occurs: (I) mRNA degradation or destabilization; or (II) translation repression (20-22). Based on these mechanisms, several algorithms for in-silico prediction of targets for a miRNA, including miRanda, Pictar, TargetScan, RNA22, PITA, and miRdb were developed. These prediction algorithms take into account the following parameters: (I) location of miRNA; (II) Watson Crick base pairing; (III) thermodynamic properties of miRNA: mRNA binding (23).
miRNAs as mediators of inflammation
Chronic inflammatory diseases, deriving from abnormal immune response, have complex mechanism that involves changes in gene expression in immune cells. The emerging role of miRNAs in innate and adaptive immunity strongly suggests an association with regulation of inflammatory diseases. To date, there are several miRNAs including miR-29, miR-133a, mir-155, miR-221, miR-223 or miR-652, with a known functional role in specific inflammatory diseases such as rheumatoid arthritis, multiple sclerosis, cancer, inflammatory bowel disease, inflammatory skin disease and inflammatory airway disease (24). Recently, a role of miRNAs in liver inflammation and hepatic fibrogenesis was also suggested by different authors (25).
miRNAs as modulators of liver inflammation
Various factors including viral and bacterial infections, alcohol abuse, toxins and dietary factors may promote liver inflammation. Immune cells and pro-inflammatory chemokines play an important role in the regulation of the inflammatory response within the fibrotic cascade. Monocyte chemotactic protein type 1 (MCP-1) and RANTES stimulate fibrogenesis whereas the opposite effect is exerted by IL-10 and IFN-γ (2,26). Pro-inflammatory cytokines including TNF, IL-6 and IL-1β are generated by leukocytes at the site of injury. Such immune responses elicited by the injured liver are also controlled by miRNAs (27,28). The compelling evidence for the importance of miRNA can be derived from studies using mice with a knock-out for Dicer1 (29). In KO-mice of 2-4 months of age, AST and ALT levels were significantly higher than in littermate controls, along with severe portal inflammation in H&E sections of knock-out mice (29). In addition, reticulin staining and TUNEL analysis revealed a loss of hepatocytes and an increase in apoptotic bodies, respectively.
miR-122 represents the most abundant liver-miRNA and was linked to hepatitis C virus infection and regulation of cholesterol synthesis in the liver. Just recently, a role of miR-122 has emerged in liver inflammation. miR-122-deficient mice displayed a phenotype of hepatic inflammation, fibrosis, and hepatocellular carcinoma, suggesting that miR-122 has an anti-inflammatory role in the liver. On a cellular level, knock-out mice showed an up-regulation of Ccl2, which resulted in the intrahepatic recruitment of CD11bhiGr1+ inflammatory cells, which are a major source of cytokines such as IL-6 and TNF during liver inflammation (30).
Besides liver specific miR-122, miRNA-132 has emerged as a mediator of inflammation in chronic liver diseases. Up-regulation of miR-132 was described in the whole liver as well as in Kupffer cells isolated from mice after chronic ethanol administration (25). Next to miR-132, induction in miR-155 expression was reported in Kupffer cells after alcohol feeding, and TNF has been identified as a target of miR-155 to promote liver inflammation. Moreover, in mouse models of NASH miR-155 levels were increased and high miR-155 expression was linked with the development of HCC in these mice by regulating C/EBP β levels. Remarkably, hepatic levels of miR-155 were also increased in patients with HCV and were shown to promote HCC through Wnt signaling (31). Finally, increased expression of miR-155 was described in peripheral monocytes from patients with chronic therapy resistant HCV-infection whereas no increase was found in patients who responded to treatment (32). Unlike miR-155, miR-146a is a negative regulator of TLR signaling; it dampens pro-inflammatory responses via TRAF6 and IRAK-1. Consistent with its negative regulatory role in inflammation, mice deficient in miR-146a acquire hyperinflammation and myeloproliferative phenotypes owing to hyperactivation of NF-κB (33).
miRNAs as modulators of liver fibrosis
Wound healing and fibrotic remodeling processes during liver fibrosis are associated with alterations in hepatic miRNA expression patterns. As mentioned above, miR-122 represents the most abundant liver-miRNA (34) and miR-122 is involved in controlling cholesterol synthesis and hepatitis C virus (HCV) replication. miR-122 is found at significantly lower levels during development of liver fibrosis (35-37). The lower levels of miR-122 expression are controlled directly by c-Myc oncogene that associates with miR-122 upstream promoter region or indirectly by HNF-3ß (38). Just recently, inhibition of miR-122 has been proposed as a new strategy for antiviral therapy of hepatitis C in fibrosis patients. Miravirsen is a 15-nucleotide phosphorothioate mixed locked nucleic acid (LNA)/DNA oligonucleotide complementary to 5’UTR of miR-122 (39). In clinical phase 2a trial, dose-dependent administration resulted in a decrease of viral RNA levels in patients with chronic HCV infection (40), supporting the use of miRNA-based strategies in treatment and prevention of liver fibrosis.
Besides miR-122 a whole panel of other miRNAs with a high expression in hepatocytes, including miR-125b and miR-22 were also described to be down-regulated in liver samples from patients with liver fibrosis or cirrhosis. Moreover, lower expression levels of miR-150 and miR-194 were associated with hepatofibrogenesis in a stage dependent manner. Very prominently, down-regulation of members of the miR-29 family as well as miR-19b was recently linked to chronic liver inflammation and fibrogenesis in different mouse models of liver cirrhosis and different species including the human (41-43). Besides these down-regulated miRNAs, miR-21, the miR-34 family, members of the miR-199- as well as the miR-200-family increased (44-46) in fibrotic liver diseases in both mouse and human. IL-6/Stat3 binds to 5’ promoter region of miR-21 and increases the expression of miR-21 in both human hepatocytes and HSC (47).
Recent data suggested that activation and trans-differentiation of HSC during the myofibroblastic transition process is associated with, and at least partially mediated by altered miRNA expression (48,49). HSCs are the major mesenchymal cells in liver whose activation drives fibrosis and the main producer of ECM like collagens, laminin, proteoglycans and fibronectin. Exposure to inflammatory cytokines, TGFβ and PDGF activate HSC which eventually transformed into myofibroblast-like cells secreting large amounts of ECM and leading finally to liver fibrosis (Tables 1,2 and Figure 1).
Table 1. Profibrotic miRNAs.
| miRNA | Models/site of action | Function | Target genes | Reference |
|---|---|---|---|---|
| miR-33a | TGFβ1-induced HSCs (human) | ↑ Proliferation of HSCs | PPARα | (50) |
| miR-200c | HCV-infected liver (human) | ↑ cSrc signaling cascade | FAP-1 | (51) |
| miR-199a/miR-199a*/miR-199b | CCl4-induced liver fibrosis (human, mouse) | ↑ ECM synthesis | (46) | |
| miR-34a | DMN-induced, CCl4-treated, ethanol-mediated ALD (human, mouse) | ↑ Proliferation | RXRα, ACSL1, CASP2, SIRT1 | (52,53) |
| miR-27a/b | Activated HSC (rat) | ↑ Lipid accumulation and proliferation of HSC | RXRα | (49) |
| miR-21 | PDGF-BB induced HSC, HCV-infected liver, CCl4-treated (human) | ↑ TGFβ signaling | PTEN, Smad7 | (54,55) |
| miR-221/222 | HCV-infected liver (F3-F4), NASH (F3-F4), TAA-induced, HSC, Biliary atresia (human, mouse) | ↑ Col1A1 and MMP-2 expression; ↑ p-AKT and cell proliferation | CDKN1B, PPP2R2A | (56) |
| miR-181a/b | TGFβ1-induced HSCs (human, rat) | ↑ Cell cycle and proliferation | p27 | (57) |
| miR-214 | HCV-infected liver(F3-F4), MCDD-fed, activated HSC (human, mouse) | ↑ α-SMA, Col1A1, PDGFR-β, FN1, MMP-2, MMP-9, TGFβ1 expression | (58) |
Table 2. Antifibrotic miRNAs.
| miRNA | Models/site of action | Function | Target genes | Reference |
|---|---|---|---|---|
| miR-29 | IFNα & TGFβ1 stimulated HSC (human, mouse) | ↓ ECM synthesis | FBN1, ELN1, MMP2, ITGB1, Col1a1, Col1a2, Col4a5, Col5a3 | (59) |
| miR-150 | Activated HSC, NASH (rat, human) | ↓ Activation and proliferation in HSC | c-myb, Col4A4, Sp1 | (60) |
| miR-132 | Activated HSC from CCl4-treated, and BDL-livers (mouse) | ↓ PPARγ transcription | MeCP2 | (61) |
| miR-122 | NASH,ALD (human) | ↓ Proliferation and activation of HSCs | P4HA1 | (62) |
| miR-335 | Activated HSC (rat) | ↓ Cell migration | TNC | (63) |
| miR-101 | Activated HSC from CCl4-treated (mouse) | ↓ TGFβ, signaling, apoptosis | CTGF, TSP-1, KLF6 | (64) |
| miR-107/miR-449a | HCV (human) | ↓ NFκB transcription | YKL40, NOTCH-1 | (65) |
| miR-200a/b | TGFβ-induced HSC, CCl4-treated (mouse, rat, human) | ↓ TGFβ dependent EMT, proliferation | ZEB1/2, TGFβ2, β-catenin | (66,67) |
| miR-126 | Activated HSC(rat) | ↓ Migration of HSC | CRK | (68) |
| miR-483 | HSC, CCl4-treated (mouse) | ↓ TGFβ-induced PDGFβ and TIMP2 expression | Socs3 | (69) |
| miR-195 | Activated HSC (human) | ↓ Cell proliferation | CyclinE1 | (70) |
| miR-15b/16 | Activated HSC (rat) | ↑ Apoptosis | Bcl-2 | (48) |
| miR-133 | TGFβ-treated HSC (mouse, human) | ↑ ECM secretion | Col1A1 | (71) |
| miR-146a | CCl4-treated, TGF-β1-treated HSCs (rat) | ↓ Proliferation of HSC | SMAD4 | (72) |
| miR-19b | Activated HSC (rat, human) | ↓ Fibrotic TGF-β signaling | TGF-βRII | (43) |
Figure 1.
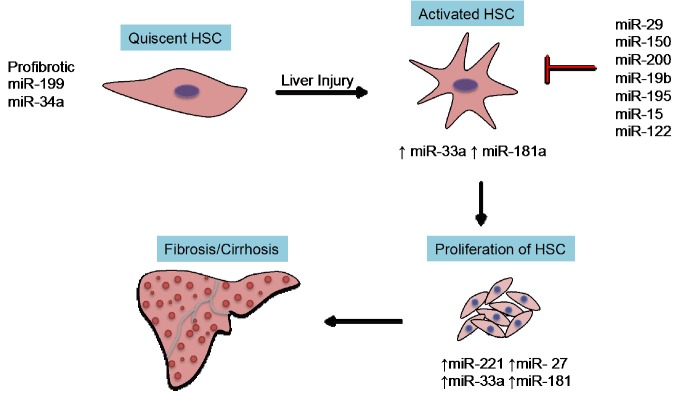
Key miRNAs in fibrogenetic events. The key event involving activation of quiescent HSC requires profibrotic miRNAs (miR-199 or miR-34a). Several antifibrotic miRNAs (miR-29, miR-133 or miR-335) inhibit the synthesis of extracellular matrix proteins. HSC, hepatic stellate cell.
Microarray-based analysis on miRNA expression in activated HSC from mice and rats revealed whole panels of miRNAs deregulated during activation and transdifferentiation of these cells. As an example, in activated HSCs from rats, 12 miRNAs (miR-874, miR-29c*, miR-501, miR-349, miR-325-5p, miR-328, miR-138, miR-143, miR-207, miR-872, miR-140, miR-193) displayed a significant up-regulation, while 9 miRNAs (miR-341, miR-20b-3p, miR-15b, miR-16, miR-375, miR-122, miR-146a, miR-92b, and miR-126) were down-regulated (48). Of note, only few miRNAs showed a consistent regulation between the different studies. Ji et al. identified the miR-27 family as part of a 15 miRNA signature associated with the activation of HSC in rats after 10 days. Inhibition of miR-27 led to a decrease in cell proliferation and an accumulation of cytoplasmic lipid droplets via targeting retinoid X receptor α (RXRα) mRNA. On the other hand, silencing of miR-27 did not affect cell apoptosis or expression of collagen type I and α-SMA, underlining the need for further studies to clarify the specific role of miR-27 in the pathophysiology of liver fibrosis (49). Besides miR-27, overexpression of miR-335 inhibited rat HSC migration and reduced collagen type I and α-SMA via inhibition of tenascin-C (TNC) (63). Cell apoptosis promotes the resolution of fibrosis and targeting apoptosis of HSC was associated with decrease in liver fibrosis. Both the miR-15 and miR-16 family were found deregulated in activated rat HSC and were shown to be involved in the regulation of HSC apoptosis by targeting the anti-apoptotic protein Bcl-2 in case of miR-15 (48) or cyclin D1 in case of miR-16 (73). We and others demonstrated a stellate cell specific expression of the miR-29 family of miRNAs. Activation of HSC was linked to a dramatic and specific down-regulation of all three members of this family and associated with an increase in ECM production (42,59), thus providing a potential target for a cell specific therapy of hepatofibrogenesis.
In the following part we summarize the role of prominent miRNAs in liver fibrosis.
miR-29 family
The miR-29 family consists of miR-29a, miR-29b, miR-29c that differs by only two or three bases. The first description of miR-29 in fibrotic diseases was delivered by van Rooij et al., demonstrating that miR-29s were down-regulated in a model of cardiac fibrosis (74). Regarding liver fibrosis, Ogawa et al. (75) provided the first line of evidence that miR-29b is down-regulated in activated mouse HSCs and regulates collagen expression via its interaction with Col1A1 3'UTR. Overexpression of miR-29b suppressed Col1A1 expression and prevented the up-regulation of collagens in TGF-β1 stimulated HSCs. By using unbiased miRNA profiling on RNA extracts from livers CCl4-treated mice we demonstrated that all three members of the miR-29 family were down-regulated and correlated with the degree of liver fibrosis. These results could be confirmed in different models of liver fibrosis and were independent of the mouse strain analyzed.
Moreover, down-regulation of miR-29 was not only limited to the above models as patients with HCV-infection and consecutive fibrosis demonstrated a significant down-regulation of miR-29 in their livers. HCV induces an inflammatory response and in line we and others found a significant down-regulation of miR-29 in LPS-stimulated HSC and primary hepatocytes, which suggests that, besides pro-fibrotic TGF-signaling also pro-inflammatory pathways contribute to the regulation of miR-29 (42,59,75). Yet another study showed the counteracting effects of pro-fibrotic TGFβ and anti-fibrotic HGF on miR-29 expression levels in HSC. Finally, other pro-fibrogenic growth factors such as PDGF-C and IGF-A are repressed in miR-29 overexpressed primary HSC as well as myofibroblastic HSC-T6 cells, placing miR-29 into a crucial position in the network of pro- and anti-inflammatory mediators regulating liver fibrosis. Besides a regulation by these classical inflammatory of fibrotic mediators, an up-regulation in miR-29-expression was described after stimulation with estradiol. Interestingly, female mice, displaying higher intrahepatic miR-29 levels are more resistant to induction of liver fibrosis in the CCl4 model when compared with male mice. Estradiol levels are regulated by pro-inflammatory transcriptional factors such NFκB and STAT-1, thus providing a functional link between inflammatory and fibrotic pathways (76). Finally, miR-29c was also found down-regulated in mouse models of NASH (45), highlighting the prominent role of this miRNA in chronic liver diseases.
In summary, miR-29 is probably the best characterized miRNA in murine liver fibrosis integrating both pro-fibrotic and inflammatory signals in HSC to modulate fibrogenesis, thus representing a promising candidate for therapeutic strategies against liver fibrosis.
miR-133a
Alterations in miR-133a, a member of the miR-1/miR-133 cluster of miRNAs, were recently described in inflammatory diseases and sepsis as well as in a setting of vascular and cardiac remodeling (77,78). Moreover, it was shown that delivery of miR-1/miR-133 into skeletal muscle promoted muscle regeneration and prevented scar formation (77). In liver fibrosis miR-133a expression did not vary when levels were compared between full livers from CCl4 treated and control animals. However, miR-133a was significantly down-regulated in human and murine HSC during fibrogenesis as well as in cardiac fibroblasts of fibrotic hearts. Treatment of primary murine and human HSC or fibroblasts with TGF-β resulted in a significant down-regulation of miR-133a in these cells. In turn, overexpression of miR-133a led to decreased expression of collagens, suggesting a direct role of miR-133a in activation of fibroblasts/HSC during organ fibrosis. Finally, miR-133a was described as a potential serum marker for development and progression of liver cirrhosis (71).
miR-150
Microarray analysis performed on HSC isolated from bile duct ligation (BDL) in rats, revealed a significant down-regulation of miR-150. In these cells, overexpression of miR-150 was linked to cell proliferation and apoptosis. Moreover, overexpression of miR-150 in human HSC cells lowered the expression of α-SMA, one of the markers of stellate cell activation along with different collagens via inhibiting its target proto-oncogene c-myb (60). Another study revealed a down-regulation of miR-150 upon stimulation of human HSC with TGF-β1 in a dose- and time-dependent manner. These data suggest a role of miR-150 as an anti-fibrotic miRNA by inhibiting activation of HSCs during chronic liver diseases (79).
miR-34a
The miR-34 family is composed of miR-34a, miR-34b and miR-34c. The first piece of evidence of the role of miR-34a in hepatic fibrosis comes from dimethylnitrosamine (DMN)-induced liver fibrosis in rats where miR-34 family was upregulated. The direct target gene of miR-34a, ASCL1 was suppressed during the development of hepatic fibrosis (80). Microarray analysis identified upregulated miR-34a expression in ethanol-exposed liver tissues compared to normal tissues. Overexpression of miR-34a increased the proliferation, migration and transformation of hepatic cells with ethanol treatment. These effects are mediated through direct target genes, CASP2 and SIRT1 that may be involved in tissue remodeling during disease progression from normal liver through cirrhosis to HCC (52). p53 activation upregulated miR-34a expression by negatively regulating RXRα in human fibrotic livers (53). The upstream transcriptional target of miR-34 is tumor suppressor p53 that may mediate cell cycle arrest (81). Furthermore, serum level of miR-34a was significantly higher in CHC and NAFLD patients, indicating novel and noninvasive biomarker of diagnosis (82).
miR-199/miR-200
The miR-199 family consists of miR-199a and miR-199b differing by only one nucleotide. Microarray analysis identified higher expression of miR-199a-5p, miR-199b and miR-199b* in CCl4 –treated mice and human chronic hepatitis C liver tissues compared to controls. Interestingly up-regulation of miR-199 was indicative for progression of chronic liver injury into liver fibrosis and advanced cirrhosis (46). In line, LX-2 cells treated with TGF-β demonstrated an up-regulation of different members of the miR-199 family (42,46). A similar phenotype was also described by Pottier and colleagues (83) demonstrating that miR-199a-5p is critically involved in the development of cardiac and lung fibrosis by down-regulating Stathmin1.
Similarly, the miR-200 family (miR-200a, miR-200b) was over-expressed in CCl4-treated mice and linked to diseases progression in humans (84). It is also shown that miR-200b was up-regulated in livers of mice fed with methyl-deficient diet with consequent down-regulation of Zeb1 mRNA level (44). This results determine the differential expression of miRNAs are susceptible to NASH development.
miRNAs as circulating biomarkers for liver inflammation and fibrosis
Besides being involved in the regulation of gene expression, increasing evidence for a role of miRNAs as blood based biomarkers for different diseases states emerged in the last years. miRNAs can also been detected and quantified in the blood, as well as in plasma and serum, saliva and urine (85). Due to their small size and their specific structure, circulating miRNAs are extraordinarily stable towards conditions that usually would degrade most RNAs. Therefore, in contrast to proteins miRNAs are more resistant against degradation and are thus regarded as “optimal” biomarkers with a high degree of stability against most biasing influence (86,87). In the liver, it was demonstrated that various miRNAs (e.g., the miR-21 and miR-29 family, miR-223 and miR-571) are strongly correlated with the degree of liver cirrhosis while other miRNAs such as miR-652 reflects the presence of liver inflammation (25,88,89) or ongoing liver damage (90). The role of circulating miRNAs as biomarkers for liver inflammation and liver fibrosis is extensively reviewed in (20,25,91).
Conclusions
The data reviewed here suggest a key position for miRNAs in the regulation of liver inflammation and liver fibrosis. Based on these findings, miRNAs may be interesting candidates to develop future strategies to identify, prevent or treat hepatic fibrosis. However, we have just started to understand the role of miRNAs networks in the process of hepatic fibrogenesis, and more functional studies are needed to identify the role of miRNAs in the pathophysiology of chronic liver injury, liver inflammation and hepatic fibrosis.
Acknowledgements
We would like to thank Dr. Mihael Vucur and Dr. Jeremie Gautheron for their valuable comments.
Disclosure: The authors declare no conflict of interest.
References
- 1.Tacke F.Functional role of intrahepatic monocyte subsets for the progression of liver inflammation and liver fibrosis in vivo. Fibrogenesis Tissue Repair 2012;5:S27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bataller R, Brenner DA. Liver fibrosis. J Clin Invest 2005;115:209-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gäbele E, Brenner DA, Rippe RA. Liver fibrosis: Signals leading to the amplification of the fibrogenic hepatic stellate cell. Front Biosci 2003;8:d69-77. [DOI] [PubMed] [Google Scholar]
- 4.Hellerbrand C.Hepatic stellate cells--the pericytes in the liver. Pflugers Arch 2013;465:775-8. [DOI] [PubMed] [Google Scholar]
- 5.Milani S, Herbst H, Schuppan D, et al. Procollagen expression by nonparenchymal rat liver cells in experimental biliary fibrosis. Gastroenterology 1990;98:175-84. [DOI] [PubMed] [Google Scholar]
- 6.Casini A, Ceni E, Salzano R, et al. Neutrophil-derived superoxide anion induces lipid peroxidation and stimulates collagen synthesis in human hepatic stellate cells: Role of nitric oxide. Hepatology 1997;25:361-7. [DOI] [PubMed] [Google Scholar]
- 7.Viñas O, Bataller R, Sancho-Bru P, et al. Human hepatic stellate cells show features of antigen-presenting cells and stimulate lymphocyte proliferation. Hepatology 2003;38:919-29. [DOI] [PubMed] [Google Scholar]
- 8.Roderburg C, Gautheron J, Luedde T.TNF-dependent signaling pathways in liver cancer: promising targets for therapeutic strategies? Dig Dis 2012;30:500-7. [DOI] [PubMed] [Google Scholar]
- 9.Flynt AS, Lai EC. Biological principles of microRNA-mediated regulation: shared themes amid diversity. Nat Rev Genet 2008;9:831-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lee RC, Ambros V. An extensive class of small RNAs in Caenorhabditis elegans. Science 2001;294:862-4. [DOI] [PubMed] [Google Scholar]
- 11.Pillai RS. MicroRNA function: multiple mechanisms for a tiny RNA? RNA 2005;11:1753-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang S, Li Q, Liu J, et al. A novel computational framework for simultaneous integration of multiple types of genomic data to identify microRNA-gene regulatory modules. Bioinformatics 2011;27:i401-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Friedman RC, Farh KK, Burge CB, et al. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res 2009;19:92-105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Selbach M, Schwanhaeusser B, Thierfelder N, et al. Widespread changes in protein synthesis induced by microRNAs. Nature 2008;455:58-63. [DOI] [PubMed] [Google Scholar]
- 15.Chen CZ, Li L, Lodish HF, et al. MicroRNAs modulate hematopoietic lineage differentiation. Science 2004;303:83-6. [DOI] [PubMed] [Google Scholar]
- 16.Cheng AM, Byrom MW, Shelton J, et al. Antisense inhibition of human miRNAs and indications for an involvement of miRNA in cell growth and apoptosis. Nucleic Acids Res 2005;33:1290-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Karp X, Ambros V.Encountering microRNAs in cell fate signaling. Science 2005;310:1288-9. [DOI] [PubMed] [Google Scholar]
- 18.Ha TY. MicroRNAs in Human Diseases: From Cancer to Cardiovascular Disease. Immune Netw 2011;11:135-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Takada S, Asahara H.Current strategies for microRNA research. Mod Rheumatol 2012;22:645-53. [DOI] [PubMed] [Google Scholar]
- 20.Haybaeck J, Zeller N, Heikenwalder M.The parallel universe: microRNAs and their role in chronic hepatitis, liver tissue damage and hepatocarcinogenesis. Swiss Med Wkly 2011;141:w13287. [DOI] [PubMed] [Google Scholar]
- 21.Carthew RW, Sontheimer EJ. Origins and Mechanisms of miRNAs and siRNAs. Cell 2009;136:642-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Winter J, Jung S, Keller S, et al. Many roads to maturity: microRNA biogenesis pathways and their regulation. Nat Cell Biol 2009;11:228-34. [DOI] [PubMed] [Google Scholar]
- 23.Zheng H, Fu R, Wang JT, et al. Advances in the Techniques for the Prediction of microRNA Targets. Int J Mol Sci 2013;14:8179-87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Plank M, Maltby S, Mattes J, et al. Targeting translational control as a novel way to treat inflammatory disease: the emerging role of MicroRNAs. Clin Exp Allergy 2013;43:981-99. [DOI] [PubMed] [Google Scholar]
- 25.Szabo G, Bala S.MicroRNAs in liver disease. Nat Rev Gastroenterol Hepatol 2013;10:542-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Paik YH, Schwabe RF, Bataller R, et al. Toll-like receptor 4 mediates inflammatory signaling by bacterial lipopolysaccharide in human hepatic stellate cells. Hepatology 2003;37:1043-55. [DOI] [PubMed] [Google Scholar]
- 27.Liu Y, Munker S, Mullenbach R, et al. IL-13 Signaling in Liver Fibrogenesis. Front Immunol 2012;3:116-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pellicoro A, Ramachandran P, Iredale JP, et al. Liver fibrosis and repair: immune regulation of wound healing in a solid organ. Nat Rev Immunol 2014;14:181-94. [DOI] [PubMed] [Google Scholar]
- 29.Hand NJ, Master ZR, Le Lay J, et al. Hepatic Function Is Preserved in the Absence of Mature MicroRNAs. Hepatology 2009;49:618-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wen J, Friedman JR. miR-122 regulates hepatic lipid metabolism and tumor suppression. J Clin Invest 2012;122:2773-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang Y, Wei W, Cheng N, et al. Hepatitis C virus-induced up-regulation of microRNA-155 promotes hepatocarcinogenesis by activating Wnt signaling. Hepatology 2012;56:1631-40. [DOI] [PubMed] [Google Scholar]
- 32.Bala S, Tilahun Y, Taha O, et al. Increased microRNA-155 expression in the serum and peripheral monocytes in chronic HCV infection. J Transl Med 2012;10:151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhao JL, Rao DS, Boldin MP, et al. NF-kappaB dysregulation in microRNA-146a-deficient mice drives the development of myeloid malignancies. Proc Natl Acad Sci U S A 2011;108:9184-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jopling CL, Norman KL, Sarnow P. Positive and negative modulation of viral and cellular mRNAs by liver-specific microRNA miR-122. Cold Spring Harb Symp Quant Biol 2006;71:369-76. [DOI] [PubMed] [Google Scholar]
- 35.Cheung O, Puri P, Eicken C, et al. Nonalcoholic steatohepatitis is associated with altered hepatic MicroRNA expression. Hepatology 2008;48:1810-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Morita K, Taketomi A, Shirabe K, et al. Clinical significance and potential of hepatic microRNA-122 expression in hepatitis C. Liver Int 2011;31:474-84. [DOI] [PubMed] [Google Scholar]
- 37.Henke JI, Goergen D, Zheng J, et al. microRNA-122 stimulates translation of hepatitis C virus RNA. EMBO J 2008;27:3300-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang B, Hsu SH, Wang X, et al. Reciprocal regulation of microRNA-122 and c-Myc in hepatocellular cancer: role of E2F1 and transcription factor dimerization partner 2. Hepatology 2014;59:555-66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lindow M, Kauppinen S.Discovering the first microRNA-targeted drug. J Cell Biol 2012;199:407-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Janssen HL, Reesink HW, Lawitz EJ, et al. Treatment of HCV infection by targeting microRNA. N Engl J Med 2013;368:1685-94. [DOI] [PubMed] [Google Scholar]
- 41.Kwiecinski M, Noetel A, Elfimova N, et al. Hepatocyte growth factor (HGF) inhibits collagen I and IV synthesis in hepatic stellate cells by miRNA-29 induction. PloS One 2011;6:e24568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Roderburg C, Urban GW, Bettermann K, et al. Micro-RNA Profiling Reveals a Role for miR-29 in Human and Murine Liver Fibrosis. Hepatology 2011;53:209-18. [DOI] [PubMed] [Google Scholar]
- 43.Lakner AM, Steuerwald NM, Walling TL, et al. Inhibitory effects of microRNA 19b in hepatic stellate cell-mediated fibrogenesis. Hepatology 2012;56:300-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Alisi A, Da Sacco L, Bruscalupi G, et al. Mirnome analysis reveals novel molecular determinants in the pathogenesis of diet-induced nonalcoholic fatty liver disease. Lab Invest 2011;91:283-93. [DOI] [PubMed] [Google Scholar]
- 45.Pogribny IP, Starlard-Davenport A, Tryndyak VP, et al. Difference in expression of hepatic microRNAs miR-29c, miR-34a, miR-155, and miR-200b is associated with strain-specific susceptibility to dietary nonalcoholic steatohepatitis in mice. Lab Invest 2010;90:1437-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Murakami Y, Toyoda H, Tanaka M, et al. The Progression of Liver Fibrosis Is Related with Overexpression of the miR-199 and 200 Families. PLoS One 2011;6:e16081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Francis H, McDaniel K, Han Y, et al. Regulation of the extrinsic apoptotic pathway by microRNA-21 in alcoholic liver injury. J Biol Chem 2014;289:27526-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Guo CJ, Pan Q, Li DG, et al. miR-15b and miR-16 are implicated in activation of the rat hepatic stellate cell: An essential role for apoptosis. J Hepatol 2009;50:766-78. [DOI] [PubMed] [Google Scholar]
- 49.Ji J, Zhang J, Huang G, et al. Over-expressed microRNA-27a and 27b influence fat accumulation and cell proliferation during rat hepatic stellate cell activation. FEBS Lett 2009;583:759-66. [DOI] [PubMed] [Google Scholar]
- 50.Li ZJ, Ou-Yang PH, Han XP. Profibrotic effect of miR-33a with Akt activation in hepatic stellate cells. Cell Signal 2014;26:141-8. [DOI] [PubMed] [Google Scholar]
- 51.Ramachandran S, Basha HI, Sarma NJ, et al. Hepatitis C Virus Induced miR200c Down Modulates FAP-1, a Negative Regulator of Src Signaling and Promotes Hepatic Fibrosis. PLoS One 2013;8:e70744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Meng F, Glaser SS, Francis H, et al. Epigenetic Regulation of miR-34a Expression in Alcoholic Liver Injury. Am J Pathol 2012;181:804-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Oda Y, Nakajima M, Tsuneyama K, et al. Retinoid X receptor alpha in human liver is regulated by miR-34a. Biochem Pharmacol 2014;90:179-87. [DOI] [PubMed] [Google Scholar]
- 54.Wei J, Feng L, Li Z, et al. MicroRNA-21 activates hepatic stellate cells via PTEN/Akt signaling. Biomed Pharmacother 2013;67:387-92. [DOI] [PubMed] [Google Scholar]
- 55.Marquez RT, Bandyopadhyay S, Wendlandt EB, et al. Correlation between microRNA expression levels and clinical parameters associated with chronic hepatitis C viral infection in humans. Lab Invest 2010;90:1727-36. [DOI] [PubMed] [Google Scholar]
- 56.Shen WJ, Dong R, Chen G, et al. microRNA-222 modulates liver fibrosis in a murine model of biliary atresia. Biochem Biophys Res Commun 2014;446:155-9. [DOI] [PubMed] [Google Scholar]
- 57.Wang B, Li W, Guo K, et al. miR-181b Promotes hepatic stellate cells proliferation by targeting p27 and is elevated in the serum of cirrhosis patients. Biochem Biophys Res Commun 2012;421:4-8. [DOI] [PubMed] [Google Scholar]
- 58.Iizuka M, Ogawa T, Enomoto M, et al. Induction of microRNA-214-5p in human and rodent liver fibrosis. Fibrogenesis Tissue Repair 2012;5:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bandyopadhyay S, Friedman RC, Marquez RT, et al. Hepatitis C Virus Infection and Hepatic Stellate Cell Activation Downregulate miR-29: miR-29 Overexpression Reduces Hepatitis C Viral Abundance in Culture. J Infect Dis 2011;203:1753-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Venugopal SK, Jiang J, Kim TH, et al. Liver fibrosis causes downregulation of miRNA-150 and miRNA-194 in hepatic stellate cells, and their overexpression causes decreased stellate cell activation. Am J Physiol Gastrointest Liver Physiol 2010;298:G101-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mann J, Chu DC, Maxwell A, et al. MeCP2 controls an epigenetic pathway that promotes myofibroblast transdifferentiation and fibrosis. Gastroenterology 2010;138:705-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Li J, Ghazwani M, Zhang Y, et al. miR-122 regulates collagen production via targeting hepatic stellate cells and suppressing P4HA1 expression. J Hepatol 2013;58:522-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chen C, Wu CQ, Zhang ZQ, et al. Loss of expression of miR-335 is implicated in hepatic stellate cell migration and activation. Exp Cell Res 2011;317:1714-25. [DOI] [PubMed] [Google Scholar]
- 64.Tu X, Zhang H, Zhang J, et al. MicroRNA-101 suppresses liver fibrosis by targeting the TGF beta signalling pathway. J Pathol 2014;234:46-59. [DOI] [PubMed] [Google Scholar]
- 65.Sarma NJ, Tiriveedhi V, Subramanian V, et al. Hepatitis C Virus Mediated Changes in miRNA-449a Modulates Inflammatory Biomarker YKL40 through Components of the NOTCH Signaling Pathway. PLoS One 2012;7:e50826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yang JJ, Tao H, Hu W, et al. MicroRNA-200a controls Nrf2 activation by target Keap1 in hepatic stellate cell proliferation and fibrosis. Cell Signal 2014;26:2381-9. [DOI] [PubMed] [Google Scholar]
- 67.Sun X, He Y, Ma TT, et al. Participation of miR-200a in TGF-beta 1-mediated hepatic stellate cell activation. Mol Cell Biochem 2014;388:11-23. [DOI] [PubMed] [Google Scholar]
- 68.Gong XH, Chen C, Hou P, et al. Overexpression of miR-126 Inhibits the Activation and Migration of HSCs through Targeting CRK. Cell Physiol Biochem 2014;33:97-106. [DOI] [PubMed] [Google Scholar]
- 69.Li F, Ma N, Zhao R, et al. Overexpression of miR-483-5p/3p cooperate to inhibit mouse liver fibrosis by suppressing the TGF-beta stimulated HSCs in transgenic mice. J Cell Mol Med 2014;18:966-74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sekiya Y, Ogawa T, Iizuka M, et al. Down-Regulation of Cyclin E1 Expression by MicroRNA-195 Accounts for Interferon-beta-Induced Inhibition of Hepatic Stellate Cell Proliferation. J Cell Physiol 2011;226:2535-42. [DOI] [PubMed] [Google Scholar]
- 71.Roderburg C, Luedde M, Vargas Cardenas D, et al. miR-133a mediates TGF-beta-dependent derepression of collagen synthesis in hepatic stellate cells during liver fibrosis. J Hepatol 2013;58:736-42. [DOI] [PubMed] [Google Scholar]
- 72.He Y, Huang C, Sun X, et al. MicroRNA-146a modulates TGF-beta1-induced hepatic stellate cell proliferation by targeting SMAD4. Cell Signal 2012;24:1923-30. [DOI] [PubMed] [Google Scholar]
- 73.Guo CJ, Pan Q, Jiang B, et al. Effects of upregulated expression of microRNA-16 on biological properties of culture-activated hepatic stellate cells. Apoptosis 2009;14:1331-40. [DOI] [PubMed] [Google Scholar]
- 74.van Rooij E, Sutherland LB, Thatcher JE, et al. Dysregulation of microRNAs after myocardial infarction reveals a role of miR-29 in cardiac fibrosis. Proc Natl Acad Sci U S A 2008;105:13027-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ogawa T, Enomoto M, Fujii H, et al. MicroRNA-221/222 upregulation indicates the activation of stellate cells and the progression of liver fibrosis. Gut 2012;61:1600-9. [DOI] [PubMed] [Google Scholar]
- 76.Zhang Y, Wu L, Wang Y, et al. Protective Role of Estrogen-induced miRNA-29 Expression in Carbon Tetrachloride-induced Mouse Liver Injury. J Biol Chem 2012;287:14851-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Salic K, De Windt LJ. MicroRNAs as biomarkers for myocardial infarction. Curr Atheroscler Rep 2012;14:193-200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Tacke F, Roderburg C, Benz F, et al. Levels of circulating miR-133a are elevated in sepsis and predict mortality in critically ill patients. Crit Care Med 2014;42:1096-104. [DOI] [PubMed] [Google Scholar]
- 79.Zheng J, Lin Z, Dong P, et al. Activation of hepatic stellate cells is suppressed by microRNA-150. Int J Mol Med 2013;32:17-24. [DOI] [PubMed] [Google Scholar]
- 80.Li WQ, Chen C, Xu MD, et al. The rno-miR-34 family is upregulated and targets ACSL1 in dimethylnitrosamine-induced hepatic fibrosis in rats. FEBS J 2011;278:1522-32. [DOI] [PubMed] [Google Scholar]
- 81.Okada N, Lin CP, Ribeiro MC, et al. A positive feedback between p53 and miR-34 miRNAs mediates tumor suppression. Genes Dev 2014;28:438-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Cermelli S, Ruggieri A, Marrero JA, et al. Circulating MicroRNAs in Patients with Chronic Hepatitis C and Non-Alcoholic Fatty Liver Disease. PloS One 2011;6:e23937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Lino Cardenas CL, Henaoui IS, Courcot E, et al. miR-199a-5p Is upregulated during fibrogenic response to tissue injury and mediates TGFbeta-induced lung fibroblast activation by targeting caveolin-1. PLoS Genet 2013;9:e1003291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Dai BH, Geng L, Wang Y, et al. microRNA-199a-5p protects hepatocytes from bile acid-induced sustained endoplasmic reticulum stress. Cell Death Dis 2013;4:e604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Roberts TC, Coenen-Stass AM, Betts CA, et al. Detection and quantification of extracellular microRNAs in murine biofluids. Biol Proced Online 2014;16:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kosaka N, Iguchi H, Yoshioka Y, et al. Secretory mechanisms and intercellular transfer of microRNAs in living cells. J Biol Chem 2010;285:17442-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Benz F, Roderburg C, Vargas Cardenas D, et al. U6 is unsuitable for normalization of serum miRNA levels in patients with sepsis or liver fibrosis. Exp Mol Med 2013;45:e42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Roderburg C, Mollnow T, Bongaerts B, et al. Micro-RNA profiling in human serum reveals compartment-specific roles of miR-571 and miR-652 in liver cirrhosis. PloS One 2012;7:e32999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Anadol E, Schierwagen R, Elfimova N, et al. Circulating MicroRNAs as a marker for liver injury in human immunodeficiency virus patients. Hepatology 2015;61:46-55. [DOI] [PubMed] [Google Scholar]
- 90.Roderburg C, Benz F, Vargas Cardenas D, et al. Elevated miR-122 serum levels are an independent marker of liver injury in inflammatory diseases. Liver Int 2014. [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 91.Elfimova N, Schlattjan M, Sowa JP, et al. Circulating microRNAs: promising candidates serving as novel biomarkers of acute hepatitis. Front Physiol 2012;3:476. [DOI] [PMC free article] [PubMed] [Google Scholar]