Natural History of Denervation in SMA: Relation to Age, SMN2 Copy Number, and Function (original) (raw)
. Author manuscript; available in PMC: 2015 Feb 19.
Published in final edited form as: Ann Neurol. 2005 May;57(5):704–712. doi: 10.1002/ana.20473
Abstract
Denervation was assessed in 89 spinal muscular atrophy (SMA) 1, 2, and 3 subjects via motor unit number estimation (MUNE) and maximum compound motor action potential amplitude (CMAP) studies, and results correlated with SMN2 copy, age, and function. MUNE and maximum CMAP values were distinct among SMA subtypes (p < 0.05). Changes in MUNE and maximum CMAP values over time were dependent on age, SMA type, and SMN2 copy number. SMN2 copy number less than 3 correlated with lower MUNE and maximum CMAP values (p < 0.0001) and worse functional outcomes. As SMN2 copy number increases, so does functional status (p < 0.0001). Change in MUNE longitudinally over the time intervals examined in this study was not statistically significant for any SMA cohort. However, a decline in maximum CMAP over time was apparent in SMA2 subjects (p = 0.049). Age-dependent decline in MUNE and maximum CMAP was apparent in both SMA 1 (p < 0.0001) and SMA 2 (p < 0.0001) subjects, with age as an independent factor regardless of type. Maximum CMAP at the time of the initial assessment was most predictive of functional outcome (p < 0.0001). Prospective longitudinal studies in four prenatally diagnosed infants demonstrated significant progressive denervation in association with symptomatic onset or functional decline. These data highlight the potential value of such measures in increasing our understanding of pathophysiological factors involved in denervation in SMA.
Spinal muscular atrophy (SMA, OMIM 253300) is a recessive motor neuron disease characterized by diffuse weakness and muscular atrophy. It is one of the most common neuromuscular conditions with an incidence of approximately 1 in 8,000 live births and a leading cause of hereditary infant mortality.1–6 The disease most frequently manifests in infancy and early childhood. The clinical spectrum of onset and severity is broad, ranging from the infant with congenital arthrogryposis requiring ventilator support to the individual with onset of proximal leg weakness in adulthood. For ease of clinical classification, SMA patients are most commonly divided into types 1 to 3,7 with further sub-classification.8 SMA 1 patients manifest weakness before 6 months and are defined by their inability to sit without support. Without substantial respiratory support, most of these children die before their second birthday. SMA 2 patients usually demonstrate weakness by 18 months. They are able to sit unsupported at some point in their clinical course, although they often later lose this ability. They suffer significant respiratory morbidity and early mortality. SMA 3 patients achieve the ability to ambulate independently. They generally have a milder course, with the potential for a normal life expectancy.9
Homozygous deletion of the survival motor neuron 1 (SMN1) gene is found in more than 94% of SMA cases regardless of type and plays a primary role in disease pathogenesis.10,11 The SMN1 gene resides in a duplicated inverted region on chromosome 5q. A near identical copy of the SMN1 gene, SMN2, contains a single nucleotide change which alters splicing, drastically reducing the efficiency of exon 7 inclusion and resulting in truncated mRNA and protein products.12 However, SMN2 transcription results in approximately 10% full-length SMN protein. An increased number of SMN2 gene copies, with the resulting increase in full-length mRNA levels and protein expression, is associated with a less severe disease phenotype in humans and phenotypic rescue in an SMN knockout mouse model.13–17 The SMN protein appears to play an important role in spliceosomal snRNP biogenesis, although the precise means by which a deficiency of the SMN protein affects neuronal viability remains unclear.18 The identification of the unique mechanism by which the phenotype is modified in SMA has led to efforts to identify specific compounds which could shift splicing in the SMN2 message toward a higher rate of exon 7 inclusion or upregulate SMN protein expression via an increase in SMN2 gene expression. Several compounds have been identified that upregulate SMN gene expression, some which are readily available for use in patients and may be of potential value in treatment.19–22
Design and implementation of effective therapeutic trials in SMA necessitates a better understanding of the pathophysiology underlying denervation, the impact of lowered SMN levels on neuronal health with regard to age and developmental status, and the potential reversibility of the direct and indirect effects of lowered SMN levels at various time points. Clinical classification in SMA implies greater lower motor neuron loss in types 1 and 2, and least in type 3. However, the magnitude and time course of such loss is not apparent from the usual clinical evaluation. Genetic testing has appropriately replaced electrophysiological testing for diagnostic purposes. However, a quantitative knowledge of the natural history of lower motor neuron loss is essential in guiding the design of clinical trials. In infants and young children, obtaining reliable and sensitive outcome measures of strength and functional status is a challenge. Assessments of strength may prove an insensitive measure for progression in a condition such as SMA in which clinical symptoms become apparent early but appear to stabilize over time. Preservation of strength in the setting of loss of function has been well documented.23 Presumably, reinnervation of muscle fibers by the remaining motor neurons concurrent with progressive motor neuron loss allows for the relative maintenance of strength for prolonged periods of time despite disease progression. Functional scales may be better but are influenced by factors including developmental maturation, such that a child may continue to gain motor milestones or remain stable despite progressive denervation. For example, children with SMA 2 often lose the ability to roll, then regain it several months later as they learn strategies to overcome their strength limitations. In light of these factors, it is critical to determine ways in which to assess the health of remaining motor neurons, their capacity for axonal sprouting and reinnervation, the size of the remaining motor units, and ultimately the effect on muscle mass, strength, and function.
We prospectively evaluated the use of two electrophysiological measures in a distally innervated muscle group in SMA. We report natural history data for 89 SMA subjects with a wide range of age and clinical severity. In each subject, we obtained an estimate of the number of motor units innervating a distal muscle group (MUNE), maximum compound motor action potential amplitude (CMAP), general functional motor status, and SMN2 copy number. These data support the applicability of assessment of MUNE and maximum CMAP amplitudes in children with SMA. Such data provide a measure of the overall health of motor neurons and their distally innervated muscle groups which may facilitate a better understanding of the natural history of SMA with regard to single motor unit size and stability. These data have been collected as part of a more comprehensive assessment protocol which includes repeated formal functional assessments, DEXA imaging of lean body mass and bone density, pulmonary function testing in children older than 5 years of age, analysis of nutritional status via 3-day dietary survey and growth parameters, and quantitative assays of SMN mRNA and protein levels from blood samples. Reliability assessment and correlation of these measures with MUNE, CMAP, and SMN2 copy number data are ongoing but ultimately will provide additional information regarding the relevance of such measures to functional outcomes in SMA subjects.
Subjects and Methods
Informed consent (and informed assent in children older than 12 years of age) was obtained from each subject or their parents. This protocol was approved independently by the institutional review and general clinical research center review boards at the University of Utah School of Medicine and Primary Children's Medical Center.
Motor Unit Number Estimation and Maximum Ulnar Compound Motor Action Potential Amplitude
We used the multiple point stimulation (MPS) technique to estimate the number of motor units innervating hypothenar muscles, as previously described, using a proprietary software program on the Advantage A200 machine.24,25 Although this machine is no longer available, similar software is available on the Comperio from Compumedics, from which we have obtained comparable results. The G1 electrode is moved for a minimum of three placements to ensure that maximum CMAP is selected. Individual single motor unit potentials (SMUPs) are identified by obtaining all-or-none responses to low-intensity stimulation. Ten consecutive observations of an individual SMUP are obtained to decrease the probability of multiplex units, and we attempt to identify at least 10 unique SMUPs. The MUNE value is the maximum CMAP negative peak area (NParea) divided by the mean SMUP NParea. Values are expressed for maximum CMAP and SMUP by both NParea and negative peak amplitude (NPamp).
Functional Assessment
Functional status was assessed at each visit to ascertain ability to sit with or without support, cruise, and walk independently and use of ventilatory support. An ordinal scale was applied to establish functional status at the time of data analysis, because frequently, SMA type as defined by maximal achievement of gross motor skills does not reflect current gross motor functional status. This ordinal scale allows documentation of critical outcomes including serious respiratory compromise or death during the course of the study. It is simplistic and follows the general delineations in function used for designating SMA type in patients. Scores were assigned as follows at the time of data analysis for this subject cohort: 0 = deceased or ventilator dependent more than 16 hours/day, 1 = unable to sit unsupported, 2 = sits independently when placed, 3 = cruises, walks with assistance or independently, and 4 = without evident muscle weakness.
Survival Motor Neuron Copy Number
Confirmation of homozygous SMN1 deletion and assessment of SMN copy number was performed as previously described26.
Conscious Sedation
Intranasal or oral midazolam was used in children in whom anxiety or discomfort would interfere with reliable testing and to facilitate repeated evaluations. Dosing was 0.2 to 0.5mg/kg, maximum 5 mg per dose, repeated as needed up to 10mg. SMA 1 subjects typically have respiratory compromise which precludes such sedation. However, their low MUNE values enhance reliability and ease of testing.
Statistical Analysis
Maximum CMAP and MUNE values were normally distributed. Analysis of variance was used to determine differences between SMA types 1, 2, and 3 for CMAP and MUNE values. Because there are multiple comparisons the Ryan-Einot-Gabriel-Welsch multiple range test was used to maintain a statistical significance level of 0.05.
The intraclass correlation coefficient (ICC) was used to measure reliability of MUNE and CMAP measurements, based on a data pair (test and retest) for each patient. Test-retest pairs were performed on different days in 21 subjects, with retest occurring within 3 months of the first assessment. Assumptions were that the reviewers were fixed and that the sample was random.
Change Over Time Analysis
MUNE and CMAP values were repeated over time in individual subjects. Age of subject, time intervals between studies, and total number of measures varied for practical reasons. Data were analyzed using generalized estimating equations (GEE)27. The validity of the GEE in evaluating sample sizes similar to this study has been established previously.28 Given the normal distribution of MUNE and maximum CMAP values, the GEE with normal distribution, exchangeable correlation structure, and an identity link function was employed. The exchangeable correlation was used because the correlation from visit to visit was the same and not time dependent. Age, SMA type, and SMN2 all were included in the model. To investigate the impact of each SMA type, we assigned a dummy variable to represent each type, and the reference point was type 3, because we expected the least change over time in that group. Age was included as a continuous variable and an age by SMA type interaction was assessed. To assess the robustness of the results a log transformation of age, MUNE and maximum CMAP were assessed. The results were the same as the non-transformed data. Outcome of the nontransformed analyses are reported for simplicity.
The Fisher-Freeman-Halton test was used to determine if functional status was independent of SMN2 copy number. Evaluation of the linear relationship between the categories of functional status and number of SMN2 copies was performed using the nonparametric linear by linear test.
Statistical analysis was not performed on prenatally identified cases, because the results were self-evident. In all instances, these subjects were ascertained in the context of an affected sibling.
Results
Motor Unit Number Estimation and Maximum Compound Motor Action Potential Amplitude Reliability
ICC to assess reliability for MUNE and CMAP, based on test–retest data pairs in 21 subjects obtained on different days within a 3-month time period confirmed excellent reliability (p < 0.001) for all measures. ICC for MUNE NPamp was 0.993, for MUNE negative peak area (NParea) 0.965, for CMAP NPamp 0.973, and for CMAP NParea 0.958.
Motor Unit Number Estimation and Compound Muscle Potential Amplitude Data
Cross-sectional data were available on 29 SMA 1, 44 SMA 2, and 14 SMA 3 subjects (one severely affected SMA 1 infant with congenital arthryogryposis was excluded, because CMAP was unelicitable). Serial longitudinal studies available in 19 SMA 1, 36 SMA 2, and 10 SMA 3 subjects, including three type 1 and one type 2 prospectively identified subjects. Excluding prenatally identified cases, mean MUNE and CMAP values differed substantially among the three types, with the lowest values observed in SMA 1 subjects, intermediate values in SMA 2 subjects, and the highest values in SMA 3 subjects (p < 0.05, Fig 1 and Table 1). However, there is overlap among types, and values varied widely with age and gross motor functional status. Cross-sectional data from 29 SMA 1, 44 SMA 2, and 14 SMA 3 subjects plotting age versus MUNE and CMAP values demonstrate an overall age-dependent decline in values, with SMA 2 subjects reaching values that overlap with the SMA 1 population (Figs 2 and 3). These data are consistent with observations from the change over time analysis in individual subjects (Fig 4).
Fig 1.
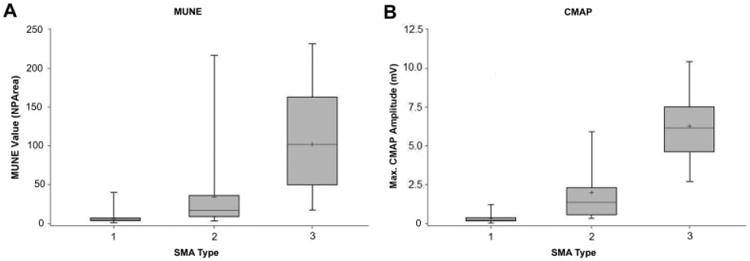
(A, B) MUNE and CMAP data, respectively, by SMA type in boxplot format. The shaded gray boxes represent the interquartile range (75th%–25th%) of the data. The whiskers demonstrate a measure of data variability, and the horizontal black lines within the gray boxes indicate median values, and plus signs indicated mean values, in each category. SMA = spinal muscular atrophy; MUNE = motor unit number estimation; CMAP = compound motor action potential amplitude.
Table 1. MUNE and CMAP Data from First Visit, Excluding Prenatally Identified Cases.
| Characteristic | SMA 1(N = 26), Mean (SE) | SMA 2(N = 43), Mean (SE) | SMA 3(N = 14), Mean (SE) | p |
|---|---|---|---|---|
| Age (mo) (range) | 18.9 (9.9) (1.08–263) | 87.1 (20.8) (0.43–589) | 146.6 (43.8) (28.4–604.8) | <0.05 (all distinct) |
| CMAP NPamp, mV (range) | 0.34 (0.06) (0.03–1.20) | 1.86 (0.24) (0.32–5.9) | 6.24 (0.68) (2.7–10.4) | <0.05 (all distinct) |
| MUNE NParea, mVms (range) | 6.3 (1.4) (1–40) | 34.5 (6.9) (3–216) | 102.1 (17.1) (17–231) | <0.05 (all distinct) |
Fig 2.
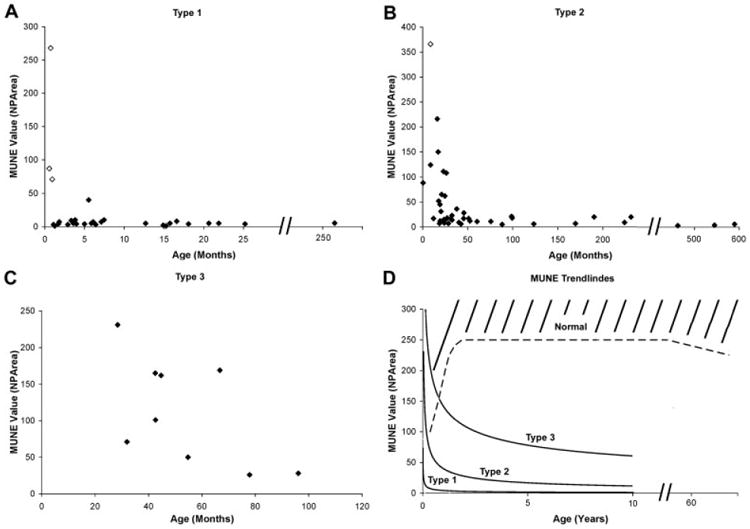
(A–C) Cross-sectional MUNE data obtained at first visit for spinal muscular atrophy type 1, 2, 3 subjects, respectively. Open diamonds indicate subjects identified prenatally via genetic testing because of a previously affected sibling. Solid diamonds indicate subjects with confirmation via genetic testing following symptom onset. (D) Power trendlines representing data for all three types. Estimated minimum normative values indicated by dashed line. MUNE = motor unit number estimation; NPArea = negative peak area.
Fig 3.
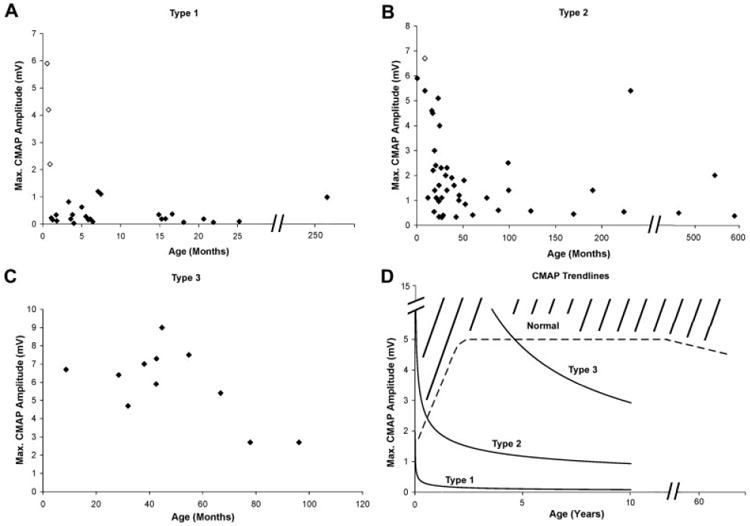
(A–C) Cross-sectional maximum CMAP data obtained at first visit for spinal muscular atrophy type 1, 2, and 3 subjects, respectively. Open diamonds indicate subjects identified presymptomatically via genetic testing because of a previously affected sibling. Solid diamonds indicate subjects with confirmation via genetic testing after symptom onset. (D) Power trendlines representing data for all three types. Estimated minimum normative values indicated by dashed line.29 CMAP = compound motor action potential amplitude.
Fig 4.
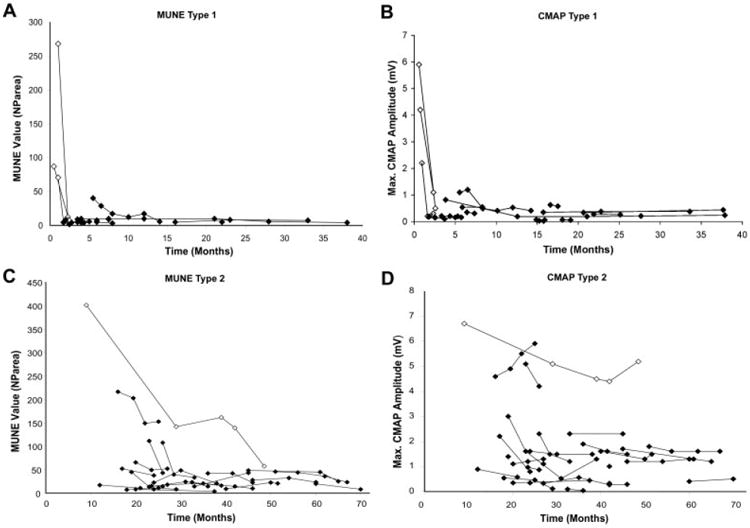
(A–C) Longitudinal MUNE and CMAP data, respectively, in type 1 subjects with more than one assessment. (B, D) Longitudinal MUNE and CMAP data, respectively, in type 2 subjects with more than one assessment. CMAP = compound motor action potential amplitude; MUNE = motor unit number estimation; NParea = negative peak area.
Prenatally Diagnosed, Prospectively Identified Subjects
Longitudinal data were obtained in three SMA 1 and one SMA 2 subject identified in the context of an affected siblings. These subjects, identified as open diamonds in Figures 2 and 3, demonstrate severe and substantial postnatal progression of motor denervation as reflected by CMAP and MUNE values. This decline was temporally associated with a precipitous decrease in motor function in all three SMA 1 infants, with progression to generalized hypotonia and quadriparesis over a 1- to 2-week period, and death from respiratory failure by 6 months of age. This early and precipitous decline in SMA 1 subjects is in contrast with the normal MUNE and CMAP values in the SMA 2 subject observed as late as 9 months of age. She had a more subtle presentation between 9 and 12 months manifest as a failure to bear weight and a failure to show continued weight gain as expected. After a decline from 9 to 28 months, MUNE and CMAP values stabilized until her most recent assessment at 49 months, when MUNE values again decreased in the face of a stable CMAP value. She has remained stable with regard to gross motor functional abilities since formal testing was possible at 22 months of age.
Change Over Time Analysis and Effects of Age on Motor Unit Number Estimation and Compound Motor Action Potential Amplitude Data
We analyzed change in MUNE and CMAP values over time in subjects with SMA types 1, 2, and 3, excluding prenatally diagnosed subjects. SMA type and age were related to change in MUNE values over time for SMA 1, 2, and 3 subjects. Age and SMA type did not have a statistically significant interaction (Table 2).
Table 2. MUNE: Change with Time Analysis.
| Parameter | Estimate | Standard Error | p |
|---|---|---|---|
| Type 1 | 98.9 | 21.4 | <0.0001 |
| Type 2 | 67.6 | 22.4 | 0.0026 |
| Age × type 1 | − 0.046 | 0.09 | 0.614 |
| Age × type 2 | − 0.0085 | 0.02 | 0.667 |
Although change over time within individual SMA cohorts was not significant, age was a significant and independent factor regardless of SMA type (p < 0.0001), with a corresponding decrease in MUNE value of −0.074/month of additional age. However, the overall contribution of SMA type was greater than age (Table 3).
Table 3. MUNE vs Age Analysis.
| Parameter | Estimate | Standard Error | p |
|---|---|---|---|
| Type 1 | 100.4 | 15.4 | <0.0001 |
| Type 2 | 71.9 | 15.2 | <0.0001 |
| Age | −0.074 | 0.032 | 0.023 |
| Intercept | −64.7 | 15.2 | <0.0001 |
Analysis of CMAP data, in contrast, demonstrates a significant change over time which is clearly dependent on SMA type. SMA 1 and 3 subjects are essentially stable, whereas SMA 2 subjects demonstrate a modest decline of − 0.007mV/month (p = 0.0492), even over the relatively short time frame of this study (Table 4).
Table 4. CMAP: Change with Time Analysis.
| Parameter | Estimate | Standard Error | p |
|---|---|---|---|
| Type 1 | 4.9 | 0.70 | <0.0001 |
| Type 2 | 3.3 | 0.74 | <0.0001 |
| Age × type 2 or 3 (type 1 absent) | 0.0049 | 0.0031 | 0.1148 |
| Age × type 1 or 3 (type 2 absent) | 0.002 | 0.0008 | 0.0079 |
| Age × type 2 | − 0.0068 | 0.0034 | 0.0492 |
| Intercept | − 3.03 | 0.75 | <0.0001 |
Correlation of Survival Motor Neuron 2 Copy Number with Motor Unit Number Estimation and Compound Motor Action Potential Amplitude Data and Functional Status
A modifying effect of SMN2 copy number on MUNE and CMAP values is clearly evident when contrasting subjects with two copies versus higher copy number (Table 5) are compared. Subjects with less than three SMN2 copies performed significantly worse for both MUNE and CMAP values over time (p < 0.0001). However, there is substantial overlap in subjects with three and four copies. The linear by linear test indicated that as SMN2 copy number increases so does functional status (p < 0.0001; Table 6). As pertinent examples, we have a 7.5-year-old SMA 3 subject with two SMN2 copies. His clinical course has been severe relative to other SMA3 subjects. He was able to walk behind his chair with ankle-foot orthotics until 5 years of age but later lost this ability and underwent surgery for scoliosis at 7 years of age. He has the lowest CMAP and MUNE values to date for any of the SMA 3 subjects. Two SMA 1 subjects have three SMN2 copies: a 23-year-old with a MUNE of 6, and a 22-month-old subject with a MUNE of 5. The 23-year-old subject has severe restrictive pulmonary disease, a tracheotomy with nocturnal ventilation requirement, and a gastrostomy-tube due to bulbar insufficiency. However, both subjects are clearly more robust than the SMA 1 subjects with two copies. Our weakest SMA 2 subject at just over 3 years of age has only two copies, with MUNE and CMAP values in the same range as our SMA 1 cohort. He was briefly able to sit unsupported before deterioration at around 6 months of age. At 38 months, he is quadriparetic with severe restrictive pulmonary disease. The highest functioning symptomatic subject in our study is a 50-year-old still ambulatory SMA 3 subject with five SMN2 copies. She skied competitively into her teen years before the development of proximal leg weakness. Her son, also with five copies, is substantially weaker and lost independent ambulation at 23 years of age.
Table 5. Change in MUNE and CMAP over Time: Interaction with SMN2 Copy Number.
| MUNE | CMAP | ||||||
|---|---|---|---|---|---|---|---|
| SMN2 Copy | Estimate | SE | p | SMN2 Copy | Estimate | SE | p |
| 1 vs 2 | −1.89 | 0.35 | <0.0001 | 1 vs 2 | −1.03 | 0.15 | <0.0001 |
| 2 vs 3 | −1.05 | 0.27 | 0.0001 | 2 vs 3 | − 0.78 | 0.11 | <0.0001 |
| 3 vs 4 | 0.17 | 0.48 | 0.727 | 3 vs 4 | − 0.05 | 0.14 | 0.739 |
| 4 vs 5 | −0.36 | 0.48 | 0.45 | 4 vs 5 | − 0.07 | 0.27 | 0.80 |
Table 6. Evaluation of the Linear Relationship between the Categories of Functional Status and Number of SMN2 Copies Using the Nonparametric Linear by Linear Test.
| SMN2 Copy No. | Functional Status | |||
|---|---|---|---|---|
| 0 | 1 | 2 | 3 | |
| 1 | 1 (100%) | 0 | 0 | 0 |
| 2 | 9 (41%) | 12 (55%) | 1 (4%) | 0 |
| 3 | 0 | 7 (17%) | 28 (68%) | 6 (15%) |
| 4 | 0 | 2 (14%) | 11 (79%) | 1 (7%) |
| 5 | 0 | 0 | 1 (33%) | 2 (67%) |
Role of Conscious Sedation in Data Collection
Conscious sedation was extremely well tolerated in the large majority of children with SMA type 2 or 3. Adverse events included occasional subjects with significant irritability after the procedure as sedation was waning. Transient oxygen desaturation less than 93% occurred in three subjects, with quick resolution after repositioning or encouragement to cough. Careful screening for current or recent illness and appropriate use of noninvasive preventive respiratory measures before and after the procedure helps ensure patient safety. Sedation is contraindicated in any child presenting with oxygen saturation less than 93% at baseline. Use of sedation has been critical in our hands in obtaining reliable MUNE data and in enhancing the acceptability of repeated testing for both parents and children, particularly in those subjects younger than 3 to 4 years of age.
Discussion
We demonstrate the feasibility and reliability of MUNE and CMAP measures in a large cohort of SMA subjects across a wide spectrum of age and disease severity. Our data support the value of these techniques in exploring issues related to disease pathophysiology and status of distal innervation over time in response to various therapeutic interventions.
We present to our knowledge the first prospective electrophysiological evaluation of prenatally identified SMA patients, demonstrating a substantial and unexpectedly precipitous decline in MUNE and CMAP values over time. In at least two of the SMA 1 infants, MUNE values, but not necessarily CMAP values, are already low at the time of initial testing. However, one SMA 1 infant and one SMA 2 child had normal values at the time of initial testing, supporting the concept that significant disease progression occurs in the postnatal period. If a therapeutic compound is identified that either rescues at-risk motor neurons or prevents ongoing motor neuron denervation, newborn screening may be indicated. Newborn screening for cystic fibrosis, despite a lack of a curative therapy, has led to significant improvements in quality of life in affected children because of early implementation of therapies such as nutritional support.
Analysis of individual trends within SMA types identifies two groups of subjects: those with relatively stable MUNE and CMAP values and those who demonstrate a significant decline in MUNE and CMAP values. This phenomenon appears to be largely age dependent, but of variable onset. It will be important for clinical trials to control for expected variability in response to a given therapeutic intervention, that is, a slowing of decline versus an increase in innervation or sprouting. The relative stability of MUNE over time across cohorts of SMA types given the relatively short time intervals is an important observation from a clinical trial design perspective, because most individual subjects of a specific type will be expected to remain stable over the time course of observation expected during a typical clinical trial. However, given that age is a significant factor in our model, age of enrollment is clearly a critical factor to consider.
MUNE and CMAP measures may provide sensitive indicators of health of motor neurons which are complementary to functional outcome measures, because some subjects with declining values are often quite stable in terms of overall functional status. MUNE and CMAP measures may have independent value, because a potential therapy could demonstrate alternative mechanisms of improvement, including (1) distal axonal sprouting leading to increased territory and size of individual SMUPs without change in absolute MUNE value, or (2) reinnervation by axons from motor units whose axons had undergone distal degeneration but were “rescuable.” Obtaining both measures may be useful in distinguishing agents that increase the reinnervation capacity of existing motor neurons (increasing size of individual SMUPs and CMAP amplitude) versus those that facilitate recovery of motor axons that have died back, allowing them to newly reinnervate muscle (increasing both MUNE and CMAP values). Such data may help to better predict the ultimate impact of a given therapy, because distal reinnervation can require considerable time. Baseline severity of denervation, duration of disease, and secondary complications such as contractures or end-stage muscle fibrosis will almost certainly be limiting variables with regard to therapeutic interventions.
Our data support a statistically significant correlation between maximum CMAP amplitude at the time of initial assessment and functional outcomes. These data may be valuable in providing relative prognostic information regarding an individual SMA subject's general health status, assessing where they lie on the curve relative to severity of denervation in a particular SMA cohort, and defining appropriate expectations regarding clinical improvement in response to therapeutic interventions.
SMN2 copy clearly plays a modifying role on MUNE and CMAP values and overall functional status, but other modifying factors are likely contributing in a complex fashion to disease phenotype. Despite the observation that SMN2 dosage has a statistically significant modifying effect on function, predicting function or type in individual cases, particularly in the prenatal setting, remains a complex issue in light of the observed heterogeneity. However, our data support a clear distinction between those with more than two SMN2 copies in overall MUNE and CMAP values, type, and functional status. An SMN2 copy number higher than 2 may be much more relevant to long-term outcomes such as survival and maintenance of motor function over time rather than maximum motor function achieved, which is what is used currently for clinical classification into types. For instance, an SMA 1 child who has three SMN2 copies may have a distinct survival advantage over one with two copies, although neither child will ever sit independently.
Based on the above observations and those of others, we propose stratification for therapeutic trials or post hoc adjustment of data analysis based on factors not limited to clinical subtype, including SMN2 copy status less than 3, and recommend choosing patient cohorts with a narrower range of age and functional ability in any prospective clinical trial design. Although a quantitative knowledge of the natural history of lower motor neuron loss may be helpful in guiding the design of clinical trials, this does not imply that MUNE is necessarily the best outcome for clinical trials to assess efficacy. CMAP assessment may be sufficient for this purpose, because processes which increase either MUNE size or number will have a corresponding increase in CMAP amplitude. However, MUNE may be a valuable technique when asking specific questions about the mechanism by which a given therapy could lead to clinical improvement. Imperative in any clinical trial design is the use of the appropriate tool to answer the question of interest. For instance, a compound which has an impact on stabilizing the neuromuscular junction could have a significant effect on fatigue levels, resulting in a quality-of-life improvement. Use of MUNE and maximum CMAP values to evaluate such a compound would be irrational in this context but would be appropriate to address a question related to a potential improvement in sprouting and reinnervation capacity of remaining motor units in various SMA sub-populations. In light of new potential therapeutic avenues for motor neuron disease in general and SMA in particular, it is critical to identify additional approaches to assess the health of remaining motor neurons, their capacity for axonal sprouting and reinnervation, the size of the remaining motor units, and ultimately the impact on muscle mass, strength, and function.
Acknowledgments
This work was supported by the grants from the Families of SMA, the NIH (National Institute of Neurological Disorders and Stroke, NS41634-01, K.J.S., M.B.B., and the National Center for Research Resources K.J.S.), the Muscular Dystrophy Association of America (K.J.S.), Howard Hughes Foundation Fellowship-to-Faculty Transition Award (K.J.S.), and the SMA and American Academy of Neurology Foundations (Young Investigator Award, K.J.S.).
We are grateful to the patients and families with SMA who participated in this study
References
- 1.Roberts DF, Chavez J, Court SD. The genetic component in child mortality. Arch Dis Child. 1970;45:33–38. doi: 10.1136/adc.45.239.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pearn JH. The gene frequency of acute Werdnig-Hoffmann disease (SMA type 1). A total population survey in North-East England. J Med Genet. 1973;10:260–265. doi: 10.1136/jmg.10.3.260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pearn J. Incidence, prevalence, and gene frequency studies of chronic childhood spinal muscular atrophy. J Med Genet. 1978;15:409–413. doi: 10.1136/jmg.15.6.409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Czeizel A, Hamula J. A hungarian study on Werdnig Hoffmann disease. J Med Genet. 1989;26:761–763. doi: 10.1136/jmg.26.12.761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Emery AE. Population frequencies of inherited neuromuscular diseases—a world survey. Neuromuscul Disord. 1991;1:19–29. doi: 10.1016/0960-8966(91)90039-u. [DOI] [PubMed] [Google Scholar]
- 6.Merlini L, Stagni SB, Marri E, Granata C. Epidemiology of neuromuscular disorders in the under-20 population in Bologna Province, Italy. Neuromuscul Disord. 1992;2:197–200. doi: 10.1016/0960-8966(92)90006-r. [DOI] [PubMed] [Google Scholar]
- 7.Pearn J. Classification of spinal muscular atrophies. Lancet. 1980;1:919–922. doi: 10.1016/s0140-6736(80)90847-8. [DOI] [PubMed] [Google Scholar]
- 8.Dubowitz V. Chaos in the classification of SMA: a possible resolution. Neuromuscul Disord. 1995;5:3–5. doi: 10.1016/0960-8966(94)00075-k. [DOI] [PubMed] [Google Scholar]
- 9.Crawford TO, Pardo CA. The neurobiology of childhood spinal muscular atrophy. Neurobiol Dis. 1996;3:97–110. doi: 10.1006/nbdi.1996.0010. [DOI] [PubMed] [Google Scholar]
- 10.Gilliam TC, Brzustowicz LM, Castilla LH, et al. Genetic homogeneity between acute and chronic forms of spinal muscular atrophy. Nature. 1990;345:823–825. doi: 10.1038/345823a0. [DOI] [PubMed] [Google Scholar]
- 11.Melki J, Lefebvre S, Burglen L, et al. De novo and inherited deletions of the 5q13 region in spinal muscular atrophies. Science. 1994;264:1474–1477. doi: 10.1126/science.7910982. [DOI] [PubMed] [Google Scholar]
- 12.Monani UR, Lorson CL, Parsons DW, et al. A single nucleotide difference that alters splicing patterns distinguishes the SMA gene SMN1 from the copy gene SMN2. Hum Mol Genet. 1999;8:1177–1183. doi: 10.1093/hmg/8.7.1177. [DOI] [PubMed] [Google Scholar]
- 13.Campbell L, Potter A, Ignatius J, et al. Genomic variation and gene conversion in spinal muscular atrophy: implications for disease process and clinical phenotype. Am J Hum Genet. 1997;61:40–50. doi: 10.1086/513886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lefebvre S, Burlet P, Liu Q, et al. Correlation between severity and SMN protein level in spinal muscular atrophy. Nat Genet. 1997;16:265–269. doi: 10.1038/ng0797-265. [DOI] [PubMed] [Google Scholar]
- 15.Monani UR, Sendtner M, Coovert DD, et al. The human centromeric survival motor neuron gene (SMN2) rescues embryonic lethality in Smn(—/—) mice and results in a mouse with spinal muscular atrophy. Hum Mol Genet. 2000;9:333–339. doi: 10.1093/hmg/9.3.333. [DOI] [PubMed] [Google Scholar]
- 16.Feldkotter M, Schwarzer V, Wirth R, et al. Quantitative analyses of SMN1 and SMN2 based on real-time lightCycler PCR: fast and highly reliable carrier testing and prediction of severity of spinal muscular atrophy. Am J Hum Genet. 2002;70:358–368. doi: 10.1086/338627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mailman MD, Heinz JW, Papp AC, et al. Molecular analysis of spinal muscular atrophy and modification of the phenotype by SMN2. Genet Med. 2002;4:20–26. doi: 10.1097/00125817-200201000-00004. [DOI] [PubMed] [Google Scholar]
- 18.Fischer U, Liu Q, Dreyfuss G. The SMN-SIP1 complex has an essential role in spliceosomal snRNP biogenesis. Cell. 1997;90:1023–1029. doi: 10.1016/s0092-8674(00)80368-2. [DOI] [PubMed] [Google Scholar]
- 19.Chang JG, Hsieh-Li HM, Jong YJ, et al. Treatment of spinal muscular atrophy by sodium butyrate. Proc Natl Acad Sci USA. 2001;98:9808–9813. doi: 10.1073/pnas.171105098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Andreassi C, Jarecki J, Zhou J, et al. Aclarubicin treatment restores SMN levels to cells derived from type I spinal muscular atrophy patients. Hum Mol Genet. 2001;10:2841–2849. doi: 10.1093/hmg/10.24.2841. [DOI] [PubMed] [Google Scholar]
- 21.Brichta L, Hofmann Y, Hahnen E, et al. Valproic acid increases the SMN2 protein level: a well-known drug as a potential therapy for spinal muscular atrophy. Hum Mol Genet. 2003;12:2481–2489. doi: 10.1093/hmg/ddg256. [DOI] [PubMed] [Google Scholar]
- 22.Andreassi C, Angelozzi C, Tiziano FD, et al. Phenylbutyrate increases SMN expression in vitro: relevance for treatment of spinal muscular atrophy. Eur J Hum Genet. 2004;12:59–65. doi: 10.1038/sj.ejhg.5201102. [DOI] [PubMed] [Google Scholar]
- 23.Iannaccone ST, Russman BS, Browne RH, et al. Prospective analysis of strength in spinal muscular atrophy. DCN/Spinal Muscular Atrophy Group. J Child Neurol. 2000;15:97–101. doi: 10.1177/088307380001500207. [DOI] [PubMed] [Google Scholar]
- 24.Bromberg MB, Swoboda KJ. Motor unit number estimation in infants and children with spinal muscular atrophy. Muscle Nerve. 2002;25:445–447. doi: 10.1002/mus.10050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Doherty TJ, Brown WF. The estimated numbers and relative sizes of thenar motor units as selected by multiple point stimulation in young and older adults. Muscle Nerve. 1993;16:355–366. doi: 10.1002/mus.880160404. [DOI] [PubMed] [Google Scholar]
- 26.McAndrew PE, Parsons DW, Simard LR, et al. Identification of Proximal spinal muscular atrophy carriers and patients by analysis of SMNT and SMNC gene copy number. Am J Hum Genet. 1997;60:1411–1422. doi: 10.1086/515465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hardin JW, Hilbe JM. Generalized estimating equations. New York: Chapman & Hall/CRC; 2003. [Google Scholar]
- 28.Pan W, Wall MM. Small-sample adjustments in using the sandwich variance estimator in generalized estimating equations. Stat Med. 2002;21:1429–1441. doi: 10.1002/sim.1142. [DOI] [PubMed] [Google Scholar]
- 29.Garcia A, Calleja J, Antolin FM, Berciano J. Peripheral motor and sensory nerve conduction studies in normal infants and children. Clin Neurophysiol. 2000;111:513–520. doi: 10.1016/s1388-2457(99)00279-5. [DOI] [PubMed] [Google Scholar]