Interdependence of hypoxic and innate immune responses (original) (raw)
. Author manuscript; available in PMC: 2015 Feb 27.
Published in final edited form as: Nat Rev Immunol. 2009 Sep;9(9):609–617. doi: 10.1038/nri2607
Abstract
Hypoxia-inducible transcription factor (HIF) is a major regulator of cellular metabolism and adaptation to cellular stress caused by oxygen deficiency (hypoxia). Phagocytic cells have an essential role in innate immune defense against pathogens; and this is a battle that takes place mainly in the hypoxic microenvironments of infected tissues. It has now become clear that HIF promotes the bactericidal activities of phagocytic cells and supports the innate immune functions of dendritic cells, mast cells and epithelial cells. In response to microbial pathogens, HIF expression is upregulated through pathways involving the key immune response regulator nuclear factor-κB, highlighting an interdependence of the innate immune and hypoxic responses to infection and tissue damage. In turn, HIF-driven innate immune responses have important consequences for both the pathogen and the host, and its central role in immune defence demonstrates the importance of the tissue microenvironment in controlling infectious disease.
Keywords: HIF-1, Hypoxia, Innate Immunity, Phagocyte, Neutrophil, Macrophage, NF-κB
Acute tissue foci of inflammation, whether generated in response to infection, injury, noxious agents or autoimmunity, present a unique and challenging microenvironment. Hypoxia (low oxygen) or anoxia (complete lack of oxygen), hypoglycaemia (low blood glucose), acidosis (high H+ concentration) and abundant free oxygen radicals are characteristic features of inflamed tissues, along with the influx of specialized myeloid cells such as neutrophils and macrophages1,2. These short-lived cells are rapidly mobilized in response to any change in tissue integrity or entry of pathogenic microorganisms. They carry out phagocytosis and release a diverse array of antimicrobial molecules and pro-inflammatory mediators. Neutrophils and macrophages are crucial elements of innate immune defense, serving to localize and eradicate pathogens and prevent the systemic spread of infection. Quantitative deficiencies in the numbers of these specialized phagocytic cell types (for example, following cancer chemotherapy) or inherited defects in their core effector functions (for example, in chronic granulomatous disease) greatly increase susceptibility to recurrent or severe infections.
The intricate regulation of the microbicidal and inflammatory functions of neutrophils and macrophages is central to our understanding of mammalian innate immunity. Innate immune cells must remain quiescent at baseline to avoid unwanted inflammatory injury to normal host tissues, yet be capable of instantaneous activation when recruited to sites of infection. Extensive investigations have defined cell surface receptors and downstream signalling pathways that allow these cells to rapidly activate gene transcription and to release pre-formed antimicrobial effectors following recognition of pathogens or cytokines. Improved understanding is emerging of how another cardinal feature of the inflammatory tissue microenvironment, the scarcity of oxygen, influences the terms of engagement between phagocytic cells and pathogens. In this Review, we describe how these frontline innate immune effector cells have evolved to generate energy and carry out their microbicidal functions under hypoxic conditions. From this fundamental need emerges an interdependence of the innate immune response and the hypoxic response, revealing a central role of the hypoxia-inducible transcription factors (HIF) as regulators of mammalian innate defense.
HIF: A central regulator of the hypoxic response
The HIF protein complex was originally discovered for its contribution to the dramatic induction of erythropoietin (EPO) gene transcription under hypoxic conditions3, 4. Further characterization revealed HIF as a heterodimeric helix-loop-helix transcription factor, the expression of which is regulated by both oxygen and iron (Figure 1). HIF is active in all mammalian cells, and is now known to regulate the expression of more than 100 genes that function in a wide variety of host cellular and systemic responses to stress triggered by low oxygen levels. HIF-controlled pathways influence metabolism, angiogenesis, vascular tone, cell differentiation and apoptosis with implications for normal physiology and development but also for cancer and many other pathological conditions5, 6.
Figure 1. Mechanisms of HIF stabilization.
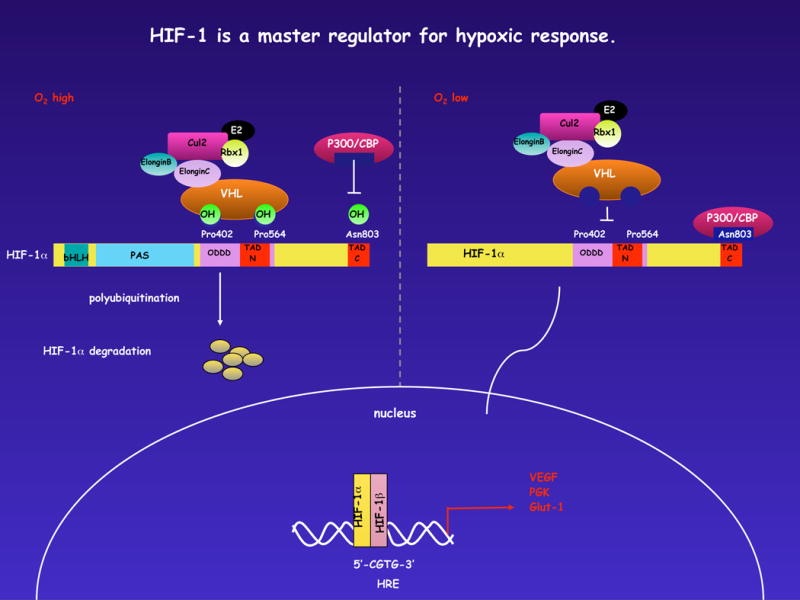
The current picture of post-translational regulation of the HIF response: Under normoxic conditions, hydroxylation by prolyl hydroxylases in an O2-dependent manner at the aa residues 402 and 564 acts to allow recognition/binding of HIF by the VHL complex which assembles in a classical SCF-like fashion around cullin-2, elongins B and C, and other elements of ubiquitination machinery. This results in a polyubiquitination of the HIF protein and its ultimate destruction by proteasomes. The asparagine hydroxylase FIH acts more or less in concert with the prolyl hydroxylation, although in this case to hydroxylate an asparagine residue in the C-terminal domain of HIF. This acts to block association with the p300/CBP protein; this in turn inhibits transcriptional enhancement by the HIF complex. As all of these post-translational events are dependent on intracellular oxygen, they are inhibited by hypoxia, and thus activation and transcription of HIF target genes facilitated when the various hydroxylations fail to occur, as depicted on the right.
The HIF DNA-binding complex is comprised of the constitutively expressed HIF1β subunit that partners with one of two hypoxia-inducible α-subunits, HIF1α or HIF2α4. The HIF α-subunits are unstable under normoxic conditions; cells continually synthesize and degrade these proteins. The short half-lives of HIF1α or HIF2α are the by-product of the activities of a family of oxygen- and iron-dependent prolyl hydroxylases (PHD1, PHD2 and PHD3)7, the actions of which direct HIF α-subunits for degradation by the ubiquitin–proteasome pathway[G] in a process that depends on interaction with von Hippel–Lindau tumour-suppressor protein (VHL)8. However, under conditions of hypoxia or iron deprivation, prolyl hydroxylase activity is inhibited, and HIF1α or HIF2α can accumulate and translocate to the nucleus, where they bind HIF1β (Figure 1). The resultant heterodimeric HIF complex binds to core pentanucleotide sequences known as hypoxic response elements (HREs) in the promoter regions of target genes. Classical HIF target genes include most of the glycolytic enzymes, glucose transporters, erythropoietin, and the angiogenic factor vascular endothelial growth factor (VEGF). The HIF-regulated genes function in concert to decrease mitochondrial oxygen consumption9, orchestrate the metabolic shift to anaerobic glycolysis10 and balance cellular pH owing to increased lactic acid production11 – thus optimizing cell energetics and homeostasis for survival and function in oxygen-poor environments.
HIF function in myeloid cells
HIF control of myeloid cell inflammatory activities
Given its central roles in development and physiology, global deletion of HIF1α in the mouse resulted in lethal embryonic defects in vascular development and morphology12–14. Recently, application of a conditional gene targeting strategy exploiting Cre-Lox recombination succeeded in lineage-specific elimination of HIF1α in macrophages and neutrophils. A mouse line was engineered to possess loxP sites flanking the Hif1a gene, and these mice were crossed into a mouse background where the myeloid-cell specific lysozyme M (lysM) promoter drives Cre recombinase expression. The resultant mice, with markedly reduced HIF1α expression levels specific to their myeloid cells, have no obvious phenotypic abnormalities at baseline, but showed marked aberrations in experimental models of acute inflammation15. In contrast to their wild-type littermates, myeloid cell Hif1a conditional knockout mice did not develop severe joint swelling and cartilage destruction in a collagen-induced arthritis model, and showed no cutaneous redness or oedema following application of a chemical irritant to the skin, suggesting impaired inflammatory responses15. The HIF1α-deficient macrophages and neutrophils had reduced levels of cellular ATP (15–40% of wild-type levels), highlighting the crucial role of the transcription factor for energy generation through glycolysis in these immune cells15. Evidence of HIF activation in the diseased tissues of patients with inflammatory disorders such as rheumatoid arthritis16, 17, dermatomyositis18, congenital lupus19 and atherosclerosis20, corroborates an important role for the hypoxic response in various human immunopathologies.
HIF control of myeloid cell defense functions
Neutrophils and macrophages are not only participants in acute and chronic inflammatory pathologies, but they are also crucial front line effectors of innate host defense against invading microbial pathogens. While the oxygen tension in healthy tissues ranges from 17.5–63 mm Hg (that is, 2.5–9% oxygen), drastically lower levels (<1% oxygen) are characteristic of wounds and sites of infection (see Text Box). Thus phagocytic cells must be adapted to generate energy and function effectively in oxygen-deprived conditions, especially as many common bacterial pathogens proliferate readily in anaerobic microenvironments. Analysis of the bactericidal capacities of phagocytes from Hif1a conditional knockout mice validated a pivotal role for the hypoxic response in innate host defense. Compared to wild-type macrophages, macrophages isolated from mice deficient in HIF1α are impaired in their capacity to kill Gram-positive and Gram-negative bacteria15, 21. When challenged subcutaneously with the pathogen group A streptococcus, myeloid cell Hif1a conditional knockout mice developed significantly larger necrotic ulcers and had higher bacterial loads in the infected tissue and blood21.
Text Box.
Normoxia
Generally defined as either:
- atmospheric oxygen at sea level for tissue culture; or as
- physiological oxygenation in a well-vascularized and perfused tissue. This latter is, of course, dependent on the tissue in question, and in the case of innate immune system cells it becomes an even more elusive quantity to define.
Hypoxia
In tissue culture, generally defined as levels that are equivalent to between 0.5% – 3% oxygen by volume in the air that perfuses the growth medium. For actual tissues in vivo, this is more difficult to define, but is generally thought to occur in any tissue where injury or other alteration in perfusion causes a significant drop in tissue oxygen levels relative to those extant normally.
Anoxia
Defined in both tissue culture and physiology as the absence of physiologically available oxygen. Can occur in tissues in areas of acute infection and severe tissue damage.
Analysis of the host–pathogen encounter by several groups has revealed several aspects of myeloid cell function that depend on HIF. In neutrophils, HIF induces leukocyte β2 integrin expression and thereby promotes neutrophil binding to epithelium22. HIF increases neutrophil expression of antimicrobial molecules such as cathelicidin peptides[G] and the granule proteases cathepsin G and elastase21. HIF also extends the functional neutrophil lifespan by inhibiting apoptotic pathways23, 24. Increased levels of HIF are also evident during the differentiation of blood monocytes into tissue macrophages25. HIF activity has been described as a key aspect of phagocytic uptake of bacteria by macrophages under hypoxic conditions26, and macrophage production of tumour necrosis factor (TNF) and nitric oxide (NO) through inducible NO synthetase (iNOS) is HIF dependent21. Since NO can stabilize HIF1α protein by redistributing intracellular oxygen and inhibiting prolyl hydroxylase activity27, an autocrine feedback loop to amplify macrophage inflammatory activation is established. HIF-responsive elements are also found in the genes for Toll-like receptors (TLRs), including TLR2 and TLR6, which are upregulated in response to hypoxia28. Finally, HIF markedly increases the release of proinflammatory cytokines and the expression of co-stimulatory molecules by murine dendritic cells (DCs), enhancing their ability to induce allogeneic lymphocyte proliferation, thus helping to bridge innate and adaptive immune responses29.
One notable exception to the HIF-1 regulation of phagocyte function may be the generation of superoxide by the oxidative burst, which appears to occur with equal efficiency in wild-type and HIF 1α-deficient macrophages21.
Synergism of the hypoxic and innate immune responses
A surprising consequence of the findings summarized above is that, because of HIF activation, myeloid cells phagocytose and kill bacteria better under hypoxic conditions than they do under normoxic conditions21, 26. Strikingly, bacteria are an even stronger stimulus for HIF protein stabilization than is hypoxia itself, and bacteria-induced HIF protein stabilization can be readily demonstrated under normoxia21. Recently, the mechanistic explanation for these phenomena has been found to reflect a close, synergistic relationship between HIF and the central regulator of innate immunity, the transcription factor nuclear factor-κB (NF-κB).
NF-κB activity is controlled by IκB kinases (IKKs), mainly IKKβ, which carry out the phosphorylation-dependent degradation of IκB inhibitors in response to infectious or inflammatory stimuli30. HIF was shown to mediate NF-κB activation in anoxic neutrophils23 and to promote the expression of NF-κB regulated cytokines in macrophages stimulated by lipopolysaccharide (LPS) in a TLR4-dependent manner31. Interestingly, hypoxia itself can stimulate NF-κB activation by inhibiting prolyl hydroxylases that negatively modulate IKKβ catalytic activity32.
NF-κB was found to contribute to increased Hif1a mRNA transcription under hypoxic conditions33, 34. The activation of HIF transcription by bacteria or LPS under normoxic as well as hypoxic conditions has been recently verified in a study using mice deficient in IKKβ35. Macrophages infected with Gram-positive or Gram-negative bacteria, or mice subjected to hypoxia, reveal a marked defect in HIF1α expression following deletion of the gene encoding IKKβ35. These results confirm that transcriptional activation of Hif1a by IKKβ-responsive NF-κB is a crucial precursor to post-transcriptional stabilization and accumulation of HIF1α protein.
Because circulating phagocytes have a unique immune defense function, and must transit through different microenvironments during rapid mobilization to infected tissues, the synergistic HIF-NFκB pathway represents an elegant control mechanism for the specialized activities of these cells (Figure 2). Phagocyte bactericidal and pro-inflammatory capacities can be maintained in an “off” state while the myeloid cells circulate in the oxygen-rich blood; but then be activated in response to the decreasing oxygen gradient that is encountered following endothelial transcytosis and entry into the infected tissues. The primed phagocyte then undergoes a more potent stimulation of HIF pathway on direct pathogen encounter and, via recognition of pathogen-associated molecular patterns by TLRs, the activation of NF-κB and upregulation of Hif1α mRNA. The maximal “on-state” of bactericidal capacities is potentiated by the NO-mediated amplification loop, release of pro-inflammatory cytokines and upregulation of TLRs. This elegant control system links HIF to the myeloid cell response, ensuring that proinflammatory mediators, degradative enzymes and antimicrobial peptides are expressed preferentially in tissue site of infection, but not in healthy tissues where they would cause unwanted damage to host cells.
Figure 2. HIF regulation of phagocyte innate immune functions.
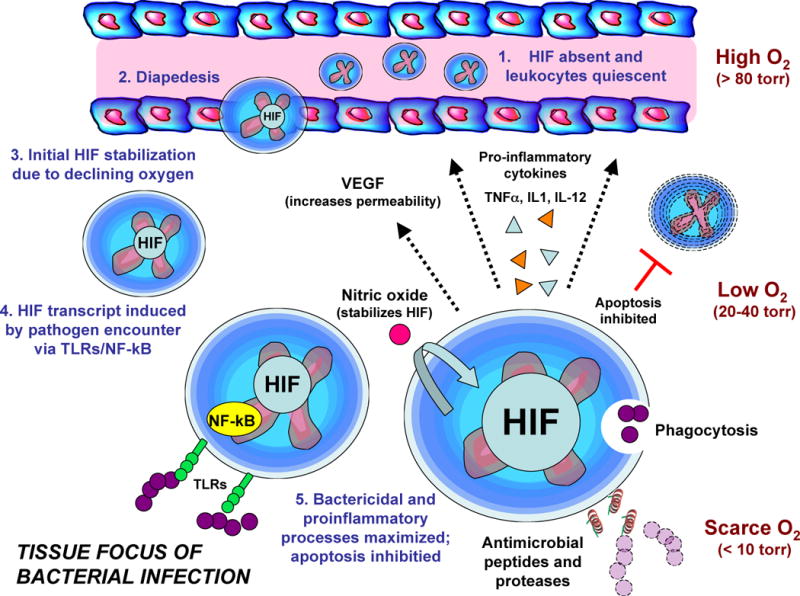
Myeloid-derived phagocytes such as neutrophils and macrophages have low hypoxia-inducible factor (HIF) levels at baseline as they circulate in the oxygen-rich blood. When recruited to tissue sites of infection, they migrate across the endothelium and immediately encounter a decreasing oxygen gradient, which leads to reduced prolyl hydroxylase activity and increased stabilization of HIF1α protein, which translocates to the nucleus and forms a functional heterodimeric transcription factor with HIF1β. Expression of innate immune response genes that contain hypoxia-responsive elements (HREs) in their promoters is increased, but maximal activation occurs only through Toll-like receptors (TLRs) and nuclear factor-KB (NF-κB) activation following pathogen encounter, which functions to boost HIF1α transcription. HIF activity promotes phagocytosis, inhibits apoptosis to increase phagocyte lifespan, stimulates the release of antimicrobial peptides, granule proteases, and pro-inflammatory cytokines such as tumour necrosis factor (TNF), interleukin-1 (IL-1) and IL-12, upregulates TLR expression, and activates the production of nitric oxide (NO) via inducible NO synthase (iNOS). NO interferes with HIF degradation, thereby creating an amplification loop for rapid phagocyte activation.
Role of HIF in other cell types
Beyond the functions of myeloid cells including neutrophils, macrophages and DCs, epithelial cells and other specialized leukocytes also have important roles in innate host defense. Evidence is mounting showing that HIF is a key regulator of the intrinsic immune and inflammatory responses in a variety of non-myeloid cell types. For example, the skin provides a highly effective physical, cellular and chemical barrier against microbial penetration36. In response to bacterial pathogens, keratinocytes produce peptides of the cathelicidin and β-defensin family that can directly kill microbial pathogens37. HIF is expressed at high levels in the skin, which is a hypoxic organ even at baseline. Following targeted deletion of Hif1a in keratinocytes, mice show a defect in controlling necrotic skin infection by group A streptococcus38. Specific RNA interference studies confirmed that HIF-mediated regulation of keratinocyte cathelicidin production is crucial for cutaneous defense against infection with this invasive bacterium38.
HIF expression is upregulated in the gut epithelium during ischemia–reperfusion injury[G], and the level of HIF activation during the reperfusion phase is strongly increased by exposure to LPS or the bacterium Pseudomonas aeruginosa39. Targeted manipulation of HIF1α expression in colonic epithelium also revealed an important role in the regulation of mucosal inflammatory responses to the irritant dextran-sulphate sodium (DSS). Mice with intestinal-specific disruption of HIF1β were protected against DSS-induced colitis, whereas those with constitutive HIF expression (as a result of VHL deletion) had increased expression of pro-inflammatory mediators, including macrophage migration inhibitor factor 1 (MIF1), leading to markedly increased oedema and cellular infiltrates40.
Mast cells are specialized granulocytic cells resident in the skin and the mucosa of the respiratory and gastrointestinal tracts. Well-studied for their roles in allergy, mast cells are increasingly recognized to function in both innate and adaptive immune responses41. Activation of HIF in human mast cells leads to release of pro-inflammatory cytokines such as interleukin-8 (IL-8) and TNF42. HIF activation in mast cells of the bronchial epithelium stimulates VEGF expression, leading to increased vascular permeability, protein extravasation into the alveolar space, and the hallmark airway oedema of asthma43. Moreover, HIF activation stimulates histidine decarboxylase expression by human mast cells, catalysing the formation of histamine, a potent inflammatory mediator44.
HIF inflammatory responses during sepsis
Sepsis, the leading cause of death in intensive care units, reflects an aberrant and potentially lethal host response to overwhelming infection, in which bacteria or LPS provoke uncontrolled pro-inflammatory cytokine release from immune cells including monocytes and macrophages. LPS raises HIF levels in macrophages through activities of the mitogen-activated protein kinase (MAPK) and NF-κB signal transduction pathways33, 35, leading to increased levels of Hif1a mRNA transcripts, which are accompanied by a TLR4-dependent decrease levels of mRNA encoding prolyl hydroxylases31. Studies of LPS challenge of mice with HIF 1α-deficient myeloid cells identified HIF as a crucial determinant of the sepsis phenotype, promoting high-level release of the pro-inflammatory cytokines TNF, IL-1 and IL_-_12. As a consequence, Hif1a deletion in the macrophage lineage protects animals against LPS-induced mortality and blocks the clinical manifestations of sepsis including hypotension, tachycardia and hypothermia31.
HIF transcriptional regulation also modulates pro-inflammatory cytokine production by CD4+ and CD8+ T cells, but in contrast to findings in myeloid cells, epithelial cells and mast cells, its net effect may be inhibitory. For example, following T cell receptor activation, the release of TNF and interferon-γ (IFNγ) by T cells with targeted deletion of Hif1a was higher than wild-type cells45. The activated HIF-deficient T cells showed enhanced proliferation and contributed to blocking bacterial proliferation in a caecal ligation and puncture[G] model of sepsis46. The paradoxical behaviour of HIF in activated T cells may derive from alternative splicing and expression of a novel isoform of HIF1α that inhibits cellular activation in a delayed feedback manner47. Data from studies involving the deletion of Hif1a specifically in T cells indicates that tight regulation of HIF function in these cells is crucial to prevent cell death, and implies that T cells may have a minor role in hypoxic tissues due to hypoxia-mediated and HIF-dependent cell death48.
Dynamics of the HIF response to infectious pathogens
Viruses
Acute infection with viruses is generally found to induce HIF protein stabilization in target cells, thus provoking local inflammation. For example, the common respiratory syncytial virus (RSV) induces HIF expression by human bronchial epithelial cells through a NO-dependent pathway, stimulating VEGF production and airway oedema characteristic of acute RSV infection49. In some cases, the HIF-dependent innate immune response to viral infection may help to mitigate cytolytic injury and viral replication. Vesicular stomatitis virus (VSV) infection is an acute illness resulting in mouth ulcers in cattle and occasionally in humans. HIF activation by hypoxia or pharmacological agents can increase the expression of IFNβ and other antiviral genes and promote cellular resistance to VSV infection50.
By contrast, with certain persistent viral infections, the induction of HIF fails to result in eradication of the virus. An unfortunate consequence of HIF activation in these circumstances is that increased VEGF and the accompanying pro-angiogenic programme can contribute to oncogenic transformation. This seems to be the case in chronic infections with the hepatitis B and C viruses (HBV and HCV), which are epidemiologically associated with development of hepatocellular carcinoma, a highly vascularized solid tumour. HIF levels are increased in liver cells transfected with the oncogenic X protein of HBV (HBx) or in the livers of HBx-transgenic mice51. HBx interacts directly with HIF1α to block its association with VHL and degradation by the ubiquitin–proteasome pathway52. Recently, HCV infection has also been found to stabilize HIF1α protein with involvement of the NF-κB and MAPK signalling pathways, stimulating VEGF production and neovascularization53.
Human papillomavirus 16 (HPV-16) is an aetiological agent of cervical interstitial neoplasia that, if undetected, can progress to cervical carcinoma. Transfection of human cervical cells with the HPV-16 oncoproteins E6 and E7 induces VEGF expression and capillary formation in a HIF-dependent manner54, and HPV-16 and HIF1α act synergistically to promote cancer lesions when expressed transgenically in the cervical epithelium of mice55. Increased HIF levels are correlated with poor prognosis of patients with advanced cervical cancer lesions, many of which are demonstrably hypoxic56. HIF activation is also apparent during other chronic viral infections that are associated with risk of neoplastic transformation, including human T-cell leukemia virus type I57 and Epstein–Barr virus58, 59.
Crosstalk between viral genes and HIF activation has recently been shown to have an intriguing role in regulating latency and reactivation of human herpesvirus 8 (HHV-8). HHV-8 infection is associated with the endothelial tumour Kaposi’s sarcoma in patients with AIDS. Biopsies of Kaposi’s sarcoma lesions demonstrate high levels of HIF, and HHV-8 infection of endothelial cells can stabilize HIF1α subunits and increase HIF target gene expression60. The HHV-8 latency-associated nuclear antigen stimulates Hif1a mRNA transcription and also physically interacts with the HIF heterodimer to enhance its promoter binding activities61. Induction and co-activation during HHV-8 infection allows HIF to bind HREs in the viral genome itself; these include one in the promoter of the gene encoding Rta, a key protein involved in reactivation of the virus from latency to a lytic replication phase61.
Bacteria
Increased levels of HIF are observed in macrophages or neutrophils that are stimulated by a wide variety of bacterial species including groups A and B streptococci, Staphylococcus aureus, Salmonella typhimurium and Pseudomonas aeruginosa15, 21, 31, 62, indicating that HIF responds in a general manner to bacterial infection. The myeloid cell HIF response pathway is beneficial to innate immune defense, promoting bacterial killing in vitro15, 21, 31 and restricting the spread of infection in vivo21). Bacterial products such as LPS and peptidoglycan can activate TLRs and NF-kB signaling to increase transcription of HIF1a {Frede, 2006 #108,35. Post-translational HIF1α protein stabilization is provided by the exhaustion of oxygen at the tissue site of infection, and perhaps in certain cases, through iron sequestration by bacterial siderophores[G]. Indeed, siderophore-deficient Yersinia enterocolitica fail to induce HIF activation in intestinal Peyer’s patches63 and the virulence of Y. enterocolitica is increased in mice that lack intestinal HIF1α expression, suggesting that the HIF response is important for mucosal innate defense.
Bartonella henselae is a facultative intracellular bacterium that causes an angioproliferative disorder known as bacillary angiomatosis in immunocompromised patients. bacillary angiomatosis lesions demonstrate high levels of HIF expression, and B. henselae activation of HIF-induced VEGF release is likely to provoke a pro-angiogenic programme that contributes to the characteristic tissue pathology64. Another intracellular bacterial pathogen, Chlamydia pneumoniae, has evolved a mechanism to counteract HIF protein stabilization, thereby inhibiting innate immune activation and promoting its survival in host cells. Secretion of a chlamydial protease-like activity factor into the cytoplasm degrades accumulated HIF1α, facilitating continued C. pneumoniae replication during hypoxia65.
Parasites
Toxoplasma gondii is an obligate intracellular parasite that produces opportunistic infections in fetuses and in immunocompromised individuals. T. gondii rapidly induces HIF expression by infected fibroblasts66, leading to upregulation of genes encoding glycolytic enzymes, glucose transporters and VEGF67. Under hypoxic conditions present in the tissues (brain, muscle and retina) in which the parasite produces serious clinical pathology, T. gondii replication and organelle maintenance was severely impaired in host cells that lack HIF1α. It is thought that T. gondii has evolved to induce HIF expression because a particular HIF target gene is essential to parasite growth, or alternatively, because HIF activation is necessary to preserve the health of the host cell in which the parasite has become established66. Cutaneous lesions can be generated in BALB/c mice by infection with Leishmania amazonensis, and in the later stages of infection, HIF is induced in the cytoplasm and parasitophorous vacuoles of macrophages recruited to the skin lesions68.
HIF-1 and SLC11A1 allele expression phenotypes
SLC11A1 (also known as Nramp1) is a protein-coupled divalent ion transporter involved in iron metabolism, resistance to pathogens, and inflammatory responses. SLC11A1 was the first infectious disease susceptibility gene identified by positional cloning, and allelic variation in SLC11A1 alters the risk for development of leishmaniasis69 and tuberculosis70, as well as autoimmune conditions such as juvenile rheumatoid arthritis71 and type 1 diabetes72. Interestingly, HIF differentially binds and activates Z-DNA-forming microsatellite polymorphisms[G] in the SLC11A1 promoter region, thereby shaping allele expression phenotypes62, a newly described function for a transcription factor. Through its differential interaction with variant SLC11A1 promoters, HIF transcriptional regulation has the potential to influence heritable differences in infectious and inflammatory disease susceptibility among and between human populations62.
HIF: a pharmacological target to augment innate immunity
Given the accumulating evidence that HIF acts as a “master regulator” of the innate immune function of phagocytes73, it is possible that boosting HIF activity through pharmacological strategies might provide a novel approach to aid in the treatment of certain infectious disease conditions74. This concept is supported by the observation that macrophages that lack VHL, thus leading to constitutively high levels of HIF, are markedly more efficient then wild-type macrophages at killing Gram-positive and Gram-negative bacteria in vitro21. This implies that our phagocytic cells are not as good as they could be at killing bacteria, mainly because their activation is tightly regulated to limit unnecessary inflammatory injury. However, in the common clinical scenario of invasive bacterial infection, a failure of innate immunity occurs, and it is possible that such patients might benefit from augmentation of phagocytic cell bactericidal activity by HIF.
As proof-of-principle, addition of a series of pharmacological agonists of HIF, each a so-called “hypoxia mimetic” that restrict prolyl hydroxylase access to iron, directly enhanced murine macrophage bactericidal activity in vitro21. Similarly, a dose-dependent enhancement of the bactericidal activities of human whole blood, neutrophils and the macrophage cell line U937 against the pathogen S. aureus was achieved using one such hypoxia-mimetic, L-mimosine75. Local treatment with L-mimosine significantly delayed progression of S. aureus abscesses in a mouse subcutaneous challenge model75.
HIF agonists that are designed to activate phagocyte bactericidal mechanisms could conceivably be used alongside conventional antibiotics in localized infections, and would be predicted to function effectively against drug-resistant bacteria such as methicillin-resistant S. aureus (MRSA). The selective pressure for pathogens to evolve resistance to HIF agonist therapy may be negligible, since the drug targets the host and not the pathogen, deploying a multifaceted combination therapy of natural antimicrobial molecules76. However, an important cautionary note should be emphasized: based on evidence from studies of mice31, HIF agonists are probably inappropriate for systemic therapy of patients with disseminated infections and symptoms of sepsis, as macrophage pro-inflammatory cytokine and NO release could be precipitously increased and symptoms could worsen. Last, strategies to inhibit HIF for the treatment of chronic inflammatory disorders, such as rheumatoid arthritis, may provide a safer therapeutic margin than cytotoxic agents and high-dose steroids, as post-translational regulation of HIF1α levels may allow rapid restoration of innate immune function of phagocytes following drug withdrawal in the event of opportunistic infection.
Conclusions and Future Directions
A wealth of emerging information shows that HIF and the hypoxic response are deeply intercalated into the regulatory pathways of innate immune defence. The key implication of these findings is that the nature and magnitude of host bactericidal and inflammatory activities are highly dependent upon factors in the local tissue microenvironment, e.g. oxygen tension and iron availability, and cannot be simply extrapolated from in vitro model systems under ambient conditions. Through HIF control of immune cell energetics and gene expression pathways, antimicrobial activities can be focused and amplified where they are needed most, namely tissue foci of infection, harsh and threatening microenvironments where oxygen and nutrients are limiting and cytotoxic molecules abound. We anticipate that future studies will reveal that major human pathogens modulate HIF activation pathways to subvert innate immunity or provoke dysregulated inflammation, contributing to cardinal clinical manifestations. A detailed understanding of the relationships between HIF, pathways of innate immune signal transduction such as TLRs/NF-κB, and the deployment of various immune effector molecules will provide a clearer and more physiologic understanding of infectious and inflammatory disease pathogenesis. Because of the short-half life and well understood mechanism for post-translational regulation of HIF levels, it represents an exciting pharmacologic target to fine tune immune cell functions up (antibiotic), or down (anti-inflammatory), in human disease therapeutics.
Table 1.
Effects of infectious pathogens on HIF levels and contribution to disease pathogenesis
| Name of Pathogen | Effect on HIF stabilization | Effect of altered HIF on disease pathogenesis | References |
|---|---|---|---|
| Bacteria | |||
| Streptococcus pyogenes | Increases | Activation of phagocyte killing | 31 |
| Streptococcus agalactiae | Increases | Activation of phagocyte killing | 15 |
| Pseudomonas aeruginosa | Increases | Activation of phagocyte killing | 31 |
| Salmonella typhimurium | Increases | Activation of phagocyte killing | 62 |
| Bartonella henselae | Increases | Angiogenesis, vascular lesions | 64 |
| Chlamydia pneumoniae | Decreases | Promotes intracellular survival | 65 |
| Bacterial components | |||
| Lipopolysaccharide | Increases | Phagocyte activation, but with high systemic doses, cytokine storm and sepsis syndrome | 31, 34 |
| Siderophores | Increases | Activation of mucosal defenses | 63 |
| Viruses | |||
| Respiratory syncytial virus | Increases | VEGF release, airway oedema | 49 |
| Vesicular stomatitis virus | Increases | Interferon production and cellular resistance to virus | 50 |
| Hepatitis B virus | Increases | Neovascularization, oncogenesis | 51 |
| Hepatitis C virus | Increases | Neovascularization, oncogenesis | 53 |
| Human papillomavirus | Increases | Neovascularization, oncogenesis | 54, 55 |
| Human herpesvirus 8 | Increases | Reactivation of virus from latency | 60, 61 |
| Parasites | |||
| Toxoplasma gondii | Increases | Preserves viability of host cell in which pathogen is established | 66 |
| Leishmania amazonensis | Increases | Development of skin ulcers | 68 |
Acknowledgments
The authors’ studies in the area of HIF and innate immunity have been supported by NIH grant AI060840 (to RSJ and VN).
Glossary
ubiquitin–proteasome pathway
An important proteolytic pathway that involves the tagging of unwanted proteins with ubiquitin, which allows their recognition by the proteasome — a large, multi-component protein-degrading complex
Cre recombinase
Cre is a site-specific recombinase that recognizes and binds specific sites known as loxP. Two loxP sites recombine in the presence of Cre, allowing DNA that is cloned between two such sites to be removed by Cre-mediated recombination
cathelicidin
Mammalian cationic microbicidal peptides expressed by epithelial cells and phagocytes that share a highly conserved ‘cathelin’ 12 kDa pro-sequence at the amino terminus, followed by diversified mature sequences at the carboxy terminus. Activation of most cathelicidin precursors requires proteolytic cleavage to release the C-terminal domain, which has microbicidal and immunomodulatory activities
ischaemia-reperfusion injury
An injury in which the tissue first suffers from hypoxia as a result of severely decreased, or completely arrested, blood flow. Restoration of normal blood flow then triggers inflammation, which exacerbates the tissue damage
caecal ligation and puncture
An experimental model of peritonitis in rodents, in which the caecum is ligated and then punctured, thereby forming a small hole. This leads to leakage of intestinal bacteria into the peritoneal cavity and subsequent peritoneal infection
siderophores
Low molecular-weight compounds that are secreted by numerous types of bacteria and that have a high affinity for iron and other metal ions. These molecules chelate metal ions and carry them into the cell through specific receptors, promoting bacterial survival in the host
Z-DNA-forming microsatellite polymorphisms
Microsatellites such as the SLC11A1 (GT/CA)n dinucleotide repeat have a propensity to form Z-DNA, an unstable left-handed form of DNA transiently induced during gene transcription by a moving RNA polymerase, and stabilized by negative supercoiling. Polymorphisms in such microsatellites have been shown to either activate or repress gene transcription in a context-dependent manner
References
- 1.Saadi S, Wrenshall LE, Platt JL. Regional manifestations and control of the immune system. FASEB J. 2002;16:849–56. doi: 10.1096/fj.01-0690hyp. [DOI] [PubMed] [Google Scholar]
- 2.Schor H, Vaday GG, Lider O. Modulation of leukocyte behavior by an inflamed extracellular matrix. Dev Immunol. 2000;7:227–38. doi: 10.1155/2000/51902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Semenza GL, Wang GL. A nuclear factor induced by hypoxia via de novo protein synthesis binds to the human erythropoietin gene enhancer at a site required for transcriptional activation. Mol Cell Biol. 1992;12:5447–54. doi: 10.1128/mcb.12.12.5447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang GL, Jiang BH, Rue EA, Semenza GL. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci U S A. 1995;92:5510–4. doi: 10.1073/pnas.92.12.5510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Weidemann A, Johnson RS. Biology of HIF-1α. Cell Death Differ. 2008;15:621–7. doi: 10.1038/cdd.2008.12. [DOI] [PubMed] [Google Scholar]
- 6.Maxwell PH. The HIF pathway in cancer. Semin Cell Dev Biol. 2005;16:523–30. doi: 10.1016/j.semcdb.2005.03.001. [DOI] [PubMed] [Google Scholar]
- 7.Bruick RK, McKnight SL. A conserved family of prolyl-4-hydroxylases that modify HIF. Science. 2001;294:1337–40. doi: 10.1126/science.1066373. [DOI] [PubMed] [Google Scholar]
- 8.Jaakkola P, et al. Targeting of HIF-1α to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science. 2001;292:468–72. doi: 10.1126/science.1059796. [DOI] [PubMed] [Google Scholar]
- 9.Fukuda R, et al. HIF-1 regulates cytochrome oxidase subunits to optimize efficiency of respiration in hypoxic cells. Cell. 2007;129:111–22. doi: 10.1016/j.cell.2007.01.047. [DOI] [PubMed] [Google Scholar]
- 10.Semenza GL, Roth PH, Fang HM, Wang GL. Transcriptional regulation of genes encoding glycolytic enzymes by hypoxia-inducible factor 1. J Biol Chem. 1994;269:23757–63. [PubMed] [Google Scholar]
- 11.Shimoda LA, Fallon M, Pisarcik S, Wang J, Semenza GL. HIF-1 regulates hypoxic induction of NHE1 expression and alkalinization of intracellular pH in pulmonary arterial myocytes. Am J Physiol Lung Cell Mol Physiol. 2006;291:L941–9. doi: 10.1152/ajplung.00528.2005. [DOI] [PubMed] [Google Scholar]
- 12.Iyer NV, et al. Cellular and developmental control of O2 homeostasis by hypoxia-inducible factor 1α. Genes Dev. 1998;12:149–62. doi: 10.1101/gad.12.2.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ryan HE, Lo J, Johnson RS. HIF-1α is required for solid tumor formation and embryonic vascularization. EMBO J. 1998;17:3005–15. doi: 10.1093/emboj/17.11.3005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kotch LE, Iyer NV, Laughner E, Semenza GL. Defective vascularization of HIF-1α-null embryos is not associated with VEGF deficiency but with mesenchymal cell death. Dev Biol. 1999;209:254–67. doi: 10.1006/dbio.1999.9253. [DOI] [PubMed] [Google Scholar]
- 15.Cramer T, et al. HIF-1α is essential for myeloid cell-mediated inflammation. Cell. 2003;112:645–57. doi: 10.1016/s0092-8674(03)00154-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hollander AP, Corke KP, Freemont AJ, Lewis CE. Expression of hypoxia-inducible factor 1alpha by macrophages in the rheumatoid synovium: implications for targeting of therapeutic genes to the inflamed joint. Arthritis Rheum. 2001;44:1540–4. doi: 10.1002/1529-0131(200107)44:7<1540::AID-ART277>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 17.Westra J, et al. Regulation of cytokine-induced HIF-1alpha expression in rheumatoid synovial fibroblasts. Ann N Y Acad Sci. 2007;1108:340–8. doi: 10.1196/annals.1422.035. [DOI] [PubMed] [Google Scholar]
- 18.Konttinen YT, et al. Disease-associated increased HIF-1, αvβ3 integrin, and Flt-1 do not suffice to compensate the damage-inducing loss of blood vessels in inflammatory myopathies. Rheumatol Int. 2004;24:333–9. doi: 10.1007/s00296-003-0379-z. [DOI] [PubMed] [Google Scholar]
- 19.Clancy RM, et al. Role of hypoxia and cAMP in the transdifferentiation of human fetal cardiac fibroblasts: implications for progression to scarring in autoimmune-associated congenital heart block. Arthritis Rheum. 2007;56:4120–31. doi: 10.1002/art.23061. [DOI] [PubMed] [Google Scholar]
- 20.Vink A, et al. HIF-1α expression is associated with an atheromatous inflammatory plaque phenotype and upregulated in activated macrophages. Atherosclerosis. 2007;195:e69–75. doi: 10.1016/j.atherosclerosis.2007.05.026. [DOI] [PubMed] [Google Scholar]
- 21.Peyssonnaux C, et al. HIF-1α expression regulates the bactericidal capacity of phagocytes. J Clin Invest. 2005;115:1806–15. doi: 10.1172/JCI23865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kong T, Eltzschig HK, Karhausen J, Colgan SP, Shelley CS. Leukocyte adhesion during hypoxia is mediated by HIF-1-dependent induction of β2 integrin gene expression. Proc Natl Acad Sci U S A. 2004;101:10440–5. doi: 10.1073/pnas.0401339101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Walmsley SR, et al. Hypoxia-induced neutrophil survival is mediated by HIF-1α-dependent NF-kappaB activity. J Exp Med. 2005;201:105–15. doi: 10.1084/jem.20040624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Walmsley SR, et al. Neutrophils from patients with heterozygous germline mutations in the von Hippel Lindau protein (pVHL) display delayed apoptosis and enhanced bacterial phagocytosis. Blood. 2006;108:3176–8. doi: 10.1182/blood-2006-04-018796. [DOI] [PubMed] [Google Scholar]
- 25.Oda T, et al. Activation of hypoxia-inducible factor 1 during macrophage differentiation. Am J Physiol Cell Physiol. 2006;291:C104–13. doi: 10.1152/ajpcell.00614.2005. [DOI] [PubMed] [Google Scholar]
- 26.Anand RJ, et al. Hypoxia causes an increase in phagocytosis by macrophages in a HIF-1alpha-dependent manner. J Leukoc Biol. 2007;82:1257–65. doi: 10.1189/jlb.0307195. [DOI] [PubMed] [Google Scholar]
- 27.Hagen T, Taylor CT, Lam F, Moncada S. Redistribution of intracellular oxygen in hypoxia by nitric oxide: effect on HIF1alpha. Science. 2003;302:1975–8. doi: 10.1126/science.1088805. [DOI] [PubMed] [Google Scholar]
- 28.Kuhlicke J, Frick JS, Morote-Garcia JC, Rosenberger P, Eltzschig HK. Hypoxia inducible factor (HIF)-1 coordinates induction of Toll-like receptors TLR2 and TLR6 during hypoxia. PLoS One. 2007;2:e1364. doi: 10.1371/journal.pone.0001364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jantsch J, et al. Hypoxia and hypoxia-inducible factor-lα modulate lipopolysaccharide-induced dendritic cell activation and function. J Immunol. 2008;180:4697–705. doi: 10.4049/jimmunol.180.7.4697. [DOI] [PubMed] [Google Scholar]
- 30.Hacker H, Karin M. Regulation and function of IKK and IKK-related kinases. Sci STKE. 2006;2006:re13. doi: 10.1126/stke.3572006re13. [DOI] [PubMed] [Google Scholar]
- 31.Peyssonnaux C, et al. Cutting edge: Essential role of hypoxia inducible factor-1alpha in development of lipopolysaccharide-induced sepsis. J Immunol. 2007;178:7516–9. doi: 10.4049/jimmunol.178.12.7516. [DOI] [PubMed] [Google Scholar]
- 32.Cummins EP, et al. Prolyl hydroxylase-1 negatively regulates IkappaB kinase-beta, giving insight into hypoxia-induced NFκB activity. Proc Natl Acad Sci U S A. 2006;103:18154–9. doi: 10.1073/pnas.0602235103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Frede S, Stockmann C, Freitag P, Fandrey J. Bacterial lipopolysaccharide induces HIF-1 activation in human monocytes via p44/42 MAPK and NF-κB. Biochem J. 2006;396:517–27. doi: 10.1042/BJ20051839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Belaiba RS, et al. Hypoxia up-regulates hypoxia-inducible factor-1α transcription by involving phosphatidylinositol 3-kinase and nuclear factor κB in pulmonary artery smooth muscle cells. Mol Biol Cell. 2007;18:4691–7. doi: 10.1091/mbc.E07-04-0391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rius J, et al. NF-κB links innate immunity to the hypoxic response through transcriptional regulation of HIF-1α. Nature. 2008;453:807–11. doi: 10.1038/nature06905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Goodarzi H, Trowbridge J, Gallo RL. Innate immunity: a cutaneous perspective. Clin Rev Allergy Immunol. 2007;33:15–26. doi: 10.1007/s12016-007-0037-4. [DOI] [PubMed] [Google Scholar]
- 37.Braff MH, Zaiou M, Fierer J, Nizet V, Gallo RL. Keratinocyte production of cathelicidin provides direct activity against bacterial skin pathogens. Infect Immun. 2005;73:6771–81. doi: 10.1128/IAI.73.10.6771-6781.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Peyssonnaux C, et al. Critical role of HIF-1alpha in keratinocyte defense against bacterial infection. J Invest Dermatol. 2008;128:1964–8. doi: 10.1038/jid.2008.27. [DOI] [PubMed] [Google Scholar]
- 39.Koury J, et al. Persistent HIF-1α activation in gut ischemia/reperfusion injury: potential role of bacteria and lipopolysaccharide. Shock. 2004;22:270–7. doi: 10.1097/01.shk.0000135256.67441.3f. [DOI] [PubMed] [Google Scholar]
- 40.Shah YM, et al. Hypoxia-inducible factor augments experimental colitis through an MIF-dependent inflammatory signaling cascade. Gastroenterology. 2008;134:2036–48. 2048e1–3. doi: 10.1053/j.gastro.2008.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rao KN, Brown MA. Mast cells: multifaceted immune cells with diverse roles in health and disease. Ann N Y Acad Sci. 2008;1143:83–104. doi: 10.1196/annals.1443.023. [DOI] [PubMed] [Google Scholar]
- 42.Jeong HJ, et al. Expression of proinflammatory cytokines via HIF-1α and NF-κB activation on desferrioxamine-stimulated HMC-1 cells. Biochem Biophys Res Commun. 2003;306:805–11. doi: 10.1016/s0006-291x(03)01073-8. [DOI] [PubMed] [Google Scholar]
- 43.Lee KS, et al. Mast cells can mediate vascular permeability through regulation of the PI3K-HIF-1α -VEGF axis. Am J Respir Crit Care Med. 2008;178:787–97. doi: 10.1164/rccm.200801-008OC. [DOI] [PubMed] [Google Scholar]
- 44.Jeong HJ, et al. Activation of hypoxia-inducible factor-1 regulates human histidine decarboxylase expression. Cell Mol Life Sci. 2009 doi: 10.1007/s00018-009-9001-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lukashev D, et al. Cutting edge: hypoxia-inducible factor 1α and its activation-inducible short isoform I.1 negatively regulate functions of CD4+ and CD8+ T lymphocytes. J Immunol. 2006;177:4962–5. doi: 10.4049/jimmunol.177.8.4962. [DOI] [PubMed] [Google Scholar]
- 46.Thiel M, et al. Targeted deletion of HIF-1α gene in T cells prevents their inhibition in hypoxic inflamed tissues and improves septic mice survival. PLoS ONE. 2007;2:e853. doi: 10.1371/journal.pone.0000853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lukashev D, Sitkovsky M. Preferential expression of the novel alternative isoform I.3 of hypoxia-inducible factor 1α in activated human T lymphocytes. Hum Immunol. 2008;69:421–5. doi: 10.1016/j.humimm.2008.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Biju MP, et al. VhlH gene deletion induces HIF-1-mediated cell death in thymocytes. Mol Cell Biol. 2004;24:9038–47. doi: 10.1128/MCB.24.20.9038-9047.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kilani MM, Mohammed KA, Nasreen N, Tepper RS, Antony VB. RSV causes HIF-1α stabilization via NO release in primary bronchial epithelial cells. Inflammation. 2004;28:245–51. doi: 10.1007/s10753-004-6047-y. [DOI] [PubMed] [Google Scholar]
- 50.Hwang II, Watson IR, Der SD, Ohh M. Loss of VHL confers hypoxia-inducible factor (HIF)-dependent resistance to vesicular stomatitis virus: role of HIF in antiviral response. J Virol. 2006;80:10712–23. doi: 10.1128/JVI.01014-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yoo YG, et al. Hepatitis B virus X protein enhances transcriptional activity of hypoxia-inducible factor-1α through activation of mitogen-activated protein kinase pathway. J Biol Chem. 2003;278:39076–84. doi: 10.1074/jbc.M305101200. [DOI] [PubMed] [Google Scholar]
- 52.Moon EJ, et al. Hepatitis B virus X protein induces angiogenesis by stabilizing hypoxia-inducible factor-1α. FASEB J. 2004;18:382–4. doi: 10.1096/fj.03-0153fje. [DOI] [PubMed] [Google Scholar]
- 53.Nasimuzzaman M, Waris G, Mikolon D, Stupack DG, Siddiqui A. Hepatitis C virus stabilizes hypoxia-inducible factor 1α and stimulates the synthesis of vascular endothelial growth factor. J Virol. 2007;81:10249–57. doi: 10.1128/JVI.00763-07. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 54.Tang X, et al. Overexpression of human papillomavirus type 16 oncoproteins enhances hypoxia-inducible factor 1α protein accumulation and vascular endothelial growth factor expression in human cervical carcinoma cells. Clin Cancer Res. 2007;13:2568–76. doi: 10.1158/1078-0432.CCR-06-2704. [DOI] [PubMed] [Google Scholar]
- 55.Lu ZH, Wright JD, Belt B, Cardiff RD, Arbeit JM. Hypoxia-inducible factor-1 facilitates cervical cancer progression in human papillomavirus type 16 transgenic mice. Am J Pathol. 2007;171:667–81. doi: 10.2353/ajpath.2007.061138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Birner P, et al. Overexpression of hypoxia-inducible factor 1α is a marker for an unfavorable prognosis in early-stage invasive cervical cancer. Cancer Res. 2000;60:4693–6. [PubMed] [Google Scholar]
- 57.Tomita M, et al. Activation of hypoxia-inducible factor 1 in human T-cell leukaemia virus type 1-infected cell lines and primary adult T-cell leukaemia cells. Biochem J. 2007;406:317–23. doi: 10.1042/BJ20070286. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 58.Wakisaka N, et al. Epstein-Barr virus latent membrane protein 1 induces synthesis of hypoxia-inducible factor 1α. Mol Cell Biol. 2004;24:5223–34. doi: 10.1128/MCB.24.12.5223-5234.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kondo S, et al. EBV latent membrane protein 1 up-regulates hypoxia-inducible factor-1α through Siah1-mediated down-regulation of prolyl hydroxylases 1 and 3 in nasopharyngeal epithelial cells. Cancer Res. 2006;66:9870–7. doi: 10.1158/0008-5472.CAN-06-1679. [DOI] [PubMed] [Google Scholar]
- 60.Carroll PA, Kenerson HL, Yeung RS, Lagunoff M. Latent Kaposi’s sarcoma-associated herpesvirus infection of endothelial cells activates hypoxia-induced factors. J Virol. 2006;80:10802–12. doi: 10.1128/JVI.00673-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cai Q, et al. Kaposi’s sarcoma-associated herpesvirus latent protein LANA interacts with HIF-1α to upregulate RTA expression during hypoxia: Latency control under low oxygen conditions. J Virol. 2006;80:7965–75. doi: 10.1128/JVI.00689-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bayele HK, et al. HIF-1 regulates heritable variation and allele expression phenotypes of the macrophage immune response gene SLC11A1 from a Z-DNA forming microsatellite. Blood. 2007;110:3039–48. doi: 10.1182/blood-2006-12-063289. [DOI] [PubMed] [Google Scholar]
- 63.Werth N, Hartmann H, Wurz H, Amr A, Kempf VA. Role of HIF-1 in bacterial infections. Acta Physiologica. 2006;188:P23. [Google Scholar]
- 64.Kempf VA, et al. Activation of hypoxia-inducible factor-1 in bacillary angiomatosis: evidence for a role of hypoxia-inducible factor-1 in bacterial infections. Circulation. 2005;111:1054–62. doi: 10.1161/01.CIR.0000155608.07691.B7. [DOI] [PubMed] [Google Scholar]
- 65.Rupp J, et al. Chlamydia pneumoniae directly interferes with HIF-1α stabilization in human host cells. Cell Microbiol. 2007;9:2181–91. doi: 10.1111/j.1462-5822.2007.00948.x. [DOI] [PubMed] [Google Scholar]
- 66.Spear W, et al. The host cell transcription factor hypoxia-inducible factor 1 is required for Toxoplasma gondii growth and survival at physiological oxygen levels. Cell Microbiol. 2006;8:339–52. doi: 10.1111/j.1462-5822.2005.00628.x. [DOI] [PubMed] [Google Scholar]
- 67.Blader IJ, Manger ID, Boothroyd JC. Microarray analysis reveals previously unknown changes in Toxoplasma gondii-infected human cells. J Biol Chem. 2001;276:24223–31. doi: 10.1074/jbc.M100951200. [DOI] [PubMed] [Google Scholar]
- 68.Arrais-Silva WW, Paffaro VA, Jr, Yamada AT, Giorgio S. Expression of hypoxia-inducible factor-1α in the cutaneous lesions of BALB/c mice infected with Leishmania amazonensis. Exp Mol Pathol. 2005;78:49–54. doi: 10.1016/j.yexmp.2004.09.002. [DOI] [PubMed] [Google Scholar]
- 69.Mohamed HS, et al. SLC11A1 (formerly NRAMP1) and susceptibility to visceral leishmaniasis in The Sudan. Eur J Hum Genet. 2004;12:66–74. doi: 10.1038/sj.ejhg.5201089. [DOI] [PubMed] [Google Scholar]
- 70.Li HT, Zhang TT, Zhou YQ, Huang QH, Huang J. SLC11A1 (formerly NRAMP1) gene polymorphisms and tuberculosis susceptibility: a meta-analysis. Int J Tuberc Lung Dis. 2006;10:3–12. [PubMed] [Google Scholar]
- 71.Runstadler JA, et al. Association of SLC11A1 (NRAMP1) with persistent oligoarticular and polyarticular rheumatoid factor-negative juvenile idiopathic arthritis in Finnish patients: haplotype analysis in Finnish families. Arthritis Rheum. 2005;52:247–56. doi: 10.1002/art.20772. [DOI] [PubMed] [Google Scholar]
- 72.Takahashi K, et al. Promoter polymorphism of SLC11A1 (formerly NRAMP1) confers susceptibility to autoimmune type 1 diabetes mellitus in Japanese. Tissue Antigens. 2004;63:231–6. doi: 10.1111/j.1399-0039.2004.000172.x. [DOI] [PubMed] [Google Scholar]
- 73.Strieter RM. Mastering innate immunity. Nat Med. 2003;9:512–3. doi: 10.1038/nm0503-512. [DOI] [PubMed] [Google Scholar]
- 74.Zarember KA, Malech HL. HIF-1α: a master regulator of innate host defenses? J Clin Invest. 2005;115:1702–4. doi: 10.1172/JCI25740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zinkernagel AS, Peyssonnaux C, Johnson RS, Nizet V. Pharmacologic augmentation of hypoxia-inducible factor-1α with mimosine boosts the bactericidal capacity of phagocytes. J Infect Dis. 2008;197:214–7. doi: 10.1086/524843. [DOI] [PubMed] [Google Scholar]
- 76.Nizet V. Understanding how leading bacterial pathogens subvert innate immunity to reveal novel therapeutic targets. J Allergy Clin Immunol. 2007;120:13–22. doi: 10.1016/j.jaci.2007.06.005. [DOI] [PubMed] [Google Scholar]