PARK2 patient neuroprogenitors show increased mitochondrial sensitivity to copper (original) (raw)
. Author manuscript; available in PMC: 2016 Jan 1.
Published in final edited form as: Neurobiol Dis. 2014 Oct 12;73:204–212. doi: 10.1016/j.nbd.2014.10.002
Abstract
Poorly-defined interactions between environmental and genetic risk factors underlie Parkinson’s disease (PD) etiology. Here we tested the hypothesis that human stem cell derived forebrain neuroprogenitors from patients with known familial risk for early onset PD will exhibit enhanced sensitivity to PD environmental risk factors compared to healthy control subjects without a family history of PD. Two male siblings (SM and PM) with biallelic loss-of-function mutations in PARK2 were identified. Human induced pluripotent stem cells (hiPSCs) from SM, PM, and four control subjects with no known family histories of PD or related neurodegenerative diseases were utilized. We tested the hypothesis that hiPSC-derived neuroprogenitors from patients with PARK2 mutations would show heightened cell death, mitochondrial dysfunction, and reactive oxygen species generation compared to control cells as a result of exposure to heavy metals (PD environmental risk factors). We report that PARK2 mutant neuroprogenitors showed increased cytotoxicity with copper (Cu) and cadmium (Cd) exposure but not manganese (Mn) or methyl mercury (MeHg) relative to control neuroprogenitors. PARK2 mutant neuroprogenitors also showed a substantial increase in mitochondrial fragmentation, initial ROS generation, and loss of mitochondrial membrane potential following Cu exposure. Our data substantiate Cu exposure as an environmental risk factor for PD. Furthermore, we report a shift in the lowest observable effect level (LOEL) for greater sensitivity to Cu-dependent mitochondrial dysfunction in patients SM and PM relative to controls, correlating with their increased genetic risk for PD.
Keywords: PARK2, neurotoxicty, environmental risk factors, Parkinson’s disease, Copper
INTRODUCTION
Parkinson’s disease (PD) is characterized by progressive motor function decline due to loss of dopaminergic neurons in the substantia nigra (SN). A minority of PD patients (familial PD) have single gene mutations associated with disease etiology (~10% of cases). The remaining idiopathic PD cases are influenced by interactions of genetic and environmental risk factors, including exposure to heavy metals and pesticides (Betarbet et al., 2000; Gorell et al., 1999; Gorell et al., 1998; Jomova et al., 2010). The role of environmental factors is underscored by a high discordance between monozygotic twins (Tanner et al., 1999).
Mutations of PARK2 are one of the most common causes of early onset PD (EOPD) (Kitada et al., 1998). Despite this association, patients with biallelic loss-of-function PARK2 mutations can have incomplete penetrance with high intra-familial and inter-familial variability in ages of onset (Abbas et al., 1999; Deng et al., 2006; Khan et al., 2003). For example, one study found a 56-year old subject with compound heterozygous mutations in PARK2 exhibiting no evidence of PD despite having four siblings with the same PARK2 mutation that were diagnosed with EOPD at ages from 30 to 38 (Deng et al., 2006). Therefore, we hypothesized that biallelic loss-of-function PARK2 mutations would increase sensitivity to PD-associated environmental risk factors.
Recently, differentiated neural lineages from hiPSCs of patients with PARK2 mutations have been reported to exhibit mitochondrial dysfunction and increased oxidative stress (Imaizumi et al., 2012). Both these phenotypes have separately been associated with exposures to heavy metals (Jomova et al., 2010). Here we performed a proof-of-principle assessment of PD-relevant toxicant vulnerability in two subjects (SM and PM) with compound heterozygous loss-of-function mutations in PARK2 versus control subjects. We utilized hiPSCs that are differentiated into neuroprogenitors that retain the unique genetic information of each human subject. Toxicant sensitivities at this early neurodevelopmental time point are pertinent given the strong evidence for in utero and early life environmental insults contributing to subsequent risk for PD (de la Fuente-Fernández and Calne, 2002; Landrigan et al., 2005).
METHODS
Human subjects, clinical findings, generation of hiPSC lines
Primary dermal fibroblasts were obtained by skin biopsy from healthy adult subjects (CA, CE, CF) with no known family history or genetic risk factors for PD and two PD patients (PM and SM) after appropriate patient consent/assent under the guidelines of an approved IRB protocol (Vanderbilt #080369). The MRC-5 fibroblasts (obtained from Coriell Institute for Medical Research and designated CB) were originally derived from a 14-week fetus aborted for maternal psychiatric reasons from a 27 year-old physically healthy woman with no known family history or genetic risk factors for PD. The studied patient PM was diagnosed with EOPD in his 30s and had dystonia from age 12 and resting tremor at age 17. He began having gait difficulties at age 23. Medical genetic analysis was performed, and biallelic mutations in the PARK2 locus were identified. The studied patient SM, brother of PM, was diagnosed at the Vanderbilt University Medical Center Movement Disorders Clinic with exercise-induced dystonia. Given his family history, SM also underwent genetic testing revealing the same compound heterozygous mutations in PARK2 as his brother PM. SM had a normal neurological exam by a movement disorders specialist at age 40 - the time of the skin biopsy - with no evidence of baseline dystonia or parkinsonism at that time. However, a clinical DaTScan via SPECT imaging later revealed bilateral dopaminergic denervation in the putamen with relative preservation of caudate and led to his current diagnosis of preclinical PD. Patients SM and PM reported no known exposures to heavy metals or other PD-relevant environmental risk factors such as pesticides. Furthermore, there was no evidence of any additional occupational-related exposures. Fibroblasts and hiPSCs were cultured as described previously (Neely et al., 2012). hiPSC lines CA4, CA6, CB5, SM3, SM4, and SM5 were reprogrammed using viral vectors whereas hiPSC lines CA11, CE6, CF1, SM14, PM12 and PM17 were reprogrammed using an episomal-based method (Okita et al., 2011; Takahashi et al., 2007). Line nomenclature follows an alphanumeric sequence, wherein the first two letters designate the human subject they are derived from. The sequential numbers identify individual clones picked from the original reprogramming plate (e.g. SM14 comes from the 14th iPSC colony isolated from this subject). The two-letter subject code does not contain patient identifying characteristics. Validation of most hiPSC lines used in this study has been previously published (Neely et al., 2012; Srinivasakumar et al., 2013). Additional lines underwent and passed the same validation. For example, see the Supplemental Material (Figure S1). Like our previously published lines, new hiPSC lines used in this study had normal euploid karyotypes and validated expression levels of pluripotency markers as assessed by immunocytochemistry, qPCR, and/or by the PluriTest, a bioinformatics assay developed by Muller and colleagues to quantify pluripotency (Muller et al., 2008). Multiple clonal lines for each subject were used in our experiments to decrease the influence of line-specific variability. See Supplemental Materials for more details.
Differentiation of early forebrain neuroprogenitors
Dual-SMAD inhibition-based forebrain neuroprogenitor differentiation of hiPSC lines was performed using our previously published protocols and based on the work of Studer and colleagues (Chambers et al., 2009; Neely et al., 2012). These forebrain neuroprogenitors are positive for expression of PAX6, FOXG1, OTX2, and SOX1 (Neely et al., 2012). See Supplemental Materials for more details.
Cell Viability/Cytotoxicity Assay
Neuroprogenitors were exposed to the metal toxicants on day 6 of neural induction for a period of 24 or 48 hours as indicated. For fibroblast experiments, cells were plated at a density of 5×104/ml and exposed to toxicants the day after plating. Toxicants included heavy metal cations of Cd (Cd2+ as CdCl2), Cu (Cu2+ as CuSO4), Mn (Mn2+ as MnCl2), and MeHg (CH3Hg+ as CH3HgCl2). Cell viability was assessed by (3-(4,5-Dimethylthiazol-2-yl)-2,5- diphenyltetrazolium bromide (MTT) assay as previously described with 2 hr incubation in neuralization medium containing MTT (Williams et al., 2010).
Alternatively, CellTiter-Blue assay (Promega) was used to measure cell viability. At 2 hours prior to the completion of the 24-hour exposure period, 20 μL of CellTiter-Blue reagent (Promega) was added to each well. Prior to this addition, cell lysis buffer was added to several wells to provide an accurate fluorescence background for 0% viable cells. The plates were then incubated for 2 hours at 37° C. Fluorescence was measured using excitation of 570 nm and emission of 600 nm on a POLARstar Omega microplate reader (BMG Labtech). Data were fitted to a normalized inhibitor curve with variable slope using Prism5 (GraphPad).
We also measured the percentage of non-apoptotic cells via forward and side scatter measurements by flow cytometry. Single cell events were also identified and gated by forward and side scatter. For each experiment, the percentage of non-apoptotic cells was calculated by excluding apoptotic cells based on their reduced forward scatter due to cell shrinkage (Healy et al., 1998). Gating was set based on the non-apoptotic cells of the vehicle-treated sample for each genotype and applied to the Cu-exposed cells of that genotype for each experiment (Figure S4). The percentage of non-apoptotic cells is expressed as the percentage of total gated events (Figure S4) in each sample that satisfy the single cell gating parameters.
Semi-quantitative mitochondrial fragmentation analysis
Semi-quantitative mitochondrial fragmentation analysis was done similar to our previously published method, based on previously published methods (Lutz et al., 2009). In brief, on day 6 of neuralization, neuroprogenitors were passaged as described in the Supplemental Methods and plated onto 8-well glass slides (Lab-Tek) at a cell density of 1×105 cells/mL. Two mitochondrial staining methods were used in this work: Mitotracker red dye and CellLight Mitochondria-GFP. In the former method, neuroprogenitors were exposed on day 7 of neuralization to either vehicle or Cu for 24 hr. Following metal exposure, neuroprogenitors were incubated with 100 nM Mitotracker red dye (Invitrogen) in neuralization medium for 45 minutes at 37°C. The dye-containing medium was subsequently taken off and cells were incubated in dye-free neuralization medium for 10 minutes, washed once with PBS, and fixed with 4% paraformaldehyde for 10–15 minutes at room temperature. Alternatively, the mitochondrial-specific viral transduction system (CellLight Mitochondria-GFP, BacMam 2.0; Invitrogen) was used to label mitochondria following the manufacturer recommended protocol. Briefly, the reagent was added to neuroprogenitors on day 6 of neuralization at time of replating at a multiplicity of infection (MOI) of 15. Neuroprogenitors were exposed 24 hours later to either vehicle or Cu for another 24 hours then fixed as specified above. Following fixation, immunofluorescence staining was performed with PAX6 antibody (Covance, 1:200 dilution). Then cells were incubated for ten minutes with 2 μM TO-PRO-3 for nuclear counterstaining. Mitochondrial fragmentation was analyzed with a confocal microscope (LSM 151 Meta confocal laser scanning microscopy system; Carl Zeiss) using 63 x 1.4 NA objective. Mitochondrial fragmentation in each of forty randomly chosen Pax6-expressing cells (Pax6+) for each exposure paradigm was assessed at the confocal microscope by a trained observer blinded to cell lineage and exposure based on published methods (Lutz et al., 2009). The number and size of mitochondrial fragments, as well as the interconnectivity of the mitochondrial network, were considered in sum by the human observer to classify mitochondria of each neuroprogenitor into the semi-quantitative scale. Using this method, cells were assigned to one of three categories of mitochondrial fragmentation: mild, moderate, or severe (an example of each category is shown, Figure 4). Apoptotic and mitotic cells were identified by morphology (shrunken cells with condensed nuclei and cells with nuclear fission, respectively) and excluded from analysis. Repeated measures general linear model two-way ANOVA (fixed factors of Cu exposure and disease group) was performed using SPSS (IBM), with the fragmentation severity categories as the repeated measure for each independent sample. Post-hoc analysis was performed by two-tail _t_-test or Bonferroni binary comparison (when the number of comparisons exceeded two) to identify significant differences between groups in each fragmentation severity class.
Figure 4. Enhanced Cu-dependent mitochondrial fragmentation in SM/PM neuroprogenitors.
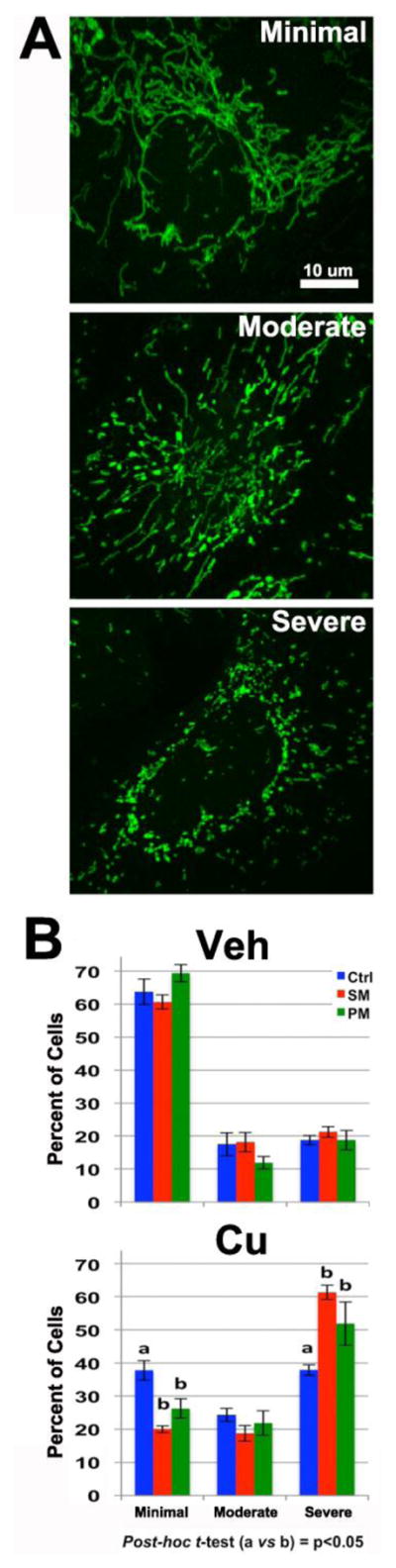
Forty individual Pax6+ neuroprogenitors were scored into three severity classes of mitochondrial fragmentation (minimal, moderate and severe) for each independent sample, and the percentage of cells in each severity category was determined. (A) Representative images of mild, moderate, and severe categories of mitochondrial fragmentation for semi-quantitative analysis. (B) Quantification of mitochondrial fragmentation in control (CA11, CE6, CF1), SM (SM3, SM14) and PM (PM12 and PM17) neuroprogenitors after a 24 hr exposure to vehicle or 100 μM Cu. Two independent experiments were performed with each line for a total of n=6 control experiments; n=8 SM/PM experiments (n=4 SM; n=4 PM). Analysis was performed using repeated measures ANOVA (across the three severity categories) and post-hoc t-test (columns are labeled with ‘a’ or ‘b’ to designate significant differences, p < 0.05). Error bars represent ± SEM.
Quantitative mitochondrial morphology analysis
Mitochondrial morphology was also assessed in a quantitative manner using Mito-Morphology macro as previously described (Dagda et al., 2009). The GFP channel of Pax6+ cells of 10 confocal images from two independent experiments (acquired as above) were converted to RGB images in ImageJ (National Institutes of Mental Health). Images were then thresholded to a binary image mask using the “Process RGB Image” function of the macro then analyzed using the “Measure” function of the macro. Circularity, particle count, and total area of the mitochondria were quantified to described mitochondrial morphological changes as previously described (Dagda et al., 2009; Xie and Chung, 2012).
Mitochondrial membrane potential analysis
Neuroprogenitors were exposed on day 7 of neuralization for 24 hr and then dissociated using Accutase, centrifugated, and resuspended in neuralization medium containing 5 nM of DiIC1(5) dye for 15 minutes. Cells were washed once in PBS and then analyzed using flow cytometry (638/658 nm excitation/emission). The samples were analyzed on a Custom Becton Dickson five-laser For tessa analytical cytometer using BD FACSDiva acquisition (BD Biosciences) and FlowJo analysis software (Tree Star, Inc.). A total of 10,000 events were acquired, and analysis was restricted to non-apoptotic single cell events as determined by light scatter properties (side and forward scatters). Apoptotic cells were identified by their relative higher side scatter and lower forward scatter due to cell shrinkage and were excluded from the analysis. Experiments were done in pairs (CA line versus an SM line, as indicated), and the average fluorescence intensity of all viable cells in the Cu-treated cells was normalized to the average intensity for the viable vehicle-treated cells for each line.
Reactive oxygen species (ROS) analysis
ROS analysis was done as previously described (Aboud et al., 2012). In brief, neuroprogenitors were passaged on day 6 of neuralization as described in the Supplemental Methods. On day 7, the cells were loaded with 2 μM CM-2′,7′-dichlorodihydrofluorescein diacetate (CM-H2DCFDA) dye for 30 minutes in exposure buffer [25 mM HEPES buffer (pH 7.2), 140 mM NaCl, 5.4 mM KCl, and 5 mM D-glucose] in the dark at room temperature. The cells were washed with exposure buffer to remove excess dye and then exposed to either vehicle (exposure buffer), Cu, or H2O2 in exposure buffer. Fluorescence intensity was measured at 37°C every 5 minutes using a Beckman Coulter DTX 880 microplate reader with excitation at 485 nm (filter bandwidth ± 20 nm) and emission at 535 nm (filter bandwidth ±25 nm). Hydrogen peroxide exposure was used as a positive control in each experiment, and a significant concentration-dependent increase by hydrogen peroxide in fluorescence was seen across the experimental set (repeated measures ANOVA, p < 0.0001, data not shown).
Additional reagent and method information is available in the Supplemental Materials.
RESULTS
Sequencing of PARK2 cDNA in the patient derived cells
Prior clinical genetic testing of patients SM and PM had revealed the inheritance of maternal and paternal PARK2 deletion alleles with one allele missing a segment of exon 3 and the other allele missing exons 5 and 6. Amplification of cDNA made from fibroblast mRNA with primers flanking the deleted exons from both alleles revealed mutant transcripts consistent with the genomic deletions identified by clinical genetic testing (Figure S2). Gene sequencing of the mutant alleles revealed the presence of both PARK2 deletion mutations in cDNA from the patient’s (SM) primary dermal fibroblasts (40 bp deletion in exon 3; and 200 bp deletion of exons 5 and 6; Figure S2). This sequence analysis confirmed the loss of the exons in the genomic DNA of SM, originally identified by clinical genetic testing. Additionally, the analysis showed that mRNA splicing occurred between the exons adjacent to the deletions in each allele. Both allelic deletions result in a frameshift predicted to cause premature termination of translation. These frameshift mutations are N-terminal to the conserved RING domain, and, thus, would be anticipated to completely abolish ubiquitination function of any potentially translated protein (Deshaies and Joazeiro, 2009). Therefore, SM and PM cells are predicted to be functional null alleles for PARK2.
Differential heavy metal sensitivity of SM and CA neuroprogenitors
We sought to test the hypothesis that PARK2 mutant neuroprogenitors (e.g. patient SM) would show elevated sensitivity to PD-relevant environmental risk factors. We tested the viability of control (e.g. subject CA) versus patient SM derived neuroprogenitors by MTT assay after 48 hour exposure to Mn, Cu, Cd, and MeHg, all of which have been implicated in PD-relevant neurodegenerative processes and/or as PD environmental risk factors (Buzanska et al., 2009; Gorell et al., 1999; Götz et al., 2002; Jomova et al., 2010; Landrigan et al., 2005; Rivera-Mancía et al., 2010; Squitti et al., 2009; Weiss et al., 2002; Willis et al., 2010; Xu et al., 2011). To control for cell line dependent variability, we used multiple clonal hiPSC lines (lines CA4 and CA6 for subject CA; lines SM3, SM4, and SM5 for subject SM). Repeated measures ANOVA across the concentration response curves demonstrated a heightened sensitivity to Cu and Cd but not to Mn or MeHg in SM neuroprogenitors (lines SM3, SM4 and SM5) when compared to CA neuroprogenitors (lines CA4 and CA6) (Figure 1A). To determine whether the differential Cu and Cd sensitivity was also observed in non-neural cell types between the same human subjects we generated 48 hour Cu and Cd cytotoxicity concentration-response curves by MTT assay in the primary epidermal fibroblasts used to generate the hiPSC lines. No significant sensitivity differences to Cu or Cd (Figure 1B) viability by MTT assay were observed, suggesting at least some cell type dependence of the differential metal sensitivity between these subjects.
Figure 1. SM neuroprogenitors but not fibroblasts show increased sensitivity to Cd and Cu toxicity.

(A) Neuroprogenitor survival curves measured by MTT assay after 48 hr exposure to Mn, MeHg, Cu, and Cd (CA: CA4, CA6 at N = 6 or 8; SM: SM3, SM4, SM5 at N = 7 or 8). Error bars represent ± SEM.
(B) The primary dermal fibroblast lines were exposed to Cd or Cu, and cell viability was measured by MTT assay (Cu: CA, n = 7; SM, n = 7; Cd: CA, n = 3; SM, n = 3). Statistical analysis for genotype effects was performed by two-way repeated measures ANOVA. Pairwise post-hoc analysis with Bonferroni correction is indicated between CA and SM hiPSC lines as * p < 0.05, *** p < 0.001. Error bars represent ± SEM
We decided to further examine the genotype-dependent/subject-dependent differences in Cu cytotoxicity, rather than Cd, given the broader concentration response curve and the redox potential of Cu that allows it to participate in Fenton chemistry. We saw similar genotype-dependent differences in viability following 24-hour Cu exposure between CA control cells and the PARK2 mutant neuroprogenitors from both SM and PM (Figure S3) by the CellTiter-Blue assay, which quantifies viable cells by measuring total cytoplasmic, microsomal, and mitochondrial reductase activity (Gonzalez and Tarloff, 2001). Since both MTT and CellTiter-Blue viability assays depend, at least in part, on mitochondrial reductase activity, we sought to confirm if the effects observed by these assays included decreased cell viability, rather than just changes in total mitochondrial reductase activity. Preliminary studies ruled out the use of the lactose dehydrogenase (LDH) assay for this purpose because the presence of Cu in the media (as low as 25 μM) significantly impaired detection of LDH as previously shown in the literature (Pamp et al., 2005). Therefore, we utilized measurement of non-apoptotic cells by flow cytometry as a non-mitochondrial dependent assessment of cytotoxicity following 24-hour vehicle or 50 μM Cu exposure in control CA and PARK2 mutant SM neuroprogenitors (Figure 2 and Figure S4). This was performed by measuring the percentage of non-apoptotic cells (apoptotic cells have reduced forward scatter) from all single cell events. Two-way ANOVA revealed significant differences in viability due to Cu exposure (F1,16=41.76, p<0.001), genotype (F1,16=14.33, p=0.002), and a Cu x genotype interaction effect (F1,16=15.39, p=0.001). Post-hoc analysis demonstrated a significant decrease in percentage of non-apoptotic Cu-exposed SM neuroprogenitors versus both vehicle (p<0.001) and Cu-exposed CA neuroprogenitors (p<0.001); as well as a significant decrease in viability of Cu-exposed CA neuroprogenitors compared to the vehicle-exposed CA group (Figure 2). No difference in baseline percentage of non-apoptotic cells was observed between CA and SM. Overall we found a consistent increase in sensitivity of SM neuroprogenitors to Cu cytotoxicity that includes increased apoptosis and loss of cellular reductase activity.
Figure 2. SM neuroprogenitors show a stronger reduction in the non-apoptotic cell population following Cu exposure.
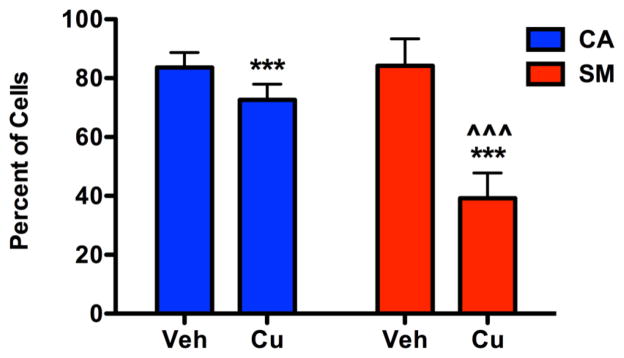
Gating for non-apoptotic cells were performed based on the forward and side scatter properties of the vehicle-exposed control group in each independent experiment as shown in Figure S4. Percentage of non-apoptotic cells is plotted. Apoptotic cells have decreased forward scatter properties due to cell shrinkage. Data collected across 5 independent paired experiments (CA6:SM4 n=2, CA6:SM5 n=2, CA11:SM14 n=1, total n=5). Statistical analysis was performed by two-way ANOVA and paired two-tailed _t_-test for post-hoc analysis (*** or ^^^ indicates p < 0.001, absence of a symbol indicates no significant difference p > 0.05). * indicates a significant difference within genotype between exposures, and ^ indicates a significant difference between genotypes with the same exposure. Error bars represent + SEM.
One possible hypothesis to explain the increased sensitivity to Cu cytotoxicity is elevated cellular uptake of Cu in the SM neuroprogenitors. Therefore, we measured the levels of intracellular Cu following a 24-hour exposure to 50 μM Cu by graphite furnace atomic absorption spectroscopy (GFAAS). No differences in intracellular Cu levels in vehicle or Cu treated SM (SM5) and CA (CA6) neuroprogenitors were observed (Figure 3).
Figure 3. Total Cu accumulation is not different between SM and CA neuroprogenitors.
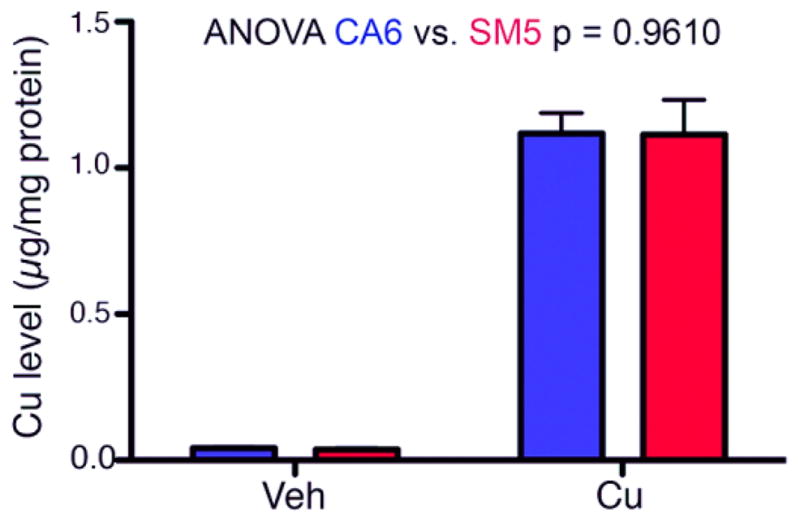
CA (CA6) and SM (SM5) neuroprogenitors were treated with either vehicle (Veh) or 50 μM Cu (Cu) for 24 hrs, and total cellular Cu levels were assessed by GFAAS (Vehicle n=3; Cu n=5). Two-way ANOVA was used for statistical analysis. Error bars represent + SEM.
Elevated mitochondrial fragmentation due to Cu exposure in SM and PM neuroprogenitors
We hypothesized that the increased sensitivity of SM and PM neuroprogenitors to Cu was due to an enhanced susceptibility of the mitochondria to oxidative damage. To evaluate this, we performed a large study with hiPSC lines from each of three control subjects (CA11, CE6 and CF1) and two lines each from SM and PM (SM3, SM14, PM12, and PM17). We assessed mitochondrial fragmentation by semi-quantitative assessment of severity using a blinded observer in two independent experiments each with 3 control lines (total n=6 across 2 experimental days) and 4 PARK2 mutant lines (total n=8 across 2 experimental days) in 24-hour vehicle or 100 μM Cu-exposed Pax6+ neuroprogenitors (Figure 4). Cellular mitochondrial fragmentation of individual Pax6+ neuroprogenitors was classified into a 3-level semi-quantitative scale including minimal, moderate, and severe by a trained observer (see examples, Figure 4A). Pax6+ cells were selected prior to visualizing the mitochondria, and then, stained mitochondria of the selected neuroprogenitor were visualized by scanning through the z-axis to inform the scoring decision. Two-way repeated measures (RM)-ANOVA revealed significant differences in severity of fragmentation due to Cu exposure (F2,23=86.753, p<0.001), genotype (F2,23=7.604, p=0.003), and a Cu x genotype interaction effect (F2,23=6.448, p=0.006) (Figure 4B). Repeated measures ANOVA of vehicle treated neuroprogenitors found no differences between SM/PM versus control (CA11, CE6, CF1) (F2,11=0.314, p=0.737), while neuroprogenitors exposed to Cu had a significant difference in the severity of fragmentation (F2,11=9.630, p=0.004) between SM/PM versus control. Post-hoc examination of the data demonstrated that both SM (p<0.05) and PM (p<0.05) neuroprogenitors showed significantly more mitochondrial fragmentation than control neuroprogenitors following Cu exposure (fewer cells with minimal fragmentation, and more cells with severe fragmentation). No significant differences in fragmentation were observed under vehicle exposure conditions between control and PD lines.
We then applied a quantitative analysis of mitochondrial morphology on a subset of mitochondrial images using Mito-Morphology macro, an ImageJ based program, as previously described (Dagda et al., 2009). 10 images were randomly selected from two independent experiments (5 images from each) for each exposure condition and processed into a binary image mask for analysis by the macro (four representative processed images for each experimental group are provided in Figure 5A). Automated analysis of the mitochondrial morphology was performed for circularity, segment/particle number, and total area. Two-way ANOVA revealed a significant difference in circularity due to Cu exposure (F1,36=14.46, p<0.001), genotype (F1,36=13.35, p<0.001), and a Cu x genotype interaction effect (F1,36=5.946, p=0.02). Post-hoc analysis demonstrated a significant increase in circularity in Cu-exposed SM neuroprogenitors compared to both vehicle-exposed SM neuroprogenitors (p<0.001) and Cu-exposed CA neuroprogenitors (p=0.02) (Figure 5B). Two-way ANOVA also revealed a significant difference in the number of mitochondrial segments due to Cu exposure (F1,36=17.21, p<0.001) and a Cu x genotype interaction effect (F1,36=6.31, p=0.02). However, genotype did not have a significant effect. Post-hoc analysis demonstrated a significant increase in the number of mitochondrial segments in Cu-exposed SM neuroprogenitors compared to both vehicle-exposed SM neuroprogenitors (p<0.001) and Cu-exposed CA neuroprogenitors (p=0.006) (Figure 5C). No significant differences were found in circularity or mitochondrial count between vehicle-exposed and Cu-exposed CA neuroprogenitors. No significant change in total mitochondrial area was observed between the groups either (Figure 5D). Taken together the automated Mito-Morphology analysis corroborated our findings by semi-quantitative analysis of increased severity of mitochondrial fragmentation by Cu exposure in SM neuroprogenitors relative to control neuroprogenitors.
Figure 5. Quantitative mitochondrial analysis of Cu-exposed neuroprogenitors.

Quantitative analysis of mitochondrial morphology was performed using the ImageJ Mito-Morphology macro. Randomly selected Pax6+ neuroprogenitors were imaged by an observer blinded to genotype or exposure condition from two independent experiments (5 images from each, total n=10) for both vehicle and Cu exposed CA6 and SM3 neuroprogenitors. (A) Four representative, binary image masks from each treatment category are shown for visual comparison. Quantitative measures were assessed by the macro including (B) mitochondrial circularity, (C) total number of mitochondrial segments/particles, and (D) total mitochondrial surface area of the binary image masks. Statistical analysis was performed by two-way ANOVA and two-tailed _t_-test for post-hoc analysis (*** or ^^^ indicates p < 0.001; ^^ indicates p < 0.01). * symbol indicates significant difference as compared to the vehicle-treated group of the same genotype, and ^ symbol represents significant difference compared to Cu-exposed group of the other genotype. Error bars represent + SEM.
Lower threshold for Cu-dependent mitochondrial fragmentation in SM neuroprogenitors
We hypothesized that the mitochondria of the PD risk group neuroprogenitors are sensitive to lower concentrations of Cu than the control group neuroprogenitors. To evaluate this hypothesis, we assessed mitochondrial fragmentation in Pax6+ neuroprogenitors derived from 3 SM lines (SM3, SM4 and SM5) and 2 control lines (CA6 and CB5) with concentrations of Cu at 10 μM, 25 μM, and 50 μM (Figure 6 and S5). We performed 8 independent paired experiments, each one with a single SM line and control line. Two-way repeated measures ANOVA of the complete data set revealed significant differences in severity of fragmentation due to Cu- exposure (F6,112=17.912, p<0.001), genotype (F2,55=110.259, p<0.001) and a Cu x genotype interaction effect (F6,112=6.973, p<0.001) similar to what was seen at 100 μM Cu in the experiment above (Figure 4). Analysis of the 10 μM Cu exposure data revealed significantly increased fragmentation severity in the SM neuroprogenitors but not the controls (Figure 6). Both 25 and 50 μM Cu caused increased fragmentation in both groups with greater fragmentation severity in SM (Figure S5). Therefore, the lowest observable exposure level (LOEL) to cause alteration in mitochondrial structure is at most 10 μM for SM and ~25 μM for controls. In addition, a subtle but significant difference (p=0.017) in baseline (vehicle exposure) fragmentation was observed between SM and control neuroprogenitors.
Figure 6. Decreased Cu-dependent LOEL for mitochondrial fragmentation in SM neuroprogenitors.
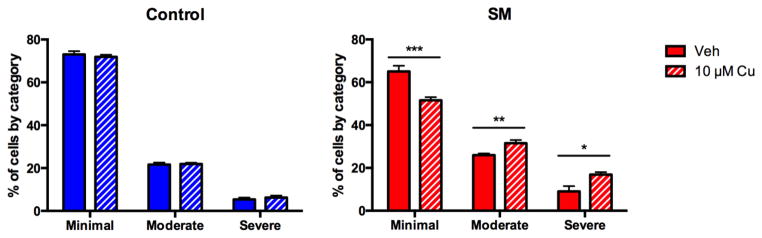
Eight independent experiments were performed with pairs of control versus SM neuroprogenitors (CA6:SM3 n=2, CA6: SM4 n=2, CA6:SM5 n=2, CB5:SM5 n=2, total n=8) exposed to vehicle or 10 μM Cu. Mitochondrial fragmentation was scored blind into three severity categories (minimal, moderate, or severe) for 40 PAX6+ neuroprogenitors for each sample. For concentration comparisons * for p < 0.05, ** p < 0.01, *** p < 0.001 by t-test. Error bars represent + SEM.
Cu toxicity is associated with perturbed mitochondrial membrane potential
As an independent assessment of mitochondrial function, we assessed the mitochondrial membrane potential in CA (lines CA6 and CA11) and SM (lines SM4, SM5, and SM14) neuroprogenitors using flow cytometry. Only live single cells were included in the analysis (Figure S4). We observed that Cu exposure resulted in a decreased mitochondrial membrane potential in the majority of SM neuroprogenitors but only in a minority of CA control neuroprogenitors (see representative experiment, Figure 7 upper panels). Similar effects were observed in 5 of 5 independent experiments. To quantify this difference in mitochondrial membrane potential, we compared the DiIC1(5) signal intensities and observed significantly decreased membrane potential in Cu-exposed SM neuroprogenitors compared to control neuroprogenitors (Figure 7 lower panel) (paired _t_-test p = 0.011).
Figure 7. Decreased mitochondrial membrane potential by Cu-exposure is more severe in SM neuroprogenitors than control.
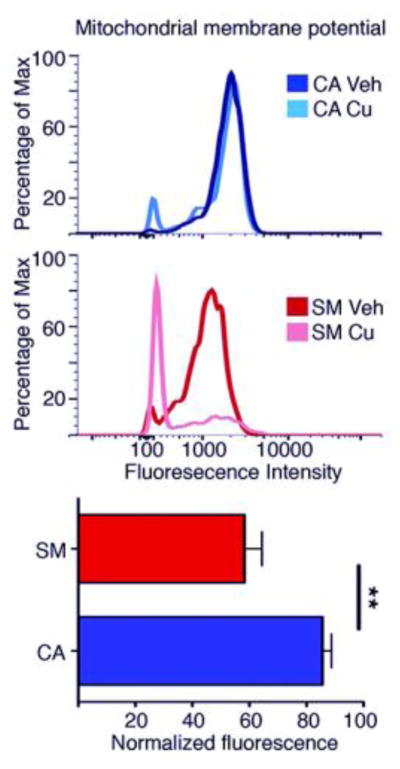
(Upper two panels) Representative flow cytometry experiment using DiIC1(5) fluorescence as an indicator of mitochondrial membrane potential after a 24 hr exposure to 50 μM Cu in CA6 and SM5. (Lower graph) Mean normalized fluorescence intensity of Cu-exposed neuroprogenitors. Data represent single live cell events assessed by flow cytometry across 5 independent paired experiments (CA6:SM4 n=2, CA6:SM5 n=2, CA11:SM14 n=1, total n=5). Statistical analysis was performed by a paired two-tailed _t_-test (p = 0.011). Error bars represent + SEM.
Increased ROS production in SM neuroprogenitors prior to mitochondrial fragmentation
We hypothesized that the increased mitochondrial fragmentation of SM neuroprogenitors may be caused by increased Cu-dependent generation of reactive oxygen species (ROS). Exposure to Cu at 50, 100, and 200 μM for 30 minutes resulted in significantly higher levels of ROS in SM (lines SM3 and SM14) compared to CA neuroprogenitors (lines CA6 and CA11) whereas baseline ROS levels were not significantly different between cells from the two subjects (Figure 8). Because these measurements were taken after only 30 minutes of Cu exposure, the elevated ROS must occur prior to Cu-induced mitochondrial fragmentation seen at 24 hours. This suggests that elevated Cu-induced oxidative stress is a potential mechanism by which SM has increased Cu-dependent mitochondrial fragmentation. The baseline mitochondrial structure differences between SM/PM and control may be the cause for this differential ROS generation.
Figure 8. SM neuroprogenitors have greater ROS production in the presence of Cu.

ROS generation in neuroprogenitors was measured using a DCF-dye based assay after a 30 minute exposure to Cu. Paired CA/SM experiments were performed (CA6:SM3 n=2, CA6:SM14 n=3, CA11:SM3 n=2, total n=7). Statistical analysis for genotype effects was by two-way repeated measures ANOVA. Pairwise post-hoc analysis with Bonferroni correction is indicated between CA and SM hiPSC lines as * p < 0.05, *** p < 0.001. Error bars represent + SEM.
DISCUSSION
Cu has been implicated in the etiology of PD, and its concentration is altered in the blood and substantia nigra of PD patients (Boll et al., 2008; Dexter et al., 1991; Gorell et al., 1999). Our data show that increased sensitivity to Cu exposure in neuroprogenitors with high genetic risk for PD is associated with elevated ROS generation and a failure to maintain mitochondrial integrity, known toxicological mechanisms of Cu toxicity (Gyulkhandanyan et al., 2003). Our data are consistent with recent reports examining hiPSCs from PD patients or isogenic lines with mutations in PD-related genes that report mitochondrial deficits and increased sensitivity to other disease-relevant oxidative stressors (e.g. 6-hydroxydopamine, paraquat, maneb, and rotenone) in hiPSC derived neural cells (Byers et al., 2011; Cooper et al., 2012; Imaizumi et al., 2012; Nguyen et al., 2011; Ryan et al., 2013; Sánchez-Danés et al., 2012; Seibler et al., 2011). The heightened vulnerability to Cu we report strongly implicates exposure to this metal (and perhaps Cd as well) as a potential compounding risk factor for patients with a family history of PD. These data, along with recent papers showing alterations in zinc homeostasis coupled with mitochondrial dysfunction in patient derived neuroprogenitors with ATP13A2/PARK9 mutations (Kong et al., 2014; Park et al., 2014; Tsunemi and Krainc, 2014), strongly implicate alterations in metal biology as a major environmental modifier of genetic risk alleles for PD.
Our data support a hypothesis in which failure to maintain mitochondrial function and integrity during heavy metal exposure may contribute to gene environment interactions underlying PD associated with mutations in PARK2. These findings are consistent with previous research demonstrating that loss of PARK2 function causes mitochondrial dysfunction and sensitivity to oxidative stress (Grünewald et al., 2010; Gyulkhandanyan et al., 2003; Mortiboys et al., 2008; Pacelli et al., 2011; Pilsl and Winklhofer, 2012; Saini et al., 2010; Ved et al., 2005). Two studies have investigated the effects of PARK2 mutations in genetic model organisms and did not observe genotype-dependent survival differences due to Cu-supplemented food (Saini et al., 2010; Ved et al., 2005). However, Saini and colleagues did report exasperation of a neuronal Cu toxicity assay in D. melanogaster (rough eye phenotype) carrying PARK2 mutations. This difference, relative to our observed increased vulnerability to Cu cytotoxicty in the context of PARK2 mutant human neuroprogenitors, may very well be explained by systemic Cu homeostatic processes including gastrointestinal regulation of Cu uptake that would not be accounted for in a cellular model system. Future studies will be needed to resolve the complex role that systemic metal homeostatic control plays in alteration of environmental risk by PARK2 and other PD-related genetic risk factors.
We previously reported a decrease in net cellular Mn uptake following Mn exposure in SM neuroprogenitors versus control, despite equivalent Mn cytotoxicity (Aboud et al., 2012). This lack of difference in Mn cytotoxicity between control and SM neuroprogenitors appears to be due to compensatory neuroprotective changes that decrease Mn uptake in the PARK2 mutant neuroprogenitors (Aboud et al., 2012). As no differences in the accumulation of intracellular Cu were observed, the heightened sensitivity of PARK2 mutant neuroprogenitors to Cu likely reflects either decreased capacity to tolerate Cu toxicity (e.g. failure of antioxidant defense mechanisms) or alterations in the biological processes that augment mechanisms of Cu cytotoxicity (e.g. decreased Cu chaperone capacity leading to increases in free Cu levels). Further study will be needed to investigate whether this gene by toxicant interaction is specific to Cu or if it occurs for a broad range of oxidative stressors, such as iron or mitochondrial inhibitors, which are also implicated as environmental modifiers or risk factors for parkinsonism (Peng et al., 2010; Saini et al., 2010; Ved et al., 2005).
CONCLUSIONS
We report that neuroprogenitors from an EOPD patient and a preclinical PD patient both carrying biallelic loss-of-function mutations in PARK2 are highly susceptible to mitochondrial toxicity by a subset of heavy metal toxicants compared to neuroprogenitors from 4 different control human subjects. This is the first report of altered cytotoxic vulnerability of PD patient-derived neuroprogenitors with PARK2 mutations to widespread environmentally relevant neurotoxicants. Specifically, we found that hiPSC-derived neuroprogenitors from a preclinical PD subject with PARK2 loss of function alleles (SM) exhibit heightened vulnerability to Cu and Cd cytotoxicity but no difference in sensitivity to Mn or MeHg relative to control neuroprogenitors. The heightened sensitivity to Cu in this patient was also observed in hiPSCs derived neuroprogenitors from his brother (PM), who shares the same PARK2 susceptibility loci and other familial risk factors and who was diagnosed with EOPD. We further report that the heightened sensitivity of SM neuroprogenitors to Cu resulting in a decreased lowest observable effect level (LOEL) for mitochondrial toxicity by Cu exposure (Figure 6). Our data support the hypothesis that genetic predisposition to PD can decrease the “safe” exposure threshold to environmental risk factors. Our data also support the potential utility of hiPSC-based neuroprogenitor model systems to define individualized environmental risk. Further research is warranted to determine whether individuals with other genetic PD risk factors may have increased risk if exposed to Cu, Cd, or other heavy metals in their environment.
Supplementary Material
supplement
Highlights.
- PARK2 mutants have heightened sensitivity to Cu and Cd but not Mn or MeHg
- Cu causes greater mitochondrial fragmentation in PARK2 mutant lines
- PARK2 mutations reduce lowest observed effect level for Cu mitochondrial toxicity
- Cu causes greater reactive oxygen species production in PARK2 mutants
Acknowledgments
Funding Sources: This study was sponsored by supported by Doris Duke Charitable Foundation and Hazinski-Turner Award (KCE), Peterson Foundation for Parkinson’s (ABB), PK Hope is Alive! (ABB), Subaward RR166-737/4787736 (ABB and KCE) under NIH/NIGMS 5PO1 GM08535403, NIH/NINDS 1 RO1 NS078289 (KCE), and Vanderbilt Center for Molecular Toxicology Pilot Project (ABB, MDN) under NIH/NIEHS 5P30 ES000267, NIH/NINDS K02NS057666 (PH), and NIH/NIEHS ES016931 and ES016931-02S1 (ABB), additional support by NIH/NINDS F31 NS077632 (AMT), the Vanderbilt Kennedy Center NIH/NICHD P30HD15052 (ABB) and the Vanderbilt Medical Scientist Training Program NIH/NIGMS T32 GM07347 (KKK).
We thank Drs. Michail Zaboikin and Srinivas Kumar (Saint Louis University, MO) for collaborating in the generation of the CB5 control line; Drs. Nathalie Maitre (Vanderbilt University) for critical review of our manuscript as well as Dr. Thomas Davis (Vanderbilt University) for valuable discussions and insight into the clinical perspective of our work; Gary Li (Vanderbilt University) for technical assistance; also Steve Fordahl (UNC Greensboro, NC) for technical assistance measuring Cu levels. Flow cytometry experiments were performed in the Vanderbilt Medical Center Flow Cytometry Shared Resource, which is supported by Vanderbilt Ingram Cancer Center (P30CA68485) and the Vanderbilt Digestive Disease Research Center (DK058404). The authors of the manuscript declare no competing financial interests related to this paper. This study was sponsored by supported by Doris Duke Charitable Foundation and Hazinski-Turner Award (KCE), Peterson Foundation for Parkinson’s (ABB), PK Hope is Alive! (ABB), Subaward RR166-737/4787736 (ABB and KCE) under NIH/NIGMS 5PO1 GM08535403, NIH/NINDS 1 RO1 NS078289 (KCE), and Vanderbilt Center for Molecular Toxicology Pilot Project (ABB, MDN) under NIH/NIEHS 5P30 ES000267, NIH/NINDS K02NS057666 (PH), and NIH/NIEHS ES016931 and ES016931-02S1 (ABB), additional support by NIH/NINDS F31 NS077632 (AMT), the Vanderbilt Kennedy Center NIH/NICHD P30HD15052 (ABB) and the Vanderbilt Medical Scientist Training Program NIH/NIGMS T32 GM07347 (KKK).
Footnotes
Supplemental Materials are included: Supplemental methods, Table S1, Figure S1, Figure S2, Figure S3, and Supplemental references.
Financial disclosure/conflict of interest related to manuscript: None
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abbas N, et al. A wide variety of mutations in the parkin gene are responsible for autosomal recessive parkinsonism in Europe. French Parkinson’s Disease Genetics Study Group and the European Consortium on Genetic Susceptibility in Parkinson’s Disease. Hum Mol Genet. 1999;8:567–74. doi: 10.1093/hmg/8.4.567. [DOI] [PubMed] [Google Scholar]
- Aboud AA, et al. Genetic risk for Parkinson’s disease correlates with alterations in neuronal manganese sensitivity between two human subjects. NeuroToxicology. 2012;33:1443–9. doi: 10.1016/j.neuro.2012.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betarbet R, et al. Chronic systemic pesticide exposure reproduces features of Parkinson’s disease. Nat Neurosci. 2000;3:1301–6. doi: 10.1038/81834. [DOI] [PubMed] [Google Scholar]
- Boll M-C, et al. Free copper, ferroxidase and SOD1 activities, lipid peroxidation and NO(x) content in the CSF. A different marker profile in four neurodegenerative diseases. Neurochem Res. 2008;33:1717–23. doi: 10.1007/s11064-008-9610-3. [DOI] [PubMed] [Google Scholar]
- Buzanska L, et al. A human stem cell-based model for identifying adverse effects of organic and inorganic chemicals on the developing nervous system. Stem Cells. 2009;27:2591–601. doi: 10.1002/stem.179. [DOI] [PubMed] [Google Scholar]
- Byers B, et al. SNCA triplication Parkinson’s patient’s iPSC-derived DA neurons accumulate α-synuclein and are susceptible to oxidative stress. PLoS ONE. 2011;6:e26159. doi: 10.1371/journal.pone.0026159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers SM, et al. Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nat Biotechnol. 2009;27:275–80. doi: 10.1038/nbt.1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper O, et al. Pharmacological rescue of mitochondrial deficits in iPSC-derived neural cells from patients with familial Parkinson’s disease. Sci Transl Med. 2012;4:141ra90. doi: 10.1126/scitranslmed.3003985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dagda RK, et al. Loss of PINK1 function promotes mitophagy through effects on oxidative stress and mitochondrial fission. Journal of Biological Chemistry. 2009;284:13843–13855. doi: 10.1074/jbc.M808515200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Fuente-Fernández R, Calne DB. Evidence for environmental causation of Parkinson’s disease. Parkinsonism Relat Disord. 2002;8:235–41. doi: 10.1016/s1353-8020(01)00055-4. [DOI] [PubMed] [Google Scholar]
- Deng H, et al. Heterogeneous phenotype in a family with compound heterozygous parkin gene mutations. Arch Neurol. 2006;63:273–7. doi: 10.1001/archneur.63.2.273. [DOI] [PubMed] [Google Scholar]
- Deshaies RJ, Joazeiro CA. RING domain E3 ubiquitin ligases. Annual review of biochemistry. 2009;78:399–434. doi: 10.1146/annurev.biochem.78.101807.093809. [DOI] [PubMed] [Google Scholar]
- Dexter DT, et al. Alterations in the levels of iron, ferritin and other trace metals in Parkinson’s disease and other neurodegenerative diseases affecting the basal ganglia. Brain. 1991;114:1953–75. doi: 10.1093/brain/114.4.1953. [DOI] [PubMed] [Google Scholar]
- Gonzalez R, Tarloff J. Evaluation of hepatic subcellular fractions for Alamar blue and MTT reductase activity. Toxicology in vitro. 2001;15:257–259. doi: 10.1016/s0887-2333(01)00014-5. [DOI] [PubMed] [Google Scholar]
- Gorell JM, et al. Occupational exposure to manganese, copper, lead, iron, mercury and zinc and the risk of Parkinson’s disease. Neurotoxicology. 1999;20:239–47. [PubMed] [Google Scholar]
- Gorell JM, et al. The risk of Parkinson’s disease with exposure to pesticides, farming, well water, and rural living. Neurology. 1998;50:1346–1350. doi: 10.1212/wnl.50.5.1346. [DOI] [PubMed] [Google Scholar]
- Götz ME, et al. Methylmercury induces neurite degeneration in primary culture of mouse dopaminergic mesencephalic cells. J Neural Transm. 2002;109:597–605. doi: 10.1007/s007020200049. [DOI] [PubMed] [Google Scholar]
- Grünewald A, et al. Mutant Parkin impairs mitochondrial function and morphology in human fibroblasts. PLoS ONE. 2010;5:e12962. doi: 10.1371/journal.pone.0012962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gyulkhandanyan AV, et al. Modulation of mitochondrial membrane potential and reactive oxygen species production by copper in astrocytes. J Neurochem. 2003;87:448–60. doi: 10.1046/j.1471-4159.2003.02029.x. [DOI] [PubMed] [Google Scholar]
- Healy E, et al. Apoptosis and necrosis: mechanisms of cell death induced by cyclosporine A in a renal proximal tubular cell line. Kidney international. 1998;54:1955–1966. doi: 10.1046/j.1523-1755.1998.00202.x. [DOI] [PubMed] [Google Scholar]
- Imaizumi Y, et al. Mitochondrial dysfunction associated with increased oxidative stress and alpha-synuclein accumulation in PARK2 iPSC-derived neurons and postmortem brain tissue. Mol Brain. 2012;5:35. doi: 10.1186/1756-6606-5-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jomova K, et al. Metals, oxidative stress and neurodegenerative disorders. Mol Cell Biochem. 2010;31:95–107. doi: 10.1007/s11010-010-0563-x. [DOI] [PubMed] [Google Scholar]
- Khan NL, et al. Parkin disease: a phenotypic study of a large case series. Brain. 2003;126:1279–92. doi: 10.1093/brain/awg142. [DOI] [PubMed] [Google Scholar]
- Kitada T, et al. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature. 1998;392:605–8. doi: 10.1038/33416. [DOI] [PubMed] [Google Scholar]
- Kong SM, et al. Parkinson’s disease-linked human PARK9/ATP13A2 maintains zinc homeostasis and promotes alpha-Synuclein externalization via exosomes. Hum Mol Genet. 2014;23:2816–33. doi: 10.1093/hmg/ddu099. [DOI] [PubMed] [Google Scholar]
- Landrigan PJ, et al. Early environmental origins of neurodegenerative disease in later life. Environ Health Perspect. 2005;113:1230–3. doi: 10.1289/ehp.7571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lutz AK, et al. Loss of parkin or PINK1 function increases Drp1-dependent mitochondrial fragmentation. Journal of Biological Chemistry. 2009;284:22938–22951. doi: 10.1074/jbc.M109.035774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortiboys H, et al. Mitochondrial function and morphology are impaired in parkin-mutant fibroblasts. Ann Neurol. 2008;64:555–65. doi: 10.1002/ana.21492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller FJ, et al. Assessment of human pluripotent stem cells with PluriTest. StemBook; Cambridge (MA): 2008. [PubMed] [Google Scholar]
- Neely MD, et al. DMH1, a Highly Selective Small Molecule BMP Inhibitor Promotes Neurogenesis of hiPSCs: Comparison of PAX6 and SOX1 Expression during Neural Induction. ACS Chem Neurosci. 2012;3:482–491. doi: 10.1021/cn300029t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen HN, et al. LRRK2 mutant iPSC-derived DA neurons demonstrate increased susceptibility to oxidative stress. Cell Stem Cell. 2011;8:267–80. doi: 10.1016/j.stem.2011.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okita K, et al. A more efficient method to generate integration-free human iPS cells. Nat Methods. 2011;8:409–412. doi: 10.1038/nmeth.1591. [DOI] [PubMed] [Google Scholar]
- Pacelli C, et al. Mitochondrial defect and PGC-1α dysfunction in parkin-associated familial Parkinson’s disease. Biochim Biophys Acta. 2011;1812:1041–53. doi: 10.1016/j.bbadis.2010.12.022. [DOI] [PubMed] [Google Scholar]
- Pamp K, et al. NAD (H) enhances the Cu (II)-mediated inactivation of lactate dehydrogenase by increasing the accessibility of sulfhydryl groups. Free radical research. 2005;39:31–40. doi: 10.1080/10715760400023671. [DOI] [PubMed] [Google Scholar]
- Park JS, et al. Parkinson’s disease-associated human ATP13A2 (PARK9) deficiency causes zinc dyshomeostasis and mitochondrial dysfunction. Hum Mol Genet. 2014;23:2802–15. doi: 10.1093/hmg/ddt623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng J, et al. Synergistic effects of environmental risk factors and gene mutations in Parkinson’s disease accelerate age-related neurodegeneration. Journal of neurochemistry. 2010;115:1363–1373. doi: 10.1111/j.1471-4159.2010.07036.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilsl A, Winklhofer KF. Parkin, PINK1 and mitochondrial integrity: emerging concepts of mitochondrial dysfunction in Parkinson’s disease. Acta Neuropathol. 2012;123:173–188. doi: 10.1007/s00401-011-0902-3. [DOI] [PubMed] [Google Scholar]
- Rivera-Mancía S, et al. The transition metals copper and iron in neurodegenerative diseases. Chem Biol Interact. 2010;186:184–99. doi: 10.1016/j.cbi.2010.04.010. [DOI] [PubMed] [Google Scholar]
- Ryan SD, et al. Isogenic human iPSC Parkinson’s model shows nitrosative stress- induced dysfunction in MEF2-PGC1alpha transcription. Cell. 2013;155:1351–64. doi: 10.1016/j.cell.2013.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saini N, et al. Extended lifespan of Drosophila parkin mutants through sequestration of redox-active metals and enhancement of anti-oxidative pathways. Neurobiol Dis. 2010;40:82–92. doi: 10.1016/j.nbd.2010.05.011. [DOI] [PubMed] [Google Scholar]
- Sánchez-Danés A, et al. Disease-specific phenotypes in dopamine neurons from human iPS-based models of genetic and sporadic Parkinson’s disease. EMBO Mol Med. 2012;4:380–395. doi: 10.1002/emmm.201200215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seibler P, et al. Mitochondrial Parkin recruitment is impaired in neurons derived from mutant PINK1 induced pluripotent stem cells. J Neurosci. 2011;31:5970–6. doi: 10.1523/JNEUROSCI.4441-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Squitti R, et al. Implications of metal exposure and liver function in Parkinsonian patients resident in the vicinities of ferroalloy plants. J Neural Transm. 2009;116:1281–7. doi: 10.1007/s00702-009-0283-0. [DOI] [PubMed] [Google Scholar]
- Srinivasakumar N, et al. Gammaretroviral vector encoding a fluorescent marker to facilitate detection of reprogrammed human fibroblasts during iPSC generation. PeerJ. 2013;1:e224. doi: 10.7717/peerj.224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K, et al. Induction of pluripotent stem cells from fibroblast cultures. Nat Protoc. 2007;2:3081–9. doi: 10.1038/nprot.2007.418. [DOI] [PubMed] [Google Scholar]
- Tanner CM, et al. Parkinson disease in twins: an etiologic study. JAMA. 1999;281:341–6. doi: 10.1001/jama.281.4.341. [DOI] [PubMed] [Google Scholar]
- Tsunemi T, Krainc D. Zn2+ dyshomeostasis caused by loss of ATP13A2/PARK9 leads to lysosomal dysfunction and alpha-synuclein accumulation. Hum Mol Genet. 2014;23:2791–801. doi: 10.1093/hmg/ddt572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ved R, et al. Similar patterns of mitochondrial vulnerability and rescue induced by genetic modification of α-synuclein, parkin, and DJ-1 in Caenorhabditis elegans. Journal of Biological Chemistry. 2005;280:42655–42668. doi: 10.1074/jbc.M505910200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss B, et al. Silent latency periods in methylmercury poisoning and in neurodegenerative disease. Environmental health perspectives. 2002;110(Suppl 5):851–4. doi: 10.1289/ehp.02110s5851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams BB, et al. Disease-toxicant screen reveals a neuroprotective interaction between Huntington’s disease and manganese exposure. J Neurochem. 2010;112:227–37. doi: 10.1111/j.1471-4159.2009.06445.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willis AW, et al. Metal emissions and urban incident Parkinson disease: a community health study of Medicare beneficiaries by using geographic information systems. Am J Epidemiol. 2010;172:1357–63. doi: 10.1093/aje/kwq303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie W, Chung KK. Alpha-synuclein impairs normal dynamics of mitochondria in cell and animal models of Parkinson’s disease. Journal of neurochemistry. 2012;122:404–414. doi: 10.1111/j.1471-4159.2012.07769.x. [DOI] [PubMed] [Google Scholar]
- Xu B, et al. Calcium signaling is involved in cadmium-induced neuronal apoptosis via induction of reactive oxygen species and activation of MAPK/mTOR network. PLoS ONE. 2011;6:e19052. doi: 10.1371/journal.pone.0019052. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
supplement