Discovery of a Selective TRPM8 Antagonist with Clinical Efficacy in Cold-Related Pain (original) (raw)
Abstract
The transient receptor potential (TRP) family of ion channels comprises nonselective cation channels that respond to a wide range of chemical and thermal stimuli. TRPM8, a member of the melastatin subfamily, is activated by cold temperatures (<28 °C), and antagonists of this channel have the potential to treat cold induced allodynia and hyperalgesia. However, TRPM8 has also been implicated in mammalian thermoregulation and antagonists have the potential to induce hypothermia in patients. We report herein the identification and optimization of a series of TRPM8 antagonists that ultimately led to the discovery of PF-05105679. The clinical finding with this compound will be discussed, including both efficacy and its ability to affect thermoregulation processes in humans.
Keywords: TRPM8, clinical tool, hypothermia, PF-05105679, thermoregulation, TRP channel, ion channel, pain
The transient receptor potential (TRP) family of ion channels are nonselective cation channels that respond to a wide range of chemical and thermal stimuli.1,2 In recent years, these ion channels have attracted attention as potential targets for the treatment of a variety of diseases, in part due to their ability to transmit noxious (or painful) stimuli, e.g., TRPA1 and TRPV1.3−6 TRPM8, a member of the melastatin subfamily, is activated by cold temperatures (8–28 °C) and cooling agents such as menthol and icilin.7,8 Preclinical studies have linked TRPM8 to pain in both oxaliplatin-induced9 and chronic nerve injury neuropathic pain models.10,11 Consequently, antagonists of this channel have the potential to treat cold induced allodynia (pain caused by normally innocuous stimuli) and hyperalgesia (a prolonged or more intense pain response) in human patients.
Beyond its role in transmitting cold induced pain signals, TRPM8 is also implicated in mammalian thermoregulation.12,13 Studies in preclinical species have demonstrated that the TRPM8 agonist menthol can induce hyperthermia in vivo.14 By analogy to TRPV1 agonists and antagonists, which cause hypothermia and hyperthermia respectively in humans,15,16 TRPM8 antagonists have the potential to induce hypothermia; an effect that has been demonstrated in preclinical species.17 A variety of small molecule TRPM8 antagonists covering a broad range of chemical and physical property space have been published in the literature.18−32 However, until very recently,33 no clinical data for these compounds had been disclosed, which might help to understand the role of TRPM8 in human physiology including the translation of these thermoregulation studies from preclinical species to humans.
In order to address this key thermoregulation concern, we adopted a project strategy that focused on rapidly identifying a compound that could be used as a clinical tool to understand the role of TRPM8 in humans from both a pain and thermoregulation standpoint. Herein we describe our efforts, which led to the identification of PF-05105679, an antagonist of TRPM8, and a key clinical tool for exploring both the role of TRPM8 in regulating core body temperature and the potential of TRPM8 antagonists for the treatment of pain in humans.
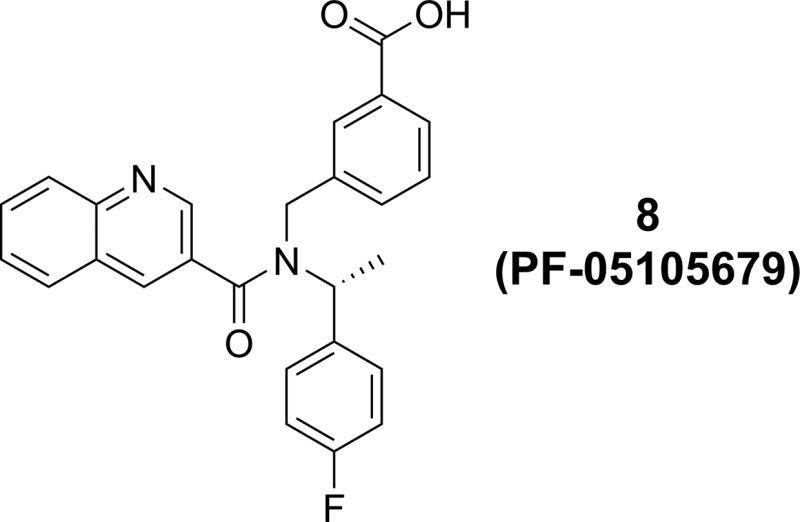
As a starting point, the project team identified compound 1 from a high throughput screen (HTS) of the Pfizer compound collection (Figure 1). Although a relatively weak inhibitor (TRPM8 FLIPR IC50 = 1173 nM), this hit was singled out as it was in good lipophilic efficiency (LipE or LLE = 4.9 as determined from logD)34−37 space and provided access to novel physicochemical space and biopharmaceutical properties, as acidic TRPM8 antagonists were unknown in the literature when we started this project.38 In addition, the chemistry available to construct this scaffold provided a convenient method for systematically varying each of the three vectors around the central nitrogen core. On the basis of these factors, the project team initiated medicinal chemistry efforts to optimize compound 1 with the goal of identifying a clinical tool.
Figure 1.

Initial HTS hit (1).
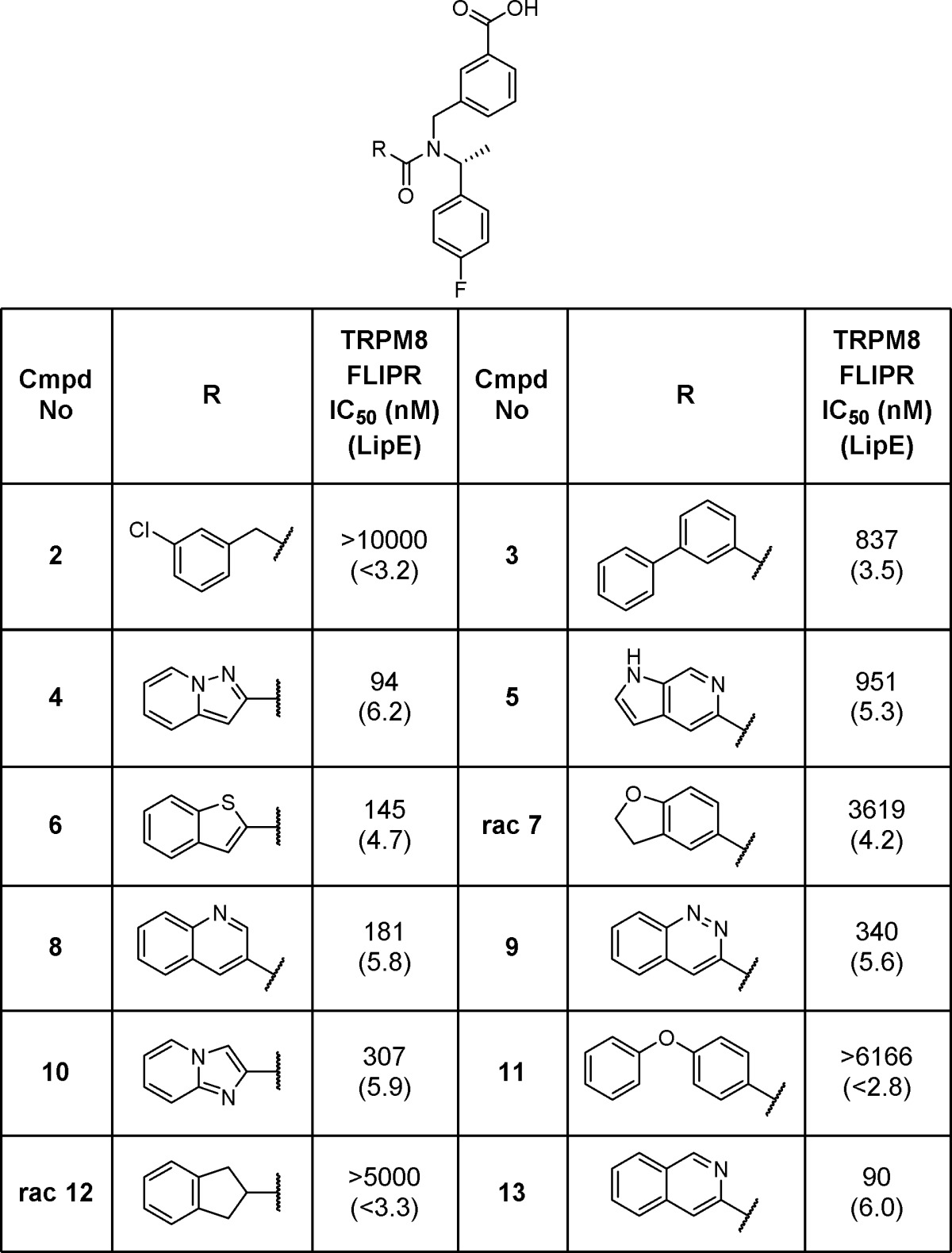
Separation of 1 into its two enantiomers indicated that potency at TRPM8 resided in the R_-enantiomer (R-1) (TRPM8 FLIPR IC50 = 702 nM), while the S_-enantiomer (S-1) was generally inactive (TRPM8 FLIPR IC50 > 10,000 nM). With this in mind, we designed a series of compounds to systematically explore the SAR of this scaffold. Optimization of the amide portion of the scaffold (Table 1) quickly demonstrated that fused, bicyclic heteroaromatic systems provided the best potency and efficiency against TRPM8, with other types of amides (2, rac-7, 12, and 11) only leading to less active compounds. Compound 3 suggested that larger groups such as biaryls could be tolerated; however, because of the poor LipE of this compound, further examples were not explored. Two of the best improvements in potency and LipE in the 6,6-biaryl amides came from replacing the 6-quinolone nitrogen (1) with the 3-quinoline (8) or the 3-isoquinoline (13). Modifying the quinoline to a 5,6-ring system (4, 6, and 10) was also tolerated. Unfortunately, attempts to introduce additional polarity within the bicyclic system proved unsuccessful (5 and 9) and did not result in any strong enhancements in potency or LipE.
Table 1. TRPM8 Potency SAR for Modification of the Amide Vector.
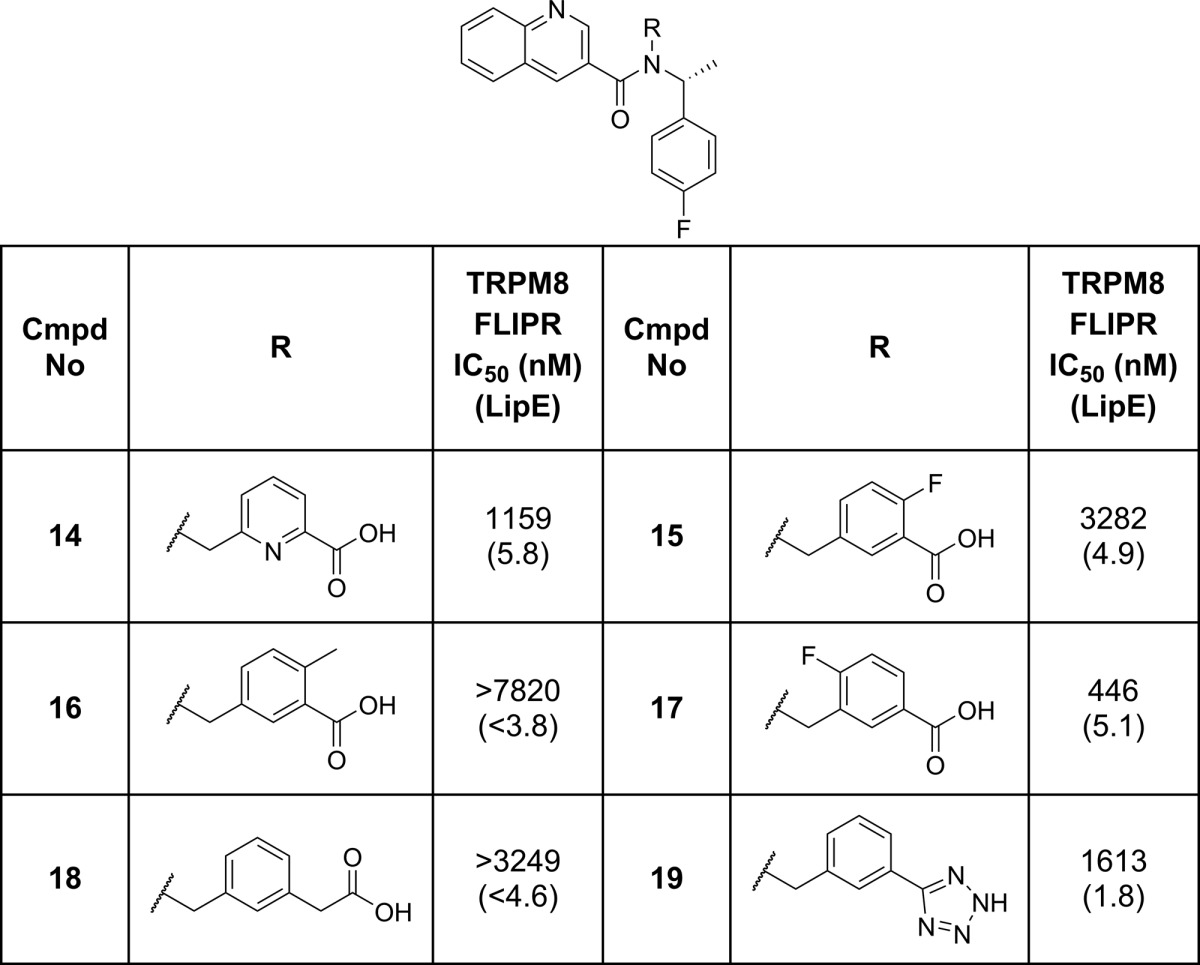
Once compound 8 had been identified, the project team next evaluated the structural requirements of the acidic portion of the scaffold on TRPM8 potency (Table 2). Modification of the key structural features of compound 1 generally led to decreases in potency. For example, the acid isostere 19 or homologation to a CH2 linked carboxylate (18) provided weaker inhibitors. More subtle modification around the benzoic acid moiety suggested that substituents ortho to the carboxylate, e.g., fluoro (15) or methyl (16), were not well tolerated. Para substituents, such as the fluorine in compound 17, were tolerated, although they did not give any potency or efficiency advantages over compound 8. Introducing polarity into the ring in the form of a pyridyl carboxylic acid (14) led to a drop in potency but maintained the compound’s overall efficiency.
Table 2. TRPM8 Potency SAR for Modification of the Acid Vector.
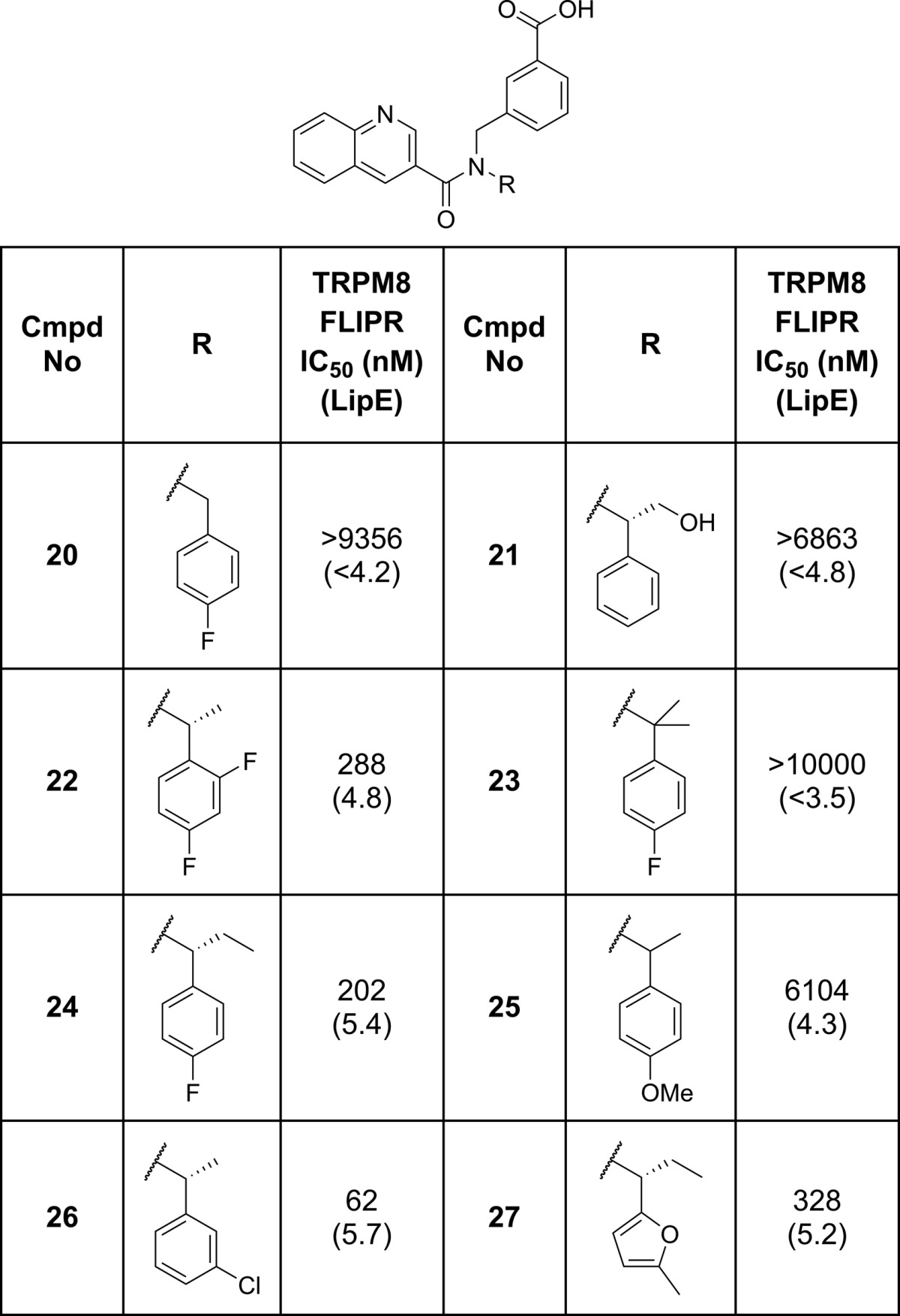
In parallel to evaluating the compounds shown in Table 2, we also explored the changes to the chiral benzyl substituent (Table 3). Subtle changes to substituents around the phenyl ring were generally well tolerated (22 and 26) but did not lead to any improvements in either potency or overall efficiency. An electron rich furan motif (27) was well tolerated, but introduction of a _para_-methoxy phenyl group (25) led to a large drop in potency. As previously mentioned, the chirality of the benzyl substituent was important for TRPM8 activity with all the activity residing in the R-enantiomer. Introduction of a gem-dimethyl group (23) significantly decreased potency, as did removal of the methyl group (20). Larger aliphatic substituents were tolerated (24) although they led to a drop in LipE, but similarly sized, more polar groups were not (21). These observations coupled with the large improvement in potency and LipE observed moving from 20 to 8 suggested the chiral methyl group may help to enforce a conformation that approaches the bound conformation of the antagonist in the TRPM8 binding site.
Table 3. TRPM8 Potency SAR for Modification of the Chiral Benzyl Substituent.
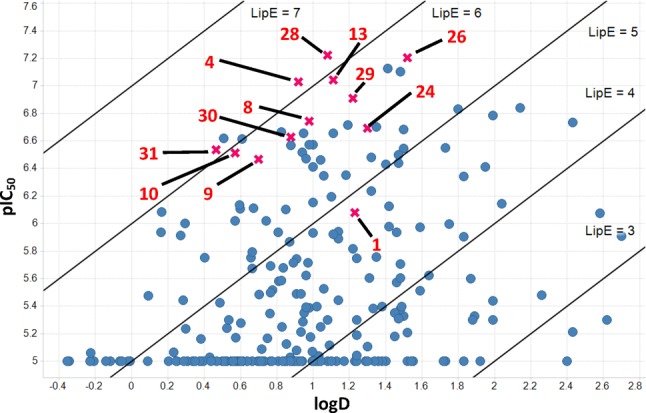
Following on from these first three rounds of design, we looked to combine some of the fragments contained in the compounds with better LipE values in order to see if we could improve on the overall efficiency of our compounds (Table 4). Gratifyingly, each of these recombinations provided potent TRPM8 antagonists in good LipE space. For example, combining the 3-isoquinoline motif from compound 13 with the pryidyl carboxylic acid moiety in 14 resulted in 31, which suggested the pyridyl acid motif could be a viable replacement for the benzoic acid moiety based on the similar LipE values between compounds 28 and 31. In addition, compounds 28 and 29 both exhibited potency and efficiency values that were on par with the best compounds identified in our first rounds of design. However, as a whole, these modifications did not provide any large improvements in efficiency.
Table 4. Recombination of the Best LipE Fragments.
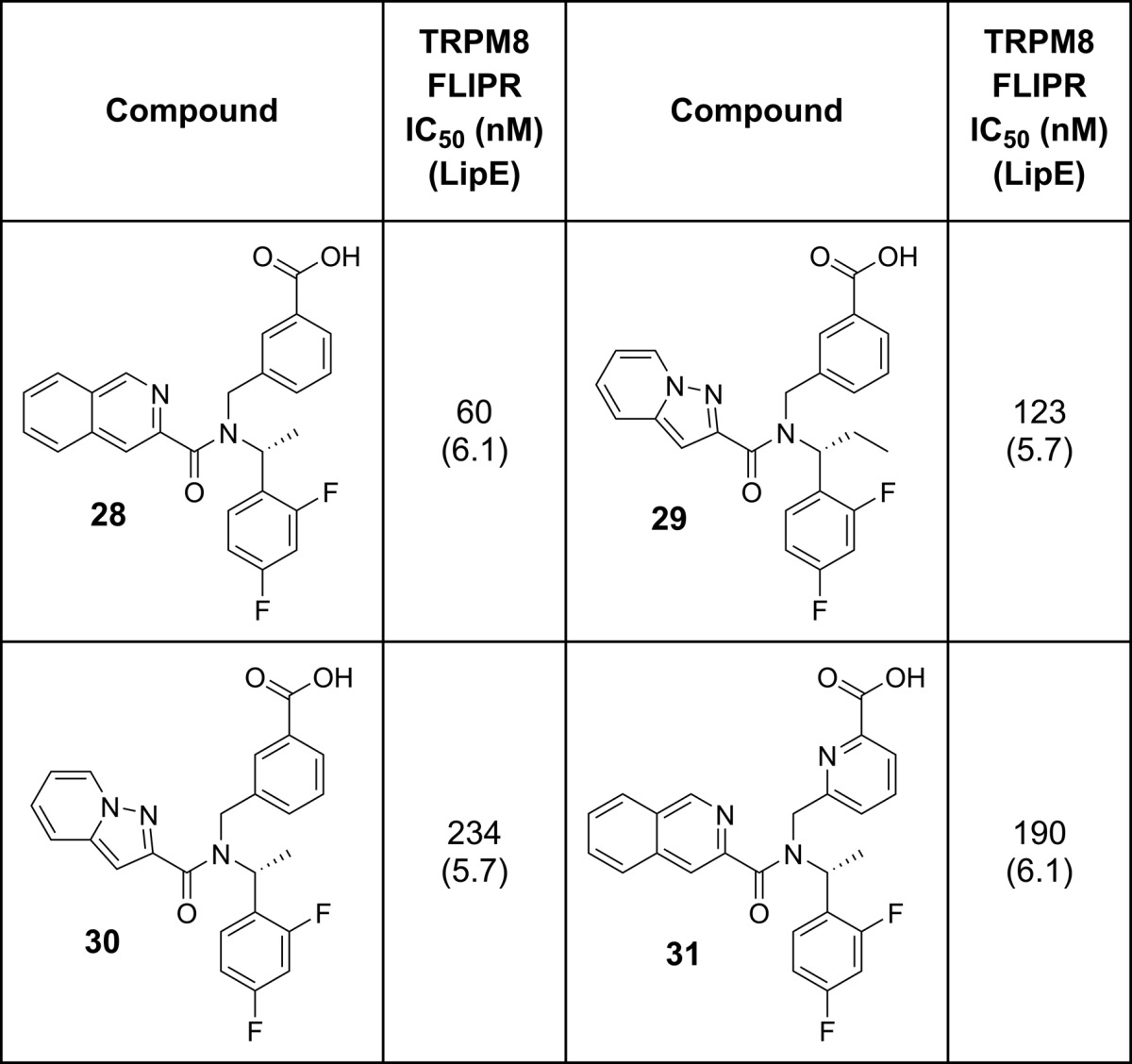
Examining the full set of compounds made by this stage of the project (Figure 2) demonstrated that, in a few rounds of design, the project had successfully taken the initial HTS hit (1) and optimized to a good cluster of efficient compounds of around LipE = 6. This analysis also suggested that the project had reached a plateau in terms of its ability to improve the efficiency of the series above a LipE ≈ 6.1. As a result, we focused on generating additional data on the best compounds identified from our design and screening efforts in the hope of identifying a suitable compound to advance to the clinic.
Figure 2.
LipE overview for project compounds. Key project compounds are marked with a red “X”; see Table 5.
The results from a standard set of in vitro ADME assays39−41 are summarized in Table 5. All compounds tested did not show any potential risks with respect to either hERG inhibition (as measured in a dofetilide binding assay) or drug–drug interactions, e.g., <20% inhibition at 3 μM for CYP3A4, CYP2D6, CYP1A2, CYP2C8, and CYP2C9. As the project was looking to quickly progress a compound to the clinic, our ideal tool compound required good antagonist potency at TRPM8 along with excellent membrane permeation and in vitro metabolic stability in both human and rat in order to facilitate pre-clinical toxicity studies. Although a number of compounds were superior to compound 8 in specific aspects of their in vitro profiles, overall 8 provided the best combination of potency and in vitro metabolic stability measurements in both human and rat and was therefore selected for further evaluation.
Table 5. Key TRPM8 Compounds Evaluated in an in Vitro ADME Panel39−41,a.
| cmpd no. | IC50 | logD7.4 | HLM | HHEP | RLM | Dof _K_i (nM) | CYP % inhib @ 3 μM |
|---|---|---|---|---|---|---|---|
| 4 | 94 | 0.9 | 42 | 23 | 325 | >9182 | ND |
| 8 | 181 | 1.0 | <8 | <15 | 17 | >15857 | 0 > 20% |
| 9 | 340 | ND | <8 | ND | 76 | ND | ND |
| 10 | 307 | 0.6 | ND | ND | 113 | >9182 | ND |
| 13 | 90 | 1.1 | 16 | 21 | 166 | >9182 | ND |
| 24 | 202 | 1.3 | <8 | 28 | <14 | >15219 | ND |
| 26 | 62 | 1.5 | 10 | 45 | 102 | ND | 0 > 20% |
| 28 | 60 | 1.1 | 11 | <2 | 31 | ND | 0 > 20% |
| 29 | 123 | 1.2 | 28 | 48 | 41 | ND | 0 > 20% |
| 30 | 234 | 0.9 | 23 | 31 | 85 | ND | ND |
| 31 | 291 | 0.4 | 11 | <2.2 | 31 | ND | 0 > 20% |
Single cell patch clamp electrophysiology (Ephys) studies, confirmed the potency of 8 against TRPM8 (Table 6).33 In addition, 8 demonstrated >100-fold selectivity across a range of different receptors, ion channels, and enzymes including the closely related TRPV1 and TRPA1 channels. As a P-gp substrate, compound 8 also exhibited some CNS restriction (CSF to free plasma ratio of 0.3 in rat and guinea pigs) but was still progressed as the project believed that clinical efficacy would be obtained by targeting peripheral TRPM8.
Table 6. Further in Vitro and in Vivo PK Evaluation of Compound 8 (PF-05105679)a.
| TRPM8 Ephys IC50 (nM) | CEREP polypharmacology | hERG (μM) | permeability | protein binding (fu) |
|---|---|---|---|---|
| 103 | >100-fold selectivity | >30 | RRCK: 6.8 × 10–6 | human: 0.012 |
| rat: 0.033 | ||||
| MDCK ER: 4.8 | guinea pig: 0.07 | |||
| dog: 0.067 |
| | rat | dog | | | --------------------- | ------------------- | ---------------------- | | Cl (mL/min/kg) (Clu) | 19.8 (618) | 31 (462) | | _V_dss (L/kg) | 6.2 | 7.4 | | _T_1/2 (h) | 3.6 | 3.9 | | %F (absorption) | 29% @ 2 mg/kg (46%) | 92% @ 20 mg/kg (∼100%) | | 62% @ 10 mg/kg (100%) | | |
In order to obtain an estimate of the efficacious concentration (_C_eff) for clinical studies, compound 8 was tested in a cold-induced bladder contractility model in guinea pigs.33,42 While not a direct measure of the compounds ability to act as an analgesic, the assay did provide a direct measure of TRMP8-mediated pharmacology. Results from these studies indicated that a maximum unbound concentration (_C_max) of drug between 254–450 nM (∼3 × IC50) would be sufficient to test the mechanism in clinical studies. Preclinical PK studies demonstrated that compound 8 should exhibit moderate blood clearance with respect to hepatic blood flow and essentially complete oral absorption in both rat and dog. Volume of distribution values (_V_dss) were much higher than would be expected for an acidic compound and may be driven by enterophepatic recirculation of either compound 8 or its primary metabolite, the acyl glucuronide, which was observed in both in vitro and bile-duct cannulated rat studies. On the basis of the worse case assumption that enterohepatic recirculation may not be a prevalent process in humans, human dose predictions using a more typical _V_dss for acids of 0.2 L/kg43 indicated that a dose of 1 g would provide sufficient exposure of 8 to reach the predicted _C_eff in the clinic. In addition, preclinical toxicity studies allowed for progression of 8 to the clinic with sufficient toxicology margins relative to the predicted _C_eff to safely test the effect of TRPM8 antagonism in humans.
Evaluation of compound 8 (PF-05105679) in humans demonstrated that a single 900 mg dose provided efficacy in a cold pressor test that was comparable to oxycodone. At this dose, compound 8 reached circulating concentrations of about 3-fold the TRPM8 IC50 at _C_max, supporting the predicted _C_eff from preclinical species and the dose prediction strategy described above.33 Importantly, doses up to 900 mg did not result in significant changes to core body temperature. However, this efficacious dose did result in unexpected adverse events, including a nontolerated, hot sensation localized to the mouth, face, upper body, arms, and hands.
In summary, we have described the rapid identification of compound 8(44) as a clinical tool for elucidating the role of TRPM8 signaling in humans. Although blockade of TRPM8 in humans did not appear to have an effect on core body temperature at efficacious concentrations, the lack of a therapeutic index between an adverse event (feeling hot) and efficacy and its short half-life in humans limit the further progression of 8 in the clinic. Further studies are required to understand if TRPM8 remains a viable target for the treatment of cold-related pain in patients.
Acknowledgments
The authors wish to acknowledge the contributions of A. Cronin, W. Klute, D. Lovering, W. McCarte, A. Nortcliffe, S. Wheeler, L. Watson, and A. Warren towards the synthesis of compounds described in this manuscript. The authors also wish to thank W. Winchester, D. Blakemore, J. Warmus, C. Rose, and J. Bian for assistance in assembling this manuscript and M. J. Palmer for his assistance in working up the project HTS.
Glossary
ABBREVIATIONS
HTS
high throughput screen
_C_eff
efficacious concentration
CYP
cytochrome P450
HLM
human liver microsomes
RLM
rat liver microsomes
HHEP
human hepatocytes
Dof
dofetilide
hERG
human ether-a-go-go related gene
LipE
lipophilic efficiency
MDCK ER
MDCK efflux ratio
Supporting Information Available
Synthetic details for all compounds described. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Ramsey I. S.; Delling M.; Clapham D. E. An introduction to TRP channels. Annu. Rev. Physiol. 2006, 68, 619–647. [DOI] [PubMed] [Google Scholar]
- Szallasi A.; Biro T.; Eds. TRP Channels in Drug Discovery; Springer: New York, 2012; Vol. 1. [Google Scholar]
- Moran M. M.; McAlexander M. A.; Biro T.; Szallasi A. Transient receptor potential channels as therapeutic targets. Nat. Rev. Drug Discovery 2011, 10, 601–620. [DOI] [PubMed] [Google Scholar]
- Bagal S. K.; Brown A. D.; Cox P. J.; Omoto K.; Owen R. M.; Pryde D. C.; Sidders B.; Skerratt S. E.; Stevens E. B.; Storer R. I.; Swain N. A. Ion Channels as Therapeutic Targets: A Drug Discovery Perspective. J. Med. Chem. 2013, 56, 593–624. [DOI] [PubMed] [Google Scholar]
- Kaneko Y.; Szallasi A. Transient receptor potential (TRP) channels: a clinical perspective. Br. J. Pharmacol. 2014, 171, 2474–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brederson J.-D.; Kym P. R.; Szallasi A. Targeting TRP channels for pain relief. Eur. J. Pharmacol. 2013, 716, 61–76. [DOI] [PubMed] [Google Scholar]
- McKemy D. D. Therapeutic potential of TRPM8 modulators. Open Drug Discovery J. 2010, 2, 81–88. [Google Scholar]
- DeFalco J.; Duncton M. A. J.; Emerling D. TRPM8 biology and medicinal chemistry. Curr. Top. Med. Chem. 2011, 11, 2237–2252. [DOI] [PubMed] [Google Scholar]
- Descoeur J.; Pereira V.; Pizzoccaro A.; Francois A.; Ling B.; Maffre V.; Couette B.; Busserolles J.; Courteix C.; Noel J.; Lazdunski M.; Eschalier A.; Authier N.; Bourinet E. Oxaliplatin-induced cold hypersensitivity is due to remodelling of ion channel expression in nociceptors. EMBO Mol. Med. 2011, 3, 266–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su L.; Wang C.; Yu Y.-H.; Ren Y.-Y.; Xie K.-L.; Wang G.-L. Role of TRPM8 in dorsal root ganglion in nerve injury-induced chronic pain. BMC Neurosci. 2011, 12, 120–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing H.; Chen M.; Ling J.; Tan W.; Gu J. G. TRPM8 mechanism of cold allodynia after chronic nerve injury. J. Neurosci. 2007, 27, 13680–13690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavva N. R.; Davis C.; Lehto S. G.; Rao S.; Wang W.; Zhu D. X. D. Transient receptor potential melastatin 8 (TRPM8) channels are involved in body temperature regulation. Mol. Pain 2012, 8, 36–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKemy D. D. The Molecular and Cellular Basis of Cold Sensation. ACS Chem. Neurosci. 2013, 4, 238–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruskin D. N.; Anand R.; LaHoste G. J. Menthol and nicotine oppositely modulate body temperature in the rat. Eur. J. Pharmacol. 2007, 559, 161–164. [DOI] [PubMed] [Google Scholar]
- Gavva N. R.; Treanor J. J. S.; Garami A.; Fang L.; Surapaneni S.; Akrami A.; Alvarez F.; Bak A.; Darling M.; Gore A.; Jang G. R.; Kesslak J. P.; Ni L.; Norman M. H.; Palluconi G.; Rose M. J.; Salfi M.; Tan E.; Romanovsky A. A.; Banfield C.; Davar G. Pharmacological blockade of the vanilloid receptor TRPV1 elicits marked hyperthermia in humans. Pain 2008, 136, 202–210. [DOI] [PubMed] [Google Scholar]
- Romanovsky A. A.; Almeida M. C.; Garami A.; Steiner A. A.; Norman M. H.; Morrison S. F.; Nakamura K.; Burmeister J. J.; Nucci T. B. The transient receptor potential vanilloid-1 channel in thermoregulation: a thermosensor it is not. Pharmacol. Rev. 2009, 61, 228–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almeida M. C.; Hew-Butler T.; Soriano R. N.; Rao S.; Wang W.; Wang J.; Tamayo N.; Oliveira D. L.; Nucci T. B.; Aryal P.; Garami A.; Bautista D.; Gavva N. R.; Romanovsky A. A. Pharmacological blockade of the cold receptor TRPM8 attenuates autonomic and behavioral cold defenses and decreases deep body temperature. J. Neurosci. 2012, 32, 2086–2099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeFalco J.; Steiger D.; Dourado M.; Emerling D.; Duncton M. A. J. 5-Benzyloxytryptamine as an antagonist of TRPM8. Bioorg. Med. Chem. Lett. 2010, 20, 7076–7079. [DOI] [PubMed] [Google Scholar]
- Inoue T.; Ohmi M.; Kawamura K.; Ando K.; Shishido Y.. Sulfamoylbenzoic acid derivatives and related compounds as TRPM8 antagonists and their preparation and use for the treatment of diseases. WO2010125831A1, 2010.
- Irlapati N. R.; Thomas A.; Kurhe D. K.; Shelke S. Y.; Khairatkar J. N.; Viswanadha S.; Mukhopadhyay I.. Preparation of fused oxazole and thiazole derivatives as TRPM8 modulators. WO2010010435A2, 2010.
- Parks D. J.; Parsons W. H.; Colburn R. W.; Meegalla S. K.; Ballentine S. K.; Illig C. R.; Qin N.; Liu Y.; Hutchinson T. L.; Lubin M. L.; Stone D. J. Jr.; Baker J. F.; Schneider C. R.; Ma J.; Damiano B. P.; Flores C. M.; Player M. R. Design and Optimization of Benzimidazole-Containing Transient Receptor Potential Melastatin 8 (TRPM8) Antagonists. J. Med. Chem. 2011, 54, 233–247. [DOI] [PubMed] [Google Scholar]
- Matthews J. M.; Qin N.; Colburn R. W.; Dax S. L.; Hawkins M.; McNally J. J.; Reany L.; Youngman M. A.; Baker J.; Hutchinson T.; Liu Y.; Lubin M. L.; Neeper M.; Brandt M. R.; Stone D. J.; Flores C. M. The design and synthesis of novel, phosphonate-containing transient receptor potential melastatin 8 (TRPM8) antagonists. Bioorg. Med. Chem. Lett. 2012, 22, 2922–2926. [DOI] [PubMed] [Google Scholar]
- Tamayo N. A.; Bo Y.; Gore V.; Ma V.; Nishimura N.; Tang P.; Deng H.; Klionsky L.; Lehto S. G.; Wang W.; Youngblood B.; Chen J.; Correll T. L.; Bartberger M. D.; Gavva N. R.; Norman M. H. Fused Piperidines as a Novel Class of Potent and Orally Available Transient Receptor Potential Melastatin Type 8 (TRPM8) Antagonists. J. Med. Chem. 2012, 55, 1593–1611. [DOI] [PubMed] [Google Scholar]
- Brown A.; Ellis D.; Favor D. A.; Kirkup T.; Klute W.; MacKenny M.; McMurray G.; Stennett A. Serendipity in drug-discovery: A new series of 2-(benzyloxy)benzamides as TRPM8 antagonists. Bioorg. Med. Chem. Lett. 2013, 23, 6118–6122. [DOI] [PubMed] [Google Scholar]
- Chaudhari S. S.; Kadam A. B.; Khairatkar-Joshi N.; Mukhopadhyay I.; Karnik P. V.; Raghuram A.; Rao S. S.; Vaiyapuri T. S.; Wale D. P.; Bhosale V. M.; Gudi G. S.; Sangana R. R.; Thomas A. Synthesis and pharmacological evaluation of novel N-aryl-3,4-dihydro-1′H-spiro[chromene-2,4′-piperidine]-1′-carboxamides as TRPM8 antagonists. Bioorg. Med. Chem. 2013, 21, 6542–6553. [DOI] [PubMed] [Google Scholar]
- Moriconi A.; Bianchini G.; Colagioia S.; Brandolini L.; Aramini A.; Liberati C.; Bovolenta S.. Preparation of 2-aryl oxazole and thiazole compounds as TRPM8 antagonists for therapy. WO2013092711A1, 2013.
- Zhu B.; Xia M.; Xu X.; Ludovici D. W.; Tennakoon M.; Youngman M. A.; Matthews J. M.; Dax S. L.; Colburn R. W.; Qin N.; Hutchinson T. L.; Lubin M. L.; Brandt M. R.; Stone D. J.; Flores C. M.; Macielag M. J. Arylglycine derivatives as potent transient receptor potential melastatin 8 (TRPM8) antagonists. Bioorg. Med. Chem. Lett. 2013, 23, 2234–2237. [DOI] [PubMed] [Google Scholar]
- Kato T.; Sakamoto T.; Kubo A.; Sawamoto D.. Preparation of sulfonamides having TRPM8-blocking effects. WO2014042238A1, 2014.
- Ohmi M.; Shishido Y.; Inoue T.; Ando K.; Fujiuchi A.; Yamada A.; Watanabe S.; Kawamura K. Identification of a novel 2-pyridyl-benzensulfonamide derivative, RQ-00203078, as a selective and orally active TRPM8 antagonist. Bioorg. Med. Chem. Lett. 2014, 24, 5364–5368. [DOI] [PubMed] [Google Scholar]
- Horne D. B.; Tamayo N. A.; Bartberger M. D.; Bo Y.; Clarine J.; Davis C. D.; Gore V. K.; Kaller M. R.; Lehto S. G.; Ma V. V.; Nishimura N.; Nguyen T. T.; Tang P.; Wang W.; Youngblood B. D.; Zhang M.; Gavva N. R.; Monenschein H.; Norman M. H. Optimization of Potency and Pharmacokinetic Properties of Tetrahydroisoquinoline Transient Receptor Potential Melastatin 8 (TRPM8) Antagonists. J. Med. Chem. 2014, 57, 2989–3004. [DOI] [PubMed] [Google Scholar]
- Biswas K.; Brown J.; Chen J. J.; Gore V. K.; Harried S.; Horne D. B.; Kaller M. R.; Ma V. V.; Nguyen T. T.; Sham K.; Zhong W.. Chroman derivatives as TRPM8 inhibitors and their preparation. WO2014025651A1, 2014.
- Lampe T.; Alonso-Alija C.; Stelte-Ludwig B.; Sandner P.; Bauser M.; Beck H.; Lustig K.; Rosentreter U.; Stahl E.; Takagi H.. Preparation of substituted N-(4-benzyloxyphenyl)methylamide derivs. as cold menthol receptor-1 (CMR-1) antagonists for treatment of urological disorders. WO2006040136A1, 2006.
- Winchester W.; Gore K.; Glatt S.; Saintot P.-P.; Reynolds D. S.; Petit W.; Gardiner J. C.; Conlon K.; Postlethwaite M.; Roberts S.; Gosset J. R.; Matsuura T.; Beaumont K.; Andrews M. D.; Glossop P. A.; Palmer M. J.; Clear N.; Collins S. Inhibition of TRPM8 channels reduces pain in the cold pressor test in humans. J. Pharmacol. Exp. Ther. 2014, 351, 259–269. [DOI] [PubMed] [Google Scholar]
- All LipE values were calculated from measured logD values or when unavailable, calculated LogD values derived from an internal model.
- Keefer C. E.; Kauffman G. W.; Gupta R. R. Interpretable, Probability-Based Confidence Metric for Continuous Quantitative Structure-Activity Relationship Models. J. Chem. Inf. Model. 2013, 53, 368–383. [DOI] [PubMed] [Google Scholar]
- Hopkins A. L.; Keseru G. M.; Leeson P. D.; Rees D. C.; Reynolds C. H. The role of ligand efficiency metrics in drug discovery. Nat. Rev. Drug Discovery 2014, 13, 105–121. [DOI] [PubMed] [Google Scholar]
- Ryckmans T.; Edwards M. P.; Horne V. A.; Correia A. M.; Owen D. R.; Thompson L. R.; Tran I.; Tutt M. F.; Young T. Rapid assessment of a novel series of selective CB(2) agonists using parallel synthesis protocols: A Lipophilic Efficiency (LipE) analysis. Bioorg. Med. Chem. Lett. 2009, 19, 4406–4409. [DOI] [PubMed] [Google Scholar]
- Subsequent to the initial work, multiple acidic TRPM8 antagonists have been disclosed in the literature. Please see refs (19), (28), (29), and (30).
- Price D. A.; Armour D.; De Groot M.; Leishman D.; Napier C.; Perros M.; Stammen B. L.; Wood A. Overcoming HERG affinity in the discovery of the CCR5 antagonist maraviroc. Bioorg. Med. Chem. Lett. 2006, 16, 4633–4637. [DOI] [PubMed] [Google Scholar]
- Allan G.; Davis J.; Dickins M.; Gardner I.; Jenkins T.; Jones H.; Webster R.; Westgate H. Pre-clinical pharmacokinetics of UK-453,061, a novel non-nucleoside reverse transcriptase inhibitor (NNRTI), and use of in silico physiologically based prediction tools to predict the oral pharmacokinetics of UK-453,061 in man. Xenobiotica 2008, 38, 620–640. [DOI] [PubMed] [Google Scholar]
- Di L.; Whitney-Pickett C.; Umland J. P.; Zhang H.; Zhang X.; Gebhard D. F.; Lai Y.; Federico J. J.; Davidson R. E.; Smith R.; Reyner E. L.; Lee C.; Feng B.; Rotter C.; Varma M. V.; Kempshall S.; Fenner K.; El-kattan A. F.; Liston T. E.; Troutman M. D. Development of a new permeability assay using low-efflux MDCKII cells. J. Pharm. Sci. 2011, 100, 4974–4985. [DOI] [PubMed] [Google Scholar]
- Gardiner J. C.; Kirkup A. J.; Curry J.; Humphreys S.; O’Regan P.; Postlethwaite M.; Young K. C.; Kitching L.; Ethell B. T.; Winpenny D.; McMurray G. The role of TRPM8 in the Guinea-pig bladder-cooling reflex investigated using a novel TRPM8 antagonist. Eur. J. Pharmacol. 2014, 740, 398–409. [DOI] [PubMed] [Google Scholar]
- Smith D. A.; Jones B. C.; Walker D. K. Design of drugs involving the concepts and theories of drug metabolism and pharmacokinetics. Med. Res. Rev. 1996, 16, 243–266. [DOI] [PubMed] [Google Scholar]
- Compound 8 (PF-05105679) is now commercially available from Sigma Aldrich (Catalog # PZ0245).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.