Cortical dysplasia resembling human type 2 lissencephaly in mice lacking all three APP family members (original) (raw)
Abstract
The Alzheimer's disease β-amyloid precursor protein (APP) is a member of a larger gene family that includes the amyloid precursor-like proteins, termed APLP1 and APLP2. We previously documented that APLP2−/−APLP1−/− and APLP2−/−APP−/− mice die postnatally, while APLP1−/−APP−/− mice and single mutants were viable. We now report that mice lacking all three APP/APLP family members survive through embryonic development, and die shortly after birth. In contrast to double-mutant animals with perinatal lethality, 81% of triple mutants showed cranial abnormalities. In 68% of triple mutants, we observed cortical dysplasias characterized by focal ectopic neuroblasts that had migrated through the basal lamina and pial membrane, a phenotype that resembles human type II lissencephaly. Moreover, at E18.5 triple mutants showed a partial loss of cortical Cajal Retzius (CR) cells, suggesting that APP/APLPs play a crucial role in the survival of CR cells and neuronal adhesion. Collectively, our data reveal an essential role for APP family members in normal brain development and early postnatal survival.
Keywords: Alzheimer's disease, APLP, β-amyloid precursor protein, lissencephaly, triple knockout
Introduction
Alzheimer's disease (AD) is pathologically characterized by the presence of neurofibrilary tangles and neuritic plaques. The predominant constituent of neuritic plaques are β-amyloid peptides (Aβ), derived by proteolytic processing from the larger amyloid precursor protein (APP). The proteolysis of APP occurs either by α-secretase within the Aβ region, or by β-secretase at the N-terminus of Aβ, leading to the secretion of large soluble ectodomains, termed sAPPα and sAPPβ, respectively. Subsequent intramembranous processing of the membrane-tethered APP C-terminal fragments by γ-secretase cleavage results in the production of Aβ and C-terminal stubs. APP is a member of a larger gene family, including in mammals the two amyloid precursor-like proteins APLP1 and APLP2. Like APP, both APLPs seem to be processed by α-, β- and γ-secretase, in a similar way, despite the absence of the Aβ region within either of the APLPs (Gu et al, 2001; Scheinfeld et al, 2002; Li and Südhof, 2003; Walsh et al, 2003; Eggert et al, 2004). APP and APLP2 are ubiquitously expressed in a largely overlapping pattern during embryonic development and in most adult tissues (Slunt et al, 1994; Lorent et al, 1995). In contrast, APLP1 expression is restricted to the nervous system (Lorent et al, 1995; Thinakaran et al, 1995).
Biochemical and genetic interaction screens have led to the identification of both extracellular and multiple intracellular binding partners, which seem to anchor the APP/APLP C-termini to a complex protein network at the cell surface, which may transduce various cellular responses (reviewed by De Strooper and Annaert, 2000; Koo, 2002). Notably, a highly conserved cytoplasmic YENPTY motif is present in all APP/APLP family members, which confers clathrin-mediated endocytosis and was shown to bind several multidomain adaptor proteins, including X11/Mints, Fe65 family proteins and mDab1.
The physiological and cell biological roles of APP/APLP family proteins, or their respective cleavage products, are still only poorly understood. To address these functions directly, we and others have previously generated mice that either carry a hypomorphic mutation of APP (APPΔ, Müller et al, 1994) or are completely deficient of APP (Zheng et al, 1995; Li et al, 1996). A host of in vitro and in vivo studies on these mice, or cells derived from them, have suggested that APP may play a role in neurite outgrowth and the formation of forebrain commissures, axonal kinesin-mediated transport, postnatal somatic growth and neurobehavioral development, locomotor activity and grip strength, copper homeostasis, and susceptibility to epileptic seizures and excitotoxic agents (Zheng et al, 1995; Li et al, 1996; Perez et al, 1997; Steinbach et al, 1998; Tremml et al, 1998; White et al, 1998, 1999; Magara et al, 1999; Kamal et al, 2001). APLP1-deficient mice showed a postnatal growth deficit, as the only obvious abnormality (Heber et al, 2000), whereas APLP2−/− mice were apparently normal (von Koch et al, 1997). In contrast to the minor phenotype of single mutants, APLP2−/−APLP1−/− and APLP2−/−APP−/− double mutants were lethal early postnatally: proving that APP family proteins serve essential, but partially redundant, functions (von Koch et al, 1997; Heber et al, 2000). Surprisingly, APLP1−/−APP−/− mice were viable and apparently normal, suggesting functional compensation by the still intact APLP2 gene.
We now generated mice lacking all three APP family members in order to eliminate any residual functional complementation. Triple knockout animals were obtained at about normal frequencies at late embryonic stages (E17-P0). Thus, the APP/APLP family members are apparently not essential for early embryonic development. Whereas lethal double mutants showed no morphological abnormalities, the brains of triple mutants displayed a phenotype resembling human type II lissencephaly with ectopic clusters of neuroblasts that had migrated through the basal lamina and pial membrane. In addition, we observed a reduced survival of cortical Cajal Retzius (CR) cells in triple mutants, which might contribute to ectopia formation. Thus, APP family proteins play a pivotal role in processes underlying normal brain development and postnatal survival.
Results
Haploinsufficiency of APP−/−/APLP1−/−/APLP2+/− mice
The viability of APP−/−APLP1−/− double mutants led us to hypothesize whether this might be due to functional compensation by the remaining third family member, APLP2. In order to generate triple knockouts, which should theoretically be feasible by intercrossing APP−/−APLP1−/−APLP2+/− mice, we intended to first generate animals of this genotype. However, we observed postnatal lethality for APP−/−APLP1−/−APLP2+/− mice. At weaning, the frequency of APP−/−APLP1−/−APLP2+/− offspring was reduced from the expected 25% to only 0.6% (see Table I). Death occurred postnatally, since normal numbers of APP−/−APLP1−/−APLP2+/− mice were born at P0 (27% of the 202 mice, Table I). As previously reported for double mutants, no histopathological abnormalities were apparent in APP−/−APLP1−/−APLP2+/− mice analyzed at P0, or when surviving mutants were killed at 24 months of age (data not shown). These data thus confirm and extend our previous findings (Heber et al, 2000) and indicate haploinsufficiency for a single APLP2 allele in the absence of both other APP family proteins.
Table 1.
Survival rates of APP−/−APLP1−/−APLP2+/− mice
| | Postnatal lethality of APP−/−APLP1−/−APLP2+/− mice | | | |
| --------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------- | --------------------- | -------- | -------- |
| Genotype of parents | Genotype of offspring | Obtained | Expected |
|  | | | |
| Shortly after birth, a total of 202 animals, obtained from parents of the indicated genotypes, were examined. At this time point, APP−/−APLP1−/−APLP2+/− animals were obtained at about the expected frequency (27%). However, at weaning, we found among 527 animals analyzed only three (0.6%) APP−/−APLP1−/−APLP2+/− pups, indicating postnatal lethality. Since individual newborns were not marked, the precise time point of death could not be determined, but most likely occurred within the first few day(s) after birth, since no dead pups were found at later time points. | | | |
| Bold characters denote the most important genotypes. | | | |
| | | |
| Shortly after birth, a total of 202 animals, obtained from parents of the indicated genotypes, were examined. At this time point, APP−/−APLP1−/−APLP2+/− animals were obtained at about the expected frequency (27%). However, at weaning, we found among 527 animals analyzed only three (0.6%) APP−/−APLP1−/−APLP2+/− pups, indicating postnatal lethality. Since individual newborns were not marked, the precise time point of death could not be determined, but most likely occurred within the first few day(s) after birth, since no dead pups were found at later time points. | | | |
| Bold characters denote the most important genotypes. | | | |
Generation of triple mutants lacking the entire APP gene family
To generate triple mutants lacking the entire APP gene family, we made use of the three adult APP−/−APLP1−/−APLP2+/− mice that were either intercrossed (mating type A, Table II) or crossed to APP−/−APLP1+/−APLP2+/− animals (mating type B, Table II). In addition, we intercrossed APP−/−APLP1+/−APLP2+/− mice, which should yield triple mutants with a much lower frequency of 6.25% (mating type C, Table II). Genotypic analysis of animals from late embryonic stages (time window E17.5-P0, Table II) revealed triple mutants with a frequency not significantly different from the theoretically expected values, indicating that APP family members are apparently not essential for early embryonic development.
Table 2.
Frequency of triple mutants at late embryonic stages until birth
| | Mating scheme | Genotype of offspring | % exp. | Number of animals obtained E17.5-P0 |
| ---------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------- | --------------------- | ------ | ----------------------------------- |
|  | | | |
| The table shows the theoretically expected frequencies for animals of the indicated genotypes (columns 2 and 3) as compared to the actual number and frequency of animals obtained (last column). Triple mutants were generated by three different mating schemes (type A, B, C) with an expected frequency of 25% for type A, 12.5% for type B and only 6.25% for type C, respectively. Overall, triple mutants were obtained at late embryonic stages (E17.5) until birth (P0), with a frequency not significantly different from the theoretically expected values. | | | |
| | | |
| The table shows the theoretically expected frequencies for animals of the indicated genotypes (columns 2 and 3) as compared to the actual number and frequency of animals obtained (last column). Triple mutants were generated by three different mating schemes (type A, B, C) with an expected frequency of 25% for type A, 12.5% for type B and only 6.25% for type C, respectively. Overall, triple mutants were obtained at late embryonic stages (E17.5) until birth (P0), with a frequency not significantly different from the theoretically expected values. | | | |
Mice lacking the entire APP gene family exhibit cortical malformations and neuronal ectopia similar to cobblestone lissencephaly
Triple knockout mice, lacking all APP family members, exhibited a high incidence of cortical dysplasias (of the 31 triple mutants analyzed, 25=81% had cranial abnormalities) when examined between E17.5 and birth. In 68% of triple mutants (21 of 31 examined at E17.5-P0), ectopic clusters of cells were found within the marginal zone (MZ) and the subarachnoid space as a result of overmigration of cortical plate cells, a form of ectopia that closely resembles human cobblestone lissencephaly (also known as type 2 lissencephaly). Ectopias were predominantly of neuronal origin, as evidenced by staining with antibodies directed against the pan-neuronal marker NeuN (data not shown). Key features of this phenotype are exemplified in Figure 1, depicting Nissl-stained coronal brain sections from E14.5, E17.5 and E18.5 triple mutant embryos. In sections through the center of dysplasias, the cortical architecture underneath ectopic neuronal clusters was completely disrupted, indicating that a cohort of neurons had moved to these ectopic positions (Figure 1B and enlargement in Figure 2B). The basement membranes underlying the leptomeninges were found to be fragmented, or completely missing in the area of the protrusions (Figure 2D). Moreover, clusters of neuroblasts appeared to have penetrated from the protrusion along the subarachnoidal space (Figure 1F and H, arrowheads). In most instances, only single, focal neuronal ectopias were found located at dorsal aspects of the fronto-parietal cortex (Figure 1A), whereas medial cortical fields close to the interhemispheric fissure and ventral areas were unaffected. Overall, ectopias were confined to the cortex and were absent from other brain regions, or spinal cord, of triple knockouts. No additional histological abnormalities were found in any other brain region, besides a high incidence of corpus callosum deficits as already noted for APP-deficient mice (Magara et al, 1999). Small protrusions and areas of ectopic neurons were also found in two out of 14 APP−/−APLP1+/−APLP2−/− mice (data not shown), but these were absent in all 21 APP−/−APLP1+/−APLP2+/− mice analyzed, as well as in all double mutants.
Figure 1.
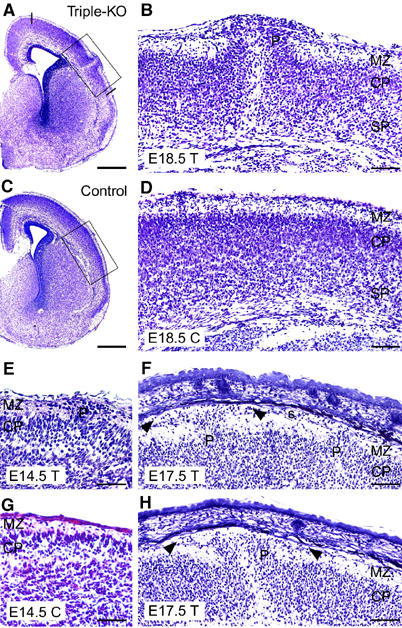
Cortical dysplasia in APP−/−APLP1−/−APLP2−/− triple knockout mice of various ages. (A, B) Frontal sections (cresyl violet staining) of a triple knockout mouse (T) at E18.5, exhibiting a prominent protrusion (P) of the right hemispheric cortical plate. The occurrence of ectopias was restricted to dorsal cortical areas as indicated by lines in (A). (B) Upon higher magnification (boxed area in A), it becomes apparent that ectopic neurons completely disrupt the subplate (SP) and cortical plate (CP); the cells have migrated into and beyond the MZ. No alterations were observed in the ventricular zone (not shown) (C, D) Depict low- and high-power images of a littermate control (C, genotype: APP−/−APLP1+/−APLP2−/−). (F) Example of an E17.5 triple knockout brain (T) showing two smaller cerebral protrusions (P). Arrowheads mark cells invading the subarachnoidal space (s). (E) Example of an E14.5 triple knockout exhibiting a small protrusion in comparison to a littermate control (C, genotype: APP−/−APLP1−/−APLP2+/−) depicted in (G). (H) Example of ectopia in a triple knockout embryo at E17.5. Note that a complete disruption of cortical layers is typically found within the center of dysplasias (B, H). In more lateral aspects (sections), the layering of deeper cortical structures appears much less affected (see, for example, F). Scale bars=500 μm (A, C), 100 μm (B, D, F, H), 50 μm (E, G).
Figure 2.
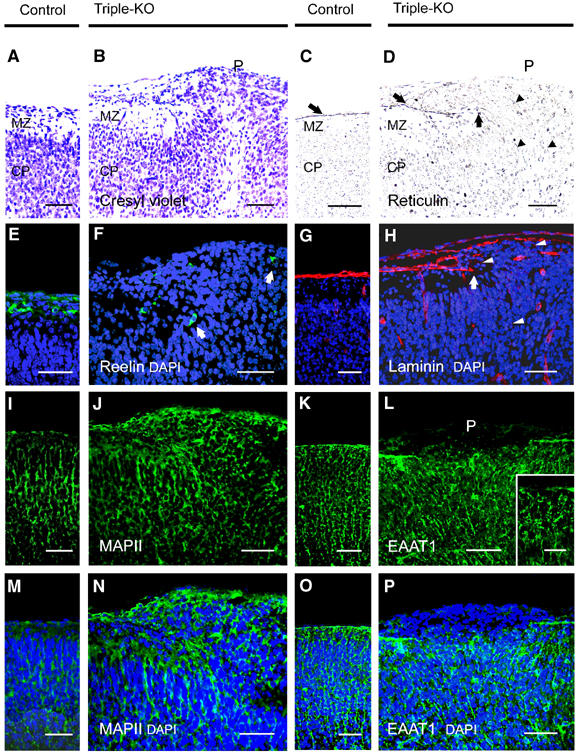
Immunohistochemical characterization of ectopias in APP−/−APLP1−/−APLP2−/− triple knockout mice at E18.5. (A, B) High-power views of cresyl violet-stained coronal sections of control (A) and (B) triple knockout cortex. (B) Shows a high power view of Figure 1B, depicting the border of a neuronal outgrowth with a well-defined MZ on the left and clustered ectopic neurons on the right (P, protrusion). (C, D) Staining of control (C) and triple knockout (D) sections for reticulin, a marker for collagen fibers in the basal lamina (BL) and pial layer revealed that the BL (arrow) was severely disrupted at the protrusion. Arrowheads indicate reticulin-stained thread-like abnormal structures. (E, F) CR cells in control (E) and triple mutant (F) sections stained for reelin (green, arrows). In controls and adjacent to ectopias, CR cells were properly located at the leptomeningeal side of the MZ. Note that, within ectopias, positioning of CR cells was severely disordered (as indicated by arrows) (G, H) Anti-laminin staining (red) of control (G) and triple mutant (H) sections. Triple mutants revealed normal laminin expression along the BL, which was, however, abrogated at the border neighboring the outgrowths (arrow). Aberrant punctate laminin staining is indicated by arrowheads. (K, L, O, P) Staining of radial glia (using an antibody against EAAT1, green) in the cerebral cortex of a control (K, O) and triple knockout. (L, P) Nuclei were stained with DAPI (see overlay depicted in P). (K, O) In control mice, radial glial fibers were found to extend through the cortical plate and MZ to the meningeal cell layer. (L) Within the protrusion, very little EAAT1 staining was detectable and fibers terminated prematurely. However, in unaffected regions of triple mutant cortex (inset in L depicts a high-power magnification), radial glial foot processes terminated normally at the pial–glial interface. (I, J, M, N) Immunostaining for MAPII (green) and counterstaining of nuclei with DAPI (blue) of control (I, M) and triple mutant (J, N) sections. Note that, within ectopias, MAPII staining is grossly disorganized. MZ, marginal zone; CP, cortical plate; scale bars=50 μm for all panels, except for the 15 μm scale bar within the inset in (L). Genotype of littermate controls: APP−/−APLP1+/−APLP2−/−.
In an effort to study the developmental ontogeny of ectopia formation, we analyzed seven triple knockout embryos at E14.5, which were obtained from 190 embryos generated by type C matings (triple frequency at E14.5: 3.7%). Triple knockout embryos were completely processed by frontal sectioning using a cryostat (8 μm) and every fifth section (approximately every 40 μm) was stained by hematoxylin/eosin (HE) to search for ectopia. Only one out of seven embryos analyzed this way displayed a protrusion as depicted in Figure 1E. We cannot exclude, however, that, due to the small size and very focal nature of the ectopias, small protrusions located in areas not covered by our stepwise staining protocol might have escaped our analysis. Overall, we noticed that protrusions became progressively bigger during later stages of embryonic development (compare stage E14.5 depicted in Figure 1E to stage E18.5 depicted in Figure 1B).
Here, in contrast to patients suffering from muscle eye brain disease MEB or Walker–Warburg syndrome (that exhibit similar cortical ectopias), no retinal abnormalities were observed in triple mutants (data not shown). Very rarely, other types of brain malformations were observed: from 31 triple mutants analyzed, one P0 animal and one E18.5 embryo showed prominent polymicrogyry and two E18.5 embryos displayed exencephaly (data not shown). Finally, and as we had previously reported for lethal APP−/−APLP2−/− and APLP1−/−APLP2−/− double mutants (Heber et al, 2000), abnormalities in the major extracranial organ systems of triple knockout animals (from E12.5 to P0) were not evident by either gross or histological evaluation at the light microscopic level.
Immunohistochemical characterization of neuronal ectopias in triple knockout mice
One possible explanation for the appearance of the neuronal ectopias in triple knockout mice would be an alteration in the basal membrane (BM), which underlies the pia mater that is in direct contact with the brain surface. Such BM defects could be due to either a failure in the proper assembly of the basal lamina, or an impairment in the attachment of glial endfeet to the basal lamina. Reticulin staining, used to stain collagen fibers in the BM and pial layer, was performed to evaluate the integrity of the basal lamina in triple knockout and control mice. As shown in Figure 2D, a gross disruption of the reticulin-labeled BM in the region of the protrusion could be observed. To determine whether the disruption of the BM results from a primary defect in basal laminin expression, known to cause neuronal ectopias (Georges-Labouesse et al, 1998), we stained brains of different ages for Engelbreth–Holm–Swarm (EHS) laminin. Laminin staining was found to be unaltered compared to wt controls along the BM in E14.5 to P0 animals (data not shown), except within the protrusions (Figure 2H, stage E18.5). Where the BM had been disrupted, or completely disappeared, laminin expression was perturbed (Figure 2H). Interestingly, prominent punctate laminin staining (Figure 2H, arrowheads) and reticulin-stained thread-like structures (Figure 2D, arrowheads) were observed within neuronal outgrowths and in the cortical plate underlying the protrusions, features which point to defects in the ECM organization. However, there is no evidence for a general primary defect of laminin expression in triple knockout embryos.
To assess a possible defect in radial glial morphology and the attachment of glial endfeet to the BM (as reported for β1-integrin-deficient mice, Graus-Porta et al, 2001), we stained frozen sections with an antibody against the excitatory amino-acid transporter 1 EAAT1 (Figure 2K, L, O and P), as a marker for radial glial cells. In unaffected cortical fields of triple knockouts, the morphology of radial glia appeared unaltered (Figure 2L, small inset), when compared to littermate controls (Figure 2K and O). In E14.5 (data not shown) and E18.5 triple mutants, radial glia spanned the cortical wall, with no obvious difference in alignment or density and with glial foot processes terminating at the pial–glial interphase (Figure 2L, inset). Neuronal ectopias, however, were characterized by grossly disrupted EAAT1 immunoreactivity and within neuronal outgrowths glial endfeet seemed to terminate prematurely at various distances from the meningeal surface (Figure 2L and P). We also examined synaptophysin immunoreactivity (present in synaptic vesicles) as a presynaptic marker for neuronal differentiation (data not shown) and MAPII as a dendritic marker. Within protrusion zones of triple mutants, we observed excessive and completely disordered MAPII (Figure 2J and N) and synaptophysin immunostaining (not shown), reminiscent of the pattern observed for neuronal ectopias described in MARCKS-deficient mice (Blackshear et al, 1997). However, in unaffected cortical fields of triple mutants (E14.5–19.5), we did not observe any abnormalities in the staining pattern of several marker proteins, such as the neuronal marker NeuN, synaptophysin, MAPII, neurofilament and GFAP (data not shown).
Cortical cell migration is not grossly abnormal in triple knockouts
To detect potential defects in the migration of cortical neurons directly, and to determine the developmental birthday of ectopic neurons, we labeled proliferating cells by injecting these mice with BrdU at E12.5, E13.5 (data not shown) and E15.5, and analyzed the extent of migration at late embryonic stages (Figure 3). In cortical fields without protrusions, no significant differences in either the total number (data not shown) or the overall percentage of BrdU-positive cells were found between triple knockouts and littermate controls, indicating similar proliferation and/or survival rates. In addition, the extent of migration, as judged by the distribution of BrdU+ cells at various depths from the brain surface (Figure 3A–D), was statistically indistinguishable between triple knockouts and littermate controls (Student's _t_-test, _P_>0.05). Likewise, the laminated structure of the hippocampus and the immature cerebellar anlage was found to be normally structured. A high percentage of cells, which were BrdU-labeled at either E12.5 (Figure 3F), or at E13.5 and E15.5 (not shown), was detected in neuronal outgrowths, suggesting that the defect might originate at around E12.5. In summary, these data indicate that a complete lack of APP family members is associated with a gross disruption of cortical layering underneath focal leptomeningeal neuronal ectopias, however, without causing major layering defects in unaffected cortical regions.
Figure 3.
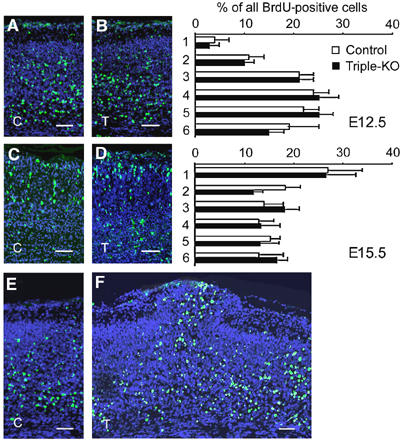
Neuronal migration in the cerebral cortex is unaltered in triple mutants. (A–F) BrdU birthdating analysis. Pregnant mice were injected with BrdU either at E12.5 or at E15.5 and the distribution of BrdU+ neurons (green) was determined at E18.5 and E19.5, respectively. (A, C) Sections of littermate controls (C) and (B, D) triple mutants (T) injected at E12.5 (A, B), or E15.5 (C, D). Panels on the right show a quantification of BrdU+ cells at various distances from the MZ. BrdU+ cells were counted in six stripes (consisting of five squares of 50 × 50 μm2) positioned underneath the MZ along the vertical axis. Note that statistical analysis (Student's _t_-test) did nor reveal any significant changes (_P_>0.05) in the distribution of BrdU+ neurons between triple knockouts (black bars) and littermate controls (open bars). The distribution of neurons labeled at E12.5 peaks within deeper layers of the cortical plate, whereas the majority of neurons labeled at E15.5 were found underneath the MZ. Values represent mean±s.d. of the percentage BrdU+ cells/stripe area relative to all positive cells from all six stripes obtained from four to five animals. (F) A high proportion of E12.5-injected BrdU+ cells are found throughout the protrusion of a triple knockout embryo sectioned at E18.5. (E) Unaffected littermate control for comparison. Genotypes of littermate controls: APP−/−APLP1−/−APLP2+/−. Scale bars=50 μm.
Reduced number of CR cells in the cortices of triple mutants
The proper architectural development of the cortical plate is known to depend on reelin secreting Cajal-Retzius (CR) cells (Hartmann et al, 1999; Tissir and Goffinet, 2003) and local extracellular matrix molecules, such as chondroitin sulfate proteoglycans (CSPGs), which have been proposed as ‘stop' signals for neuronal migration (Grumet et al, 1996). To assess whether alterations within the MZ might lead to ectopia formation, we studied the distribution and number of CR cells within neuronal outgrowths and within the MZ adjacent to ectopias by anti-reelin and anti-calretinin staining (data not shown). Within protrusions, the number of CR cells appeared reduced and positioned in a disorganized, scattered pattern (Figure 2F). CR cells located in ectopias were, however, still able to secrete reelin, as shown by anti-reelin immunohistochemistry (Figure 2F). In unaffected cortical regions of triple knockout mice, reelin-positive neurons were found to be properly positioned within the leptomeningeal side of the MZ, as seen for wild-type cortex (Figure 4E). However, quantification of CR cell number at E18.5 revealed a 37% reduction within the parietal cortices of triple mutants (Figure 4C–E). In contrast, no significant changes in CR cell number were detectable at E14.5 (data not shown), suggesting reduced survival of CR cells at late embryonic stages in the absence of APP/APLPs. As a control, we also determined the number of cortical plate cells (within a 250 × 300 μm2 area directly underlying the MZ) in HE-stained sections from E18.5 triple mutants exhibiting ectopias. No significant difference (_P_>0.05) was found between the numbers of cortical plate cells from triple knockouts (633±47, _n_=5) and littermate controls (682±45, _n_=5). The reduction in CR cell number was, however, not associated with a substantial reduction in CSPG expression, within unaffected cortical areas of triple knockout brains analyzed at E14.5 (data not shown), or at E18.5 (Figure 4G), when compared to littermate controls (Figure 4F). Within ectopias, the pattern of CSPG immunostaining was found to be grossly abnormal, appearing disorganized and scattered (Figure 4H). mDab1 immunoreactivity, previously found to be upregulated in reelin receptor-deficient mice (Trommsdorff et al, 1999), appeared unaltered in intensity and distribution in unaffected cortical fields of triple mutants at both E14.5 (data not shown), and at E18.5 (Figure 4J), suggesting that lack of binding to APP family proteins does not alter mDab1 expression/turnover. Thus, a deficiency in all three APP family members leads to reduced survival of cortical CR cells, which is associated with focal cobblestone lissencephaly in a high proportion of triple mutants.
Figure 4.
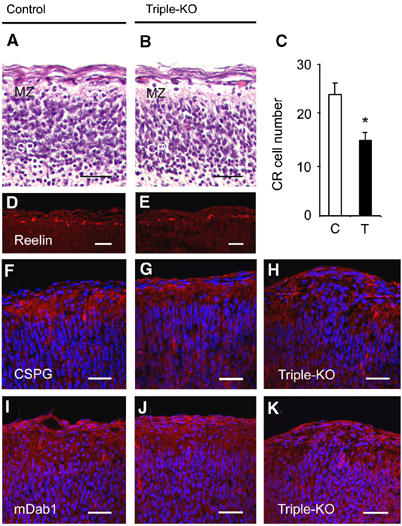
Alterations in the MZ of triple knockout mice. (A, B) Appearance of the MZ in HE-stained sections of (A) littermate control and (B) triple knockout cortices at E16.5. Note the prominent reduction in the number of nuclei in the MZ of triple mutants. (D, E) Immunohistochemistry for reelin (red) revealed a considerable reduction of CR cells in triple knockouts (E) as compared to controls (D) at E18.5. (C) The number of reelin-positive neurons within a 1200 μm wide MZ strip of the parietal cortex of E18.5 embryos was determined for both hemispheres on 8-μm-thick frozen frontal cortical sections. Values represent average cell counts±s.d. from _n_=8 stripes of four triple mutants (T, filled bar) and _n_=10 stripes of five littermate controls (C, open bar). *P<0.05, Student's _t_-test. (F–K) Frontal sections of E18.5 cortices from littermate control (F, I) and triple mutants (G, H, J, K) stained with antibodies against CSPG (F–H, red) or mDab1 (I–K, red). Cell nuclei were stained with DAPI (blue). The expression of CSPG and mDab1 within the MZ of triple knockouts (G, J) appeared unaffected in areas lacking protrusions. However, within protrusions (H, K), the normal pattern of expression of CSPG (H) and mDab1 (K) was interrupted and appeared disorganized. Scale bars=30 μm. Genotypes of littermate controls: APP−/−APLP1−/−APLP2+/− (A, D), APP−/−APLP1+/+APLP2+/+ (F), APP−/−APLP1+/+APLP2+/− (I).
Discussion
In this study, we have generated mice lacking functional genes encoding all three APP/APLP gene family members and now demonstrate that APP/APLPs play a pivotal role in establishing the cytoarchitecture of cortical structures and early postnatal survival. Within this highly redundant gene family, APLP2 seems to exert a key physiological role: both double mutants lacking APLP2 (APLP2−/−APP−/− and APLP2−/−APLP1−/− mice) exhibit postnatal lethality and inactivation of a single APLP2 allele on a viable APLP1−/−APP−/− background results in haploinsufficiency for the remaining APLP2 allele and lethality. However, no morphological abnormalities were detected in lethal double or APP−/−APLP1−/−APLP2+/− mutants (Heber et al, 2000) and cortical dysplasias are only found with high incidence upon complete inactivation of all three APP/APLP gene family members. Since we did not observe a strict correlation between cortical abnormalities and lethality (about 20% of lethal triple mutants show apparently normal brain development), we consider it unlikely that these morphological defects are the primary cause of death.
The key finding of our study is that APP family members play a crucial role in processes controlling cortical development and that lack of all three APP/APLPs results in focal neuronal ectopia within the forebrain. Two lines of evidence suggest that ectopia formation originates at around E12–14. First, protrusions were morphologically detectable at E14.5, although the incidence and size of heterotopic cell clusters was much smaller compared to later developmental time points. Second, BrdU incorporation experiments revealed a high proportion of ectopic neurons that started to migrate at E12.5. Lack of all three APP family members did not, however, interfere with a normal inside-out generation of cortical layers, suggesting that neuronal migration as such is not grossly abnormal. In view of the known interaction of APP family proteins with the adapter mDab1 (Trommsdorff et al, 1998; Homayouni et al, 1999), which also binds to the cytoplasmic domains of the reelin receptors ApoE-R2 and VLDL-R and plays a critical role in reelin signal transduction (Rice and Curran, 2001) (Magdaleno et al, 2002), it is interesting that we did not observe a reeler phenotype, as a consequence of APP/APLP deficiency. Although mDab1 immunohistochemistry appeared unaltered in unaffected cortical fields, more detailed studies (e.g. Western blot analysis using antibodies recognizing specific mDab1 phosphorylation sites) will be needed to assess whether lack of mDab1-APP/APLP interactions may result in altered mDab1 signaling and might thus contribute to ectopia formation.
Specific cell–cell recognition and adhesive interactions between neurons, radial glia and the extracellular matrix are thought to control neuronal positioning. Here, we have addressed several mechanisms that may underlie the development of ectopias: primary defects in the composition and/or structure of the basal lamina, interactions of basal lamina and radial glia, alterations in the distribution and/or function of CR cells and alterations in extracellular matrix components, such as reelin and CSPG proteins. Despite severely disorganized staining patterns for a panel of markers in protrusion zones, APP/APLP deficiency did not lead to generalized, widespread defects in BM assembly, or overall gross alterations in radial glial attachment to the BM. Thus, defects underlying ectopias appear very focal in nature, besides the overall reduced survival rate of cortical CR cells (see below).
The cortical dysplasias observed in the APP/APLP triple knockout mice closely resemble the phenotypes seen in human type II lissencephaly (for reviews, see Muntoni et al, 2002, 2004; Gleeson and Walsh, 2000). In mice, ectopia formation is genetically complex and similar cortical dysplasias have been observed for mice, with genetic ablations of the transcription factors Lmx1a (Costa et al, 2001) and Emx2 (Ligon et al, 2003), α6-integrin (Georges-Labouesse et al, 1998), β-integrin (Graus-Porta et al, 2001) and MARCKS (Stumpo et al, 1995; Blackshear et al, 1997). Intriguingly, mice lacking presenilin 1 (PS1), an essential component of the APP γ-secretase, also exhibit leptomeningeal neuronal ectopia and a loss of CR neurons, apart from other severe abnormalities (Hartmann et al, 1999). In PS1−/− mice, this phenotype has been attributed to a potential defect of meningeal fibroblasts (cells that normally express PS1), to secrete trophic factors required for CR cell survival. In this regard, it is interesting that APP family proteins are not only expressed in the developing cortex, but have also been detected in meninges at E15.5 (Lorent et al, 1995). As APP/APLPs are major substrates of PS1, one might hypothesize that the trophic functions performed by meningeal cells might also be impaired as a consequence of APP/APLP deficiency, leading to a partial loss of CR cells. A reduction in the number of CR cells may in turn alter the local ECM and thus contribute to ectopia formation. It should be noted that the neuronal ectopia phenotype is restricted to the forebrain and is incompletely penetrant in APP/APLP triple mutants, which suggests that other proteins exerting redundant functions may partially compensate for APP/APLP deficiency.
A potential explanation for the puzzling overlap in phenotype observed for PS1−/− mice and APP/APLP triple knockouts is that the generation of intracellular, γ-secretase-generated APP/APLP fragments, termed ‘AICDs' (Cao and Südhof, 2001), does not occur in mice with deficiencies of either PS1 or APP/APLP genes. Like APP, both APLPs are subject to γ-secretase cleavage (Gu et al, 2001; Sato et al, 2003; Walsh et al, 2003) and their ICDs translocate to the nucleus, where they activate Fe65-dependent transcription (Cao and Südhof, 2001, 2004). It is thus tempting to speculate that, in wild-type animals, the APP-intracellular domain and/or the APLP-ICDs could serve as effector molecule(s) involved in normal neuronal positioning. Alternatively, lack of APP family members may result in the formation of neuronal ectopia due to defects in the ability of migrating neurons to adhere to ECM components or disruption of neuron-glia de-adhesion signals that are activated when migrating neurons encounter the MZ. Consistent with a role of APP family proteins in neuronal adhesion is the reported colocalization of APP with integrins in focal adhesions of fibroblast (Sabo et al, 2001), at the surface of axons (Yamazaki et al, 1997) and within adhesion patch components of cortical neurons (Storey et al, 1996). Further, APP has been shown to biochemically interact with ECM molecules such as collagen, heparin and laminin (Kibbey et al, 1993; Beher et al, 1996; Clarris et al, 1997). Whatever the precise mechanism of dysplasia formation, whether through ICD-mediated signaling or via the C-terminus of membrane-attached APP/APLPs, Fe65 interactions seem to be pivotal in this process, as recently generated double knockout mice, deficient for Fe65 and the related protein Fe65L1, showed a strikingly similar phenotype of neuronal heterotopias within their MZs (Guenette et al, 2003). Thus, APP and Fe65 family proteins may function in a common pathway regulating cortical cytoarchitecture. Ultimately, it will be necessary to identify in vivo ligands of APP/APLP family members and to further delineate the downstream components of the signaling cascade in order to clarify the precise mechanisms that link APP/APLP deficiency to neuronal ectopia.
Materials and methods
Animals and genotyping
APP−/−, APLP1−/− and APLP2−/− single and combined mutants were generated and genotyped as described previously (Heber et al, 2000). Triple knockouts were generated from matings type A, B and C, as described in the Results section. Animal housing and timed matings were performed as described (Heber et al, 2000).
Histology
For histology, whole embryos or dissected brains were fixed at 4°C in 4% paraformaldehyde (in PBS) and either embedded in paraffin or incubated for 24 h in 25% sucrose/PBS at 4°C before being frozen on dry ice. Paraffin-embedded material was sectioned at 1–2 μm, frozen brains were sectioned at 8–10 μm. Sections were stained with hematoxylin and eosin, or with cresyl violet as described (Heber et al, 2000). Reticulin staining was performed as described by Gomorri (1937).
Immunohistochemistry
Immunohistochemistry on paraffin-embedded material was performed as described previously (Heber et al, 2000), with the following antibodies: EHS-laminin (1:25, Sigma L9393), neurofilament (SMI-31 at 1:10.000 and SMI-32 at 1:2000, Sternberger Monoclonals, Baltimore), calretinin (1:1000, Swant), reelin (G10 at 1:200), MAPII (1:400, Boehringer, Mannheim, Germany), anti-synaptophysin (1:50, Dako), glial fibrillary acidic protein (GFAP, 1:50, Dako) and anti-BrdU (1:10, Bio-Science Products AG) diluted in PBS. Incubation with first antibodies was for 2 h at room temperature. For secondary antibodies, we used rabbit-anti mouse IgG (Dako, diluted 1:50 in PBS) for 45 min at room temperature. Bound secondary antibodies were detected either by using the alkaline phosphatase-anti-alkaline phosphatase complex (APAAP; mouse monoclonal, Dako, diluted 1:40 in PBS and incubated for 45 min at room temperature), or by the avidin–biotin peroxidase complex (ABC, Vector laboratories). The alkaline phosphatase activity was visualized by using Astranenfuchsin (Aldrich, Milwaukee, WI); in case of the avidin–biotin peroxidase complex, sections were developed in AEC (3-amino-9-ethylcarbazone) and counterstained with hemalaun. Alternatively, fluorescently labeled secondary antibodies were used (see below).
Immunohistochemistry on frozen sections was performed upon fixation in 4% PFA (w/v in PBS (pH 7.4)) for 2 min and followed by 30 min incubation in blocking buffer (10% normal goat serum (NGS), 1% BSA, 0.5% Triton X-100 in PBS). First antibody incubation was carried out overnight at 4°C at the dilutions indicated below, followed by incubation (1 h) with the secondary antibodies, which were conjugated either to Alexa TM 594 (red fluorescence) or Alexa TM 488 (green fluorescence) (Molecular Probes, Eugene, OR). The following monoclonal and polyclonal antibodies were used: reelin (G10 at 1:200, kind gift from AM Goffinet, Namur, Belgium), CSPG (clone CS-56 at 1:1000, from Sigma), EAAT1 (1:50, Novocastra), mDab1 (CT38 at 1:500, a kind gift from T Curran), NeuN (1:200, Chemicon), MAPII (1:400, Boehringer Mannheim) and laminin (1:100, Sigma). Nuclei were counterstained with DAPI (Hoechst No. 33342; Sigma). Fluorescent specimens were viewed with a Zeiss (Oberkochen, Germany) confocal laser-scanning microscope (Zeiss 510 Meta).
Quantification of CR cells
Quantification of CR cell number was carried out upon anti-reelin staining of 8 μm frontal frozen sections. Immunoreactive neurons were counted within a 1200 μm wide strip of the MZ in the parietal cortex of both hemispheres using an × 20 objective equipped with a graticule (SQ515 eyepiece micrometer, Olympus, Japan; Aherne, 1975).
BrdU birthdating and morphometric analysis
Pregnant females were injected at 11:00 either on E12.5, E13.5, or on E15.5, with a single dose of BrdU (Roche) at a concentration of 50 μg/g of body weight. Females were killed between 11:00 and 12:00 on E18.5 (for E12.5 and E13.5 injections) and E19.5 (for E15.5 injections, respectively) and pups were genotyped by PCR as described (Heber et al, 2000). Brains from pups were processed for cryosectioning (E18.5 samples), or paraffin embedding (E19.5 samples), and incorporated BrdU was detected by immunohistochemistry (see above). Cells with dense staining of more than half of the nucleus were considered BrdU positive. Positive nuclei were counted using a graticule (see above) in six horizontal stripes positioned underneath the MZ along the vertical axis at various depths from the surface. Each stripe consisted of five squares (50 μm × 50 μm) provided by the graticule. Values represent average±s.d. of BrdU+ neurons per stripe area. Data from each bin were tested for significance by Student's _t_-test. Triple knockouts were always compared to unaffected littermate controls (APP−/−APLP1−/−APLP2+/+, APP−/−APLP1+/+APLP2+/−, APP−/−APLP1+/−APLP2+/−, APP−/−APLP1−/−APLP2+/− mice).
Acknowledgments
We are grateful to T Curran and AM Goffinet for their kind gifts of antibodies directed against mDab1 and reelin, and to R Homayouni and S Magdaleno for advice. We thank E Löwen, N Fürst, K Giehl, Brigitte Maruschak, Ursula Jung and Martina Sauerer for excellent technical assistance. We thank H Betz for critically reading the manuscript, crucial advice and continuous support to UM. This work was supported by grants from the EEC 5th Framework Program to JH, from the Bundesministerium für Bildung und Forschung (FK2: 01GS0469) to UM, from ‘The Volkswagen Foundation' to UM and from the ‘Deutsche Forschungsgemeinschaft' to both UM (priority program on Alzheimer's disease) and to JH (SFB 596). BA was supported by a scholarship from the Boehringer Ingelheim Foundation.
References
- Aherne W (1975) Some morphometric methods for the central nervous system. J Neurol Sci 24: 221–241 [DOI] [PubMed] [Google Scholar]
- Beher D, Hesse L, Masters CL, Multhaup G (1996) Regulation of amyloid protein precursor (APP) binding to collagen and mapping of the binding sites on APP and collagen type I. J Biol Chem 271: 1613–1620 [DOI] [PubMed] [Google Scholar]
- Blackshear PJ, Silver J, Nairn AC, Sulik KK, Squier MV, Stumpo DJ, Tuttle JS (1997) Widespread neuronal ectopia associated with secondary defects in cerebrocortical chondroitin sulfate proteoglycans and basal lamina in MARCKS-deficient mice. Exp Neurol 145: 46–61 [DOI] [PubMed] [Google Scholar]
- Cao X, Südhof TC (2001) A transcriptively active complex of APP with Fe65 and histone acetyltransferase Tip60. Science 293: 115–120 [DOI] [PubMed] [Google Scholar]
- Cao X, Südhof TC (2004) Dissection of amyloid b-precursor protein-dependent transcriptional activation. J Biol Chem 279: 2460–24611 [DOI] [PubMed] [Google Scholar]
- Clarris HJ, Cappai R, Heffernan D, Beyreuther K, Masters CL, Small DH (1997) Identification of heparin-binding domains in the amyloid precursor protein of Alzheimer's disease by deletion mutagenesis and peptide mapping. J Neurochem 68: 1164–1172 [DOI] [PubMed] [Google Scholar]
- Costa C, Harding B, Copp AJ (2001) Neuronal migration defects in the Dreher (Lmx1a) mutant mouse: role of disorders of the glial limiting membrane. Cereb Cortex 11: 498–505 [DOI] [PubMed] [Google Scholar]
- De Strooper B, Annaert W (2000) Proteolytic processing and cell biological functions of the amyloid precursor protein. J Cell Sci 113: 1857–1870 [DOI] [PubMed] [Google Scholar]
- Eggert S, Paligas K, Soba P, Evin G, Masters CL, Weidemann A, Beyreuther K (2004) The proteolytic processing of the amyloid precursor protein gene family members APLP-1 and APLP-2 involves a-, b-, g-, and e-like cleavages. J Biol Chem 279: 18146–18156 [DOI] [PubMed] [Google Scholar]
- Georges-Labouesse E, Mark M, Messaddeq N, Gansmüller A (1998) Essential role of alpha 6 integrins in cortical and retinal lamination. Curr Biol 8: 983–986 [DOI] [PubMed] [Google Scholar]
- Gleeson JG, Walsh CA (2000) Neuronal migration disorders: from genetic diseases to developmental mechanisms. Trends Neurosci 23: 352–359 [DOI] [PubMed] [Google Scholar]
- Gomorri G (1937) Silver impregnation of reticulin in paraffin sections. Am J Pathol 13: 993–1002 [PMC free article] [PubMed] [Google Scholar]
- Graus-Porta D, Blaess S, Senften M, Littlewood-Evans A, Damsky C, Huang Z, Orban P, Klein R, Schittny JC, Müller U (2001) Beta1-class integrins regulate the development of laminae and folia in the cerebral and cerebellar cortex. Neuron 31: 367–379 [DOI] [PubMed] [Google Scholar]
- Grumet M, Friedlander DR, Sakurai T (1996) Functions of brain chondroitin sulfate proteoglycans during developments: interactions with adhesion molecules. Perspect Dev Neurobiol 3: 319–330 [PubMed] [Google Scholar]
- Guenette SY, Chang Y, Hiesberger T, Richardson JA, Herz J (2003) Mice deficient for the Fe65 and Fe65L1 proteins have neurological defects, Society for Neuroscience Abstract No. 336.10 [Google Scholar]
- Gu Y, Misonou H, Sato T, Dohmae N, Takio K, Ihara Y (2001) Distinct intramembrane cleavage of the beta-amyloid precursor protein family resembling gamma-secretase-like cleavage of Notch. J Biol Chem 276: 35235–35238 [DOI] [PubMed] [Google Scholar]
- Hartmann D, De Strooper B, Saftig P (1999) Presenilin-1 deficiency leads to loss of Cajal-Retzius neurons and cortical dysplasia similar to human type 2 lissencephaly. Curr Biol 9: 719–727 [DOI] [PubMed] [Google Scholar]
- Heber S, Herms J, Gajic V, Hainfellner J, Aguzzi A, Rulicke T, Kretzschmar H, von Koch C, Sisodia S, Tremml P, Lipp HP, Wolfer DP, Müller U (2000) Mice with combined gene knock-outs reveal essential and partially redundant functions of amyloid precursor protein family members. J Neurosci 20: 7951–7963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Homayouni R, Rice DS, Sheldon M, Curran T (1999) Disabled-1 binds to the cytoplasmic domain of amyloid precursor-like protein 1. J Neurosci 19: 7507–7515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamal A, Almenar-Queralt A, LeBlanc JF, Roberts EA, Goldstein LSB (2001) Kinesin-mediated axonal transport of a membrane compartment containing β-secretase and presenilin-1 requires APP. Nature 414: 643–648 [DOI] [PubMed] [Google Scholar]
- Kibbey MC, Jucker M, Weeks BS, Neve RL, Van Nostrand WE, Kleinman HK (1993) beta-Amyloid precursor protein binds to the neurite-promoting IKVAV site of laminin. Proc Natl Acad Sci USA 90: 10150–10153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koo EH (2002) The beta-amyloid precursor protein (APP) and Alzheimer's disease: does the tail wag the dog? Traffic 3: 763–770 [DOI] [PubMed] [Google Scholar]
- Li Q, Südhof TC (2003) Cleavage of APP and APLP's by BACE 1. J Biol Chem 279: 10542–10550 [DOI] [PubMed] [Google Scholar]
- Li ZW, Stark G, Götz J, Rülicke T, Gschwind M, Huber G, Müller U, Weissmann C (1996) Generation of mice with a 200-kb amyloid precursor protein gene deletion by Cre recombinase-mediated site-specific recombination in embryonic stem cells. Proc Natl Acad Sci USA 93: 6158–6162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ligon KL, Echelard Y, Assimacopoulos S, Danielian PS, Kaing S, Grove EA, McMahon AP, Rowitch DH (2003) Loss of Emx2 function leads to ectopic expression of Wnt1 in the developing telencephalon and cortical dyplasia. Development 130: 2275–2287 [DOI] [PubMed] [Google Scholar]
- Lorent K, Overbergh L, Moechars D, De Strooper B, Van Leuven F, Van den Berghe H (1995) Expression in mouse embryos and in adult mouse brain of three members of the amyloid precursor protein family, of the alpha-2-macroglobulin receptor/low density lipoprotein receptor-related protein and of its ligands apolipoprotein E, lipoprotein lipase, alpha-2-macroglobulin and the 40,000 molecular weight receptor-associated protein. Neuroscience 65: 1009–1025 [DOI] [PubMed] [Google Scholar]
- Magara F, Müller U, Li ZW, Lipp HP, Weissmann C, Stagljar M, Wolfer DP (1999) Genetic background changes the pattern of forebrain commissure defects in transgenic mice underexpressing the beta-amyloid-precursor protein. Proc Natl Acad Sci USA 96: 4656–4661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magdaleno S, Keshvara L, Curran T (2002) Rescue of ataxia and preplate splitting by ectopic expression of Reelin in reeler mice. Neuron 33: 573–586 [DOI] [PubMed] [Google Scholar]
- Müller U, Cristina N, Li ZW, Wolfer DP, Lipp HP, Rülicke T, Brandner S, Aguzzi A, Weissmann C (1994) Behavioral and anatomical deficits in mice homozygous for a modified beta-amyloid precursor protein gene. Cell 79: 755–765 [DOI] [PubMed] [Google Scholar]
- Muntoni F, Brockington M, Blake DJ, Torelli S, Brown SC (2002) Defective glycosylation in muscular dystrophy. Lancet 360: 1419–1421 [DOI] [PubMed] [Google Scholar]
- Muntoni F, Brockington M, Torelli S, Brown SC (2004) Defective glycosylation in congenital muscular dystrophies. Curr Opin Neurol 17: 205–209 [DOI] [PubMed] [Google Scholar]
- Perez RG, Zheng H, Van der Ploeg LH, Koo EH (1997) The beta-amyloid precursor protein of Alzheimer's disease enhances neuron viability and modulates neuronal polarity. J Neurosci 17: 9407–9414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice DS, Curran T (2001) Role of the reelin signaling pathway in central nervous system development. Annu Rev Neurosci 24: 1005–1039 [DOI] [PubMed] [Google Scholar]
- Sabo SL, Ikin AF, Buxbaum JD, Greengard P (2001) The Alzheimer amyloid precursor protein (APP) and FE65, an APP-binding protein, regulate cell movement. J Cell Biol 153: 1403–1414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato T, Dohmae N, Qi Y, Kakuda N, Misonou H, Mitsumori R, Maruyama H, Koo EH, Haass C, Takio K, Morishima-Kawashima M, Ishiura S, Ihara Y (2003) Potential link between amyloid beta-protein 42 and C-terminal fragment gamma 49–99 of beta-amyloid precursor protein. J Biol Chem 278: 24294–24301 [DOI] [PubMed] [Google Scholar]
- Scheinfeld MH, Ghersi E, Laky K, Fowlkes BJ, D'Adamio L (2002) Processing of beta-amyloid precursor-like protein-1 and -2 by gamma-secretase regulates transcription. J Biol Chem 277: 44195–44201 [DOI] [PubMed] [Google Scholar]
- Slunt HH, Thinakaran G, Von KC, Lo AC, Tanzi RE, Sisodia SS (1994) Expression of a ubiquitous, cross-reactive homologue of the mouse beta-amyloid precursor protein (APP). J Biol Chem 269: 2637–2644 [PubMed] [Google Scholar]
- Steinbach JP, Müller U, Leist M, Li ZW, Nicotera P, Aguzzi A (1998) Hypersensitivity to seizures in beta-amyloid precursor protein deficient mice. Cell Death Differ 5: 858–866 [DOI] [PubMed] [Google Scholar]
- Storey E, Beyreuther K, Masters CL (1996) Alzheimer's disease amyloid precursor protein on the surface of cortical neurons in primary culture co-localizes with adhesion patch components. Brain Res 735: 217–231 [DOI] [PubMed] [Google Scholar]
- Stumpo DJ, Bock CB, Tuttle JS, Blackshear PJ (1995) MARCKS deficiency in mice leads to abnormal brain development and perinatal death. Proc Natl Acad Sci USA 92: 944–948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thinakaran G, Slunt HH, Spitzer L, Lee MK, Sisodia SS (1995) Tissue distribution and developmental expression of the amyloid precursor protein homolog, APLP1. Soc Neurosci Abstr 21: 205 [Google Scholar]
- Tissir F, Goffinet AM (2003) Reelin and brain development. Nat Rev Neurosci 4: 496–505 [DOI] [PubMed] [Google Scholar]
- Tremml P, Lipp HP, Müller U, Ricceri L, Wolfer DP (1998) Neurobehavioral development, adult openfield exploration and swimming navigation learning in mice with a modified beta-amyloid precursor protein gene. Behav Brain Res 95: 65–76 [DOI] [PubMed] [Google Scholar]
- Trommsdorff M, Borg J-P, Margolis B, Herz J (1998) Interaction of cytosolic adaptor proteins with neuronal apolipoprotein E receptors and the amyloid precursor protien. J Biol Chem 273: 33556–33560 [DOI] [PubMed] [Google Scholar]
- Trommsdorff M, Gotthardt M, Hiesberger T, Shelton J, Stockinger W, Nimpf J, Hammer RE, Richardson JA, Herz J (1999) Reeler/disabled-like disruption of neuronal migration in knockout mice lacking the VLDL receptor and ApoE receptor 2. Cell 97: 689–701 [DOI] [PubMed] [Google Scholar]
- von Koch CS, Zheng H, Chen H, Trumbauer M, Thinakaran G, van der Ploeg LH, Price DL, Sisodia SS (1997) Generation of APLP2 KO mice and early postnatal lethality in APLP2/APP double KO mice. Neurobiol Aging 18: 661–669 [DOI] [PubMed] [Google Scholar]
- Walsh DM, Fadeeva JV, LaVoie MJ, Paliga K, Eggert S, Kimberly WT, Wasco W, Selkoe DJ (2003) gamma-Secretase cleavage and binding to FE65 regulate the nuclear translocation of the intracellular C-terminal domain (ICD) of the APP family of proteins. Biochemistry 42: 6664–6673 [DOI] [PubMed] [Google Scholar]
- White AR, Multhaup G, Maher F, Bellingham S, Camakaris J, Zheng H, Bush AI, Beyreuther K, Masters CL, Cappai R (1999) The Alzheimer's disease amyloid precursor protein modulates copper-induced toxicity and oxidative stress in primary neuronal cultures. J Neurosci 19: 9170–9179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- White AR, Zheng H, Galatis D, Maher F, Hesse L, Multhaup G, Beyreuther K, Masters CL, Cappai R (1998) Survival of cultured neurons from amyloid precursor protein knock-out mice against Alzheimer's amyloid-beta toxicity and oxidative stress. J Neurosci 18: 6207–6217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamazaki T, Koo EH, Selkoe DJ (1997) Cell surface amyloid beta-protein precursor colocalizes with beta 1 integrins at substrate contact sites in neural cells. J Neurosci 17: 1004–1010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng H, Jiang M, Trumbauer ME, Sirinathsinghji DJ, Hopkins R, Smith DW, Heavens RP, Dawson GR, Boyce S, Conner MW, Stevens KA, Slunt HH, Sisodia SS, Chen HY, Van der Ploeg LH (1995) beta-Amyloid precursor protein-deficient mice show reactive gliosis and decreased locomotor activity. Cell 81: 525–531 [DOI] [PubMed] [Google Scholar]