Effects of PCSK9 Inhibition With Alirocumab on Lipoprotein Metabolism in Healthy Humans (original) (raw)
Supplemental Digital Content is available in the text.
Keywords: apoliprotein, LDL receptor, lipoproteins, low-density lipoprotein, Lp(a), PCSK9
Abstract
Background:
Alirocumab, a monoclonal antibody to proprotein convertase subtilisin/kexin type 9 (PCSK9), lowers plasma low-density lipoprotein (LDL) cholesterol and apolipoprotein B100 (apoB). Although studies in mice and cells have identified increased hepatic LDL receptors as the basis for LDL lowering by PCSK9 inhibitors, there have been no human studies characterizing the effects of PCSK9 inhibitors on lipoprotein metabolism. In particular, it is not known whether inhibition of PCSK9 has any effects on very low-density lipoprotein or intermediate-density lipoprotein (IDL) metabolism. Inhibition of PCSK9 also results in reductions of plasma lipoprotein (a) levels. The regulation of plasma Lp(a) levels, including the role of LDL receptors in the clearance of Lp(a), is poorly defined, and no mechanistic studies of the Lp(a) lowering by alirocumab in humans have been published to date.
Methods:
Eighteen (10 F, 8 mol/L) participants completed a placebo-controlled, 2-period study. They received 2 doses of placebo, 2 weeks apart, followed by 5 doses of 150 mg of alirocumab, 2 weeks apart. At the end of each period, fractional clearance rates (FCRs) and production rates (PRs) of apoB and apo(a) were determined. In 10 participants, postprandial triglycerides and apoB48 levels were measured.
Results:
Alirocumab reduced ultracentrifugally isolated LDL-C by 55.1%, LDL-apoB by 56.3%, and plasma Lp(a) by 18.7%. The fall in LDL-apoB was caused by an 80.4% increase in LDL-apoB FCR and a 23.9% reduction in LDL-apoB PR. The latter was due to a 46.1% increase in IDL-apoB FCR coupled with a 27.2% decrease in conversion of IDL to LDL. The FCR of apo(a) tended to increase (24.6%) without any change in apo(a) PR. Alirocumab had no effects on FCRs or PRs of very low-density lipoproteins-apoB and very low-density lipoproteins triglycerides or on postprandial plasma triglycerides or apoB48 concentrations.
Conclusions:
Alirocumab decreased LDL-C and LDL-apoB by increasing IDL- and LDL-apoB FCRs and decreasing LDL-apoB PR. These results are consistent with increases in LDL receptors available to clear IDL and LDL from blood during PCSK9 inhibition. The increase in apo(a) FCR during alirocumab treatment suggests that increased LDL receptors may also play a role in the reduction of plasma Lp(a).
Clinical Trial Registration:
URL: http://www.clinicaltrials.gov. Unique identifier: NCT01959971.
Editorial, see p 363
Proprotein convertase subtilisin/kexin type 9 (PCSK9) is a secreted member of the subtilase family of serine proteases. PCSK9 binds to hepatic low-density lipoprotein receptors (LDLRs) and targets them, together with bound LDLs for degradation rather than recycling. As a result, PCSK9 reduces the number of functional LDLRs on cell surfaces, thereby increasing levels of LDL-cholesterol (LDL-C) in the blood.1,2 Alirocumab is a fully human monoclonal antibody that specifically binds to PCSK9 and inhibits PCSK9 binding to, and PCSK9-mediated degradation of, LDLRs. As a result, alirocumab treatment substantially decreases blood levels of LDL-C and apolipoprotein B (apoB).3–5
Based on the mechanism of PCSK9 action, it has been postulated that alirocumab lowers blood levels of LDL-C and apoB by increasing LDLRs in the liver, leading to increased efficiency of the removal of LDL particles from the circulation. However, apoB enters the circulation from the liver on very low-density lipoproteins (VLDLs), which are converted first to intermediate-density lipoproteins (IDLs) and, subsequently, to LDL. During this process, both VLDL and IDL particles may leave the bloodstream by interacting with LDLRs, particularly in the liver.6–8 Thus, drugs that reduce blood levels of LDL-C can mediate their effects through pathways other than the classical LDLR-mediated uptake of LDL, such as by reducing the initial secretion of VLDL or the conversion of VLDL and IDL to LDL. The purpose of this study was to evaluate the effects of alirocumab on the flux of apoB from its entry into the circulation as VLDL, through IDL and LDL. In addition, we determined the effects of PCSK9 inhibition on the metabolism of VLDL triglyceride (TG) and levels of TG-rich lipoproteins during the postprandial (PP) period.
An unexpected finding in early clinical trials was a reduction in lipoprotein (a) levels with inhibition of PCSK9 activity.9,10 The role of the LDLR in the removal of Lp(a) from the circulation remains controversial. For example, statins, which increase LDLRs significantly, do not reduce or may even increase plasma Lp(a) levels.11 We also, therefore, conducted an exploratory study of the effects of inhibition of PCSK9 on the metabolism of Lp(a).
Methods
Study Subjects
We enrolled 20 healthy volunteers, 18 to 65 years of age, with LDL-C levels >90 mg/dL. Subjects were recruited from internal clinical trial databases, advertisements, and postings at the Columbia University Medical Center (CUMC). Participants were not receiving lipid-lowering medications. All subjects of child-bearing capacity agreed to use birth control for the duration of the study and had a negative serum pregnancy test when applicable. This study was conducted in accordance with the principles of Good Clinical Practice and was approved by the CUMC Institutional Review Board. All study subjects provided written informed consent.
Study Design
This was a Phase 1, placebo-controlled, single-blind, single-sequence, 2-period study (http://www.clinicaltrials.gov. Unique identifier: NCT01959971; for the study design, see online-only Data Supplement Figure I). All subjects were instructed on adherence to the American Heart Association Heart Healthy Diet guidelines during the study and were regularly checked for dietary compliance. Participants received 2 subcutaneous injections of placebo every 2 weeks followed immediately by 5 subcutaneous injections of alirocumab, 150 mg every 2 weeks. Studies of PP lipemia were conducted 2 weeks after the first placebo injection and again immediately after 5 doses of alirocumab. Lipoprotein kinetic studies were performed 2 weeks after completion of placebo treatment and 2 weeks after the last alirocumab dose. For each participant, a comparison of study outcomes across the 2 treatment periods (placebo vs alirocumab) was performed. Thus, each person served as his or her own control.
Safety
Alirocumab is a US Food and Drug Administration–approved lipid-altering treatment. The safety of alirocumab and its effects on blood lipids have been previously reported for men and women.5,10 Safety data were closely monitored with clinical and laboratory assessments throughout the study (see online-only Data Supplement for further details).
Biochemical Assays
Blood was collected at the end of each treatment period after a 12-hour fast. Plasma total cholesterol, TG, high-density lipoprotein cholesterol (HDL-C), apoC-III, and apoE were measured by Human Elisa Kits (ab154131 [apoC3]; ab108813 [apoE], Abcam, Cambridge, MA). Plasma LDL-C levels were estimated using the Friedewald formula. Cholesterol and TG were also measured enzymatically in VLDL, IDL, LDL, and HDL isolated by ultracentrifugation.12 ApoB in plasma and in VLDL, IDL, and LDL was measured using an apoB enzyme-linked immunosorbent assay (ELISA) kit (AlerCheck, Inc.). Plasma Lp(a) levels were measured using a monoclonal antibody-based ELISA method validated to accurately measure Lp(a) independently of the number of repeats in Kringle IV type 2.13 Apo(a) isoforms were assayed with methodology previously described,14 in which the isoform size visualized on agarose gel electrophoresis is directly proportional to the number of Kringle IV type 2 repeats. In some subjects, only 1 apo(a) isoform is expressed. In most individuals, however, apo(a) is expressed by both alleles, with 1 isoform more prevalent than the other. Particle sizes and number were determined by ion mobility analysis.15 Lathosterol, campesterol, and β-sitosterol were measured by gas chromatography as previously described.16
Serum levels of anti-alirocumab antibodies (ADA) and free PCSK9 were assayed by Regeneron Pharmaceuticals, Inc., Tarrytown, NY (see online-only Data Supplement). Serum samples were assayed for ADAs using a validated immunoassay developed by Regeneron Pharmaceuticals, Inc. In brief, this titer-based, bridging immunoassay used a Meso Scale Discovery electrochemiluminescence instrument (Rockville, MD). The assay comprised 3 different stages: an initial screening assay to identify potentially ADA-positive samples, a confirmation assay based on competition with exogenously added alirocumab, and a determination of the titer of ADA for confirmed positive samples.
Free PCSK9 levels in serum were determined using specific validated enzyme-linked immunosorbent assays developed by Regeneron Pharmaceuticals, Inc. The lower limit of detection of free PCSK9 was 31.2 ng/mL.
Hepatic Lipase (HL) and Lipoprotein Lipase (LpL) Activity Measurements
Post-heparin plasma was collected 15 minutes after an i.v. injection of heparin (60 U/kg body weight [BW]) on 2 occasions: after subjects had received 2 doses of placebo and after they had received 5 doses of alirocumab (see online-only Data Supplement).
Stable Isotope Kinetic Studies
Participants were admitted to the inpatient unit of the Irving Institute for Clinical and Translational Research (IICTR) at CUMC for 2.5 days. On day 1, fasting bloods were obtained, and meals for the day were provided. At 1 am on the morning of day 2, constant feedings of an isocaloric, 18% fat diet began and continued every 2 hours for 32 hours. At 9 am, boluses of 2H3-L-leucine (10 μmol/kg BW), Ring-13C6-L-phenylalanine (29.4 μmol/kg BW), and 2H5-glycerol (100 μmol/kg BW) were administered over a 10-minute period, followed by a constant infusion of 2H3-L-leucine (10 μmol/kg BW/hr) over 15 hours. Blood samples were collected at 0 (prebolus), 20, and 40 minutes and at 1, 2, 4, 6, 8, 10, 12, 14, 15, 15.5, 16, 18, 21, 24, and 48 hours and processed to isolate VLDL, IDL, LDL, and HDL.12,17
Determination of Stable Isotopic Enrichment of apoB and TG
The lipoprotein fractions isolated during each turnover study were used to determine stable isotopic enrichments of apoB in VLDL, IDL, and LDL with 2H3-L-leucine (10 μmol/kg BW) and Ring-13C6-L-phenylalanine (29.4 μmol/kg BW). Additionally, kinetic analysis of TG in VLDL was performed with2H5-glycerol (100 μmol/kg BW). ApoB was isolated from VLDL, IDL, and LDL by SDS gel electrophoresis. Enrichment of leucine and phenyalanine in apoB lipoproteins and plasma-free leucine and phenylalanine enrichment was measured by gas chromatography–mass spectrometry using an Agilent 6890 gas chromatograph and a 5973 mass spectrometer using negative chemical ionization.12 The enrichment of TG-glycerol was determined by gas chromatography–mass spectrometry using positive chemical ionization after derivatization of glycerol to triacetin.12
Mathematical Modeling of the apoB and TG Enrichment Data
A multicompartmental model was used to estimate apoB and TG rate constants and fluxes from the enrichment data.18 ApoB and TG were required to have the same pool structure for VLDL and the same rate constants for the VLDL pools but with different mass distributions. The minimum number of pools needed to fit the 9 sets of data (2 tracers, leucine and phenylalanine, in VLDL-, IDL-, and LDL-apoB and plasma amino acids, and 1 tracer, glycerol, in VLDL-TG simultaneously was chosen for the final model).17 The data were fitted by least squares using a computer program, Poolfit,19 which solves the differential equations in closed form and computes the fits and parameter sensitivities as sums of exponentials. The fits yielded FCRs of apoB in VLDL, IDL, and LDL and of TG in VLDL. The model also estimated rates of conversion of apoB between VLDL and IDL, and IDL and LDL. PRs were calculated by multiplying FCRs by the appropriate lipoprotein pool sizes of apoB, which were estimated as each lipoprotein’s concentration of apoB in mg/dL multiplied by plasma volume (taken as 45 mL/kg BW).
Determination of Stable Isotope Enrichment of apo(a)
Apo(a) enrichment with 2H3-L-leucine was measured as described by Zhou et al.20 In brief, equal volumes (100 µL) of ultracentrifuged fractions of LDL and HDL, which together hold 95% of plasma Lp(a), were combined, desalted, reduced with DTT, alkylated with iodoacetamide, and digested using trypsin. A multiple reaction monitoring method was used to monitor the following precursor product ion transitions to a peptide specific to apo(a) (LFLEPTQADIALLK): 786.7>1069.7 (M0) and 788.2>1069.7 (M3); 2 µL of the digested samples were analyzed using a nanoAcquity ultraperformance liquid chromatography system coupled with an ionKey source integrated to a Xevo TQS triple quad tandem mass spectrometer (Waters, Milford, MA). The separation was achieved using an iKey Peptide BEH C18 separation device (130 Å, 1.7 µm, 150 µm X 100 mm) maintained at 60°C. The gradient was 90% A (0.1% formic acid in water)/10% B (0.1% formic acid in acetonitrile) ramped linearly to 10% A at 6 minutes, held for 3 minutes, and then re-equilibrated to initial conditions (total run time: 12 minutes; flow rate: 3 µL/min). The multiple reaction monitoring transitions were monitored with a collision energy of 24 eV.
Apo(a) Modeling
The apo(a) enrichment data were modeled as previously described for apoC-III.17 Briefly, we estimated apo(a) FCR by fitting the leucine enrichment in the apo(a) protein with a single-pool model, with the precursor enrichment set at the same level as found with the apoB model described earlier. Apo(a) PR in nM/kg/d was calculated as the product of apo(a) FCR and the molar pool size per kg BW of apo(a), which is the apo(a) concentration (nM) multiplied by the plasma volume (taken as 45 mL/kg BW).
Post-Prandial Lipemia Studies
PP TG and apoB48 levels were measured after consumption of a standard high-fat liquid meal.21 Details of the protocol are found in the online-only Data Supplement.
Statistical Analysis
The corresponding authors had full access to all the data and take responsibility for its integrity and the data analysis. The primary end point was the change in LDL-apoB FCR from the end of placebo treatment to the end of alirocumab treatment. With 18 subjects completing the protocol, the study had 80% power to see a 35% change in LDL-apoB FCR, assuming an FCR of 0.42 pools/day at the end of the placebo treatment period using a 2-sided paired t test with an alpha of 0.05. Prespecified secondary analyses included FCRs of VLDL- and IDL-apoB, PRs of VLDL-, IDL-, and LDL-apoB, conversion of VLDL to LDL and IDL to LDL, FCR and PR of VLDL-TG, plasma concentrations of Lp(a), FCR and PR of apo(a), lipoprotein size and number, and postheparin plasma LpL and HL activities at the end of placebo and alirocumab treatment periods. Exploratory analyses included levels of plant sterols, apoCIII, and apoE at the end of each treatment period. All secondary and exploratory analyses are presented with nominal _P_-values uncorrected for multiple comparisons. Exploratory outcomes analyses were performed at CUMC and were not included in the original Sanofi study statistical analysis plan.
Results
Eight males (mean age 39.9±9.9 years) and 10 females (mean age 47.0±12.2 years) completed the study. Two subjects decided to leave the study for personal reasons. One dropped out during the placebo period and 1 at the end of the placebo period; neither received alirocumab (online-only Data Supplement Figure II). No serious adverse events occurred during the study (see online-only Data Supplement). Baseline characteristics are shown Table 1. Participants had normal plasma glucose levels and normal hepatic and renal function. Five doses of alirocumab treatment resulted in significant reductions of fasting plasma levels of total cholesterol (34.4%), calculated LDL-C (57.9%), and apoB (46.6%), with no significant effects on plasma HDL-C and TG levels (Table 2).
Table 1.
Baseline Characteristics: 8 Healthy Men and 10 Healthy Women Completed the Study
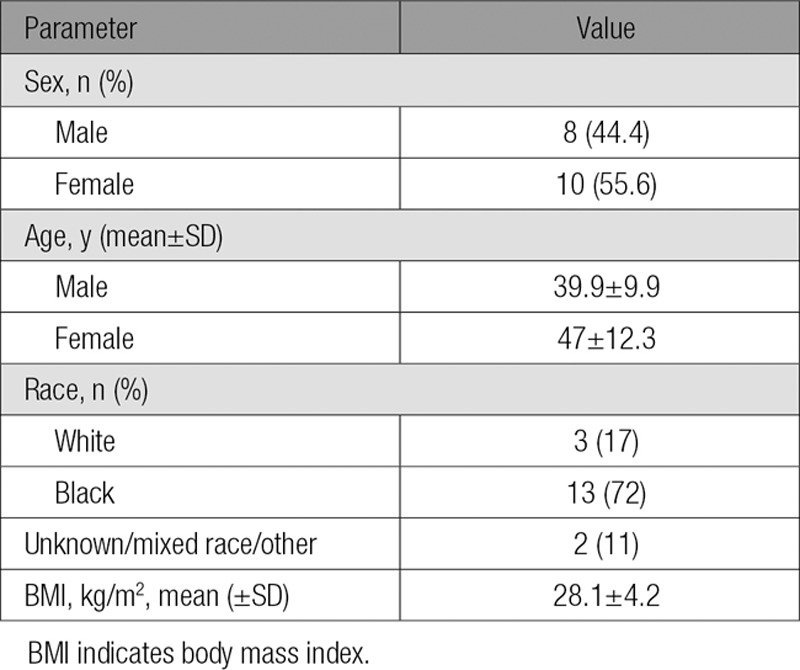
Table 2.
Effects of 10 Weeks of Alirocumab Treatment* on Plasma Lipids and Lipoproteins
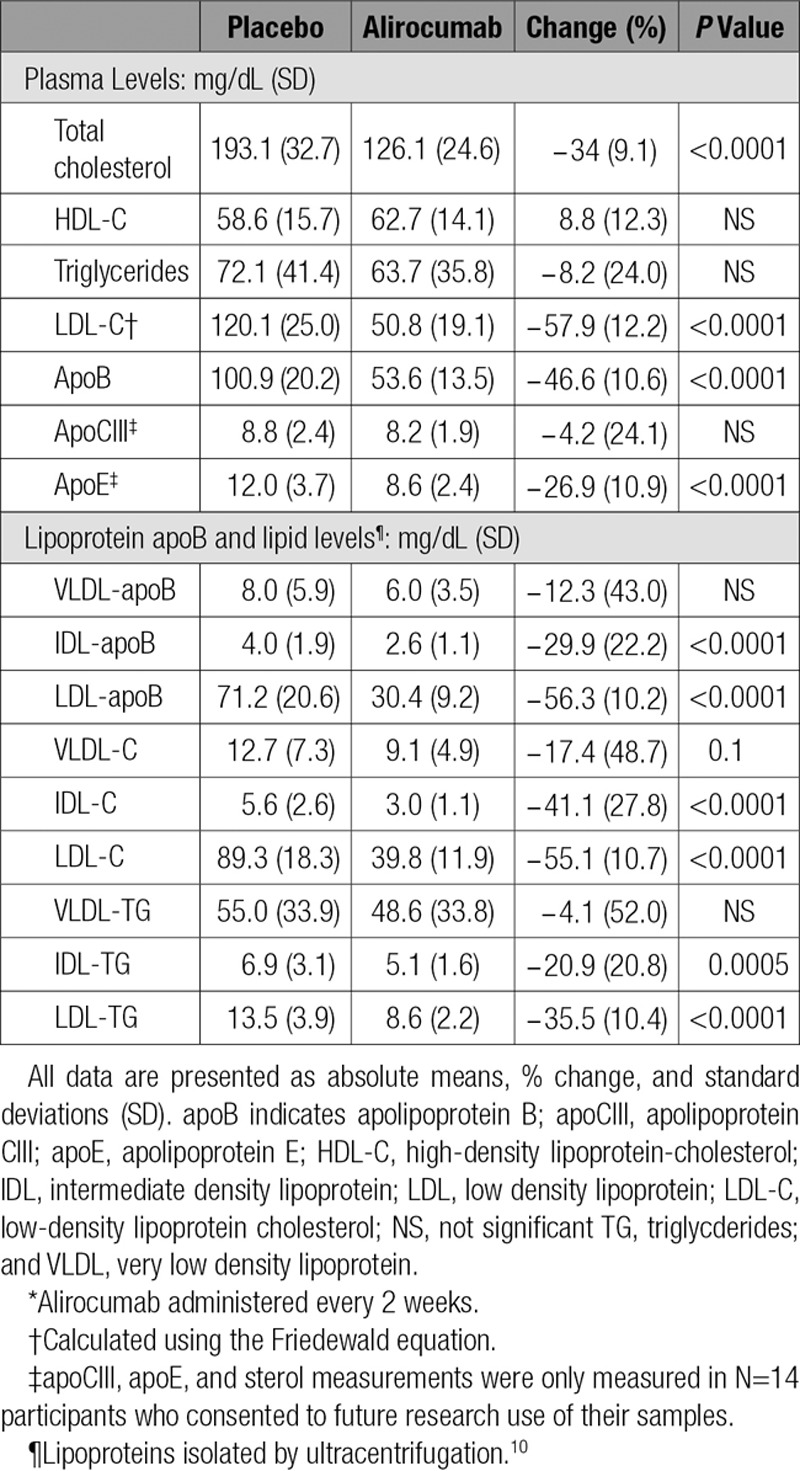
Alirocumab reduced mean apoB levels in ultracentrifugally isolated IDL (29.9% _P_≤0.0001) and LDL (56.3% _P_≤0.0001); no significant effect was found on the concentration of VLDL-apoB (Table 2). In addition, alirocumab treatment resulted in decreased mean levels of cholesterol and TG concentrations in IDL (41.1%, _P_≤0.0001 and 20.9%, _P_=0.0005, respectively) and LDL (55.1%, _P_≤0.0001 and 35.5%, _P_≤0.0001, respectively) without affecting lipid levels in VLDL (Table 2). The reductions in IDL- and LDL-apoB levels measured by ELISA were in accord with similar reductions in IDL and LDL particle numbers determined by ion mobility (online-only Data Supplement Table I). The reduction in LDL particle number was associated with a shift toward smaller particles after alirocumab treatment (online-only Data Supplement Table II).
Reductions in IDL- and LDL-apoB concentrations were caused by increases in the mean FCRs of these lipoproteins (46.1%, P<0.001 and 80.4%, _P_≤0.0001, respectively) (Figure 1). No significant change occurred in the mean VLDL-apoB FCR during alirocumab administration. In addition to the marked increase in LDL-apoB FCR, mean LDL-apoB PR was reduced by 23.9% (P<0.0001). The decrease in the PR of LDL-apoB was caused by the significant increase in the FCR of IDL-apoB noted earlier coupled with a mean 27.2% decrease in the conversion of IDL-apoB to LDL-apoB (P<0.005). No significant changes were found in the mean VLDL- or IDL-apoB PRs. Consistent with alirocumab lack of effects on VLDL-TG levels or VLDL-apoB metabolism, no changes occurred in mean FCR or PR of VLDL-TG (Figure 2).
Figure 1.
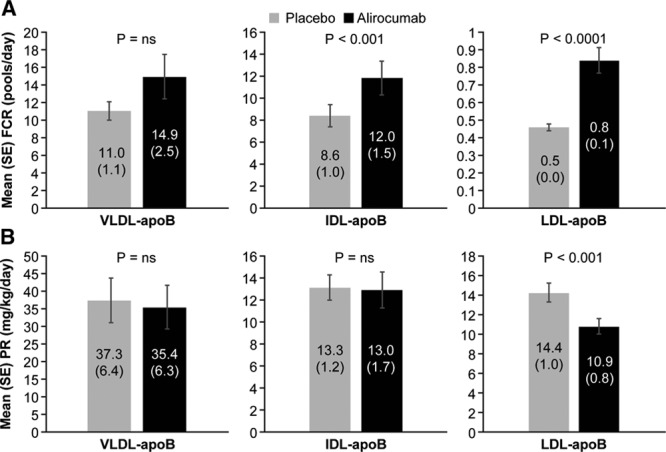
Effects of alirocumab on VLDL-, IDL-, and LDL–apoB fractional clearance rates (A) and production rates (B). FCRs of VLDL, IDL, and LDL-apoB (mean±SE) were determined by stable isotopic enrichment of apoB in each lipoproteins. PRs (mean±SE) were calculated using the FCRs and plasma pool size of apoB in each lipoprotein. Alirocumab treatment significantly increased the FCRs of IDL and LDL apoB and reduced the PR of LDL apoB compared with placebo. No significant effects of alirocumab on VLDL apoB metabolism were found. apoB indicates apolipoprotein B; FCR, fractional clearance rate; IDL, intermediate-density lipoprotein; LDL, low-density lipoprotein; ns, not significant; PR, production rate; SE, standard error; and VLDL, very low-density lipoprotein.
Figure 2.
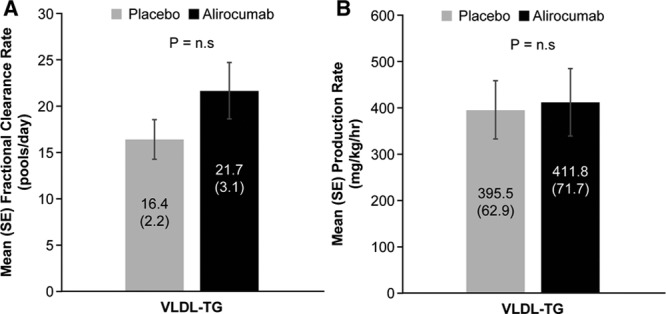
Effect of alirocumab on VLDL-TG fractional clearance rates (A) and production rates (B). FCR of VLDL-TG (mean±SE) was determined by stable isotopic enrichment of glycerol in each sample of isolated VLDL over 48 hours. VLDL-TG PR (mean±SE) was calculated using the FCR and the plasma pool size of TG in VLDL. Alirocumab treatment did not affect either the FCR or the PR of VLDL-TG compared with placebo. FCR indicates fractional clearance rate; PR, production rate; TG, triglycerides; and VLDL, very low-density lipoprotein.
Significant correlations were found between baseline PCSK9 levels and changes in levels of both LDL-C (_R_2 =0.42; _P_=0.003) and LDL-apoB (_R_2=0.22; _P_=0.05) (Figure 3).
Figure 3.
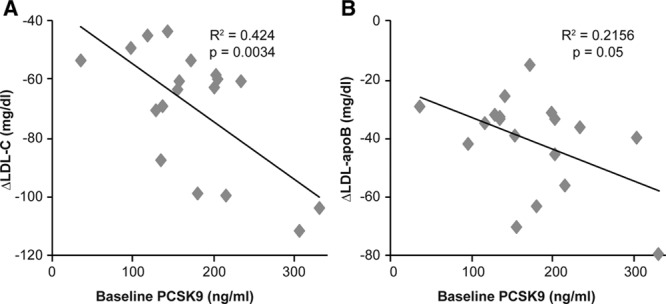
Correlation between baseline serum-free PCSK9 levels and changes in (A) LDL-C and (B) LDL-apoB levels. Baseline levels of serum-free PCSK9 were examined for correlation with alirocumab-induced changes in LDL-C and LDL-apoB in all 18 subjects. Changes in both LDL-C and LDL-apoB were related to PCSK9 levels at baseline. apoB indicates apolipoprotein B; LDL, low-density lipoprotein; LDL-C, low-density lipoprotein cholesterol; and PCSK9, proprotein convertase subtilisin/kexin type 9.
Apo CIII levels were not different between the 2 study periods (8.8±2.4 vs 8.2±1.9 mg/dL), but a 26.9±10.9% decrease (_P_=0.0034) in plasma apo E levels (12.0±3.7 vs 8.6±2.4 mg/dL) did occur at the end of alirocumab administration (Table 2). Plasma levels of sitosterol tended to increase (113.0±37.9 vs 128.4±31.4 µmol X 102/mmol cholesterol; _P_=0.07), whereas levels of lathosterol (81.0±33.5 vs 90.4±39.6 µmol X 102/mmol cholesterol) and campesterol (167.3±54.9 vs 182.8±53.9 µmol X 102/mmol cholesterol) were unaffected by alirocumab treatments.
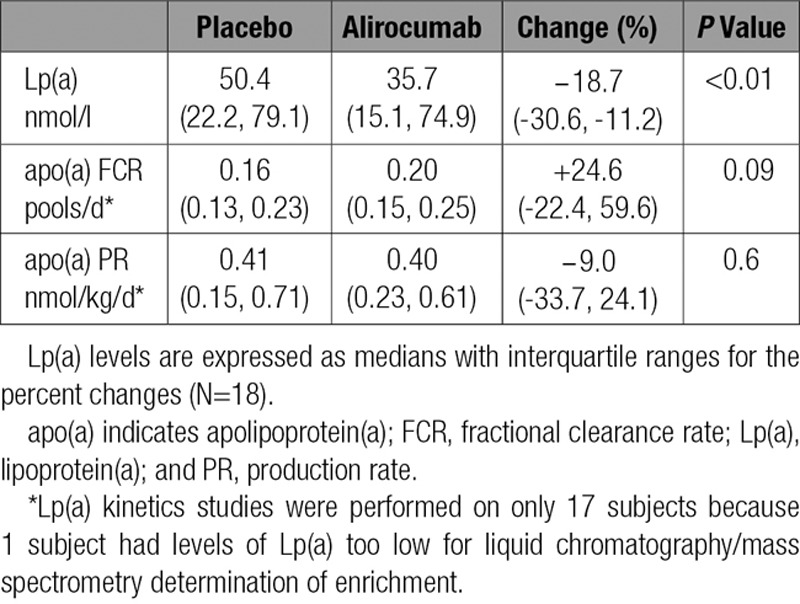
Inhibition of PCSK9 caused a reduction in the median level of plasma Lp(a) of 18.7% compared with placebo (P<0.01). These reductions were associated with a trend for an increase in the median FCR for apo(a) (24.6%; _P_=0.09) with no change in the median apo(a) PR (−9.0%; _P_=0.6) (Table 3). No significant correlations were found between baseline Lp(a) levels and changes in Lp(a) levels or apo(a) FCR after alirocumab treatment. However, a positive correlation did occur between Lp(a) isoform size and changes in apo(a) FCR on alirocumab (_R_2=0.25; _P_=0.04) (data not shown).
Table 3.
Effects of Alirocumab on Lp(a) Plasma Levels and the Kinetics of apo(a)
The last 10 subjects enrolled participated in a substudy of the effects of alirocumab on PP lipemia. Inhibition of PCSK9 activity by alirocumab had no effects on the mean incremental area under the curve above baseline of either TG or apoB48 (a marker of chylomicrons) during the 8 hours after consumption of a high-fat meal (Figure 4). In accord with the absence of effects of PCSK9 inhibition on either fasting or PP TG metabolism, treatment with alirocumab did not alter the mean levels of postheparin HL (0.96±0.6 vs 1.05±0.62 µmol ffa/mL plasma/hr) or LpL activities (1.26±0.93 vs1.35±0.90 µmol ffa/mL plasma/hr) compared with placebo.
Figure 4.
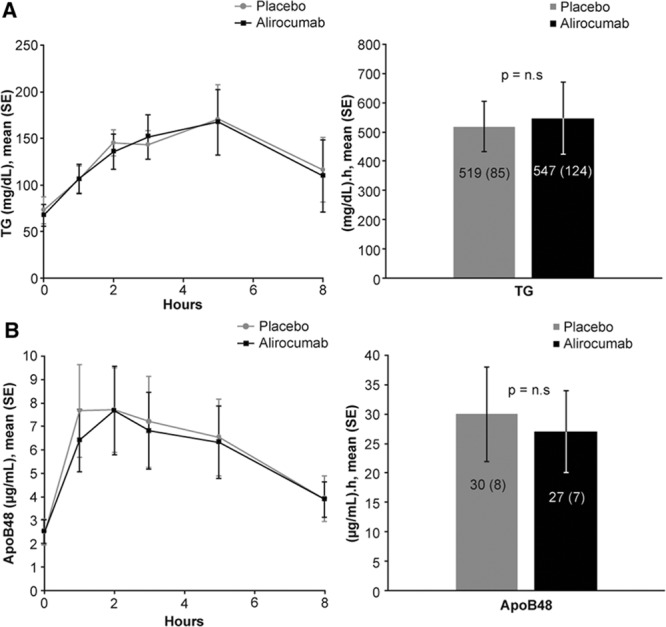
Postprandial levels of TG (A) and ApoB48 (B) after a high-fat meal. (Left) Mean concentrations over time. (Right) Incremental area-under-the-curve (IAUC). Ten participants had blood samples just before and at 1, 2, 3, 5, and 8 hours after consuming a high-fat liquid meal providing 1237 Kcal per 2 m2 body surface area from 75% fat, 10% protein, and 15% carbohydrate The data are presented as individual timepoints (mean±SE) and area under the curve above baseline (IAUC) (mean±SE) of plasma TG and apoB48 concentrations. Alirocumab had no effects on postprandial levels of TG and apoB48 compared with placebo. apoB indicates apolipoprotein B; and TG, triglycerides.
Discussion
In this study of the effects of alirocumab, a fully human monoclonal antibody against PCSK9, we demonstrated that 55% reductions in LDL-C and LDL-apoB were caused by a dramatic increase in the efficiency of clearance of LDL particles, as evidenced by an 80% increase in the FCR of LDL-apoB. We also found a 24% reduction in LDL-apoB PR caused by increased efficiency of removal of IDL particles and, therefore, less conversion of IDL to LDL. Both of these results are fully consistent with marked increases in LDLR mediated by inhibition of PCSK9.
The efficacy of alirocumab and other antibodies directed against PCSK922 is compatible with the ability of the protein to target the complex of the LDL/LDLR to the lysosome for degradation, thereby reducing the recycling of the LDLR that normally occurs after delivery of LDL to that organelle.1,2 Earlier studies in cells and animals supported a model in which inhibition of PCSK9 activity leads to increased LDLRs on the surface of the liver and increased efficiency of LDL particles removal from the circulation (Figure 5). Our results provide evidence in humans confirming those preclinical investigations.
Figure 5.
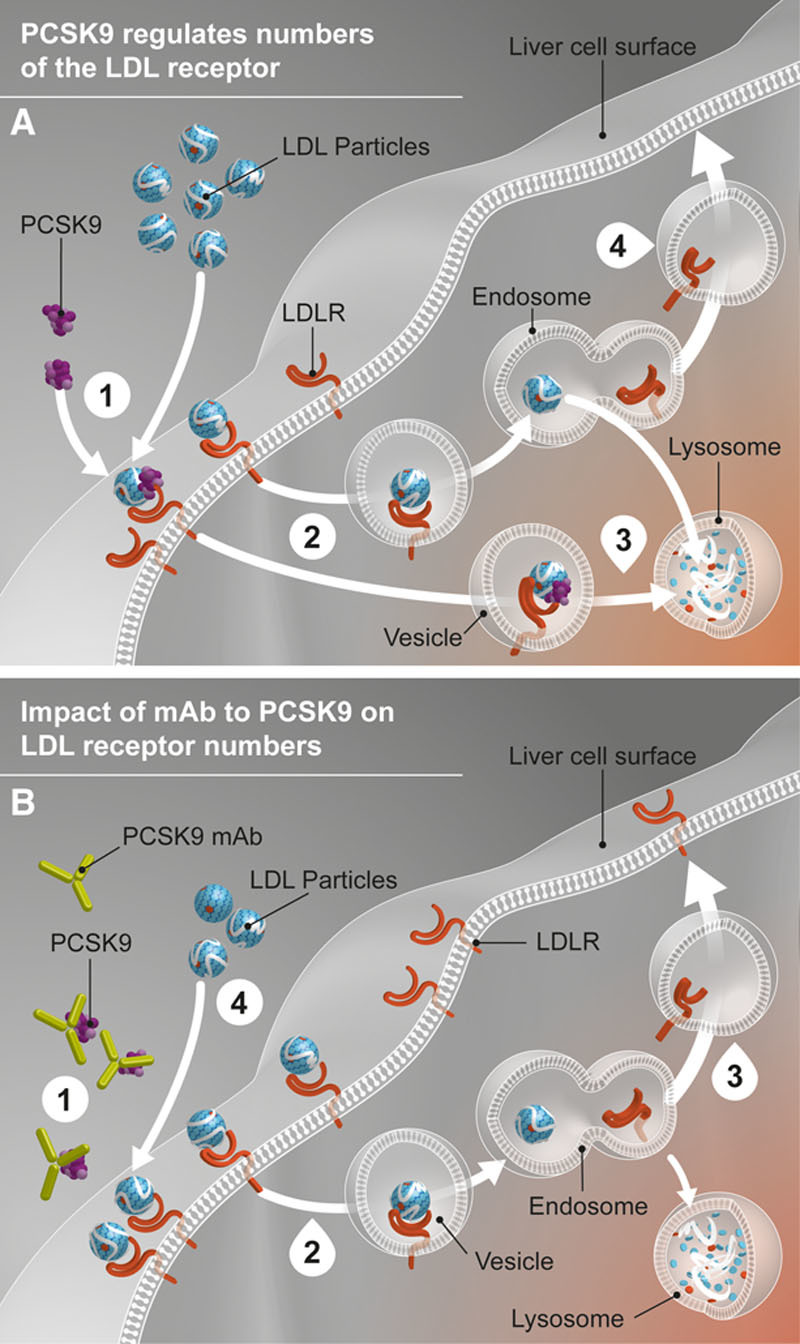
Role of PCSK9 in LDL metabolism and impact of PCSK9 monoclonal antibody. (A) (1) LDLR binds to LDL particle at the liver cell surface. PCSK9 can also bind to the LDLR. (2) The LDL particle-LDLR complexes with or without PCSK9 bound are internalized in the liver cell by endocytosis. (3) LDLR not bound to PCSK9 release the LDL particle, which goes to a lysosome for digestion, whereas the LDLR is recycled to the cell surface. (4) LDLR bound to PCSK9 is digested in the lysosome along with the LDL particle. (B) (1) PCSK9 mAb binds PCSK9 in the circulation, preventing it from binding the LDLR. (2) The LDL particle-LDLR complexes are internalized in the liver cell. (3) In the absence of PCSK9 binding, increased LDLR receptor recycling and more LDLR on the liver cell surface to bind result. (4) Circulating LDL particle levels are reduced. LDL indicates low-density lipoprotein; LDLR, low-density lipoprotein receptor; mAb, monoclonal antibody; and PCSK9, proprotein convertase subtilisin/kexin type 9.
The importance of the present results derives from the possibility that mechanisms other than more efficient LDLR-mediated removal of LDL could account for some of the significant reductions in plasma LDL-C and LDL-apoB levels observed during alirocumab treatment. This study explored several potential alternative effects of alirocumab, including decreased secretion of VLDL (the major precursor of LDL), increased removal of VLDL or its remnants without conversion to IDL and then LDL, or, more likely, increased removal of IDL, a lipoprotein close to LDL in size and composition, without conversion to LDL. Importantly, statins, which increase the number of LDLRs by inhibiting hydroxymethylglutaryl-CoA reductase, the rate-limiting enzyme of cholesterol synthesis, have been demonstrated to have multiple effects on the metabolism of VLDL, IDL, and LDL.23,24 Thus, it seemed possible that, depending on the background lipid metabolism present in any individual, one or more of the scenarios described previously might be important in the lowering of LDL-C by inhibition of PCSK9.
In this study of healthy volunteers, it is clear that the major effect of alirocumab was to enhance the efficiency of LDL particle clearance from the circulation; the FCR for LDL-apoB increased by 80.4%. Because only 1 apoB per LDL particle exists, this increase in the FCR translates to nearly twice as many LDL particles cleared per day during alirocumab treatment compared with placebo. Because LDLRs account for at least 80% of the clearance of LDL particles from the circulation, the near doubling of the FCR for LDL-apoB almost certainly resulted from a similar increase in available LDLRs on the surface of the liver.25 We also demonstrated a significant increase in the FCR of IDL-apoB, compatible with the known ability of LDLRs to bind and internalize a range of apoB-lipoproteins. This increase in IDL-FCR was accompanied by greater direct removal of IDL-apoB, which resulted in a reduced LDL-apoB PR. Thus, two related mechanisms for the marked reductions in LDL-C and LDL-apoB levels occurred during alirocumab therapy. An additional or alternative explanation for the increase in LDL-apoB FCR after alirocumab treatment could have been that changes in the physical-chemical characteristics of LDL particles would increase their affinity for the LDLR. For example, alirocumab might directly increase LDL particle size, and larger LDL particles have a greater affinity for the LDLR.26 However, our finding that alirocumab treatment resulted in a shift in the distribution of LDL toward smaller particles is compatible with greater removal of larger LDL particles by increased numbers of LDLR rather than a primary treatment-mediated shift in LDL toward larger particles.
In contrast to the findings for IDL and LDL, we saw no significant effect of alirocumab on VLDL-apoB and VLDL-TG metabolism. Because VLDL-apoB or its remnants can interact with the LDLR, resulting in direct removal of VLDL particles from the circulation without conversion to LDL,6,8,27 the possibility existed that PCSK9 inhibition would be associated with an increased flux of VLDL particles through that pathway. The probability that VLDL will be converted to IDL and LDL by lipolysis rather than being internalized by LDLRs depends on the quantity of TG in each VLDL particle, with TG-rich VLDL particles more likely to be removed directly from plasma compared with smaller, TG-poor VLDL particles.28 We studied healthy individuals with normal plasma TG levels, and, therefore, lipolytic conversion to IDL and LDL was probably rapid, reducing the possibility of an interaction between VLDL and LDLRs in the hepatic bed. Lipolysis, which would be the dominant determinant of the fate of relatively TG-poor VLDL particles entering the circulation in our healthy volunteers, was not affected by alirocumab, as indicated by unchanged postheparin lipolytic activities and apoCIII levels. The decrease in apoE levels was most likely the result of increased fractional removal of apoE-containing IDL and LDL particles from the blood. However, lower apoE levels could have contributed to the lack of an increase in direct removal of VLDL despite increased LDLR availability.
In addition to the potential for increased removal of VLDL by hepatic LDLRs, the effects of PCSK9 on the secretion of apoB-lipoproteins have also been demonstrated. Overexpression of PCSK9 in either the liver or the intestine increases secretion of apoB-lipoproteins,29,30 whereas knockdown of PCSK9 has the opposite effect.30,31 Additionally, individuals with gain-of-function mutations in PCSK9 were found to have increased secretion of VLDL-, IDL-, and LDL-apoB.32 Our finding that alirocumab did not affect either VLDL PR or PP lipemia is, therefore, in contrast to these reports, but it is possible that these differences also reflect the absence of underlying abnormalities in either lipolysis- or receptor-mediated removal of apoB48 particles in the individuals we studied. Alternatively, pharmacological interference with the action of circulating PCSK9 does not affect intracellular actions of PCSK9 on VLDL or chylomicron secretion. Finally, studies with alirocumab and other monoclonal antibodies directed against PCSK9 in populations with elevated fasting TG levels may produce different results.
A theoretical concern related to the alirocumab-mediated increase in LDL clearance from the circulation is an increase in the flux of LDL-C into the liver, with the potential for steatosis or cholesterol gallstone formation. However, after an initial increase in hepatic uptake of IDL and LDL particles immediately after the first dose of alirocumab, a stable increase in the FCR of a smaller circulating pool of LDL particles will result in little or no change in the absolute rate of cholesterol uptake by the liver in a new steady state. The absence of significant changes in lathosterol, a measure of cholesterol synthesis, or either sitosterol or campesterol, markers of cholesterol absorption, indicate hepatic cholesterol homeostasis was maintained during chronic alirocumab therapy.33,34
The metabolism of Lp(a) remains poorly defined 50 years after its discovery.35,36 Although plasma levels appear to be closely related to PR,36,37 understanding of the pathways involved in the clearance of Lp(a) remains modest at best. Indeed, some studies indicate that neither the LDLR nor apoB plays a role in the removal of Lp(a) from the circulation,38,39 whereas others support a central role for the LDLR in the uptake of Lp(a) by hepatocytes.40 Additional uncertainty arises from the observations that although Lp(a) levels are increased in individuals with familial hypercholesterolemia,41 a disorder with reduced levels of LDLR, statins, which increase the FCR of LDL-apoB by 25% to 40%, do not lower Lp(a) levels.11 It must be noted, however, that statins raise both the number of LDLR and plasma concentrations of PCSK9, with the latter effect negating, at least partially, the increase in LDLR.42 Treatment of homozygous familial hypercholesterolemia patients with evolocumab was associated with reductions in Lp(a) levels, suggesting non-LDLR clearance of Lp(a), but only 1 of the 49 subjects reported was homozygous for a null mutation in the LDLR.43
Our results for Lp(a), which are suggestive and not conclusive, certainly do not rule out other complementary or alternative pathways for Lp(a) removal from the circulation. We also acknowledge studies of the simultaneous kinetics of both apo(a) and apoB demonstrating different FCRs for apo(a) and apoB, suggesting that they are not always linked on a single Lp(a) particle.44,45 However, in a third study, it appeared that the 2 proteins were linked throughout their time in the circulation.46 Although further work is required to fully define the stability of the association between apo(a) and apoB in Lp(a), this potential complexity would not impact our findings because the FCR of apo(a) was determined in a similar fashion in our study and the others. Indeed, the FCRs of apo(a) in all 3 of those published reports were similar to those we determined in the present investigation. Finally, a study in which FCRs of apo(a) were determined by the rate of rise of plasma levels after physical removal of Lp(a) by apheresis reported rates nearly identical to the FCRs we found at baseline.47
In conclusion, inhibition of PCSK9 activity with alirocumab treatment of healthy, normolipidemic volunteers reduced LDL-C and LDL-apoB concentrations by 55% and 56%, respectively, in association with a nearly doubling of the efficiency with which LDL particles were removed from the circulation. A significant fall in LDL-apoB PR caused by increased direct removal of IDL particles from the circulation was also found. Both of these findings are consistent with increased availability of LDLR on the surface of the liver, the main organ for LDL clearance. Lp(a) concentrations were reduced significantly, with a strong trend toward an increased FCR, suggestive of a role for the LDLR in the clearance of Lp(a). Alirocumab did not have an effect on Lp(a) PR.
Of note, the potent effect of inhibition of PCSK9 activity on the FCR of LDL-apoB observed in this study indicates that individuals without dyslipidemia continually lose ≥50% of LDLRs to lysosomal degradation because of the actions of PCSK9. The number of LDLRs on cell surfaces is exquisitely regulated at the transcriptional level by effects of the concentration of hepatic-free cholesterol on the sterol response element binding protein-2,48 raising the interesting question as to why we evolved with PCSK9 as a parallel regulator of LDL uptake.
Acknowledgments
We are grateful for the editorial assistance provided by Rob Campbell and Adam Moriarty of Prime Medica, Knutsford and London, UK, funded by Sanofi and Regeneron Pharmaceuticals, Inc. Responsibility for all opinions, conclusions, and data interpretation lies with the authors. Technical assistance was provided by Anastasiya Matveyenko. We thank the Irving Institute research nurses.
Sources of Funding
This study was funded by Sanofi and Regeneron Pharmaceuticals, Inc. Additional support was provided by National Institutes of Health/National Center for Advancing Translational Science 1UL1 TR001873.
Disclosures
Dr Reyes-Soffer received research support from Sanofi and Merck Inc. and is a consultant for Merck; Mr Matta, Mr Becue, Mr Poitiers, Dr Swanson, Ms Cowan, and Dr Surks are employees of Sanofi; Ms Maroccia is a contract employee of Sanofi; Dr Sasiela is an employee of Regeneron Pharmaceuticals, Inc.; and Dr Ginsberg received research support from Merck, Sanofi and Regeneron, and Amgen and is a consultant for and on the advisory boards of Amgen, AstraZeneca, Bristol Myers Squibb, GlaxoSmithKline, Ionis, Janssen, Kowa, Merck, Novartis, Sanofi, Regeneron, Pfizer, and Resverlogix. The other authors report no conflicts of interest.
Supplementary Material
Footnotes
Clinical Perspective
What Is New?
- In this study of healthy volunteers, we demonstrated that the marked reductions in low-density lipoprotein (LDL) cholesterol levels observed when proprotein convertase subtilisin/kexin type 9 (PCSK9) is inhibited by the monoclonal antibody, alirocumab, are caused by a dramatic increase in the efficiency of removal of atherogenic LDL particles from the circulation.
- We also observed reduced production of LDL-apolipoprotein B resulting from increased efficiency of removal of intermediate density lipoproteins and, therefore, less production of LDL.
- These findings are the first human data supporting earlier studies in cells and mice that showed increased LDL receptor (LDLR) activity when PCSK9 is inhibited.
What Are the Clinical Implications?
- The role of the LDLR in regulating blood levels of atherogenic apolipoprotein B lipoproteins, particularly LDL, is clear, as are the benefits of increasing the capacity of the LDLR pathway for cardiovascular disease risk.
- The discovery that PCSK9 reduces LDLRs on the surface of cells by targeting the receptor for degradation stimulated development of PCSK9 inhibitors.
- Our study, which demonstrates that inhibition of PCSK9 with alirocumab increases the removal of LDL from the circulation by the LDLR, increases confidence that PCSK9 inhibitors will confer clinical benefit in the ongoing cardiovascular outcome trials.
References
- 1.Horton JD, Cohen JC, Hobbs HH. PCSK9: a convertase that coordinates LDL catabolism. J Lipid Res. 2009;50(Suppl):S172–S177. doi: 10.1194/jlr.R800091-JLR200. doi: 10.1194/jlr.R800091-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Seidah NG, Awan Z, Chrétien M, Mbikay M. PCSK9: a key modulator of cardiovascular health. Circ Res. 2014;114:1022–1036. doi: 10.1161/CIRCRESAHA.114.301621. doi: 10.1161/CIRCRESAHA.114.301621. [DOI] [PubMed] [Google Scholar]
- 3.Cannon CP, Cariou B, Blom D, McKenney JM, Lorenzato C, Pordy R, Chaudhari U, Colhoun HM ODYSSEY COMBO II investigators. Efficacy and safety of alirocumab in high cardiovascular risk patients with inadequately controlled hypercholesterolaemia on maximally tolerated doses of statins: the ODYSSEY COMBO II randomized controlled trial. Eur Heart J. 2015;36:1186–1194. doi: 10.1093/eurheartj/ehv028. doi: 10.1093/eurheartj/ehv028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kastelein JJ, Ginsberg HN, Langslet G, Hovingh GK, Ceska R, Dufour R, Blom D, Civeira F, Krempf M, Lorenzato C, Zhao J, Pordy R, Baccara-Dinet MT, Gipe DA, Geiger MJ, Farnier M. ODYSSEY FH I and FH II: 78 week results with alirocumab treatment in 735 patients with heterozygous familial hypercholesterolaemia. Eur Heart J. 2015;36:2996–3003. doi: 10.1093/eurheartj/ehv370. doi: 10.1093/eurheartj/ehv370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Robinson JG, Farnier M, Krempf M, Bergeron J, Luc G, Averna M, Stroes ES, Langslet G, Raal FJ, El Shahawy M, Koren MJ, Lepor NE, Lorenzato C, Pordy R, Chaudhari U, Kastelein JJ ODYSSEY LONG TERM investigators. Efficacy and safety of alirocumab in reducing lipids and cardiovascular events. N Engl J Med. 2015;372:1489–1499. doi: 10.1056/NEJMoa1501031. doi: 10.1056/NEJMoa1501031. [DOI] [PubMed] [Google Scholar]
- 6.Kita T, Brown MS, Bilheimer DW, Goldstein JL. Delayed clearance of very low density and intermediate density lipoproteins with enhanced conversion to low density lipoprotein in WHHL rabbits. Proc Natl Acad Sci U S A. 1982;79:5693–5697. doi: 10.1073/pnas.79.18.5693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Krul ES, Tikkanen MJ, Cole TG, Davie JM, Schonfeld G. Roles of apolipoproteins B and E in the cellular binding of very low density lipoproteins. J Clin Invest. 1985;75:361–369. doi: 10.1172/JCI111708. doi: 10.1172/JCI111708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mahley RW, Ji ZS. Remnant lipoprotein metabolism: key pathways involving cell-surface heparan sulfate proteoglycans and apolipoprotein E. J Lipid Res. 1999;40:1–16. [PubMed] [Google Scholar]
- 9.Gaudet D, Kereiakes DJ, McKenney JM, Roth EM, Hanotin C, Gipe D, Du Y, Ferrand AC, Ginsberg HN, Stein EA. Effect of alirocumab, a monoclonal proprotein convertase subtilisin/kexin 9 antibody, on lipoprotein(a) concentrations (a pooled analysis of 150 mg every two weeks dosing from phase 2 trials). Am J Cardiol. 2014;114:711–715. doi: 10.1016/j.amjcard.2014.05.060. doi: 10.1016/j.amjcard.2014.05.060. [DOI] [PubMed] [Google Scholar]
- 10.McKenney JM, Koren MJ, Kereiakes DJ, Hanotin C, Ferrand AC, Stein EA. Safety and efficacy of a monoclonal antibody to proprotein convertase subtilisin/kexin type 9 serine protease, SAR236553/REGN727, in patients with primary hypercholesterolemia receiving ongoing stable atorvastatin therapy. J Am Coll Cardiol. 2012;59:2344–2353. doi: 10.1016/j.jacc.2012.03.007. doi: 10.1016/j.jacc.2012.03.007. [DOI] [PubMed] [Google Scholar]
- 11.Kostner GM, Gavish D, Leopold B, Bolzano K, Weintraub MS, Breslow JL. HMG CoA reductase inhibitors lower LDL cholesterol without reducing Lp(a) levels. Circulation. 1989;80:1313–1319. doi: 10.1161/01.cir.80.5.1313. [DOI] [PubMed] [Google Scholar]
- 12.Reyes-Soffer G, Moon B, Hernandez-Ono A, Dionizovik-Dimanovski M, Dionizovick-Dimanovski M, Jimenez J, Obunike J, Thomas T, Ngai C, Fontanez N, Donovan DS, Karmally W, Holleran S, Ramakrishnan R, Mittleman RS, Ginsberg HN. Complex effects of inhibiting hepatic apolipoprotein B100 synthesis in humans. Sci Transl Med. 2016;8:323ra12. doi: 10.1126/scitranslmed.aad2195. doi: 10.1126/scitranslmed.aad2195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Marcovina SM, Albers JJ, Scanu AM, Kennedy H, Giaculli F, Berg K, Couderc R, Dati F, Rifai N, Sakurabayashi I, Tate JR, Steinmetz A. Use of a reference material proposed by the International Federation of Clinical Chemistry and Laboratory Medicine to evaluate analytical methods for the determination of plasma lipoprotein(a). Clin Chem. 2000;46:1956–1967. [PubMed] [Google Scholar]
- 14.Marcovina SM, Hobbs HH, Albers JJ. Relation between number of apolipoprotein(a) kringle 4 repeats and mobility of isoforms in agarose gel: basis for a standardized isoform nomenclature. Clin Chem. 1996;42:436–439. [PubMed] [Google Scholar]
- 15.Caulfield MP, Li S, Lee G, Blanche PJ, Salameh WA, Benner WH, Reitz RE, Krauss RM. Direct determination of lipoprotein particle sizes and concentrations by ion mobility analysis. Clin Chem. 2008;54:1307–1316. doi: 10.1373/clinchem.2007.100586. doi: 10.1373/clinchem.2007.100586. [DOI] [PubMed] [Google Scholar]
- 16.Matthan NR, Resteghini N, Robertson M, Ford I, Shepherd J, Packard C, Buckley BM, Jukema JW, Lichtenstein AH, Schaefer EJ PROSPER Group. Cholesterol absorption and synthesis markers in individuals with and without a CHD event during pravastatin therapy: insights from the PROSPER trial. J Lipid Res. 2010;51:202–209. doi: 10.1194/jlr.M900032-JLR200. doi: 10.1194/jlr.M900032-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nagashima K, Lopez C, Donovan D, Ngai C, Fontanez N, Bensadoun A, Fruchart-Najib J, Holleran S, Cohn JS, Ramakrishnan R, Ginsberg HN. Effects of the PPARgamma agonist pioglitazone on lipoprotein metabolism in patients with type 2 diabetes mellitus. J Clin Invest. 2005;115:1323–1332. doi: 10.1172/JCI23219. doi: 10.1172/JCI23219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ramakrishnan R. Studying apolipoprotein turnover with stable isotope tracers: correct analysis is by modeling enrichments. J Lipid Res. 2006;47:2738–2753. doi: 10.1194/jlr.M600302-JLR200. doi: 10.1194/jlr.M600302-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Berglund L, Witztum JL, Galeano NF, Khouw AS, Ginsberg HN, Ramakrishnan R. Three-fold effect of lovastatin treatment on low density lipoprotein metabolism in subjects with hyperlipidemia: increase in receptor activity, decrease in apoB production, and decrease in particle affinity for the receptor: results from a novel triple-tracer approach. J Lipid Res. 1998;39:913–924. [PMC free article] [PubMed] [Google Scholar]
- 20.Zhou H, Castro-Perez J, Lassman ME, Thomas T, Li W, McLaughlin T, Dan X, Jumes P, Wagner JA, Gutstein DE, Hubbard BK, Rader DJ, Millar JS, Ginsberg HN, Reyes-Soffer G, Cleary M, Previs SF, Roddy TP. Measurement of apo(a) kinetics in human subjects using a microfluidic device with tandem mass spectrometry. Rapid Commun Mass Spectrom. 2013;27:1294–1302. doi: 10.1002/rcm.6572. doi: 10.1002/rcm.6572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Reyes-Soffer G, Holleran S, Karmally W, Ngai CI, Chen NT, Torres M, Ramakrishnan R, Blaner WS, Berglund L, Ginsberg HN, Tuck C. Measures of postprandial lipoproteins are not associated with coronary artery disease in patients with type 2 diabetes mellitus. J Lipid Res. 2009;50:1901–1909. doi: 10.1194/jlr.M900092-JLR200. doi: 10.1194/jlr.M900092-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sabatine MS, Giugliano RP, Wiviott SD, Raal FJ, Blom DJ, Robinson J, Ballantyne CM, Somaratne R, Legg J, Wasserman SM, Scott R, Koren MJ, Stein EA Open-Label Study of Long-Term Evaluation against LDL Cholesterol (OSLER) investigators. Efficacy and safety of evolocumab in reducing lipids and cardiovascular events. N Engl J Med. 2015;372:1500–1509. doi: 10.1056/NEJMoa1500858. doi: 10.1056/NEJMoa1500858. [DOI] [PubMed] [Google Scholar]
- 23.Ginsberg HN. Effects of statins on triglyceride metabolism. Am J Cardiol. 1998;81(4A):32B–35B. doi: 10.1016/s0002-9149(98)00035-6. [DOI] [PubMed] [Google Scholar]
- 24.Ginsberg HN. Review: Efficacy and mechanisms of action of statins in the treatment of diabetic dyslipidemia. J Clin Endocrinol Metab. 2006;91:383–392. doi: 10.1210/jc.2005-2084. doi: 10.1210/jc.2005-2084. [DOI] [PubMed] [Google Scholar]
- 25.Brown MS, Goldstein JL. How LDL receptors influence cholesterol and atherosclerosis. Sci Am. 1984;251:58–66. doi: 10.1038/scientificamerican1184-58. [DOI] [PubMed] [Google Scholar]
- 26.Campos H, Arnold KS, Balestra ME, Innerarity TL, Krauss RM. Differences in receptor binding of LDL subfractions. Arterioscler Thromb Vasc Biol. 1996;16:794–801. doi: 10.1161/01.atv.16.6.794. [DOI] [PubMed] [Google Scholar]
- 27.Havel RJ. Functional activities of hepatic lipoprotein receptors. Annu Rev Physiol. 1986;48:119–134. doi: 10.1146/annurev.ph.48.030186.001003. doi: 10.1146/annurev.ph.48.030186.001003. [DOI] [PubMed] [Google Scholar]
- 28.Packard CJ, Munro A, Lorimer AR, Gotto AM, Shepherd J. Metabolism of apolipoprotein B in large triglyceride-rich very low density lipoproteins of normal and hypertriglyceridemic subjects. J Clin Invest. 1984;74:2178–2192. doi: 10.1172/JCI111644. doi: 10.1172/JCI111644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sun H, Samarghandi A, Zhang N, Yao Z, Xiong M, Teng BB. Proprotein convertase subtilisin/kexin type 9 interacts with apolipoprotein B and prevents its intracellular degradation, irrespective of the low-density lipoprotein receptor. Arterioscler Thromb Vasc Biol. 2012;32:1585–1595. doi: 10.1161/ATVBAHA.112.250043. doi: 10.1161/ATVBAHA.112.250043. [DOI] [PubMed] [Google Scholar]
- 30.Rashid S, Tavori H, Brown PE, Linton MF, He J, Giunzioni I, Fazio S. Proprotein convertase subtilisin kexin type 9 promotes intestinal overproduction of triglyceride-rich apolipoprotein B lipoproteins through both low-density lipoprotein receptor-dependent and -independent mechanisms. Circulation. 2014;130:431–441. doi: 10.1161/CIRCULATIONAHA.113.006720. doi: 10.1161/CIRCULATIONAHA.113.006720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Le May C, Kourimate S, Langhi C, Chétiveaux M, Jarry A, Comera C, Collet X, Kuipers F, Krempf M, Cariou B, Costet P. Proprotein convertase subtilisin kexin type 9 null mice are protected from postprandial triglyceridemia. Arterioscler Thromb Vasc Biol. 2009;29:684–690. doi: 10.1161/ATVBAHA.108.181586. doi: 10.1161/ATVBAHA.108.181586. [DOI] [PubMed] [Google Scholar]
- 32.Ouguerram K, Chetiveaux M, Zair Y, Costet P, Abifadel M, Varret M, Boileau C, Magot T, Krempf M. Apolipoprotein B100 metabolism in autosomal-dominant hypercholesterolemia related to mutations in PCSK9. Arterioscler Thromb Vasc Biol. 2004;24:1448–1453. doi: 10.1161/01.ATV.0000133684.77013.88. doi: 10.1161/01.ATV.0000133684.77013.88. [DOI] [PubMed] [Google Scholar]
- 33.Kempen HJ, Glatz JF, Gevers Leuven JA, van der Voort HA, Katan MB. Serum lathosterol concentration is an indicator of whole-body cholesterol synthesis in humans. J Lipid Res. 1988;29:1149–1155. [PubMed] [Google Scholar]
- 34.Stellaard F, von Bergmann K, Sudhop T, Lütjohann D. The value of surrogate markers to monitor cholesterol absorption, synthesis and bioconversion to bile acids under lipid lowering therapies. J Steroid Biochem Mol Biol. 2016:S0960-0760(16)30076-0. doi: 10.1016/j.jsbmb.2016.03.030. pii. doi: 10.1016/j.jsbmb.2016.03.030. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 35.Berg K. A new serum type system in man – the Lp system. Acta Pathol Microbiol Scand. 1963;59:369–382. doi: 10.1111/j.1699-0463.1963.tb01808.x. [DOI] [PubMed] [Google Scholar]
- 36.Schmidt K, Noureen A, Kronenberg F, Utermann G. Structure, function, and genetics of lipoprotein (a). J Lipid Res. 2016;57:1339–1359. doi: 10.1194/jlr.R067314. doi: 10.1194/jlr.R067314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rader DJ, Cain W, Ikewaki K, Talley G, Zech LA, Usher D, Brewer HB., Jr. The inverse association of plasma lipoprotein(a) concentrations with apolipoprotein(a) isoform size is not due to differences in Lp(a) catabolism but to differences in production rate. J Clin Invest. 1994;93:2758–2763. doi: 10.1172/JCI117292. doi: 10.1172/JCI117292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rader DJ, Mann WA, Cain W, Kraft HG, Usher D, Zech LA, Hoeg JM, Davignon J, Lupien P, Grossman M. The low density lipoprotein receptor is not required for normal catabolism of Lp(a) in humans. J Clin Invest. 1995;95:1403–1408. doi: 10.1172/JCI117794. doi: 10.1172/JCI117794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cain WJ, Millar JS, Himebauch AS, Tietge UJ, Maugeais C, Usher D, Rader DJ. Lipoprotein [a] is cleared from the plasma primarily by the liver in a process mediated by apolipoprotein [a]. J Lipid Res. 2005;46:2681–2691. doi: 10.1194/jlr.M500249-JLR200. doi: 10.1194/jlr.M500249-JLR200. [DOI] [PubMed] [Google Scholar]
- 40.Romagnuolo R, Scipione CA, Boffa MB, Marcovina SM, Seidah NG, Koschinsky ML. Lipoprotein(a) catabolism is regulated by proprotein convertase subtilisin/kexin type 9 through the low density lipoprotein receptor. J Biol Chem. 2015;290:11649–11662. doi: 10.1074/jbc.M114.611988. doi: 10.1074/jbc.M114.611988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tada H, Kawashiri MA, Yoshida T, Teramoto R, Nohara A, Konno T, Inazu A, Mabuchi H, Yamagishi M, Hayashi K. Lipoprotein(a) in familial hypercholesterolemia with proprotein convertase subtilisin/kexin type 9 (PCSK9) gain-of-function mutations. Circ J. 2016;80:512–518. doi: 10.1253/circj.CJ-15-0999. doi: 10.1253/circj.CJ-15-0999. [DOI] [PubMed] [Google Scholar]
- 42.Welder G, Zineh I, Pacanowski MA, Troutt JS, Cao G, Konrad RJ. High-dose atorvastatin causes a rapid sustained increase in human serum PCSK9 and disrupts its correlation with LDL cholesterol. J Lipid Res. 2010;51:2714–2721. doi: 10.1194/jlr.M008144. doi: 10.1194/jlr.M008144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Raal FJ, Honarpour N, Blom DJ, Hovingh GK, Xu F, Scott R, Wasserman SM, Stein EA TESLA investigators. Inhibition of PCSK9 with evolocumab in homozygous familial hypercholesterolaemia (TESLA Part B): a randomised, double-blind, placebo-controlled trial. Lancet. 2015;385:341–350. doi: 10.1016/S0140-6736(14)61374-X. doi: 10.1016/S0140-6736(14)61374-X. [DOI] [PubMed] [Google Scholar]
- 44.Diffenderfer MR, Lamon-Fava S, Marcovina SM, Barrett PH, Lel J, Dolnikowski GG, Berglund L, Schaefer EJ. Distinct metabolism of apolipoproteins (a) and B-100 within plasma lipoprotein(a). Metabolism. 2016;65:381–390. doi: 10.1016/j.metabol.2015.10.031. doi: 10.1016/j.metabol.2015.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jenner JL, Seman LJ, Millar JS, Lamon-Fava S, Welty FK, Dolnikowski GG, Marcovina SM, Lichtenstein AH, Barrett PH, deLuca C, Schaefer EJ. The metabolism of apolipoproteins (a) and B-100 within plasma lipoprotein (a) in human beings. Metabolism. 2005;54:361–369. doi: 10.1016/j.metabol.2004.10.001. doi: 10.1016/j.metabol.2004.10.001. [DOI] [PubMed] [Google Scholar]
- 46.Frischmann ME, Ikewaki K, Trenkwalder E, Lamina C, Dieplinger B, Soufi M, Schweer H, Schaefer JR, König P, Kronenberg F, Dieplinger H. In vivo stable-isotope kinetic study suggests intracellular assembly of lipoprotein(a). Atherosclerosis. 2012;225:322–327. doi: 10.1016/j.atherosclerosis.2012.09.031. doi: 10.1016/j.atherosclerosis.2012.09.031. [DOI] [PubMed] [Google Scholar]
- 47.Parhofer KG, Demant T, Ritter MM, Geiss HC, Donner M, Schwandt P. Lipoprotein (a) metabolism estimated by nonsteady-state kinetics. Lipids. 1999;34:325–335. doi: 10.1007/s11745-999-0370-z. [DOI] [PubMed] [Google Scholar]
- 48.Brown MS, Goldstein JL. The SREBP pathway: regulation of cholesterol metabolism by proteolysis of a membrane-bound transcription factor. Cell. 1997;89:331–340. doi: 10.1016/s0092-8674(00)80213-5. [DOI] [PubMed] [Google Scholar]