CSNK1a1 Regulates PRMT1 to Maintain the Progenitor State in Self-renewing Somatic Tissue (original) (raw)
. Author manuscript; available in PMC: 2018 Oct 23.
Summary
Somatic progenitors sustain tissue self-renewal while suppressing premature differentiation. Protein arginine methyltransferases (PRMTs) impact many processes, however, their role in progenitor function is incompletely understood. PRMT1 was found to be the most highly expressed PRMT in epidermal progenitors and the most downregulated PRMT during differentiation. In targeted mouse knockouts and in long-term regenerated human mosaic epidermis in vivo, epidermal PRMT1 loss abolished progenitor self-renewal and led to premature differentiation. Mass spectrometry of the PRMT1 protein interactome identified the CSNK1a1 kinase, which also proved essential for progenitor maintenance. CSNK1a1 directly bound and phosphorylated PRMT1 to control its genomic targeting to PRMT1-sustained proliferation genes as well as PRMT1-suppressed differentiation genes. Among the latter were GRHL3, whose de-repression was required for the premature differentiation seen with PRMT1 and CSNK1a1 loss. Maintenance of the progenitors thus requires cooperation by PRMT1 and CSNK1a1 to sustain proliferation gene expression and suppress premature differentiation driven by GRHL3.
Keywords: Progenitors, PRMT1, CSNK1a1, Differentiation, Keratinocyte
eTOC
Bao et al. demonstrate an essential role for the arginine methyltransferase PRMT1 in epidermal progenitor maintenance. They further identify the kinase CSNK1a1 as a key PRMT1-interacting protein. CSNK1a1 phosphorylates PRMT1 and cooperates with PRMT1 to suppress GRHL3-mediated terminal differentiation.
Introduction
Self-renewing somatic tissue is maintained by the continuous cycling of progenitors and by the restricted activation of terminal differentiation. Premature differentiation leads to progenitor depletion and tissue failure (Himes and Raetzman, 2009; Schuster-Gossler et al., 2007; Sen et al., 2010). In the case of epidermis, a stratified epithelial tissue, the progenitors reside in the basal layer adherent to the basement membrane. Upon differentiation, cells exit from the progenitor compartment to form suprabasal layers in concert with activation of terminal differentiation genes that mediate epidermal barrier function. Recent studies identified essential roles of several protein methylation regulators in progenitor maintenance in epidermal tissue. These regulators include JMJD3, Setd8, CBX4, Jarid2 and Polycomb complex proteins (Driskell et al., 2011; Ezhkova et al., 2009; Luis et al., 2011; Mejetta et al., 2011; Sen et al., 2008). However, the majority of these current studies have been focused on regulators of lysine methylation.
In addition to lysine methylation, arginine methylation is a common feature of eukaryotic proteins. This post-translational modification directly impacts protein-protein, protein-DNA, and protein-RNA interactions by removing hydrogen bonds and changing the shape of the arginine residue (Bedford and Clarke, 2009). There are 10 protein arginine methyltransferases (PRMTs) identified in mammals. They function as critical modulators of various processes, including RNA splicing, transcription regulation and DNA repair. Aberrant expression and activity of these PRMTs are implicated in oncogenesis (Yang and Bedford, 2013). The roles of these PRMTs in mammalian tissue homeostasis, and the mechanisms of their action in gene regulation are incompletely understood.
PRMT1 is highly conserved from yeast to human, and is ubiquitously expressed in a variety of mammalian tissue types. Although PRMT1 is not required for the viability of yeast and mouse ES cells, PRMT1 loss leads to lethality at E6.5 during mouse embryonic development (Pawlak et al., 2000; Yu et al., 2009), indicating that PRMT1 actions extend beyond housekeeping function to impact developmental processes. Over the last 30 years since the initial discovery of mammalian PRMT1 (Gary et al., 1996), dozens of PRMT1-interacting proteins and substrates have been isolated from various cell lines, including proteins involved in transcription regulation, DNA repair, and RNA processing. How PRMT1 might interface with its interactome to influence specific cellular states remains incompletely understood.
Using epidermis as a prototype of a self-renewing mammalian tissue, here we demonstrate a role for PRMT1 in progenitor maintenance. PRMT1 was found to be enriched in epidermal progenitors and significantly down-regulated during epidermal differentiation, suggesting a role in the undifferentiated cell state. PRMT1 loss in both murine and human epidermal tissue profoundly impaired progenitor function in a cell-autonomous fashion. Using tandem affinity purification, the CSNK1a1 kinase was identified as an essential PRMT1-interacting protein in epidermal progenitor maintenance. CSNK1a1 depletion phenocopied PRMT1 loss in impairing progenitor self-renewal and de-repressing terminal differentiation. CSNK1a1 directly interacted with PRMT1, mediated PRMT1 phosphorylation, and controlled PRMT1 genomic targeting to sustain expression of proliferation genes. PRMT1 also suppressed premature progenitor differentiation, in part, by repressing ectopic expression of GRHL3, an essential activator of terminal differentiation, with the importance of PRMT1-CSNK1a1 suppression of GRHL3 in this setting confirmed by genetic rescue. These data establish an essential role for PRMT1 and CSNK1a1 in progenitor maintenance in murine and human tissue in vivo and show that their action in this setting involves CSNK1a1 control of PRMT1 genomic targeting to genes that promote progenitor self-renewal and suppress premature differentiation driven by GRHL3.
Results
PRMT1 is enriched in progenitors and is required for progenitor function
To explore a potential role for PRMTs in human tissue homeostasis, we first examined their transcript levels during keratinocyte differentiation. PRMT1 was the most abundant PRMT in undifferentiated cells, and was also the most down-regulated PRMT during differentiation (Figure 1A–C). This mRNA down-regulation was reflected in PRMT1 protein levels as well, which were strikingly decreased during differentiation (Figure 1D). Within intact tissue, PRMT1 protein was likewise most strongly expressed in the undifferentiated progenitor-containing layer adjacent to the epidermal basement membrane (Figure 1E). PRMT1 expression is thus down-regulated in differentiating epidermal cells in vitro and in tissue.
Figure 1. PRMT1 is enriched in progenitors and is required for progenitor maintenance.

(A) Bar graph representing the mRNA abundance of PRMTs in undifferentiated primary human keratinocytes based on RNA-seq data. X-axis represents reads per kb per million reads (RPKM). (B) Heatmap comparing the relative PRMT mRNA expression levels between undifferentiated and differentiated human keratinocytes based on RNA-seq data. (C) qRT-PCR quantification of relative PRMT1 mRNA expression among undifferentiated state (UD), differentiation day 2 (DF_Day2), and differentiation day 4 (DF_Day4). PRMT1 mRNA is downregulated during keratinocyte differentiation in vitro. (D) Immunoblots demonstrating PRMT1 protein levels decrease during human keratinocyte differentiation. β-tubulin was used as loading control. (E) Immunostaining of PRMT1 in human skin section. PRMT1 protein localizes primarily to nuclei in the basal progenitor epidermal layer [PRMT1=green; collagen VII basement membrane marker=orange; nuclear stain with Hoechst 33342=blue; scale bar=100 microns. Note that rabbit anti-serum tends to cross react with stratum corneum]. (F and I) Immunoblots demonstrating the knockdown efficiency of four independent shRNAs targeting PRMT1 in primary human keratinocytes. (G and J) Clonogenic assays of human keratinocytes with PRMT1 RNAi or control. (H and K) Colonies > 1mm2 in clonogenic assays are quantified (n=2/group, p<0.001, ANOVA, error bars represent standard deviation.). (L) Progenitor competition assay. Human epidermal tissue was regenerated on immunodeficient mice with equal number of GFP-expressing keratinocytes with PRMT1 loss as well as DsRed-expressing control keratinocytes. Tissues were harvest on day 10 and day 40 post grafting. (M) Progenitor competition assay at the time point of day 40, with PRMT1 loss labeled using GFP and control cells labelled using DsRed. (N) Fold change of PRMT1i cell percentage relative to scrambled control percentage. 4 mice were grafted at each time point (Day 10 and Day 40). Significant reduction of PRMT1i cells was observed on Day 40 as compared to Day 10 (p<0.01, Two-tailed student T-test).
The enrichment of PRMT1 in progenitors raised the possibility that PRMT1 may contribute to progenitor function. To explore this, we first tested PRMT1 loss in undifferentiated human keratinocytes. We selected 4 independent shRNAs that efficiently knocked down PRMT1. PRMT1 loss mediated by each of these shRNAs led to decreased clonogenicity and impaired proliferation in keratinocytes (Figure 1F–K, Figure S2A). We next investigated the role of PRMT1 in epidermal tissue using a mosaic progenitor competition assay in vivo. In this approach, two groups of keratinocytes expressing either PRMT1 shRNA or control were respectively marked with GFP and DsRed. To exclude that differential fluorescent protein toxicity accounted for these findings, the reverse color label experiment was also performed. Equal numbers of these keratinocytes expressing PRMT1 shRNA or control were mixed to regenerate epidermal tissue on immunodeficient mice. Regenerated epidermal tissues were collected and analyzed on Day 10 post-grafting after initial tissue establishment and on Day 40 after completion of a full epidermal turnover cycle. PRMT1-depleted cells contributed roughly equal numbers of cells as control at Day 10, confirming their viability and capacity to contribute to tissue generation, however, by Day 40 these cells were largely lost (Figure 1L–N), suggesting that PRMT1 is required to sustain the presence of progenitor cells in this setting.
To confirm this observation in an orthogonal mammalian system, we performed targeted PRMT1 knockout in mice. A floxed PRMT1 allele was excised in mouse epidermis using Cre recombinase driven by the epidermal basal layer keratin 14 (K14) promoter during embryonic development (Huelsken et al., 2001). Compared to heterozygous littermate controls, epidermal PRMT1 deletion led to smaller mice with clinically thin epidermis who died before birth (Figure S1A). PRMT1-deficient mouse epidermis at E17.5 was hypoplastic, with a striking presence of differentiating cells immediately adjacent to the epidermal basement membrane (Figure S1B–D). This PRMT1 KO epidermal tissue was also characterized by dysregulated cell cycle progression as well as increased 53BP1 levels (Figure S1E–J), consistent with previous findings with PRMT1 loss in murine embryonic fibroblasts (Yu et al., 2009). Therefore, PRMT1 deletion in murine epidermis also leads to signs of impaired progenitor function.
PRMT1 sustains proliferation genes and suppresses differentiation genes
Given these observations indicating that PRMT1 is essential in epidermal tissue development and homeostasis, we set out to identify the downstream target genes controlled by PRMT1 in progenitors. mRNA expression profiling was performed on undifferentiated human epidermal keratinocytes treated with two independent PRMT1 shRNAs. PRMT1 depletion altered a total of 644 genes (326 [50.6%] induced, 318 [49.4%] repressed, Table S1). These PRMT1-regulated genes displayed a significant (p<2×10−218, Fisher’s Exact Test) overlap of 434 genes with the published calcium-induced epidermal keratinocyte differentiation profile (Sen et al., 2010) (Figure 2A). The 326 genes up-regulated with PRMT1 loss were enriched with Gene Ontology (GO) terms such as “epidermal differentiation” and “development and wound healing” while down-regulated genes were enriched with terms like “proliferation” (Figure 2B). To further confirm the findings from transcriptome profiling, we performed qRT-PCR on a representative panel of PRMT1-regulated targets involved in progenitor proliferation as well as keratinocyte differentiation genes that are normally repressed in progenitors. Both PRMT1 knockdown as well as PRMT1 pharmacologic blockade suppressed progenitor-associated proliferation genes and prominently de-repressed specific well-characterized differentiation genes (Figure 2C–F). Similar to the phenotype observed in knockout mice, PRMT1 knockdown also led to hypoplasia in regenerated human epidermal tissue (Figure S2B). However, reduced PRMT1 levels in human keratinocytes did not result in increased DNA damage or apoptosis (Figure S2C–F), indicating that the altered differentiation and proliferation gene expression was not a consequence of a DNA damage response. FACS analysis revealed no substantial alteration of cell cycle distribution (Figure S2G), consistent with reduced overall cell growth. Overexpression of PRMT1 also increased clonogenic capacity (Figure S2H–I). These data suggest that the enrichment of PRMT1 in epidermal progenitors may function to sustain proliferation and to repress premature differentiation.
Figure 2. PRMT1 sustains proliferation genes and suppresses differentiation genes.

(A) Heat map (right) and Venn diagram (left) (Rezai-Zadeh et al., 2003) illustrating the overlap between expression changes identified with PRMT1 loss and calcium-induced differentiation (P<2×10−218, Fisher’s Exact Test). Genes induced are colored in red, and repressed genes are colored in green. (B) GO (Gene Ontology) analysis of the 644 PRMT1 target genes, demonstrating that PRMT1 loss suppresses proliferation-associated genes and induces differentiation-associated genes. (C–F) qRT-PCR verification of array data showing mRNA levels of differentiation or proliferation associated genes with PRMT1 knockdown or with PRMT1 inhibition using AMI-1.
Tandem affinity purification of PRMT1-interacting proteins identified CSNK1a1
In undifferentiated human primary keratinocytes, we observed that PRMT1 protein present in higher molecular weight complexes using FPLC fractionation (Figure 3A), raising the possibility that PRMT1’s role in progenitors may require additional interacting proteins. To search for such PRMT1-interacting proteins in epidermal progenitor populations, we undertook tandem affinity purification of PRMT1 from undifferentiated keratinocytes (Figure 3B). When expressed at near-endogenous levels, tagged TAP-PRMT1 distributed in complexes with similar sizes to endogenous PRMT1 (Figure 3C). Purification enriched both TAP-PRMT1 and endogenous PRMT1 from keratinocyte lysates (Figure 3D), consistent with prior work showing that PRMT1 forms homodimers (Zhang and Cheng, 2003). TAP-PRMT1 purification captured discrete protein bands absent in control (Figure 3E). Liquid chromatography tandem mass spectrometry (LC-MS/MS) identified 38 unique PRMT1-interacting proteins. Among these were previously identified PRMT1 interactors, such as histone H4, CHTOP, TAP15, EWSR1, HNRNRA1, HNRNPR, and SYNCRIP as well as 29 other PRMT1 interactors (Table S2). PRMT1 interacting proteins are associated with biological functions such as transcription regulation, RNA binding, cell cycle, kinase, apoptosis, and chromosome organization (Figure 3F). To search for interacting proteins that cooperate with PRMT1 in progenitor maintenance, loss-of-function analysis of 9 PRMT1 interactors with no known basic impact on overall cellular viability were undertaken using multiple siRNAs/shRNAs targeting each candidate. Among these 9 PRMT1 interactors tested, only CSNK1a1 depletion phenocopied PRMT1 loss in human keratinocytes (Figure 3G).
Figure 3. Tandem affinity purification of PRMT1-interacting proteins identified CSNK1a1.

(A) Western blots detecting the distribution of endogenous PRMT1 in fractions of human keratinocyte nuclear lysate from FPLC analysis using Superose 6 10/300GL column. The 669KD marker Thyroglobulin was detected in the fraction #12.5. (B) Schematic representation of TAP-PRMT1 fusion protein. (C) Western blots detecting the distribution of TAP-PRMT1 and endogenous PRMT1 in fractions of human keratinocyte lysate from FPLC analysis using Superose 6 10/300GL column. TAP-PRMT1 distributed in the same fractions as the endogenous PRMT1. (D) Western blots showing the efficiency of PRMT1 TAP purification, comparing control (left) versus TAP-PRMT1 (right). TAP-PRMT1 was efficiently eluted by imidazole and captured by streptavidin beads. (E) Colloidal Blue staining showing the proteins co-purified with either TAP-PRMT1 or tag-only control. (F) Network of PRMT1-interacting proteins identified by mass spectrometry, grouped by functional category. (G) Table of raw LC-MS/MS spectral counts and knockdown phenotype of PRMT1-interacting proteins.
Similar to PRMT1, CSNK1a1 depletion by 4 independent shRNAs impaired clonogenic function of undifferentiated keratinocytes (Figure 4A–F, Figure S3A). Also like PRMT1, impairing CSNK1a1 function genetically and pharmacologically, via CSNK1a1 knockdown and enzymatic inhibitors of CSNK1a1 kinase activity, respectively, de-repressed differentiation genes and suppressed proliferation genes (Figure 4G–J). CSNK1a1 depletion induced modest signs of increased DNA damage and apoptosis as well as altered cell cycle progression (Figure S3B–F). CSNK1a1 target genes were then compared to the PRMT1 target gene set using transcriptome profiling. CSNK1a1 loss altered a total of 1036 genes (Table S3), among which 243 genes overlapped with the gene set controlled by PRMT1 (p<1.5×10−117). These 243 genes controlled by both PRMT1 and CSNK1a1 were highly enriched for GO terms associated with proliferation (suppressed by loss of PRMT1-CSNK1a1) and epidermal differentiation (induced by their loss) (Figure 4K–L). Among these 243 overlapped genes, 216 (89%) of these 243 genes also overlapped with the known calcium-induced epidermal keratinocyte differentiation profile (Figure S4). Thus intact function of both PRMT1 and CSNK1a1 is required to sustain genes associated with keratinocyte proliferation and to repress premature expression of genes associated with terminal differentiation.
Figure 4. CSNK1a1 loss phenocopies PRMT1 loss in epidermal progenitors.

(A and D) qRT-PCR analysis of CSNK1a1 shRNA knockdown efficiency in primary human keratinocytes. (B and E) Clonogenic assays of human keratinocytes with CSNK1a1 RNAi or control. (C and F) Colonies > 1mm2 in clonogenic assays are quantified (n=2/group, p<0.001, ANOVA, error bars represent standard deviation). (G–J) qRT-PCR verification of array data showing mRNA levels of differentiation or proliferation associated genes with CSNK1a1 loss via CSNK1a1 knockdown or by the D4476 CSNK1a1 enzymatic inhibitor. (K) Heat map (right) and Venn diagram (left) illustrating the overlap between expression changes identified with PRMT1 loss and CSNK1a1 loss (P<1.5×10−117, Fisher’s Exact Test). Genes induced are colored in red, and repressed genes are colored in green. (L) GO (Gene Ontology) analysis demonstrating that the shared genes controlled by CSNK1a1 and PRMT1 are associated with differentiation (upregulated) and proliferation (downregulated).
CSNK1a1 directly interacts with PRMT1 and controls PRMT1 phosphorylation
In light of the overlap of CSNK1a1’s effects with PRMT1 in progenitor assays and gene regulation, the nature of the CSNK1a1-PRMT1 physical interaction was next examined. First, CSNK1a1’s interaction with endogenous PRMT1 was assessed by co-immunoprecipitation. PRMT1 antibody immunoprecipitated CSNK1a1 from keratinocyte lysates and the reverse was also true in that CSNK1a1 antibody immunoprecipitated PRMT1 (Figure 5A–B). To test if these two proteins may interact directly, far western analysis was next performed using purified recombinant proteins. Purified recombinant CSNK1a1 associated specifically with MBP-PRMT1, but not with MBP (Figure 5C–D), suggesting that the interaction between CSNK1a1 and PRMT1 may be direct. To localize this interaction within intact cells, proximity ligation assay (PLA) was performed in undifferentiated keratinocytes. Strong proximity signal overlapping with DAPI nuclear staining was observed with the two antibodies recognizing PRMT1 and CSNK1a1. This PLA signal was abolished by knockdown of either PRMT1 or CSNK1a1 (Figure 5E), confirming its specificity. These data indicate that PRMT1 and CSNK1a1 associate with each other within the nucleus.
Figure 5. CSNK1a1 directly interacts with PRMT1 and controls PRMT1 phosphorylation.

(A) Co-immunoprecipitation assay using PRMT1 antibody, detected with CSNK1a1 antibody. (B) Co-immunoprecipitation assay using CSNK1a1 antibody, detected with PRMT1 antibody. (C) Far-western analysis demonstrating the direct interaction between recombinant PRMT1 and CSNK1a1 proteins in vitro. MBP-PRMT1 and MBP recombinant proteins were spotted on nitrocellulose membrane, incubated with recombinant CSNK1a1, and detected by anti-CSNK1a1 antibody. (D) Ponceau S staining demonstrating the input for Far-Western analysis. (E) Proximity Ligation analysis showing the interaction between PRMT1 and CSNK1a1 in human keratinocytes. (F) Kinase assay. Recombinant CSNK1a1 and PRMT1 were incubated together in kinase assay buffer with ATP. Anti-phosphoSerine antibody was used to detect phosphorylation on PRMT1. Anti-GST antibody was used to demonstrate protein input. (G) Phosphorylated serines and threonines on PRMT1 protein detected using Mass Spectrometry.
The observed interaction between the PRMT1 arginine methyltransferase and the CSNK1a1 kinase raised the possibility that one protein might modify the other. To test this, we first performed kinase assays using purified recombinant proteins in vitro, which demonstrated strong phosphorylation of PRMT1 by CSNK1a1 (Figure 5F). To extend this finding to intact cells, PLA was performed using a pair of antibodies recognizing PRMT1 and phospho-serine/threonine in undifferentiated keratinocytes, in an approach that permits localization of proteins with specific post-translational modifications within the cell. Specific PLA signal was observed in the nucleus, and this PLA signal was abolished by CSNK1a1 kinase inhibitor (Figure S5A). In parallel to kinase assays, we performed methylation assays for PRMT1 in vitro. Although PRMT1 methylated its known substrate histone H4, it did not methylate CSNK1a1, suggesting that CSNK1a1 is not a substrate of PRMT1. We also compared H4R3 methylation mediated by PRMT1 with or without CSNK1a1 phosphorylation. No obvious difference was observed in these two conditions (Figure S5B). We further tested if CSNK1a1 may regulate PRMT1 activity in keratinocytes using the antibody ASYM24, which recognizes asymmetric arginine dimethylation on a subset of arginine-glycine-rich proteins (Boisvert et al., 2003). While PRMT1 knockdown strongly decreased the methylation levels on many substrates detectable by ASYM24, CSNK1a1 knockdown did not alter the methylation levels (Figure S5C). Taken together, these data established PRMT1 as a substrate of CSNK1a1, although CSNK1a1 does not appear to alter PRMT1’s methylation efficiency toward a subset of known substrates.
We further identified putative serine/threonine phosphorylation sites on PRMT1 using mass spectrometry, using PRMT1 purified from keratinocytes as well as recombinant PRMT1 incubated in a kinase reaction with recombinant CSNK1a1 (Figure 5G, Table S4). Two phosphorylated regions identified from keratinocyte extracts coincide with the regions identified by vitro kinase assay with recombinant proteins, suggesting that these two regions of PRMT1 may be phosphorylated by CSNK1a1 in primary human keratinocytes. To characterize the biological function of these phosphorylation sites, we expressed either wild-type or S/T-to-A mutant PRMT1 in keratinocytes while depleting endogenous PRMT1 using shRNA. S/T-to-A mutations in three regions impaired PRMT1’s function in suppressing premature differentiation: aa 284–289 (strongest effect), aa 55–57 (mild effect) and aa 102–105 (mild effect). The T-to-A mutations at T122, T179 and T183 did not disrupt PRMT1’s function in suppressing differentiation (Figure S5D). These data suggest that phosphorylation mediated by CSNK1a1 may be required for the intact function of PRMT1 in this setting.
CSNK1a1 controls PRMT1 genomic targeting
The nuclear localization of the observed PRMT1-CSNK1a1 association suggests they might effect their impacts on progenitor gene regulation via binding to the genome, a possibility strengthened by prior reports of PRMT1 interactions with several chromatin-associated proteins, including histone H4 and nuclear receptors. To assess this, PRMT1 ChIP-seq was performed in undifferentiated keratinocytes with and without CSNK1a1 inhibition. PRMT1 ChIP-seq, using two different antibodies, identified a total of 14,392 ChIP-seq peaks shared in both conditions (Figure 6A). PRMT1 peaks were enriched in open chromatin regions, with 98% of the peaks overlapped with the ENCODE DNase I hypersensitivity sites (DHS) in human keratinocytes (Figure S6A–B). A total of 37.7% of these peaks localized to promoters and intergenic distal regulatory regions (Figure S6C). 60.9% of PRMT1 regulated genes from our transcriptome analysis are correlated with PRMT1 genomic binding using GREAT analysis (McLean et al., 2010) (Figure S6D). These data are consistent with a potential role for PRMT1 target gene regulation at these genomic features.
Figure 6. CSNK1a1 controls PRMT1 chromatin localization to co-regulate gene expression.

(A) Venn diagram showing the overlap between the PRMT1 ChIP-seq peaks using two different antibodies to epitope-tagged PRMT1. (B) Summit-centered heatmap (+/−1Kb from ChIP-seq peak summit, 50bp resoulation) comparing PRMT1 ChIP-seq enrichment between CSNK1a1 inhibition and control conditions. PRMT1 enrichment is decreased > 2 fold in 2144 regions. (C) Gene Ontology (GO) terms of the genes associated with the 2144 PRMT1 ChIP-seq peaks that were decreased >2 fold with CSNK1a1 inhibition. (D) Pie chart showing the association percentage of the 243 PRMT1 and CSNK1a1 target genes with altered PRMT1 ChIP-seq peaks. (E) GO analysis of the 47 target genes associated with CSNK1a1-dependent PRMT1 ChIP-seq peaks and the % of genes for each term. (F) UCSC genome browser tracks of the AURKB gene locus showing PRMT1 binding in both control and CSNK1 inhibition conditions with two different antibodies. (G) UCSC genome browser track of the GRHL3 gene locus showing PRMT1 binding in both control and CSNK1 inhibition conditions with two different antibodies. (H) qPCR analysis of differentiation gene expression with PRMT1-GRHL3 double RNAi. (I) qPCR analysis of differentiation gene expression with CSNK1a1-GRHL3 double RNAi.
The impact of inhibiting CSNK1a1 enzymatic activity was assessed on this pattern of PRMT1 genomic binding. CSNK1a1 inhibition selectively reduced PRMT1 binding at 2,144 genome sites to <50% (Figure 6B), but not all PRMT1-binding sites (Figure S6E). Analysis of these CSNK1a1-dependent PRMT1 binding sites using GREAT (McLean et al., 2010) demonstrated an enrichment for genes related to epidermal differentiation and development (Figure 6C). Intersection of these CSNK1a1-dependent PRMT1 binding site-associated genes with the 243 PRMT1-CSNK1a1-co-regulated genes further revealed an overlap of 47 genes that are candidates for direct PRMT1-CSNK1a1 regulation (Figure 6D–E). These 47 genes represented PRMT1-bound and regulated genes dependent on CSNK1a1 function, and included both proliferation genes, such as AURKB, as well as epidermal differentiation genes, such as the pro-differentiation transcriptional activator, GRHL3 (Figure 6F–G).
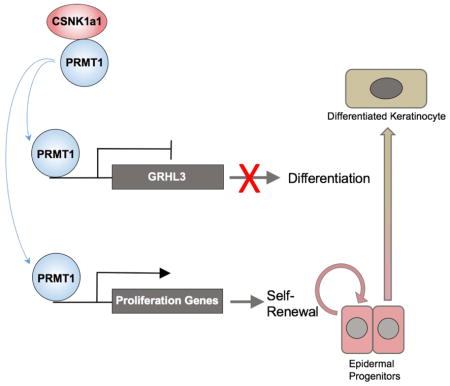
GRHL3 is normally suppressed in undifferentiated state, and was upregulated by both PRMT1 and CSNK1a1 loss. To test whether the upregulation of GRHL3 could be partially responsible for the premature differentiation observed in both PRMT1 and CSNK1a1 loss, double knockdown was performed. GRHL3 depletion in conjunction with either PRMT1 or CSNK1a1 knockdown impaired ectopic induction of differentiation genes, including S100A8 and SPRR1a (Figure 6H–I). Both genes have been demonstrated as GRHL3 target genes in published transcriptome profiling data (GSE37049). These data indicate that GRHL3 is a key downstream transcription factor whose repression is required for PRMT1 and CSNK1a1 to maintain progenitor function. Taken together, these data suggest a model in which CSNK1a1 phosphorylates PRMT1 to facilitate its genomic targeting to sustain proliferation genes involved in cell proliferation and to repress GRHL3 and other pro-differentiation genes to sustain the epidermal progenitor state (Figure 7).
Figure 7. Provisional working model of PRMT1-CSNK1a1 regulation of progenitor maintenance.

PRMT1 is enriched in progenitors and it associates with CSNK1a1 to maintain progenitor state in epidermal tissue. CSNK1a1 promotes direct PRMT1 targeting to proliferation genes to sustain self-renewal. CSNK1a1 also facilitates PRMT1 to suppress the differentiation activator GRHL3 to inhibit premature differentiation in progenitors.
Discussion
Here we present data indicating that both PRMT1 and CSNK1a1 are required for progenitor maintenance in self-renewing mammalian tissue, specifically in human and murine epidermis. PRMT1 expression is enriched in progenitor cells, where it sustains cellular proliferation genes and represses premature differentiation. In progenitors, PRMT1 directly associates with the CSNK1a1 kinase in the nucleus and loss of CSNK1a1 phenocopies PRMT1 inhibition. CSNK1a1 controls PRMT1 phosphorylation and regulates PRMT1’s genomic targeting. PRMT1’s genomic targeting to proliferation genes, such as Aurora Kinase B, and the differentiation activator, GRHL3, requires intact function of CSNK1a1. Taken together, these data support a provisional model in which CSNK1a1 directs PRMT1 to genomic targets that promote self-renewal and suppress terminal differentiation.
In the context of transcription regulation, PRMT1 has been generally considered as a transcriptional co-activator. The most well-characterized PRMT1 substrate is histone H4R3, which cooperates with histone acetylation to promote transcription (An et al., 2004; Huang et al., 2005; Strahl et al., 2001). PRMT1 can also associate with other transcription activators such as estrogen receptor alpha, PGC1a, HNF4 and p160 coactivator (Barrero and Malik, 2006; Koh et al., 2001; Métivier et al., 2003; Teyssier et al., 2005). A few recent studies in other cell types suggest that PRMT1 may also function as a repressor. PRMT1 methylation of PIAS1 is essential for PIAS1 targeting to and repressing STAT1 target genes. PRMT1 also methylates RelA on the DNA binding region and decreases RelA transcriptional activity. Hence the role of PRMT1 in transcription regulation depends on its interacting proteins.
PRMT1 loss impaired epidermal progenitor function in both mouse and human models, yet a couple of differences in gene expression were observed. First, the upregulation of 53BP1 in PRMT1 knockout mouse tissue was not detected in human keratinocytes with PRMT1 knockdown. Second, the down regulation of Ki67 mRNA expression by PRMT1 knockdown in human keratinocytes was not reflected by staining in knockout mouse tissue. The discrepancy of 53BP1 levels was likely due to the differences between knockout and knockdown. The remaining 10–20% of PRMT1 level from knockdown could still be sufficient to prevent DNA damage and the associated 53BP1 upregulation. Regarding the Ki67 level, one possibility could the differential gene regulatory mechanism between embryonic development and adult tissue homeostasis. It is also possible that the gene regulatory networks controlled by PRMT1 may not be 100% conserved between mouse and human.
38 PRMT1-interacting proteins were purified here from undifferentiated, progenitor-containing keratinocyte populations using tandem affinity purification coupled with LC-MS/MS. This purification not only confirmed multiple known interactors, it also revealed a number of undercharacterized PRMT1-interacting proteins. Although PRMT1 is required for progenitor function, 8 out of the 9 PRMT1-interacting proteins that we screened did not phenocopy PRMT1 loss. One possibility to account for this lack of phenotype is functional redundancy, another may be that total target knockout is required to unmask the impacts of some PRMT1-associated proteins. Regarding potential redundancy, YBX1 has 62% similarity to YBX3, and it may functionally compensate for the loss of YBX3. Future efforts at simultaneous multiplex disruption of multiple PRMT1-interacting proteins may identify additional functionally important interactors from among this pool of candidates.
Casein kinases are a group of highly conserved serine-threonine kinases that have been shown to modify substrates implicated in several signaling pathways, including those involving Wnt, Ras, NF-κB and p53 (Schittek et al., 2014). CSNK1a1 is essential for development. Homozygous deletion of CSNK1a1 in mice led to embryonic lethality before E6.5 (Elyada et al., 2011). Recent studies further pointed out non-redundant roles of CSNK1a1 in the progression of different cancer types including melanoma, leukemia and lung cancer (Bowman et al., 2015; Järås et al., 2014; Lantermann et al., 2015; Sinnberg et al., 2016).
The current work identifies CSNK1a1 as essential for epidermal progenitor maintenance in concert with PRMT1. The protein sizes of both CSNK1a1 and PRMT1 are under 50KDa, permitting their variable subcellular localization in different cell types to interact with different substrates (Burzio et al., 2002; Herrmann et al., 2005; Elyada et al., 2011). In human epidermal keratinocytes, CSNK1a1 and PRMT1 are both primarily enriched in the nucleus. The proximity ligation assays done here in epidermal keratinocytes also indicate that their interaction occurs in the nucleus. Since only a subset of CSNK1a1 target genes are co-controlled by PRMT1, it is possible that CSNK1a1 also interacts with additional regulators and targets to mediate its impacts on progenitor function.
Based on our ChIP-seq data, PRMT1 associates with both its activated and repressed target genes in epidermal progenitors. Both types of PRMT1 genome-binding events occur in open chromatin regions, as defined by their DNase I hypersensitivity. The cofactors that cooperate with PRMT1 to mediate gene activation and repression, however, are of significant interest for additional study.
The grainyhead family transcription factor, GRHL3, is expressed primarily in epithelial tissues. In epidermis, GRHL3 is upregulated in differentiated layers, and it directly promotes terminal differentiation marker genes (Gordon et al., 2014; Kretz et al., 2012). The dynamic regulation of GRHL3 level during the differentiation process remains incompletely understood. It has been previously demonstrated that GRHL3 transcript is fine-tuned by EXOSC9 in progenitors (Mistry et al., 2012). Our data demonstrated an independent regulatory mechanism whereby GRHL3 is also repressed by PRMT1 and CSNK1a1 at the transcriptional level to prevent premature activation of differentiation in progenitors. Other than GRHL3, a number of other transcription factors, such as ZNF750, KLF4 and OVOL1, are known to function non-redundantly as activators for keratinocyte differentiation. PRMT1, CSNK1a1 and GRHL3 therefore only control a subset of the epidermal differentiation program. The present work thus identifies a essential mechanism of progenitor maintenance involving the interaction between the arginine methyltransferase, PRMT1, and the serine/threonine kinase, CSNK1a1. CSNK1a1 modulates the genomic targeting of PRMT1 in association with its essential role in sustaining the expression of proliferation genes and repressing pro-differentiation genes, including GRHL3. Future studies on the functions of PRMT1 and its interacting proteins in different tissue types will further enhance understanding of arginine methylation in progenitor maintenance.
STAR Methods
Contact for Reagent and Resource Sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact Paul Khavari (khavari@stanford.edu).
Experimental Model and Subject Details
Cell Culture
Primary human keratinocytes were isolated from fresh surgically discarded newborn foreskin. Keratinocytes from 3–5 donors were mixed and cultured in complete Keratinocyte-SFM (Life Technologies #17005-142) and Medium 154 (Life Technologies #M-154-500). To induce differentiation, 1 million keratinocytes were seeded into each well in a 6-well plate overnight in normal medium. CaCl2 was added to the final concentration of 1.2mM the next day when the cells reached confluency.
Knockout Mice
PRMT1 floxed mice were generous gifts from Dr. Stephane Richard from McGill University. K14-Cre Mice were ordered from the Jackson Laboratory. After cross-breeding, 5 heterozygous mice and 5 homozygous knockout mice were analyzed. For genotyping, mouse toes or 3mm of tail tissue were cut with clean surgical scissors and heat in 75uL of Reagent A (25mM NaOH, 2mM EDTA) at 95°C for 1 hour. After cooling down to room temperature, tissue was mashed with pipette tip to aid the release of genomic DNA. 75uL of Reagent B (40mM of Tris-HCl, pH7.5) were then added to neutralize. 1uL of each tissue genomic DNA extraction was used in a 20uL PCR reaction for genotyping.
Method Details
Gene Transfer and Knockdown
Gene transfer by viral transduction was performed as described (Sen et al., 2010). In brief, amphotrophic phoenix cells were transfected with 3ug of DNA in 6-well plates. Viral supernatants were placed on keratinocytes for 1 hour with 5ug/mL polybrene for viral transduction. shRNAs targeting PRMT1 and CSNK1a1 were ordered from Openbiosystems. Additional PRMT1 shRNA-C and shRNA-D were designed with BLOCK-it RNAi designer (Invitrogen) and cloned into pSUPER. ON-TARGETplus siRNA smartpool targeting GRHL3 were ordered from Dharmacon. For siRNA knockdown, 1×106 cells were electroporated with 1 nmol siRNA using Amaxa Human Keratinocyte Nucleofector Kit (Lonza VPD-1002).
Progenitor Competition Assay
5×105 keratinocytes expressing control shRNA and one fluorescent protein were mixed with 5×105 keratinocytes expressing shRNA targeting PRMT1 and a different fluorescent marker. These two populations of cells were mixed to firstly regenerate epidermal in organotypic culture 1. The regenerated epidermis were further grafted on the back of immunodeficient mice 2. Tissues were harvested on day 10 and day 40 post-surgery for imaging analysis. The reverse experiment was performed by switching fluorescent proteins to exclude artifact due to the fluorescent protein used.
Protein Expression and Tissue Analysis
For immunoblot analysis, 20–50 μg of cell lysate was loaded per lane for SDS-PAGE and transferred to PVDF membranes. For immunofluorescence staining, tissue sections (7 μm thick) were fixed using either 50% acetone and 50% methanol, or 4% formaldehyde. Primary antibodies were incubated at 4°C overnight and secondary antibodies were incubated at room temperature for 1 hour. Images were taken using a Zeiss Observer Z1 fluorescence microscope, and the staining signals were quantified using Image J software. The antibodies used in this study for western blotting and immunostaining include: pAb-anti-PRMT1 (Millipore), ms-anti-PRMT1 (Santa Cruz), anti-CSNK1a1 (Santa Cruz), anti-Krt1 (Covance), anti-Krt10 (Neomarkers), anti-Loricrin (Covance), ms-anti-CollagenVII (Millipore), pAb-anti-CollagenVII (Calbiochem), GST (Cell Signaling), phosphoserine pAb (Invitrogen), phospho-threonine Ab (Invitrogen), Anti-phospho Histone H3 (Ser10) pAb (Millipore), Anti 53BP1 pAb (Novus), ASYM24 (Millipore).
ChIP-seq
Chromatin immunoprecipitation (ChIP) assays were performed essentially as previously described 3 with minor modifications. Human keratinocytes expressing HA-PRMT1 were dual cross-linked with 2mM DSG for 30min and 1% formaldehyde for 10min at room temperature. The chromatin was sonicated to achieve fragments with an average length between 200–500 bp. The sonicated chromatin was immunoprecipitated overnight at 4°C with ms-anti-HA (Santa Cruz) and pAb-anti-HA (Abcam). Following reverse-cross-linking, the samples were treated with RNase and Protease K, and the DNA was purified using the Qiagen PCR Purification Kit. For ChIP-seq, 10ng of purified ChIP DNA were used to generate sequencing library using NEBNext ChIP-Seq Library Prep Master Mix Set (New England BioLabs).
Tandem Affinity Purification and LC-MS/MS
Keratinocytes were trypsinized, washed in PBS, and resuspended in 5 cell pellet volumes of hypotonic buffer (10 mM HEPES at pH 7.6, 1.5 mM MgCl2, 10 mM KCl, 1× protease inhibitor cocktail (Roche)). Cells were lysed by addition of an equal volume of hypotonic buffer with 0.4% NP-40 for 2 min. Nuclei were pelleted by centrifugation at 4000 rpm and lysed in three cell pellet volumes of nucleus lysis buffer (50 mM Tris at pH 7.6, 0.05% igepal, 10% glycerol, 2 mM MgCl2, 250 mM NaCl, protease inhibitor cocktail). Nuclei were sheared with a 27.5-gauge needle, and lysis proceeded for 30 min. Insoluble material was removed by centrifugation at 13,000 rpm for 10 min, and nuclear supernatant was used for purification. His beads (Sigma) were added to nuclear supernatant for 1.5 hours, and were washed four times with 20mM Imidazole). Proteins were eluted off beads with EDTA for 2 hours. His eluate was diluted 4x, and was incubated with Myone Streptavidin (Invitrogen) for 1.5 hours. Purified proteins were boiled off from beads and separated on SDS-PAGE, stained with colloidal blue (Life Technologies), and analyzed by the Harvard Mass Spectrometry Facility by microcapillary reverse-phase high-pressure liquid chromatography (HPLC) nano-electrospray tandem mass spectrometry (μLC/MS/MS) on a Thermo LTQ-Orbitrap mass spectrometer.
Proximity Ligation Analysis
Primary human keratinocytes were cultured in differentiation condition on chamber slides, fixed by 4% Formaldehyde Solution (Thermo #28906) for 10 min at room temperature. Slides were blocked using blocking buffer (PBS with 0.3% TritonX-100, 3% normal horse serum) for 1 hour at room temperature, followed by primary antibody incubation overnight at 4 degree. Slides were washed by PBS three times the next day, and were subsequently incubated with the PLA probes anti-mouse and anti-rabbit (Sigma). Duolink In Situ Detection Reagents Orange (Sigma) were used for ligation and signal amplification.
Colony Formation Assay
Mouse fibroblast 3T3 cells were treated with 15 μg/mL mitomycin C (Sigma) in DMEM for 2 hours, then trypsinized and plated at 8×105 cells per well in a 6-well plate. The media was changed to KGM 24 hours after plating. 300 keratinocytes were seeded onto the feeder layer 24 hours after the media change. Media was changed every two days for 14 days. At the end of 14 days, the cells were washed with PBS to remove the 3T3 cells, then fixed in 1:1 acetone/methanol for 5 minutes. The plate was allowed to air dry for 3–5 minutes and then colonies were stained with crystal violet.
Enzymatic Inhibition Using inhibitors
CSNK1a1 inhibitor D4476 (Tocris) 4 was dissolved in DMSO, and was added at the final concentration at 60uM for 48 hours in keratinocytes. The PRMT1 inhibitor AMI-1 (Sigma) was dissolved in H2O, and was used at 120uM for 48 hours in keratinocytes.
Far Western
Recombinant MBP-PRMT1 and MBP proteins (Prospec) were spotted on nitrocellulose membrane. The membrane was blocked with 5% milk, incubated with recombinant CSNK1a1 (ThermoFisher Scientific) at 1ug/mL, and detected by anti-CSNK1a1. Ponceau S (Sigma) was used for loading control.
Kinase Assay
0.12ug of CSNK1a1 recombinant human protein (ThermoFisher Scientific) and 0.2ug of PRMT1 recombinant human protein (SignalChem) were combined in 25uL of kinase assay buffer (50 mM Tris-HCl, 10 mM MgCl2, 0.1 mM EDTA, 2 mM DTT, 0.01% Brij 35, 200 μM ATP, pH 7.5), and incubated for 60min at 30°C. The entire reactions were heat inactivated and separated on a SDS-PAGE gel. Anti-phospho-Serine antibody (ThermoFisher Scientific) were used at 2ug/mL to detect serine phosphorylation. Since both recombinant proteins have GST-tag, anti-GST (Cell Signaling 1:2000) were used to detect input.
Methylation Assay
Both wild-type and catalytic inactive (E143Q) recombinant GST-PRMT1 proteins were purchased from Active Motif. Histone H4 was purchased from NEB. CSNK1a1 was purchased from ThermoFisher Scientific. GST was purified by expressing pGEX-6p1 vector in BL21 and eluted from Glutathione beads (Sigma). For H4 methylation, 0.5ug H4 and 0.4ug PRMT1 were combined in 30uL of HMT buffer (50mM Tris 8.0, 5mM MgCl2, 20mM KCl, 3mM DTT, 1mM PMSF, 1uL SAM[3H]) at 30°C for 70min. To test the effect of CSNK1a1, 0.72ug CSNK1a1 and 0.5uL ATP were added to the reaction. GST alone incubated with H4 was used as negative control. To test if CSNK1a1 is a substrate of PRMT1, 0.72ug of CSNK1a1 and 0.4ug PRMT1 were combined in the HMT buffer. The entire reactions were heat inactivated and separated onto 15% SDS-PAGE. The gel was stained by commassie blue to visualize the input proteins, and was further dried and exposed to X-ray film for autoradiography of the reaction.
Annexin Staining
Keratinocytes were seeded at low confluence in 50/50 media and allowed to expand for 24 hours. Cells were trypsinized in 0.05% Trypsin+EDTA for 5 minutes then quenched with room temperature DMEM +10% FCS, centrifuged at 500xg and the pellet was resuspended in Annexin V staining buffer (10mM HEPES, 140mM NaCl, 2.5mM CaCl2, pH 7.4) and again centrifuged at 500xg. The pellet was resuspended in 50ul of Annexin V staining buffer supplemented with 2.5uL of Annexin V-APC (BD Phamingen) and propidium iodide (PI, BD Phamingen) for 15 minutes at room temperature as outlined in the BD Pharmingen Annexin V staining protocol. 200ul of Annexin V Staining buffer was then added to staining solution and the cells were interrogated on BD FACSCalibur. Annexin-negative/PI-Negative cells were counted as viable and Annexin-positive/PI-positive cells were counted as dead. Analysis was performed using FlowJo software (Tree Star, Oregon).
Cell Cycle Analyses
Keratinocytes were seeded at low confluence in 50/50 media and allowed to expand for 24 hours. Cells were pulsed with 10ul of 1mM BrdU per 1 mL of culture media for 6 hours. Cells were trypsinzed in 0.05% Trypsin+EDTA for 5 minutes then quenched with ice-cold DMEM + 10% FCS and centrifuged at 500xg at 4°C. The pellet was resuspended in PBS + 1% FCS and again centrifuged at 500xg at 4°C. Fixation, permeabilization and anti-BrdU staining was performed as outlined in the BD Pharmingen APC BrdU Kit (BD Pharmingen). Stained cells were interrogated on BD FACSCalibur. Analysis was performed using FlowJo software (Tree Star, Oregon).
Quantification and Statistical Analysis
qRT-PCR Expression Analysis
For qRT-PCR, total RNA was extracted using the RNeasy Plus (Qiagen) and subsequently subjected to reverse transcription using SuperScript VILO cDNA synthesis kit (Invitrogen). qRT-PCR analysis was performed using the Roche Lightcycler instrument with the SYBR Green Master Mix (Fermentas). Samples were run in duplicate and normalized to levels of GAPDH mRNA or 18S ribosomal RNA for each reaction. Primer sequences are listed in Supplemental Table S5. Statistical analysis such as ANOVA and t-test was calculated using GraphPad Prism7. Bar graphs and their associated error bars are represented as mean +/− standard deviation. All experiments were repeated minimally two independent times, and representative images were shown in the figures.
Mouse tissue staining quantification
A total of 10 images/condition were taken from control and knockout mouse tissue. Percentage of staining positive cells (positive/100 cells) were calculated based on all the cells in the epidermal tissue. Unpaired T-test were performed for statistical analysis using Prism7 (GraphPad).
DNA damage and Cell Cycle Analysis in Cultured Keratinocytes
Technical triplicates were conducted for 53BP1 and γH2Ax staining, TUNEL assay and FACS analysis for Annexin as well as Cell Cycle. Bar graphs are represented as mean +/− standard deviation. Statistical analysis using ANOVA was performed using Prism7 (GraphPad). Only p<0.05 was considered as statistically significant.
mRNA Expression Profiling Analysis
Amplification and labeling of cDNA probes and hybridization to the HG-U133 plus 2.0 microarray chip (Affymetrix) were performed by the Stanford PAN Facility. Data analysis was performed using R. Each data set for an experiment was filtered for probes that had an expression value ≥100 in at least 1 of the samples along with a p-value ≤ 0.05 based on SAM analysis. Pair-wise comparisons between the RNAi-treated samples and the control samples were performed to find probes that showed ≥ 2-fold expression change.
ChIP-seq analysis
ChIP-seq samples were sequenced by Stanford Functional Genomics Facility (SFGF) using Illumina NextSeq (1×74bp). Sequencing reads were mapped to human genome build hg19 using Bowtie, and ChIP-seq peaks were called using MACS14 (--bw 200, -m 10,50, FDR<0.05). GREAT analysis (great.stanford.edu) was used to correlate ChIP-seq peaks to their associated genes.
DATA AND SOFTWARE AVAILABILITY
ChIP-seq and transcriptome profiling data sets have been deposited with GEO accession # GSE81942.
Supplementary Material
1
Table S1. Genes differentially expressed between PRMT1 knockdown and control human keratinocytes, related to Figure 2
Table S2. Unique hits identified by TAP-PRMT1 MS-SPEC, related to Figure 3
Table S3. Genes differentially expressed between CSNK1a1 knockdown and control human keratinocytes, related to Figure 4
Table S4. Phospho peptides detected by mass spectrometry, related to Figure 5
Table S5. Primer sequences used in this study, related to the STAR Methods
2
3
4
Highlights.
- PRMT1 is enriched in progenitors and is down-regulated during differentiation.
- PRMT1 is essential for progenitor maintenance in murine and human epidermis.
- CSNK1a1 directly binds and phosphorylates PRMT1 and controls its genomic targeting.
- PRMT1 and CSNK1a1 cooperatively suppress GRHL3-mediated terminal differentiation.
Acknowledgments
We thank H Chang and A Oro for presubmission review, L Morcom and P Bernstein for administrative assistance. We are grateful for Dr. Stephane Richard for the generous gift of PRMT1 floxed mice. Profiling data has been deposited under GEO Accession Code GSE#81942. This work is supported by the U.S. Department of Veterans Affairs Office of Research and Development, by NIH R01 AR45192 (P.A.K), and by a K99/R00 Award (R00AR065480) to X. B.
Footnotes
Author Contributions
XB and PAK designed this study. XB conducted most of the experiments. ZS and RS contributed Figure S1E–J, Figure S2C–E and Figure S3B–D. BZ and AM performed PRMT1 TAP-tag purification. ER and ZS conducted FACS analysis for Figure S2 F–G and Figure S3 E–F. NN and JW helped with arginine methyltransferase assay as shown in Figure S5B. KQ and DW analyzed microarray data. DW and XB co-developed mosaic progenitor competition assay in vivo with fluorescent labeling. AR and GW helped with functional analysis of PRMT1 phosphorylation. ST provided technical support for mouse genotyping. XB and PAK wrote this paper in consultation with the other authors.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- An W, Kim J, Roeder RG. Ordered cooperative functions of PRMT1, p300, and CARM1 in transcriptional activation by p53. Cell. 2004;117:735–748. doi: 10.1016/j.cell.2004.05.009. [DOI] [PubMed] [Google Scholar]
- Bao X, Rubin AJ, Qu K, Zhang J, Giresi PG, Chang HY, Khavari PA. A novel ATAC-seq approach reveals lineage-specific reinforcement of the open chromatin landscape via cooperation between BAF and p63. Genome Biol. 2015;16:284. doi: 10.1186/s13059-015-0840-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrero MJ, Malik S. Two functional modes of a nuclear receptor-recruited arginine methyltransferase in transcriptional activation. Mol Cell. 2006;24:233–243. doi: 10.1016/j.molcel.2006.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedford MT, Clarke SG. Protein arginine methylation in mammals: who, what, and why. Mol Cell. 2009;33:1–13. doi: 10.1016/j.molcel.2008.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boisvert FM, Côté J, Boulanger MC, Richard S. A proteomic analysis of arginine-methylated protein complexes. Mol Cell Proteomics. 2003;2:1319–1330. doi: 10.1074/mcp.M300088-MCP200. [DOI] [PubMed] [Google Scholar]
- Bowman BM, Sebolt KA, Hoff BA, Boes JL, Daniels DL, Heist KA, Galbán CJ, Patel RM, Zhang J, Beer DG, et al. Phosphorylation of FADD by the kinase CK1α promotes KRASG12D-induced lung cancer. Sci Signal. 2015;8:ra9. doi: 10.1126/scisignal.2005607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burzio V, Antonelli M, Allende CC, Allende JE. Biochemical and cellular characteristics of the four splice variants of protein kinase CK1a from zebrafish (Danio Rerio) J Cell Biochem. 2002;86:805–814. doi: 10.1002/jcb.10263. [DOI] [PubMed] [Google Scholar]
- Feng J, Liu T, Qin B, Zhang Y, Liu XS. Identifying ChIP-seq enrichment using MACS. Nat Protoc. 2012;7:1728–1740. doi: 10.1038/nprot.2012.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gary JD, Gary JD, Yang MC, Clarke S, Herschman HR. The Mammalian Immediate-early TIS21 Protein and the Leukemia-associated BTG1 Protein Interact with a Protein-arginine N-Methyltransferase. J Biol Chem. 1996;271:15034–15044. doi: 10.1074/jbc.271.25.15034. [DOI] [PubMed] [Google Scholar]
- Gordon WM, Zeller MD, Klein RH, Swindell WR, Ho H, Espetia F, Gudjonsson JE, Baldi PF, Andersen B. A GRHL3-regulated repair pathway suppresses immune-mediated epidermal hyperplasia. J Clin Invest. 2014;124:5205–5218. doi: 10.1172/JCI77138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrmann F, Lee J, Bedford MT, Fackelmayer FO. Dynamics of Human Protein Arginine Methyltransferase 1(PRMT1) in Vivo. J Biol Chem. 2005;280:38005–38010. doi: 10.1074/jbc.M502458200. [DOI] [PubMed] [Google Scholar]
- Himes AD, Raetzman LT. Premature differentiation and aberrant movement of pituitary cells lacking both Hes1 and Prop1. Dev Biol. 2009;325:151–161. doi: 10.1016/j.ydbio.2008.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang S, Litt M, Felsenfeld G. Methylation of histone H4 by arginine methyltransferase PRMT1 is essential in vivo for many subsequent histone modifications. Genes Dev. 2005;19:1885–1893. doi: 10.1101/gad.1333905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Järås M, Miller PG, Chu LP, Puram RV, Fink EC, Schneider RK, Al-Shahrour F, Peña P, Breyfogle LJ, Hartwell KA, et al. Csnk1a1 inhibition has p53-dependent therapeutic efficacy in acute myeloid leukemia. J Exp Med. 2014;211:605–612. doi: 10.1084/jem.20131033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh SS, Chen D, Lee Y-H, Stallcup MR. Synergistic enhancement of nuclear receptor function by p160 coactivators and two coactivators with protein methyltransferase activities. J Biol Chem. 2001;276:1089–1098. doi: 10.1074/jbc.M004228200. [DOI] [PubMed] [Google Scholar]
- Kretz M, Webster DE, Flockhart RJ, Lee CS, Zehnder A, Lopez-Pajares V, Qu K, Zheng GXY, Chow J, Kim GE, et al. Suppression of progenitor differentiation requires the long noncoding RNA ANCR. Genes Dev. 2012;26:338–343. doi: 10.1101/gad.182121.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biology. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lantermann AB, Chen D, McCutcheon K, Hoffman G, Frias E, Ruddy D, Rakiec D, Korn J, McAllister G, Stegmeier F, et al. Inhibition of casein kinase 1 alpha prevents acquired drug resistance to Erlotinib in EGFR-mutant non–small cell lung cancer. Cancer Res. 2015;75:4937–4948. doi: 10.1158/0008-5472.CAN-15-1113. [DOI] [PubMed] [Google Scholar]
- McLean CY, Bristor D, Hiller M, Clarke SL, Schaar BT, Lowe CB, Wenger AM, Bejerano G. GREAT improves functional interpretation of cis-regulatory regions. Nat Biotechnol. 2010;28:495–501. doi: 10.1038/nbt.1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Métivier R, Penot G, Hübner MR, Reid G, Brand H, Koš M, Gannon F. Estrogen Receptor-α Directs Ordered, Cyclical, and Combinatorial Recruitment of Cofactors on a Natural Target Promoter. Cell. 2003;115:751–763. doi: 10.1016/s0092-8674(03)00934-6. [DOI] [PubMed] [Google Scholar]
- Mistry DS, Chen Y, Sen GL. Progenitor function in self-renewing human epidermis is maintained by the exosome. Cell Stem Cell. 2012;11:127–135. doi: 10.1016/j.stem.2012.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawlak MR, Scherer CA, Chen J, Roshon MJ, Ruley HE. Arginine N-methyltransferase 1 is required for early postimplantation mouse development, but cells deficient in the enzyme are viable. Mol Cell Biol. 2000;20:4859–4869. doi: 10.1128/mcb.20.13.4859-4869.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rena G, Bain J, Elliott M, Cohen P. D4476, a cell-permeant inhibitor of CK1, suppresses the site-specific phosphorylation and nuclear exclusion of FOXO1a. EMBO Rep. 2004;5:60–65. doi: 10.1038/sj.embor.7400048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reuter JA, Ortiz-Urda S, Kretz M, Garcia J, Scholl FA, Pasmooij AMG, Cassarino D, Chang HY, Khavari PA. Modeling inducible human tissue neoplasia identifies an extracellular matrix interaction network involved in cancer progression. Cancer Cell. 2009;15:477–488. doi: 10.1016/j.ccr.2009.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schittek B, Sinnberg T. Biological functions of casein kinase 1 isoforms and putative roles in tumorigenesis. Mol Cancer. 2014;13:231. doi: 10.1186/1476-4598-13-231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuster-Gossler K, Cordes R, Gossler A. Premature myogenic differentiation and depletion of progenitor cells cause severe muscle hypotrophy in Delta1 mutants. Proc Natl Acad Sci. 2007;104:537–542. doi: 10.1073/pnas.0608281104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen GL, Reuter JA, Webster DE, Zhu L, Khavari PA. DNMT1 maintains progenitor function in self-renewing somatic tissue. Nature. 2010;463:563–567. doi: 10.1038/nature08683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinnberg T, Wang J, Sauer B, Schittek B. Casein kinase 1α has a non-redundant and dominant role within the CK1 family in melanoma progression. BMC Cancer. 2016;16:594. doi: 10.1186/s12885-016-2643-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strahl BD, Briggs SD, Brame CJ, Caldwell JA, Koh SS, Ma H, Cook RG, Shabanowitz J, Hunt DF, Stallcup MR, et al. Methylation of histone H4 at arginine 3 occurs in vivo and is mediated by the nuclear receptor coactivator PRMT1. Current Biology. 2001;11:996–1000. doi: 10.1016/s0960-9822(01)00294-9. [DOI] [PubMed] [Google Scholar]
- Teyssier C, Ma H, Emter R, Kralli A, Stallcup MR. Activation of nuclear receptor coactivator PGC-1 by arginine methylation. Genes Dev. 2005;19:1466–1473. doi: 10.1101/gad.1295005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Truong AB, Kretz M, Ridky TW, Kimmel R, Khavari PA. p63 regulates proliferation and differentiation of developmentally mature keratinocytes. Genes Dev. 2006;20:3185–3197. doi: 10.1101/gad.1463206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Bedford MT. Protein arginine methyltransferases and cancer. Nat Rev Cancer. 2013;13:37–50. doi: 10.1038/nrc3409. [DOI] [PubMed] [Google Scholar]
- Yu Z, Chen T, Hébert J, Li E, Richard S. A mouse PRMT1 null allele defines an essential role for arginine methylation in genome maintenance and cell proliferation. Mol Cell Biol. 2009;29:2982–2996. doi: 10.1128/MCB.00042-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Cheng X. Structure of the predominant protein arginine methyltransferase PRMT1 and analysis of its binding to substrate peptides. Structure. 2003;11:509–520. doi: 10.1016/s0969-2126(03)00071-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
1
Table S1. Genes differentially expressed between PRMT1 knockdown and control human keratinocytes, related to Figure 2
Table S2. Unique hits identified by TAP-PRMT1 MS-SPEC, related to Figure 3
Table S3. Genes differentially expressed between CSNK1a1 knockdown and control human keratinocytes, related to Figure 4
Table S4. Phospho peptides detected by mass spectrometry, related to Figure 5
Table S5. Primer sequences used in this study, related to the STAR Methods
2
3
4