Cancer-associated fibroblasts neutralize the anti-tumor effect of CSF1 receptor blockade by inducing PMN-MDSC infiltration of tumors (original) (raw)
. Author manuscript; available in PMC: 2018 Nov 13.
Published in final edited form as: Cancer Cell. 2017 Nov 13;32(5):654–668.e5. doi: 10.1016/j.ccell.2017.10.005
Summary
Tumor associated macrophages (TAM) contribute to all aspects of tumor progression. Use of CSF1R inhibitors to target TAM is therapeutically appealing, but has had very limited antitumor effects. Here, we have identified the mechanism that limited the effect of CSF1R targeted therapy. We demonstrated that carcinoma associated fibroblasts (CAF) are major sources of chemokines that recruit granulocytes to tumors. CSF1 produced by tumor cells caused HDAC2-mediated down-regulation of granulocyte-specific chemokine expression in CAF, which limited migration of these cells to tumors. Treatment with CSF1R inhibitors disrupted this cross talk and triggered a profound increase in granulocyte recruitment to tumors. Combining CSF1R inhibitor with a CXCR2 antagonist blocked granulocyte infiltration of tumors and showed strong anti-tumor effects.
Keywords: CSF1R, M-CSF, macrophages, fibroblasts, PMN-MDSC, granulocytes
In Brief
Kumar et al. show that CSF1R inhibition alters chemokine secretion by cancer associated fibroblasts, which attracts pro-tumor PMN-MDSCs and results in poor efficacy. Combined inhibition of CSF1R and CXCR2 blocks MDSC recruitment and reduces tumor growth, which is further improved by the addition of anti-PD-1.
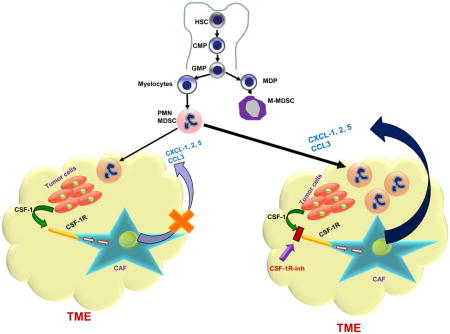
Introduction
Myeloid cells are critical components of the tumor microenvironment, and contribute to all aspects of tumor progression. These cells are comprised of tumor-associated macrophages (TAM), polymorphonuclear myeloid-derived suppressor cells, (PMN-MDSC), monocytic MDSC (M-MDSC), and dendritic cells (DC) (Gabrilovich et al., 2012; Grivennikov et al., 2010). There is now ample evidence that TAM and MDSC exert their pro-tumorigenic effects by suppressing T-cell functions and promoting tumor angiogenesis, proliferation, survival, and metastasis (Coussens and Pollard, 2011; Talmadge and Gabrilovich, 2013). CSF1(M-CSF)/CSF1R signaling is a key regulator of TAM differentiation and survival (Lin et al., 2001; Pixley and Stanley, 2004). CSF1R is a receptor tyrosine kinase that undergoes oligomerization and auto-phosphorylation upon binding to its ligands (CSF1 or IL-34). Mice lacking functional CSF1 ligands or receptors have significantly diminished macrophage populations (Guleria and Pollard, 2001). Consequently, targeting TAM by blocking the CSF1/CSF1R pathway has generated significant interest in recent years and there are a number of ongoing or recently completed clinical studies targeting this pathway (Ries et al., 2015). So far, clinical successes have been limited only to patients with diffuse-type giant cell tumor, which is characterized by translocations of chromosome 1 involving the CSF1 gene (Ries et al., 2014; Tap et al., 2015). However, in these patients, CSF1R inhibitors directly targeted tumor cells. Other clinical study examples include Hodgkin’s lymphoma patients treated with single agent PLX3397 (Butowski et al., 2016) and relapsed or refractory Hodgkin lymphoma patients who have been treated with a CSF1R inhibitor, JNJ-40346527 (Sasse et al., 2016).
In preclinical models, various small molecule inhibitors, such as Ki20227 (Ohno et al., 2006), PLX3397 (Mitchem et al., 2013; Mok et al., 2015; Sluijter et al., 2014), GW2580 (Conway et al., 2005), and BLZ945(Strachan et al., 2013), and antibodies, such as 5A1 (Lokeshwar and Lin, 1988), M279(MacDonald et al., 2010) have been studied in blocking the CSF1/CSF1R pathway. However, none of these single agent treatments showed therapeutic benefits. In the study where CSF1R inhibitor (BLZ945) did show regression of established tumors and increased survival in a mouse proneural glioblastoma multiforme model, no depletion of TAM was observed (Pyonteck et al., 2013). It remains unclear why the rather potent depletion of TAM in various tumor models has failed to deliver an antitumor effect. Recently, the focus has shifted to using combinations of CSF1R inhibitors with various other agents. Treatment with PLX3397 in combination with paclitaxel improved survival of mammary tumor-bearing mice (DeNardo et al., 2011b). In preclinical models of prostate cancer, PLX3397 treatment in combination with radiation therapy demonstrated an augmented and more durable response than irradiation alone (Xu et al., 2013). PLX3397 improved the efficacy of adoptive cell transfer immunotherapy in mouse melanoma models (Mok et al., 2014; Sluijter et al., 2014). PLX3397 treatment in pancreatic cancer models upregulated T-cell checkpoint molecules, specifically PD-L1 and CTLA-4, which restrained antitumor effect. When combined with PD1 and CTLA-4 antagonists, PLX3397 treatment elicited potent tumor regressions (Zhu et al., 2014).
Although data on combination therapy are encouraging, a lack of understanding of the mechanism that regulates tumor progression, even with substantial depletion of TAM, raised concerns regarding the potential clinical utility of these therapeutic strategies. The goal of this study was to understand the mechanism of the effects of CSF1R inhibitors on tumor progression.
Results
Inhibition of CSF1R signaling induces accumulation of PMN-MDSC in tumors
To elucidate the mechanism of CSF1R inhibition effect on microenvironment and tumor progression and we used a selective CSF1R inhibitor JNJ-40346527 (Genovese et al., 2015; Huang et al., 2013) given daily via oral administration (20 mg/kg). To test its effect on tumor growth we used subcutaneous C57BL6 mouse models of melanoma (B16F10), lung carcinoma (LLC), lymphoma (EL-4) and BALB/c models of colon carcinoma (CT26) and breast carcinoma (4T1). Treatment started one day after tumor inoculation and continued for 4–5 weeks. In addition, an orthotopic model of lung cancer (LLC), transgenic Ret melanoma and TRAMP prostate cancer models were used The transgenic Ret melanoma model is based on the expression of the human RET oncogene in melanocytes, which results in spontaneous development of melanoma metastasizing to different organs (Kato et al., 1998). Ret mice were treated starting at two months of age. In the TRAMP model of prostate cancer SV40 large T antigen is expressed in the prostatic epithelium (Greenberg et al., 1995). In this model, treatment was started at 5 months of age. In most tumor models, treatment with JNJ-40346527 did not delay tumor progression (Fig. 1A). However, this CSF1R inhibitor did have the expected effect on CD11b+F4/80+Gr-1− TAM. In all tested models, the proportion of TAM among CD45+ cells hematopoietic cells was dramatically reduced (Fig. 1B, Fig. S1A). While no significant difference was seen in the proportion of CD11b+Ly6ChiLy6G− monocytic cells (Fig. 1C), the proportion of CD11b+Ly6CloLy6G+ granulocytic cells was significantly increased (Fig. 1D). Granulocytes isolated from tumor tissues of JNJ-40346527 treated mice had potent immune suppressive activity (Fig. 1E), which characterized these cells as PMN-MDSC. Increase in PMN-MDSC was not the result of just a re-distribution between the proportions of myeloid cells, since treatment of mice with JNJ-40346527 significantly increased the absolute number of PMN-MDSC adjusted to tumor weight (Fig. 1F). In two models (LLC and EL-4) we also evaluated the presence of granulocytes by immunohistochemistry. Treatment with CSF1R inhibitor caused a significant increase in Ly6G+ granulocytes (Fig. S1B,C).
Figure 1. Effect of CSF1R inhibitor on tumor infiltration by PMN-MDSC.

A. Effect of JNJ-40346527 treatment on tumor growth in different tumor models. JNJ-40346527 was administered 6 days a week orally (20 mg/kg) from one day after tumor inoculation and continued until end of observation. Tumors size in mm2 is shown. Each group included 5–9 mice. Means and SD are shown. B–D. The proportion of cells in tumors from mice at the end of the treatment as described in Fig. 1A. Each group included 5–8 mice. Means and SD are shown. * - p<0.05, **- p<0.01, *** - p<0.001 in Student’s t-test from vehicle control group. B. CD11b+Gr-1−F4/80hi TAM. C. CD11b+Ly6ChiLy6G− monocytic cells. D. CD11b+Ly6CloLy6G+ granulocytic cells. E. Immune suppressive activity of Ly6G+ cells isolated from tumors of JNJ-40346527 treated mice. Ly6G+ cells were added at indicated ratios to splenocytes from Pmel transgenic mice and stimulated with cognate peptide. Proliferation was measured by 3H-thymidine incorporation in triplicates. Two experiments with the same results were performed. **- p<0.01, *** - p<0.001 in Student’s t-test from splenocytes alone. F. The absolute number of myeloid cells per gram tumor of JNJ-40346527 treated mice in indicated tumor models. See also Figures S1.
To evaluate the effect of JNJ-40346527 on palpable tumors, the treatment was started 10 days after tumor cell injection. After two weeks of JNJ-40346527 treatment PMN-MDSC were significantly increased in tumor-bearing (TB) mice (Fig. S1D). To make sure that the observed effect was not unique to JNJ-40346527, we treated LLC TB mice with CSF1R (CD115) antibody. CD115 antibody did not affect tumor progression (Fig. S1E), but caused a significant decrease in TAM and a significant increase of tumor PMN-MDSC (Fig. S1F). Thus, while inhibition of CSF1R caused substantial decrease in TAM, it was also associated with accumulation of PMN-MDSC in tumors.
Inhibition of CSF1R caused recruitment of PMN-MDSC to tumors
We observed only a small increase in the frequency of granulocytic cells in the bone marrow (BM), spleen (SP), or peripheral blood of mice treated with JNJ-40346527 (Fig. 2A) and CD115 antibody (Fig. S1G). No significant increase of monocytic cells was evident in BM and SP (Fig. S1H). Enriched hematopoietic progenitor cells (HPC) were cultured with GM-CSF and JNJ-40346527 with and without tumor explant supernatants (TES). Three-day treatment in the absence (Fig. S2A,B) or presence (Fig. S2C,D) of TES did not affect differentiation of myeloid cells, whereas after 6 days in culture JNJ-40346527 caused a significant decrease in the presence of macrophages, but had little effect on granulocytic cells (Fig. S2E,F).
Figure 2. CSF1R inhibitor causes recruitment of PMN-MDSC to tumors.

A. The proportion of CD11b+Ly6CloLy6G+ PMN-MDSC in different tissues of mice treated with JNJ-40346527 at the end of the treatment. Each group included 3–4 mice. * - p<0.05 in Student’s t-test from vehicle only group. B. Proportion of myeloid cells in tumors evaluated at different time during the treatment of LLC-TB mice with JNJ-40346527. Each group included 4 mice. Mean and SD are shown. * - p<0.05 from vehicle control group. C–D. The proportion and absolute number of PMN-MDSC of LLC-TB mice treated with JNJ-40346527. in spleen (C) in BM (D) E–F Proportion and absolute number of M-MDSC of LLC-TB mice treated with JNJ-40346527 in spleen (E) in BM (F) Mean and SD are shown (n=4). * - p<0.05 from vehicle control group. See also Figures S2 and S3.
We next investigated the kinetics of changes in myeloid cells in LLC TB mice. In this model, treatment with JNJ-40346527 did not significantly affect tumor growth until day 16. By day 23 tumor size in the treated group was significantly higher than in untreated mice (Fig. 1A). JNJ-40346527 caused a decrease in the presence of TAM only by day 23 (Fig. 2B). Tumor progression in untreated mice was associated with accumulation of PMN-MDSC in the tumor, SP and BM (Fig. 2C–F). Eight-day treatment with JNJ-40346527 resulted in a small but significant increase in the presence of PMN-MDSC in tumors. This increase became larger by day 16, and further increased by day 23 (Fig. 2B). In contrast, JNJ-40346527 induced accumulation of PMN-MDSC in SP and BM only by day 23 when tumors in the treated group were significantly larger than in untreated mice (Fig. 2C,D). Similarly, JNJ-40346527-induced accumulation of M-MDSC in spleens and BM was evident only on day 23 (Fig. 2E,F). JNJ-40346527 increased the number of common myeloid progenitors and granulocyte-macrophage progenitors in BM only by day 23 (Fig. S3A). There was no significant difference in BM CFU-GM colonies on day 9 between untreated and JNJ-40346527 treated mice and a small increase in colonies was observed on day 16 (Fig. S3B). Taken together, these results indicate that JNJ-40346527 induced accumulation of PMN-MDSC in the tumor site was not the result of initial expansion of these cells in BM and SP, and that this accumulation preceded accelerated tumor progression.
Mechanism of CSF1R inhibitor mediated PMN-MDSC recruitment to tumors
In an attempt to understand the mechanism of PMN-MDSC migration to tumor sites, we collected tumors from control and JNJ-40346527 treated LLC TB mice and evaluated expression of various chemokines in tumor cell lysates. We observed significant up-regulation of most chemokines responsible for granulocyte recruitment (Fig. 3A). Similar results were seen in the TRAMP model (Fig. 3B). Some of the chemokines were not specific to granulocytes and could recruit monocytic cells and macrophages. More detailed studies were performed with Cxcl-1, which plays a major role in recruitment of neutrophils and PMN-MDSC to tumor site (Noman et al., 2015; Obermajer et al., 2011). Proteins concentrations were measured in LLC tumor lysates at different times during the treatment. After 9 days, JNJ-40346527 caused modest but significant up-regulation of Cxcl-1, this difference became more pronounced by day 16. By day 23 this difference decreased, which correlated with a substantial increase in tumor size in the untreated mice (Fig. 3C). PDGFRα+ carcinoma associated fibroblasts (CAF), CD45+CD11b+ myeloid cells, and CD45−EPCAM+ tumor cells were sorted directly from LLC tumors on day 16 after inoculation and the amount of Cxcl-1 was measured in cell lysates by ELISA. CAF were found to be the major producers of Cxcl-1 (Fig. 3D). Treatment with CSF1R inhibitor did not affect the amount of Cxcl-1 secreted by tumor or myeloid cells but did cause increased release of Cxcl-1 by CAF (Fig. 3E). A similar effect was observed in CAF in CD115 antibody treated mice (Fig. 3F). Three-week treatment of LLC TB mice with JNJ-40346527 caused decrease in the expression of CXCR2 (a major Cxcl-1 receptor on granulocytes) on BM granulocytes, but the receptor expression remained unchanged in the EL-4 and B16F10 model (Fig. S3C). CSF1R inhibitor did not affect the proportion of CAF in the tumor microenvironment.
Figure 3. Granulocytic chemokines in tumors of CSF1R inhibitor treated mice.

A and B. Expression of mRNA (by qPCR) of indicated chemokines in tumor tissues treated with JNJ-40346527 for 16 days or 2 weeks respectively. LLC-TB mice (A) TRAMP mice (B) Mean and SD (n=3) are shown. * - p<0.05 in Student’s t-test from vehicle control group. C. Amount of Cxcl1 (measured in lysates by ELISA) in tumors of mice treated with JNJ-40346527 for 9, 16 or 23 days as described. Mean and SD (n=4) are shown * - p<0.05; ** - p<0.01 in Student’s t-test from vehicle control group. D. Endogenous level of Cxcl1 measured in lysates by ELISA in different cells isolated from tumors of LLC-TB mice. Fibroblasts – PDGFRa+ cells, tumor cells – CD45-EPCAM+, myeloid cells – CD45+CD11b+. Mean and SD are shown. * p<0.05; *** p<0.001 in in Student’s t-test from fibroblasts values. E. Cxcl1 in different cells isolated from tumors of LLC-TB mice treated with JNJ-40346527 for 16 days. ** - p<0.01 in Student’s t-test from vehicle control group (n=3). F. Cxcl-1 in fibroblasts isolated from tumors of LLC-TB mice treated with CD115 antibody twice a week for 3 weeks. *** - p<0.001 in Student’s t-test from vehicle control group (n=3). G. Cxcl1 expression by qPCR in pancreatic ductal adenocarcinoma treated with FAP specific CAR T-cells. Tumors were collected 3 days after the injection of second dose of CAR T-cells. * - p<0.05 from control (n=4). H. Ly6G+ cells in tumor tissues of mice treated with FAP CAR-T cells with atypical staining (red – Ly6G, blue-DAPI), scale bar = 50 μm and quantitative results shown (n=4). ** - p<0.05 in Student’s t-test from control. I. Ly6G+ cells per mm2 in tumor tissues of mice treated with FAP CAR-T cells. Tumors were collected 3 days after CAR-T cell injection. * - p<0.05 in Student’s t-test from control (n=4). J. Cxcl1 expression (by qPCR) in AE17.ova mesothelioma tumors from mice treated with FAP specific CAR-T cells. * - p<0.05 from control.
To test whether CAF could be directly involved in the recruitment of PMN-MDSC to the tumors, we evaluated archived tumor tissues from mice with 4662 pancreatic ductal adenocarcinoma (PDA) treated with fibroblast activation protein (FAP) - specific CAR-T cells, which selectively target and deplete CAF. FAP CAR-T cells had been shown to reduce tumor growth in these mice (Lo et al., 2015; Wang et al., 2014). Tumors were collected 3 days after the injection of a second dose of FAP-CAR-T cells. We found that FAP-CAR-T cell induced depletion of CAF was associated with significant reduction in Cxcl1 mRNA in tumors (Fig. 3G) and a significant decrease in the presence of PMN-MDSC (Fig. 3H). Since treatment with FAP-CAR-T cells substantially reduced tumor growth in mice, it was possible that observed changes in tumor PMN-MDSC were due to decreased tumor burden and not CAF depletion. To address this concern, we conducted a separate set of experiments in which we evaluated samples collected 3 days after initial FAP-CAR-T cells administration. We found that the number of PMN-MDSC significantly decreased (Fig. 3I). We also found significant decrease in expression of Cxcl1 in the tumors (Fig. 3J).
These data suggested that CAF could be important for PMN-MDSC migration to tumor sites. How could CSF1 regulate cytokine production by fibroblasts? First, we used primary lung fibroblasts isolated from naïve mice (Fig. S4A). We found that these cells expressed detectable CSF1R protein on the cell surface and cytoplasm (Fig. 4A), and also expressed Cxcl1 mRNA and Cxcl1 protein (Fig. 4B). Treatment of these cells with CSF1 caused up-regulation of CSF1R (Fig. 4A) and significant reduction of Cxcl1. This effect was abrogated when JNJ-40346527 was added during the last 48 hr of culture (Fig. 4B). To investigate whether this effect translated to PMN-MDSC migration, we isolated PMN-MDSC from BM of LLC TB mice. Supernatants were prepared from lung fibroblasts cultures treated with CSF1 with or without JNJ-40346527 and then were used in neutrophil migration assays. Supernatant from untreated fibroblasts caused robust migration of PMN-MDSC. CSF1 significantly reduced PMN-MDSC migration, but this effect was completely reversed by JNJ-40346527 (Fig. 4C). Treatment of BM PMN-MDSC with CSF1 did not change the expression of CXCR2 (Fig. S4B). CSF1 neutralizing antibody abrogated the inhibitory effect of CSF1 on fibroblast mediated PMN-MDSC migration (Fig. 4D). To confirm a specific role of Cxcl1 in this process, we used Cxcl-1 neutralizing antibody, which dramatically reduced PMN-MDSC migration caused by fibroblasts. Cxcl1 antibody abrogated the effect of JNJ-40346527 on PMN-MDSC migration (Fig. 4E), thus directly implicating Cxcl1 in PMN-MDSC migration induced by CSF1R inhibition. To determine the specific nature of CSF1 effect on Cxcl1 production by fibroblasts, we evaluated PMN-MDSC migration to recombinant Cxcl1 in the absence of supernatants from fibroblasts. Cxcl-1 caused a potent chemotactic effect on PMN-MDSC. However, this effect was not regulated by the addition of CSF1 or JNJ-40346527 (Fig. 4F). To test whether CSF1R inhibition affected the tumor microenvironment in vivo, LLC tumors were excised 14 days after the treatment of mice with JNJ-40346527. Tumor explant supernatants (TES) were prepared and tested in neutrophil migration assays. Despite the fact that TES from JNJ-40346527 treated mice had higher amounts of CSF1 (Fig. 4G), these TES caused significantly higher neutrophil migration than the TES from vehicle treated mice (Fig. 4H).
Figure 4. Effect of CSF1 on fibroblasts’ ability to recruit PMN-MDSC.

A. Expression of CD115 on the surface of fibroblasts tested by FACs and in cell lysates of cultured CAF tested by western blot B. Expression of Cxcl1 mRNA (qPCR) (left) and Cxcl-1 protein (ELISA) (right) in normal lung fibroblasts treated with Csf1 with and without JNJ-40346527. Mean and SD are shown (n=3). **- p<0.01, *** - p<0.001 in Student’s t-test from control group. C. Effect of Csf1 on fibroblast-mediated recruitment of BM PMN-MDSC. Chemotaxis of BM PMN-MDSC from LLC-TB mice was evaluated against supernatants obtained from fibroblasts cultured with Csf1 and Csf1R inhibitor. *- p<0.05 in Student’s t-test from control group (n=4). D. PMN-MDSC chemotaxis to supernatants from fibroblasts in presence of neutralizing Csf1 antibody (Ab). *- p<0.05 from control group in Student’s t-test (n=3). E. PMN-MDSC chemotaxis to supernatants from fibroblasts in presence of neutralizing CXCL1 Ab. **-p<0.01 from control group in Student’s t-test. F. PMN-MDSC chemotaxis to recombinant CXCL1 (100 ng/ml) in the presence of Csf1 and CSF1R inhibitor ***- p<0.001 in Student’s t-test from control group (n=3). G. Csf1 ELISA with TES collected from control or JNJ-40346527-treated LLC TB mice. *- p<0.05 in Student’s t-test from control group (n=4). H. PMN-MDSC chemotaxis in presence of TES collected from control or JNJ-40346527-treated LLC TB mice. *- p<0.05 in Student’s t-test from control group (n=4). I. LLC tumor growth kinetics in WT and CXCR2 KO mice with or without JNJ-40346527 treatment for indicated days. J. The absolute counts of PMN-MDSC and TAM in tumors of WT or CXCR2 KO TB mice treated with JNJ-40346527. *- p<0.05 in Student’s t-test from control group (n=4). See also Figure S4.
To clarify the role of PMN-MDSC migration in the JNJ-40346527 mediated effect in the tumor microenvironment in vivo, lethally irradiated recipient wild-type (WT) mice were reconstituted with BM from either WT or CXCR2 KO donor mice. Six weeks after reconstitution, the absence of CXCR2 expression on neutrophils was confirmed (Fig. S4C), and LLC cells were injected. Mice were treated with JNJ-40346527 for 12 days. In contrast to all other groups where tumors grew progressively; mice reconstituted with CXCR2-KO BM and treated with JNJ-40346527 had dramatically reduced tumor growth (Fig. 4I). While JNJ-40346527 caused significant increase in the presence of PMN-MDSC in WT BM recipients, in CXCR2 KO BM recipient, the presence of PMN-MDSC was practically undetectable (Fig. 5J) as anticipated (Chao et al., 2016). TAM were significantly decreased in all JNJ-40346527 treated mice (Fig. 5J).
Figure 5. Effect of CSF1 on fibroblasts.

A. Presence of myeloid cells in LLC tumors with deleted CSF1. WT and CSF1 deleted tumors were collected from mice 4–5 weeks after inoculation. Mean and SD are shown (n=4). *- p<0.05 from WT tumors. B. Expression (Means and SD) of chemokines mRNA (by qPCR) in these tumors. *- p<0.05, ***-p<0.001 in Student’s t-test from respective controls. C. PMN-MDSC chemotaxis in the presence of fibroblasts treated with supernatants from WT or CSF1 KO LLC. **-p<0.01 in Student’s t-test from control group (n=3). D. Supernatants were collected from fibroblasts with silenced csf1r expression (CSF1R shRNA) treated for 4 days with CSF1. Supernatants were used in chemotaxis of BM PMN-MDSC (n=3). *** - p<0.001 in Student’s t-test from control. E. Fibroblasts transduced with control or Csf1r shRNA were mixed at 1:1 ratio with LLC tumor cells and injected s.c. to mice. Twelve days later, the presence of myeloid cells in tumors was evaluated (n=4). Mean and SD are shown ** - p<0.01 in Student’s t-test from control. F. Expression of CSF1R (CD115) on cells in tumor microenvironment (CD3 T cells, FAP fibroblasts, CD163 macrophages) from 4 different patients. GeoMean is shown. G. Fibroblasts isolated from resected lung tissues of patients with NSCLC were treated for 4 days with CSF1 followed by JNJ-40346527 for additional 2 days. Expression of CXCL8 by qPCR (n=3) Mean and SD are shown. ** - p<0.01 in Student’s t-test from control. H. Migration of peripheral blood donor’s neutrophils to supernatants from fibroblasts cultured with CSF1 and CSF1R inhibitor (n=3), * - p<0.05 from control. Means and SD are shown. I. Peripheral blood donor’s neutrophils chemotaxis to supernatants from fibroblasts in presence of CXCL8 neutralizing antibody (n=3). * - p<0.05 in Student’s t-test from control. Means and SD are shown. See also Figures S4 and S5
Tumor cells expressed much higher amounts of Csf1 than CAF (Fig. S4D). Expression of Csf1 varied between different tumors with LLC cells expressing the highest level of Csf1 among tested cell lines (Fig. S4E). To verify the role of CSF1 in PMN-MDSC recruitment in vivo, we deleted Csf1 in LLC tumor cells using CRISPR/CAS9 technology (Fig. S4F). LLC clone lacking CSF1 production (Fig. S4G) was injected s.c. into the mice. Tumor cells with deleted Csf1 had delayed growth kinetics, but tumors still grew progressively (Fig. S4H). Mice with WT and Csf1 deleted tumors were sacrificed when tumors were of similar sizes and the presence of PMN-MDSC and chemokine profiles were measured. Lack of CSF1 in tumor cells resulted in a dramatic reduction of TAM but a significant increase in tumor PMN-MDSC (Fig. 5A). Deletion of Csf1 resulted in up-regulation of gene expression of several chemokines involved in granulocyte recruitment including Cxcl1 (Fig. 5B). Supernatant from WT but not from Csf1 deficient LLC inhibited migration of PMN-MDSC (Fig. 5C).
To verify the role of Csf1R in fibroblasts, we down-regulated this receptor in primary mouse lung fibroblasts using lentiviral shRNA constructs (Fig. S5A,B). Csf1 induced inhibition of PMN-MDSC migration was abrogated in fibroblasts transduced with Csf1R shRNA (Fig. 5D), and the same results were obtained with two different shRNA constructs. To test the role of CSF1R in fibroblasts in vivo, mice were injected s.c. with LLC and control or CSF1R silenced fibroblasts mixed with tumor cells at 1:1 ratio. Twelve days later, the absolute number of myeloid cells was counted per gram of tumor. CSF1R silenced fibroblasts did not affect the number of TAM and M-MDSC in tumors, but significantly increased the presence of PMN-MDSC (Fig. 5E).
We asked whether similar phenomena are also observed with CAFs. CAFs were isolated from LLC tumors and cultured with CSF1 with and without JNJ-40346527 or CSF1 antibody. CSF1 caused significant reduction in CXCL1 production, which was abrogated by both JNJ-40346527 and CSF1 antibody (Fig. S5C,D). Csf1 significantly inhibited CAF induced neutrophils migration. This inhibition was abrogated by JNJ-40346527 and by Csf1 antibody (Fig. S5E,F). We down-regulated Csf1R in CAFs using shRNA (Fig. S5G) and found that it cancelled CSF1 induced inhibition of neutrophil migration (Fig. S5H). Thus, tumor cells-derived CSF1 negatively regulated the production of chemokines recruiting PMN-MDSC to the tumor site by CAF. Inhibition of CSF1R signaling eliminated this down-regulation and caused accumulation of PMN-MDSC in tumors.
Regulation of human neutrophil’s migration by CSF-R inhibitor
To assess the above described mechanism in humans, we evaluated tumors from 4 cancer patients within 3 hr after surgical resection. Expression of CSF1R was evaluated in CD45+CD3+ T lymphocytes, CD45+CD163+ TAM, and CD45−FAP+ CAF (Fig. S5I). In all 4 patients CAF clearly demonstrated detectable surface expression of CSF1R. In contrast, T cells were negative for CSF1R. TAM had higher expression of CSF1R than CAF (Fig. 5F).
CAF were isolated from resected lung tissues of patients with non-small cell lung cancer (NSCLC) and were treated for 4 days with CSF1 followed by JNJ-40346527 for an additional 2 days. Fibroblasts expressed high levels of Cxcl8 – a major chemokine regulating migration of human neutrophils. CSF1 dramatically reduced its expression but JNJ-40346527 completely reversed this effect (Fig. 5G). Supernatant from fibroblasts treated as described above were used to stimulate the migration of neutrophils isolated from peripheral blood of healthy donors. Treatment of fibroblasts with CSF1 significantly reduced the ability of the supernatants to attract neutrophils, whereas JNJ-40346527 completely abrogated this effect (Fig. 5H). Importantly, in the absence of fibroblast supernatants neither CSF1, nor CSF1R inhibitor induced neutrophil migration (Fig. 5H). Adding a neutralizing CXCL8 antibody significantly reduced fibroblast-induced neutrophil migration and abrogated the effect of JNJ-40346527 on neutrophil migration (Fig. 5I).
To confirm the role of CSF1R in the regulation of human fibroblasts ability to attract neutrophils, the expression of the receptor was silenced using lentiviral shRNA constructs (Fig. S5J,K). Silencing of CSF1R in human fibroblasts abrogated the ability of CSF1 to suppress neutrophil migration by these cells (Fig. 6A). CXCL8 had potent activity to attract neutrophils. In the absence of fibroblast supernatants, neither CSF1 nor the combination of CSF1 with JNJ-40346527 affected neutrophil migration to CXCL8 (Fig. 6B). These data strongly suggested that in humans, CSF1 and CSF1R inhibitor could regulate fibroblast-mediated neutrophil migration. To determine a possible link between CXCL8, CSF1, and the accumulation of PMN-MDSC in the tumors of cancer patients, we collected samples from patients with colorectal cancer, breast cancer, and NSCLC. Populations of CD11b+CD14−CD15+ PMN-MDSC and CD14+HLA-DR−/lo M-MDSC in CD45+ hematopoietic cells from tumors were evaluated and CXCL8 and CSF1 proteins were measured in tumor lysates. Samples were split based on the median of CXCL8 amount. The presence of PMN-MDSC in the samples with CXCL8 level above the median was 5-times higher than in samples with CXCL8 level below the median (Fig. 6C). In contrast, no such correlation was found in the level of M-MDSC (Fig. 6D). In addition, no connection was observed between CXCL8 level in tumors and the presence of PMN or M-MDSC in peripheral blood in these patients (Fig. S6A). A similar analysis was performed for CSF1 but the results were reversed. The presence of PMN-MDSC in tumor samples with a CSF1 concentration above the median was 3 times lower than in samples with CSF1 level below the median (Fig. 6E). Again, no differences were observed in the presence of M-MDSC (Fig. 6F) and CSF1 amounts in tumor tissues did not correlate with the presence of PMN- or M-MDSC in peripheral blood (Fig. S6B). The level of CSF1 inversely related with the amount of CXCL8 in tumor tissues (Fig. 6G). These data indicate that in patients with at least 3 types of cancer lower amount of CSF1 in tumor tissues were associated with the increased production of CXCL8 and accumulation of PMN-MDSC.
Figure 6. Association between CSF1 and PMN-MDSC recruitment to human tumors.

A. Effect of CSF1R expression silencing in human fibroblasts on their ability to induce neutrophil migration. CSF1R was silenced in human fibroblasts using lentiviral shRNA construct and then cells were treated with CSF1 for 5 days. Supernatant was used in chemotaxis of donor peripheral blood neutrophils. Control – fibroblasts infected with lentivirus containing empty vector or the construct which did not down-regulate CSF1R. (n=3), ** - p<0.01 in Student’s t-test from control. Mean and SD are shown. B. Migration of peripheral blood donor’s neutrophils to recombinant CXCL8 (100 ng/ml) in the presence CSF1 and CSF1R inhibitor. Mean and SD are shown. n = 3. *- p<0.05 from control group. C–G. Evaluation of the proportion of tumor PMN-MDSC and M-MDSC and amount of cytokines in tumors of cancer patients. Values of individual tumor samples are plotted. The proportion of intratumoral CD11b+CD14−CD15+ PMN-MDSC among CD45+ cells evaluated by flow cytometry. CXCL8 in tumor cell lysates was measured by ELISA and split based on median (7.84 pg/μg) (C). CD14+HLA-DR−/lo M-MDSC among CD45+ cells (D). The proportion of PMN-MDSC in tumor samples split based on the amount of CSF1 present (median 7.5 pg/μg) (E). M-MDSC in samples split based on CSF1 amount in tumors (F). Amount of CXCL8 in tumor samples split based on CSF1 median (G). Mean and SD are shown. p values in Student’s t-test are shown on the graphs. H. Correlation between the amount of CSF1 and CXCL8 proteins in secretomes from 15 ovarian tumors (top panel). Spearman correlation coefficient was calculated. Analysis of the same samples based on median of CSF1 amount (bottom panel). Mean and SD are shown. p values in Student’s t-test I. Survival of patients with HNC based on the number of PMN-MDSC (CD15+LOX-1+) in tumor tissues. TMA contained samples from 58 patients with pathologically confirmed HNC. Median of PMN-MDSC presence per mm2 in all samples was 50. Survival log rank test was used. J. The number of PMN-MDSC in tumor tissues (per mm2) in TMA samples divided based on median score of M-CSF staining (160). K. Correlation between neutrophil and monocyte gene signatures in tumor samples (19,000 samples from Merck & Co., Inc., Kenilworth, NJ USA database) and expression of CSF1 and CSF2. **** - p<0.00001 calculated between indicated pairs. Mann-Whitney U test was used. The box-and-whisker plots are show. The box shows the median as well as 25th and 75th percentile. The whiskers stretch to the maximum and minimum values of that variable. See also Figure S6.
We verified this conclusion in different experimental systems. First, we evaluated the secretomes from 15 freshly isolated ovarian tumors, which included all major subtypes. We found a highly significant inverse correlation between the CXCL8 and CSF1 proteins (Fig. 6H). Since we have recently identified LOX-1 as a marker of human PMN-MDSC, which allows for detection of these cells in tumor tissues using immune fluorescent microscopy (Condamine et al., 2016), we then tested the clinical significance of LOX-1+CD15+ PMN-MDSC in tissue microarray (TMA) obtained from patients with head and neck cancer. The presence of PMN-MDSC was significantly associated with shorter survival in these patients (Fig. 6I). Using this TMA we also tested the link between the presence of PMN-MDSC and tissue CSF1 protein by immunohistochemistry (Fig. S6C). To quantify the association of CSF1 in tumors with the presence of PMN-MDSC, all samples were split into two groups based on the median level (160) of CSF1 expression evaluated using the H-score of intensity and percentage of positive cells. PMN-MDSC abundance in samples with above the median expression of CSF1 was significantly lower than in samples with lower than median expression of CSF1 (Fig. 6J). We evaluated de-convoluted neutrophil and monocyte signatures (Newman et al., 2015) in >19,000 samples from molecular profiling tumor database (Merck & Co., Inc., Kenilworth, NJ USA). These signatures were correlated with the expression of CSF1 and CSF2. Higher expression of CSF1 was associated with lower presence of cells with neutrophil signature (Fig. 6K, quadrants III and IV). The higher expression of CSF1 was associated with increased presence of cells with a monocyte signature (quadrants I and III). In contrast expression of CSF2 (which encodes GM-CSF) was associated with increased presence of cells with neutrophil signature (quadrants III and IV) (Fig. 6K). These results confirmed that CSF1 expression was negatively associated with the presence of PMN-MDSC in tumor tissues.
Mechanisms regulating inhibition of chemokines production by CSF1
To investigate the mechanism of CSF1R mediated regulation of Cxcl1 expression, we looked at the role of histone deacetylases (HDAC). Mouse fibroblasts were cultured for 6 days with CSF1 in the presence of entinostat, a class I HDAC inhibitor. CSF1 dramatically reduced Cxcl1 expression and Cxcl1 released by these cells. Entinostat completely abrogated this effect (Fig. 7A). To test this effect in vivo, LLC TB mice were treated with entinostat (10 mg/kg, p.o.) daily for 10 days starting from day 15 after tumor inoculation. Entinostat treatment induced substantial increase in the expression of several granulocyte-specific chemokines including Cxcl1 (Fig. 7B). Treatment of mice with entinostat despite causing increase in the amount of CSF1 in tumor cells (Fig. 7C), strongly up-regulated Cxcl1 in CAF (Fig. 7D). Entinostat did not affect the expression of CSF1R in CAF (Fig. 7E), and was associated with increased infiltration of tumor tissues by PMN-MDSC but not TAM in LLC and CT-26 tumor models (Fig. 7F).
Figure 7. The mechanisms regulating inhibition of chemokines production by CSF1.

A. mRNA for Cxcl1 (by qPCR) (left) and Cxcl1 protein by ELISA (right) in mouse fibroblasts cultured for 6 days with CSF1 in the presence (during last three days of culture) of class I HDAC inhibitor entinostat. Mean and SD (n=3) are shown. ***-p<0.001 in Student’s t test from control. B–F. The effect of entinostat in vivo. LLC TB mice were treated with entinostat (10 mg/kg, p.o.) daily for 10 days starting from day 15 after tumor inoculation. Expression of mRNA for chemokines (qPCR) in tumor tissues. **-p<0.01 in Student’s t test from control (n=3) (B). Tumor cells were isolated at the end of the treatment and the amount of CSF1 was measured by ELISA (n=6) (C). CAF were sorted from tumor tissues and the amount of Cxcl1 was measured in cell lysates by ELISA. Mean and SD are shown. ****-p<0.0001 in Student’s t test from control (n=4) (D). CSF1R amount in CAF (E). The presence of PMN-MDSC and TAM in tumors of LLC (n=10) and CT-26 (n=3) TB mice treated with entinostat. Mean and SD are shown *-p<0.05; **-p<0.01 in Student’s t test from control (F). G. Mouse fibroblasts were treated with CSF1 for 4 days, followed by 2-day treatment of entinostat. ChIP of Cxcl1 promoter was performed using acetylated histone H3 or acetylated histone H4 antibody. The results are expressed as DNA enrichment, normalized to corresponding input values. Control IgG was used as a negative control (n=3). H. ChIP of Cxcl1 promoter was performed using HDAC2 specific antibody (n=3).
To identify the specific HDAC that is recruited to the Cxcl1 promoter, we used a ChIP assay. CSF1 caused a significant decrease in the recruitment of acetylated H3 histone to cxcl-1 promoter. This decrease was completely reversed by entinostat (Fig. 7G). CSF1 induced significant increase in the recruitment of HDAC2 to Cxcl1 promoter (Fig. 7H), whereas no binding of HDAC1 was detected (data not shown). Thus, CSF1 caused recruitment of HDAC2 to Cxcl1 promoter, which silenced Cxcl1 expression.
Therapeutic targeting of PMN-MDSC and TAM
We hypothesized that the combination of CSF1R inhibitor with an inhibitor of granulocyte recruitment may have therapeutic effect. Because CXCR2 is the receptor for most chemokines up-regulated by the CSF1R inhibitor (including Cxcl-1), we used a selective inhibitor of CXCR2 (SB225002). Mice with established LLC or B16F10 tumors were treated with JNJ-40346527, SB225002 or combination of both. As expected, neither of the inhibitors alone had significant antitumor effect. However, the combination of inhibitors resulted in a highly significant delay in tumor progression in both cancer models (Fig. 8A). The presences of TAM and PMN-MDSC were evaluated in B16F10 tumors. Combined treatment abrogated JNJ-40346527 induced accumulation of PMN-MDSC, while maintaining decreased presence of TAM (Fig. 8B). We wanted to know whether two inhibitors provided additional therapeutic benefits if combined with check-point inhibitor, so we treated LLC TB mice with the combination of inhibitors and PD-1 antibody. The PD-1 antibody alone had a weak therapeutic effect and combination of PD-1 antibody with JNJ-40346527 or SB225002 did not enhance antitumor effect. However, the combination of the two inhibitors together with PD-1 antibody resulted in a dramatic antitumor effect (Fig. 8C).
Figure 8. Therapeutic targeting of granulocytes and macrophages.

A. Tumor growth in mice treated with JNJ-40346527 (CSF1R inhibitor), SB225002 (CXCR2 inhibitor), or their combination. Mean and SD are shown. Each group included 5 mice. Significance was calculated using two-way ANOVA test. B. The presence of TAM and PMN-MDSC in B16F10 tumors. **-p<0.01 from control (n=4). C. LLC TB mice were treated with inhibitors and PD-1 antibody (100 μg twice a week). Mean and SD are shown. Each group included 4–5 mice. Significance was calculated using a linear mixed-effect model with the random effect at individual animal level.
Discussion
In an attempt to elucidate a mechanism that limits the antitumor activity of CSF1R inhibitor, we have identified a crosstalk between tumor cells and CAF that limits the recruitment of PMN-MDSC to tumors. Fibroblasts are the key cells regulating multiple biological processes, including inflammation, wound healing, and tissue remodeling, and are intimately involved in tumor progression (Fearon, 2014; Gascard and Tlsty, 2016). CAF differentiate from pre-existing normal fibroblasts, myofibroblasts, preadipocytes, smooth muscle cells, or BM derived progenitors. Tumor cells activate stromal fibroblasts or recruited mesenchymal stem cells to become CAFs, which release a variety of chemokines that promote tumor growth and metastasis (Mishra et al., 2011). There is now ample evidence that CAF produce a large array of chemokines, including the ones specific for granulocytes, such as CXCL1 (Feig et al., 2013; Orimo and Weinberg, 2006). It has recently been reported that CAF also promote migration of monocytic cells to tumors (Yang et al., 2016), but the direct effect of fibroblasts on neutrophil migration has not been previously demonstrated. In our study, PMN-MDSC recruitment by CAF is consistent with the ability of CAF to produce granulocytic chemokines. However, this was not consistent with the observations that granulocytic infiltration of tumors is usually not very strong. In most types of cancer, granulocytes are present in smaller numbers than TAM and monocytic cells (DeNardo et al., 2011a; Kumar et al., 2016). Our data suggest an explanation for this phenomenon. Cxcl1 expression is down-regulated by CSF1, which is produced by many tumor cells (Lucas et al., 2008). This limits the migration of PMN-MDSC to the tumor site. In this study, we focused on Cxcl1 as the most important chemokine for granulocytic migration. However, it is likely that similar effects could be observed for other granulocytic chemokines as well.
In humans, CXCL1 is not a potent granulocytic chemokine. Instead, this role is played by CXCL8, but it appears that CSF1 has similar effects on human fibroblasts. More importantly, CSF1 levels in human tumors were inversely correlated with the presence of PMN-MDSC in tumors. This effect was not observed for M-MDSC, which demonstrates the specific nature of this interaction.
It is now known that PMN-MDSC contribute to tumor progression by inhibiting immune responses and promoting tumor angiogenesis, tumor cell invasion, metastasis, etc. (Gabrilovich, 2017). Ample evidence now supports a close association between MDSC accumulation in peripheral blood and clinical outcome in cancer patients (Messmer et al., 2015; Solito et al., 2014). A recent meta-analysis of 442 patients with various solid tumors demonstrated that peripheral blood MDSC were significantly associated with poor overall and progression-free survival (Zhang et al., 2016a). MDSC have also been implicated in resistance to anti-cancer therapies, including sunitinib (Finke et al., 2011), cisplatin, and other chemotherapeutics in lung cancer (Vetsika et al., 2014), and doxorubicin and melphalan in multiple myeloma (Ramachandran et al., 2016). Recent studies showed an association of a lower presence of MDSC with positive patient clinical responses to ipilimumab (Meyer et al., 2014) and PD-1 antibodies (Weber et al., 2016).
In human tumor tissues, CD15 or CD66b markers encompass both neutrophils and PMN-MDSC in human tumor tissues. Accumulation of these cells was associated with negative outcomes in a number of human cancers, including gallbladder carcinoma (GBC) (Zhang et al., 2016b), cholangiocarcinoma (Mao et al., 2015), cutaneous melanoma (Ladanyi, 2015), soft tissue sarcoma (Szkandera et al., 2013), and NSCLC (Carus et al., 2013). A recent analysis of 22 immune populations across human malignancies demonstrated that polymorphonuclear cell signatures emerged as the most significant adverse cancer-wide prognostic populations (Gentles et al., 2015).
These data raised an important question. If PMN-MDSCs have such strong pro-tumorigenic activity, then why is their recruitment limited? We believe that this mechanism is employed to limit strong inflammation at early stages of tumor development. PMN-MDSC, with their immune suppressive and pro-tumorigenic activity, are developed in response to tumor-derived factors and are a feature of a more advanced stage of cancer. In early stages, PMN-MDSC usually are not detected. Therefore, high expression of granulocytic chemokines may result in strong infiltration of neutrophils in the tumor microenvironment. Activated neutrophils are known to have some antitumor activity (Challacombe et al., 2006; Sionov et al., 2015), and their massive recruitment may be detrimental for tumors.
Our data suggest that the CSF1 granulocytic chemokine network can be exploited for therapeutic purposes. Combining CSF1R and CXCR2 inhibitors reduced the presence of both populations (TAM and PMN-MDSC) in tumor site. In contrast to a lack of antitumor effect of each inhibitor alone, combination treatment significantly reduced tumor growth. Moreover, it dramatically enhanced the effect of immunotherapy with PD-1 antibody. This may open an opportunity for therapeutic intervention.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for reagents may be directed to, and will be fulfilled by the corresponding author Dmitry Gabrilovich (dgabrilovich@wistar.org).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Human subjects and samples
Samples of peripheral blood and tumor tissues were collected from patients at Helen F. Graham Cancer Center and University of Pennsylvania. The study was approved by Institutional Review Boards of the Christiana Care Health System at the Helen F. Graham Cancer Center, University of Pennsylvania and The Wistar Institute. All patients signed approved consent forms. Peripheral blood (PB) and tumor tissues were collected at Helen F. Graham Cancer Center from 11 patients with previously untreated stage II-III non-small cell lung cancer (NSCLC), 8 patients with stage III-IV breast cancer, and 6 patients with stage III-IV colon adenocarcinoma. This cohort includes 14 females and 11 males, aged 48–74 years. Human fibroblasts were isolated from resected lung tissues from NSCLC patients.
Tissue Microarrays (TMA) were prepared, validated, and clinically annotated by Helen F. Graham Cancer Center tissue procurement and pathology department. Using the Quick-Ray Manual Tissue Mircoarrayer, each patient was selected and approved by a pathologist and later validated by three quality control evaluations. TMAs contained samples from 58 patients with pathologically confirmed head and neck cancer.
Tumor secretomes were prepared from fresh specimens that were stored on wet ice in RPMI cell culture media and were processed within 24 hr of collection. The tissue sample was minced into 1 × 1 mm slices and incubated in RPMI cell culture media at 37°C for 4h. After incubation, the tumor tissue was pelleted at 500 × g for 2 min at 4°C. The RPMI containing the secreted tissue proteins was removed, filtered using a 0.22 μm centrifugal filter unit, aliquoted, snap frozen, and stored at -80°C until analysis.
Mice and tumor models
All experiments with mice were approved by The Wistar Institute Animal Care and Use Committee. The mice were kept under pathogen-free conditions. C57BL/6 and Balb/c were obtained from Charles River Laboratories. In all experiments, adult (6–8 weeks old) female mice were used. EL4 thymoma, CT26 colon carcinoma, Lewis Lung Carcinoma (LLC), 4T1 mammary carcinoma, and B16F10 melanoma were obtained from ATCC. 4662 primary pancreatic tumor cells were derived from female KPC mouse model. To establish s.c. tumors, 5×105 tumor cells were injected into mice, which formed a tumor with a 1.5-cm diameter within 2–3 weeks of injection. For orthotropic models 105 LLC cells were injected i.v. Ret mice were obtained from Dr. Umansky (German Cancer Center, Heidelberg, Germany). TRAMP mice were bred in Dr. L. Languino laboratory (Thomas Jefferson University)
CSF1R inhibitor, JNJ-40346527 (Janssen) was administered at dose 20 mg/kg 6 days per week p.o. As a control, mice treated with vehicle alone (0.5% HPMC, hydroxypropyl methyl cellulose, dissolved in water) were used. CXCR2 antagonist SB225002 (Tocris Bioscience) was administered at dose 2mg/kg 6 days a week i.p. Treatment was started next day after tumor injection and continued until the mice were sacrificed. For the treatment with CD115 antibody, it was obtained from Bioxcell (Clone: AFS98) and administered twice a week for 3 weeks at dose 250 μg per mouse. The treatment with anti-PD-1 ab (100 μg; twice a week; clone RMP1–14, BioXcell) was started 1 week after tumor injection. Entinostat (10 mg/kg, p.o; Syndax Phermaceuticals) was administered daily for 10 days starting from day 15 after tumor inoculation.
METHOD DETAILS
Cell isolation and culture
Single-cell suspensions were prepared from spleen and BM followed by red blood cell removal using ammonium chloride lysis buffer. Single-cell suspensions from tumor tissues were prepared using Mouse Tumor Dissociation Kit according to the manufacturer’s recommendation (Miltenyi). HPC-enriched cells were purified from BM using kit from Miltenyi. Cells were then cultured in the presence of 20 ng/ml recombinant GM-CSF (Invitrogen) in RPMI (Biosource International) supplemented with 10% FBS, 5 nM glutamine, 25 mM HEPES and 1% antibiotics (Invitrogen). 20% v/v tumor explant supernatant from B16-F10 and LLC tumors were used in some experiments.
To obtain the single cell suspensions, human solid tumor tissues were subjected to an hour of enzymatic digestion using 0.1 mg/ml hyaluronidase (Sigma-Aldrich), 2 mg/ml collagenase (Sigma-Aldrich), 600 U/ml DNase (Sigma-Aldrich), and 0.2 mg/ml protease (Sigma-Aldrich) in RPMI 1640. The digested tissue was passed through a 70-μm mesh, and erythrocytes were removed by hypotonic lysis and washed thoroughly to remove debris.
For fibroblast isolation, resected lungs were chopped into pieces, followed by incubation with collagenase cocktail (250 ug/ml collagenase I, II, and IV in DMEM) on rotator for ~1–2 hr. The collagenase treatment was quenched by 10% FCS DMEM, cells were filtered, washed with PBS and incubated in complete media overnight. Next day, the monolayer was washed several times and fresh media was added for next 4 days until the cells were ready for treatment.
Flow cytometry
Antibodies specific for the mouse cell surface markers CD11b, Ly6C, Ly6G, Gr1, CD45, CD326 (EPCAM), and CD140a (PDGFRa) and for the human cell surface markers CD11b, CD14, CD33, CD15, CD3, and HLA-DR were purchased from BD Biosciences. Anti-human CD163 and CD115 antibodies were purchased from Biolegend, anti-human FAP and anti-mouse CXCR2 antibodies – from R&D Systems. F4/80 and CD115 were purchased from eBiosciences. The flow cytometry data were acquired using BD LSRII flow cytometer and analyzed by FlowJo Software (Tree Star).
qRT-PCR
RNA was extracted using total RNA extraction kit (Omega Bio-tek). cDNA was synthesized using cDNA reverse transcriptase kit (Applied Biosystems) and qPCR was performed in triplicates using 10 μl of SYBR Master Mixture (Applied Biosystems). The list of primers’ is attached in Supplemental Figs.
Immunofluorescence and immunohistochemistry
Mouse Tumor tissues were snap-frozen and slides were fixed with acetone and blocked for 30 min with 5% BSA containing Fc block (1:100dilution) in 1X PBST. The primary antibody (Ly6G; clone 1A8; BD Biosciences) was used at 1:100 dilutions for 1 hr at RT, whereas the secondary antibody (AF 594; goat anti-rat; Life Technologies) was used at 1:400 dilution. Nuclei were stained using DAPI (1: 5000 dilutions in PBS, Life technologies). Images were obtained using Leica TCS SP5 Confocal microscope. During imaging, the gain (voltage), laser intensity settings and imaging parameters were kept constant between all the samples. Cell number was calculated per mm2.
Paraffin-embedded human tissue microarray sections were deparaffinized and rehydrated, followed by heat-induced antigen retrieval. For immunofluorescence staining, mouse monoclonal anti-human CD15 (BD Biosciences) and rabbit polyclonal anti-human LOX-1 (Abcam) antibodies were used. Goat anti-mouse IgG Alexa Fluor 514 and goat anti-rabbit IgG Alexa Fluor 647 (Life Technologies) antibodies were used as secondary antibodies. Cell nuclei were stained with DAPI (Life Technologies). Imaging was performed using a Leica TCS SP5 confocal microscope. Sixteen frames acquired with a 63X objective lens were used to calculate the cell count per mm2. For immunohistochemistry staining of human M-CSF, tissue microarray sections were incubated with primary antibody, rabbit polyclonal anti-human M-CSF (Abcam). Biotinylated goat anti-rabbit IgG, Avidin/Biotin block kit, ABC HRP kit, and DAB substrate kit (Vector Laboratories) were used for detection and visualization.
H-score method was used to evaluate the percentage of positive stained cells and staining intensity. Average intensity of staining in positive cells was assigned as an intensity score (0 = none; 1 = weak; 2 = moderate; 3 = strong). The score was obtained by the formula: 3 x percentage of strongly stained cells + 2 x percentage of moderately stained cells + 1 x weakly stained cells.
Generation of CSF1 KO tumor line using CRISPR-Cas9
Murine csf-1 gene (gene ID number 12977) was used as target to design DNA guides. The three isoforms of the murine gene were compared and a homologous region at the beginning of the gene sequence was chosen. From this region, the 120-nucleotide sequence 3731 to 3853, that belongs to a CDS common in all three were selected: acatggctgggctcccggctgctgctggtctgtctcctcatgagcaggagtattgccaaggaggtgtcagaacactgtagccacatgattgggaatggacacctgaaggtcctgcagcag
This sequence was introduced in the Massachusetts Institute of Technology software CRISPR Design and six guides with low frequency of off-site sites were selected. These guides were synthetized as pairs of annealed oligos with overhangs cohesive to the digestion product of BmsBI restriction enzyme. Oligoduplexes were ligated to the vector pLentiCRISPRv2 and transformed to Stbl3 bacteria. pLentiCRISPRv2 vectors containing the sgRNA guides were transfected with pMD2.G and pSPAX2 to 293T cells in order to produce lentiviral vectors. After 24 hr’ incubation, supernatants were collected, filtered through a 0.45um filter and added to monolayers of LLC tumor cells. Lentiviral vectors were produced from a pLentiCRISPRv2 plasmid without the sgRNA guides. Three days later, 3 ug/ml of puromycin was added to the culture and knock out of csf-1 was detected by ELISA.
ELISA
The expression levels of CXCL1, CXCL12, CXCL8 or CSF1 in supernatants, cell lysates or tumor cell lysates were evaluated using ELISA kit as per manufacturers’ instructions (R&D Biosystems).
Primers are provided in Table S1 related to STAR Methods
Chromatin immunoprecipitation (ChIP) assay
ChIP was performed using the acetyl-histone immunoprecipitation assay kit (Millipore). Briefly, histone crosslinking to DNA was performed by incubating cells with 1% formaldehyde for 10 min at 37°C. Thereafter, cells were sonicated to shear DNA to an average length of 200–1,000 base pairs, followed by centrifugation at 13,000 r.p.m. for 10 min. The samples were precleared, with 30 μl of salmon sperm DNA/Protein A Agarose-50% Slurry for 30 min, and incubated overnight with primary antibody (acetyl H3/H4, HDAC1 or HDAC2) at 4 °C. The immunocomplexes were recovered by adding 60 μl of a salmon sperm DNA/Protein A Agarose 50% slurry, followed by washing, reverse cross-linking with NaCl and recovering DNA by phenol/chloroform extraction and ethanol precipitation. The DNA was amplified by qPCR using the primer specific for cxcl1 promoter sequence provided in Key Resources Table.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rat anti-mouse CD11b | BD Biosciences | Cat#557657 |
| Rat anti-mouse Ly6C | BD Biosciences | Cat#560593 |
| Rat anti-mouse Ly6G | BD Biosciences | Cat#553129 |
| Rat anti-mouse Gr1 | BD Biosciences | Cat#553127 |
| Mouse anti-mouse CD45.2 | BD Biosciences | Cat#552950 |
| Rat anti-mouse CD326 | BD Biosciences | Cat#563478 |
| Rat anti-mouse CD140a | BD Biosciences | Cat#562776 |
| Mouse anti-human CD11b | BD Biosciences | Cat#550019 |
| Mouse anti-human CD14 | BD Biosciences | Cat#557831 |
| Mouse anti-human CD33 | BD Biosciences | Cat#333946 |
| Mouse anti-human CD15 | BD Biosciences | Cat#560828 |
| Mouse anti-human CD3 | BD Biosciences | Cat#333946 |
| Mouse anti-human HLA-DR | BD Biosciences | Cat#556643 |
| Mouse anti-human CD163 | BioLegend | Cat#333617 |
| Mouse anti-human CD115 | BioLegend | Cat#347304 |
| Mouse anti-human FAP | R&D Systems | Cat#FAB3715A-025 |
| Mouse monoclonal CXCR2 | R&D Systems | Cat#FAB2164P-025 |
| Mouse monoclonal F4/80 | eBioscience | Cat#12-4801-82 |
| Anti-human LOX-1 | Abcam | Cat#ab126538 |
| InVivoMab anti-mouse PD-1 | Bioxcell | Clone#RMP1-14 |
| InVivoMab anti-mouse CSF1R (CD115) | Bioxcell | Clone#AFS98 |
| Mouse CXCL1/KC | R&D Systems | Cat#AF-453 |
| Human CXCL8/IL-8 | R&D Systems | Cat#MAB208-100 |
| Mouse M-CSF | R&D Systems | Cat#MAB416-100 |
| Rabbit anti-human M-CSF | Abcam | Cat#ab9693 |
| Anti-HDAC2 CHIP grade | Abcam | Cat#ab7029 |
| Bacterial and Virus Strains | ||
| Mouse: Csf1r shRNA lentivirus, see METHOD DETAILS | Sigma-Aldrich Mission shRNA library (St Louis, MO, USA) | Type: NM_001037859.2-1091s21c1; |
| Mouse: Csf1r shRNA lentivirus, see METHOD DETAILS | Sigma-Aldrich Mission shRNA library (St Louis, MO, USA) | Type: NM_001037859.2-3516s21c1; |
| Mouse: Csf1r shRNA lentivirus, see METHOD DETAILS | Sigma-Aldrich Mission shRNA library (St Louis, MO, USA) | Type: NM_001037859.2-818s21c1; |
| Mouse: Csf1r shRNA lentivirus, see METHOD DETAILS | Sigma-Aldrich Mission shRNA library (St Louis, MO, USA) | Type: NM_001037859.2-1972s21c1 |
| Human: CSF1r shRNA lentivirus, see METHOD DETAILS | Sigma-Aldrich Mission shRNA library (St Louis, MO, USA) | Type: NM_005211.3-3460s21c1, |
| Human: CSF1r shRNA lentivirus, see METHOD DETAILS | Sigma-Aldrich Mission shRNA library (St Louis, MO, USA) | Type: NM_005211.x-504s1c1, |
| Human: CSF1r shRNA lentivirus, see METHOD DETAILS | Sigma-Aldrich Mission shRNA library (St Louis, MO, USA) | Type: NM_005211.3-1893s21c1, |
| Human: CSF1r shRNA lentivirus, see METHOD DETAILS | Sigma-Aldrich Mission shRNA library (St Louis, MO, USA) | Type: NM_005211.3-3042s21c1, |
| Human: CSF1r shRNA lentivirus, see METHOD DETAILS | Sigma-Aldrich Mission shRNA library (St Louis, MO, USA) | Type: NM_005211.3-2873s21c1 |
| Empty vector control plasmid DNA, pLKO.1 | Sigma-Aldrich | Cat#SHC001 |
| Biological samples | ||
| Peripheral blood, tumor tissues, and tissue microarray | Helen F. Graham Cancer Center and University of Pennsylvania | |
| Chemicals, Peptides, and Recombinant Proteins | ||
| CSF-1R inhibitor, JNJ-40346527 | Janssen Inc. | N/A |
| Entinostat | Syndax Pharmaceuticals | N/A |
| SB225002 | Tocris Biosciences | Cat#2725 |
| Mouse tumor dissociation kit | Miltenyi Biotech | Cat#130-096-730 |
| CXCL-1 recombinant mouse | BioLegend | Cat#573702 |
| CXCL-8/IL-8 recombinant human | R&D Systems | Cat#208-IL-010/CF |
| GM-CSF recombinant mouse | Invitrogen | Cat#PMC2013 |
| DAPI | Life Technologies | Cat#D1306 |
| Critical commercial assays | ||
| Mouse CXCL1/KC DuoSet for ELISA | R&D Systems | Cat#DY453-05 |
| Human CXCL8/IL-8 DuoSet for ELISA | R&D Systems | Cat#DY208-05 |
| Mouse M-CSF DuoSet for ELISA | R&D Systems | Cat#DY416 |
| Chromatin Immunoprecipitation (ChIP) Assay | Millipore | Cat#17-295 |
| Experimental Models: Mouse Cell Lines | ||
| EL4 thymoma | ATCC | N/A |
| CT26 colon carcinoma | ATCC | N/A |
| Lewis Lung Carcinoma (LLC) | ATCC | N/A |
| 4T1 mammary carcinoma | ATCC | N/A |
| B16F10 melanoma | ATCC | N/A |
| Mouse: 4662 pancreatic ductal adenocarcinoma | KPC mouse | N/A |
| Experimental Models: Organisms/Strains | ||
| Mouse: C57BL/6 | Charles River | N/A |
| Mouse: Balb/c | Charles River | N/A |
| Mouse: Ret melanoma model | Dr. Umansky (German Cancer Center, Heidelberg, Germany) | N/A |
| Mouse: TRAMP prostate cancer model | Dr. Languino (Thomas Jefferson University, Philadelphia) | N/A |
| Oligonucleotides | ||
| Primers, see Table S1 | This paper | N/A |
| Cxcl1 promoter FW: 5′-GTGCACATAACGCGTGTGAC-3′ | This paper | N/A |
| Cxcl1 promoter RV: 5′-TGCTTCGCTGGACAAGTGAC-3′ | This paper | N/A |
| Cxcl1 promoter for ChIP assay FW: 5′-GTGCACATAACGCGTGTGAC-3′, | This paper | N/A |
| Cxcl1 promoter for ChIP assay RV: 5′-TGCTTCGCTGGACAAGTGAC-3′ | This paper | N/A |
| Software and Algorithms | ||
| GraphPad Prism 5 | GraphPad Software Inc. | N/A |
Lentivirus csf1r shRNA
Lentiviral vectors containing short hairpin RNAi against human and mouse CSF1R was obtained from Sigma-Aldrich Mission shRNA library (St Louis, MO, USA) and listed in Key Resources Table.
Empty vector pLKO.1 was used as control. Lentiviral production was carried out as described in the protocol developed by the TRC library (Broad Institute). Briefly, 293T cells were co-transfected with shRNA vector and lentiviral packaging plasmids (pCMV-dR8.74psPAX2, pMD2.G). At 36 and 60 hr, the supernatant containing virus was harvested, and filtered through a 0.45μm filter. For transduction, the cells were layered overnight with lentivirus containing 8μg/ml polybrene, allowed to recover for 24 hr and then selected using 1μg/ml puromycin.
Analysis of cell signature in tumor samples
A molecular profiling database of >16,000 primary and >3000 metastatic tumors representing 25 different cancers was developed in a collaboration between collaboration between Merck & Co., Inc., Kenilworth, NJ USA andthe H. Lee Moffitt Cancer Center, Tampa, FL. All tumor samples were profiled on a standardized platform, Affymetrix Custom GeneChip that measures the expression of ~60,000 transcripts. Tissues were processed and accessed for RNA quality according to the Moffitt Cancer Center’s Total Cancer Care (TCCTM) protocol prior to expression analysis. Samples were amplified and labeled using a custom automated version of the NuGEN Ovation WB protocol. Hybridization, labeling and scanning using Affymetrix ovens, fluidics stations and scanners following the protocols recommended (NuGEN). Hybridization of affymetrix chips was done following the standard Affymetrix protocol. Generated .CEL files were then processed using the RMA algorithm as implemented in Affymetrix Power Tools (APT) package, using default settings and standard CDF file. Generated probeset intensities were then log10-transformed. Cell fraction deconvolution for neutrophils and monocytes cell types were generated as described (Newman et al., 2015).
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analyses were performed using 2-tailed Student’s t test or Mann-Whitney U test and GraphPad Prism 5 software (GraphPad Software Inc.). Paired t test was used since data were normally distributed. All the data are presented as mean ± SD and p value less than 0.05 was considered significant. The tumor growth kinetics was assessed in two-way ANOVA test. The tumor volumes measured over time were used to reflect the tumor growth trend affected by different treatments. The velocities of tumor growth were compared between the treatments using a linear mixed-effect model with the random effect at individual animal level.
Supplementary Material
1
2
Significance.
Inhibitors of CSF1R have demonstrated the potent ability to deplete tumor associated macrophages (TAM). However, they have not had antitumor effects in mice and have failed in several clinical trials. In this study, we have identified a cross-talk in the tumor microenvironment that may explain therapeutic limitations of CSF1R inhibitors. CSF1 produced by tumor cells represses the production of granulocytic chemokines by carcinoma associated fibroblasts and thus limits granulocyte recruitment. Inhibition of CSF1R reversed this effect and caused substantial accumulation of tumor promoting polymorphonuclear myeloid-derived suppressor cells (PMN-MDSC). The combination of CSF1R inhibitor with CXCR2 inhibitor, which prevents PMN-MDSC migration, significantly reduced tumor growth. Moreover, when PD-1 antibody added to this combination, it resulted in block of tumor growth.
Highlights.
- PMN-MDSC infiltration of tumors is controlled by carcinoma associated fibroblasts;
- CSF-1 silences expression of granulocytic chemokines by CAF mediated by HDAC2;
- Inhibition of CSF-1R induced tumor infiltration by PMN-MDSC;
- Combination of CSF-1R with CXCR2 inhibitors demonstrated strong antitumor effect
Acknowledgments
This work was supported by Janssen R&D and Syndax Pharmaceuticals as well as NIH grants P01 CA 140043, P50 CA168536, and P01 CA114046.
P.O. is an employee of Syndax Pharmaceuticals; M.A.S. and L.A.S. are employees of Janssen R&D; C.Z. and A.L. are employees of Merck & Co., Inc., Kenilworth, NJ, USA.
Footnotes
Author contributions
Conceptualization, D.I.G., Methodology, E.P., Formal Analysis Q.L., Investigation, V.K. L.D., D.M., T.C., F.W., S.L., A.H., P. V., X.X. A. L., R.B., C.Z., A.L. Resources, L.L, P.O., M.A.S., L.A.S., E.P., M.A.G., C.M., B.N., N. H., F. D., S.S., A.T. W., T.C., R.H.V, D.W.S. Writing – original draft – V.K., Writing – Review & Editing, D.I.G., E.P., Supervision, D.I.G. Funding Acquisition, M.A.S., L.A.S., P.O.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Butowski N, Colman H, De Groot JF, Omuro AM, Nayak L, Wen PY, Cloughesy TF, Marimuthu A, Haidar S, Perry A, et al. Orally administered colony stimulating factor 1 receptor inhibitor PLX3397 in recurrent glioblastoma: an Ivy Foundation Early Phase Clinical Trials Consortium phase II study. Neuro Oncol. 2016;18:557–564. doi: 10.1093/neuonc/nov245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carus A, Ladekarl M, Hager H, Pilegaard H, Nielsen PS, Donskov F. Tumor-associated neutrophils and macrophages in non-small cell lung cancer: no immediate impact on patient outcome. Lung Cancer. 2013;81:130–137. doi: 10.1016/j.lungcan.2013.03.003. [DOI] [PubMed] [Google Scholar]
- Challacombe JM, Suhrbier A, Parsons PG, Jones B, Hampson P, Kavanagh D, Rainger GE, Morris M, Lord JM, Le TT, et al. Neutrophils are a key component of the antitumor efficacy of topical chemotherapy with ingenol-3-angelate. J Immunol. 2006;177:8123–8132. doi: 10.4049/jimmunol.177.11.8123. [DOI] [PubMed] [Google Scholar]
- Chao T, Furth EE, Vonderheide RH. CXCR2-Dependent Accumulation of Tumor-Associated Neutrophils Regulates T-cell Immunity in Pancreatic Ductal Adenocarcinoma. Cancer Immunol Res. 2016;4:968–982. doi: 10.1158/2326-6066.CIR-16-0188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Condamine T, Dominguez G, Youn J, Kossenkov A, Mony S, Alicea-Torres K, Tcyganov E, Hashimoto A, Nefedova Y, Lin C, et al. Lectin-type oxidized LDL receptor 1 distinguishes population of human polymorphonuclear myeloid-derived suppressor cells in cancer patients. Science Immunol. 2016:1. doi: 10.1126/sciimmunol.aaf8943. [DOI] [PMC free article] [PubMed]
- Conway JG, McDonald B, Parham J, Keith B, Rusnak DW, Shaw E, Jansen M, Lin P, Payne A, Crosby RM, et al. Inhibition of colony-stimulating-factor-1 signaling in vivo with the orally bioavailable cFMS kinase inhibitor GW2580. Proc Natl Acad Sci U S A. 2005;102:16078–16083. doi: 10.1073/pnas.0502000102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coussens LM, Pollard JW. Leukocytes in mammary development and cancer. Cold Spring Harbor perspectives in biology. 2011:3. doi: 10.1101/cshperspect.a003285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeNardo D, Brennan D, Rexhepaj E, Ruffell B, Shiao S, Madden S, Gallagher W, Wadhwani N, Keil S, Junaid S, et al. Leukocyte Complexity Predicts Breast Cancer Survival and Functionally Regulates Response to Chemotherapy. Cancer Discovery. 2011a;1:54–67. doi: 10.1158/2159-8274.CD-10-0028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeNardo DG, Brennan DJ, Rexhepaj E, Ruffell B, Shiao SL, Madden SF, Gallagher WM, Wadhwani N, Keil SD, Junaid SA, et al. Leukocyte complexity predicts breast cancer survival and functionally regulates response to chemotherapy. Cancer Discov. 2011b;1:54–67. doi: 10.1158/2159-8274.CD-10-0028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fearon DT. The carcinoma-associated fibroblast expressing fibroblast activation protein and escape from immune surveillance. Cancer Immunol Res. 2014;2:187–193. doi: 10.1158/2326-6066.CIR-14-0002. [DOI] [PubMed] [Google Scholar]
- Feig C, Jones JO, Kraman M, Wells RJ, Deonarine A, Chan DS, Connell CM, Roberts EW, Zhao Q, Caballero OL, et al. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. Proc Natl Acad Sci U S A. 2013;110:20212–20217. doi: 10.1073/pnas.1320318110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finke J, Ko J, Rini B, Rayman P, Ireland J, Cohen P. MDSC as a mechanism of tumor escape from sunitinib mediated anti-angiogenic therapy. Int Immunopharmacol. 2011;11:856–861. doi: 10.1016/j.intimp.2011.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabrilovich DI. Myeloid-Derived Suppressor Cells. Cancer Immunol Res. 2017;5:3–8. doi: 10.1158/2326-6066.CIR-16-0297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabrilovich DI, Ostrand-Rosenberg S, Bronte V. Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol. 2012;12:253–268. doi: 10.1038/nri3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gascard P, Tlsty TD. Carcinoma-associated fibroblasts: orchestrating the composition of malignancy. Genes Dev. 2016;30:1002–1019. doi: 10.1101/gad.279737.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genovese MC, Hsia E, Belkowski SM, Chien C, Masterson T, Thurmond RL, Manthey CL, Yan XD, Ge T, Franks C, Greenspan A. Results from a Phase IIA Parallel Group Study of JNJ-40346527, an Oral CSF-1R Inhibitor, in Patients with Active Rheumatoid Arthritis despite Disease-modifying Antirheumatic Drug Therapy. J Rheumatol. 2015;42:1752–1760. doi: 10.3899/jrheum.141580. [DOI] [PubMed] [Google Scholar]
- Gentles AJ, Newman AM, Liu CL, Bratman SV, Feng W, Kim D, Nair VS, Xu Y, Khuong A, Hoang CD, et al. The prognostic landscape of genes and infiltrating immune cells across human cancers. Nat Med. 2015;21:938–945. doi: 10.1038/nm.3909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenberg NM, DeMayo F, Finegold MJ, Medina D, Tilley WD, Aspinall JO, Cunha GR, Donjacour AA, Matusik RJ, Rosen JM. Prostate cancer in a transgenic mouse. Proc Natl Acad Sci U S A. 1995;92:3439–3443. doi: 10.1073/pnas.92.8.3439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell. 2010;140:883–899. doi: 10.1016/j.cell.2010.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guleria I, Pollard JW. Aberrant macrophage and neutrophil population dynamics and impaired Th1 response to Listeria monocytogenes in colony-stimulating factor 1-deficient mice. Infect Immun. 2001;69:1795–1807. doi: 10.1128/IAI.69.3.1795-1807.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang TS, Shyu YC, Chen HY, Yuan SS, Shih JN, Chen PJ. A systematic review and meta-analysis of adjuvant interferon therapy after curative treatment for patients with viral hepatitis-related hepatocellular carcinoma. Journal of viral hepatitis. 2013;20:729–743. doi: 10.1111/jvh.12096. [DOI] [PubMed] [Google Scholar]
- Kato M, Takahashi M, Akhand AA, Liu W, Dai Y, Shimizu S, Iwamoto T, Suzuki H, Nakashima I. Transgenic mouse model for skin malignant melanoma. Oncogene. 1998;17:1885–1888. doi: 10.1038/sj.onc.1202077. [DOI] [PubMed] [Google Scholar]
- Kumar V, Cheng P, Condamine T, Mony S, Languino LR, McCaffrey JC, Hockstein N, Guarino M, Masters G, Penman E, et al. CD45 Phosphatase Inhibits STAT3 Transcription Factor Activity in Myeloid Cells and Promotes Tumor-Associated Macrophage Differentiation. Immunity. 2016;44:303–315. doi: 10.1016/j.immuni.2016.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ladanyi A. Prognostic and predictive significance of immune cells infiltrating cutaneous melanoma. Pigment cell & melanoma research. 2015;28:490–500. doi: 10.1111/pcmr.12371. [DOI] [PubMed] [Google Scholar]
- Lin EY, Nguyen AV, Russell RG, Pollard JW. Colony-stimulating factor 1 promotes progression of mammary tumors to malignancy. J Exp Med. 2001;193:727–740. doi: 10.1084/jem.193.6.727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo A, Wang LC, Scholler J, Monslow J, Avery D, Newick K, O’Brien S, Evans RA, Bajor DJ, Clendenin C, et al. Tumor-Promoting Desmoplasia Is Disrupted by Depleting FAP-Expressing Stromal Cells. Cancer Res. 2015;75:2800–2810. doi: 10.1158/0008-5472.CAN-14-3041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lokeshwar BL, Lin HS. Development and characterization of monoclonal antibodies to murine macrophage colony-stimulating factor. J Immunol. 1988;141:483–488. [PubMed] [Google Scholar]
- Lucas T, Abraham D, Aharinejad S. Modulation of tumor associated macrophages in solid tumors. Front Biosci. 2008;13:5580–5588. doi: 10.2741/3101. [DOI] [PubMed] [Google Scholar]
- MacDonald KP, Palmer JS, Cronau S, Seppanen E, Olver S, Raffelt NC, Kuns R, Pettit AR, Clouston A, Wainwright B, et al. An antibody against the colony-stimulating factor 1 receptor depletes the resident subset of monocytes and tissue- and tumor-associated macrophages but does not inhibit inflammation. Blood. 2010;116:3955–3963. doi: 10.1182/blood-2010-02-266296. [DOI] [PubMed] [Google Scholar]
- Mao ZY, Zhu GQ, Xiong M, Ren L, Bai L. Prognostic value of neutrophil distribution in cholangiocarcinoma. World J Gastroenterol. 2015;21:4961–4968. doi: 10.3748/wjg.v21.i16.4961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messmer MN, Netherby CS, Banik D, Abrams SI. Tumor-induced myeloid dysfunction and its implications for cancer immunotherapy. Cancer Immunol Immunother. 2015;64:1–13. doi: 10.1007/s00262-014-1639-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer C, Cagnon L, Costa-Nunes CM, Baumgaertner P, Montandon N, Leyvraz L, Michielin O, Romano E, Speiser DE. Frequencies of circulating MDSC correlate with clinical outcome of melanoma patients treated with ipilimumab. Cancer Immunol Immunother. 2014;63:247–257. doi: 10.1007/s00262-013-1508-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra P, Banerjee D, Ben-Baruch A. Chemokines at the crossroads of tumor-fibroblast interactions that promote malignancy. J Leukoc Biol. 2011;89:31–39. doi: 10.1189/jlb.0310182. [DOI] [PubMed] [Google Scholar]
- Mitchem JB, Brennan DJ, Knolhoff BL, Belt BA, Zhu Y, Sanford DE, Belaygorod L, Carpenter D, Collins L, Piwnica-Worms D, et al. Targeting tumor-infiltrating macrophages decreases tumor-initiating cells, relieves immunosuppression, and improves chemotherapeutic responses. Cancer Res. 2013;73:1128–1141. doi: 10.1158/0008-5472.CAN-12-2731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mok S, Koya RC, Tsui C, Xu J, Robert L, Wu L, Graeber TG, West BL, Bollag G, Ribas A. Inhibition of CSF-1 receptor improves the antitumor efficacy of adoptive cell transfer immunotherapy. Cancer Res. 2014;74:153–161. doi: 10.1158/0008-5472.CAN-13-1816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mok S, Tsoi J, Koya RC, Hu-Lieskovan S, West BL, Bollag G, Graeber TG, Ribas A. Inhibition of colony stimulating factor-1 receptor improves antitumor efficacy of BRAF inhibition. BMC Cancer. 2015;15:356. doi: 10.1186/s12885-015-1377-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman AM, Liu CL, Green MR, Gentles AJ, Feng W, Xu Y, Hoang CD, Diehn M, Alizadeh AA. Robust enumeration of cell subsets from tissue expression profiles. Nat Methods. 2015;12:453–457. doi: 10.1038/nmeth.3337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noman MZ, Janji B, Hu S, Wu JC, Martelli F, Bronte V, Chouaib S. Tumor-Promoting Effects of Myeloid-Derived Suppressor Cells Are Potentiated by Hypoxia-Induced Expression of miR-210. Cancer Res. 2015;75:3771–3787. doi: 10.1158/0008-5472.CAN-15-0405. [DOI] [PubMed] [Google Scholar]
- Obermajer N, Muthuswamy R, Odunsi K, Edwards RP, Kalinski P. PGE(2)-induced CXCL12 production and CXCR4 expression controls the accumulation of human MDSCs in ovarian cancer environment. Cancer research. 2011;71:7463–7470. doi: 10.1158/0008-5472.CAN-11-2449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno H, Kubo K, Murooka H, Kobayashi Y, Nishitoba T, Shibuya M, Yoneda T, Isoe T. A c-fms tyrosine kinase inhibitor, Ki20227, suppresses osteoclast differentiation and osteolytic bone destruction in a bone metastasis model. Mol Cancer Ther. 2006;5:2634–2643. doi: 10.1158/1535-7163.MCT-05-0313. [DOI] [PubMed] [Google Scholar]
- Orimo A, Weinberg RA. Stromal fibroblasts in cancer: a novel tumor-promoting cell type. Cell cycle (Georgetown, Tex) 2006;5:1597–1601. doi: 10.4161/cc.5.15.3112. [DOI] [PubMed] [Google Scholar]
- Pixley FJ, Stanley ER. CSF-1 regulation of the wandering macrophage: complexity in action. Trends Cell Biol. 2004;14:628–638. doi: 10.1016/j.tcb.2004.09.016. [DOI] [PubMed] [Google Scholar]
- Pyonteck SM, Akkari L, Schuhmacher AJ, Bowman RL, Sevenich L, Quail DF, Olson OC, Quick ML, Huse JT, Teijeiro V, et al. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat Med. 2013;19:1264–1272. doi: 10.1038/nm.3337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramachandran IR, Condamine T, Lin C, Herlihy SE, Garfall A, Vogl DT, Gabrilovich DI, Nefedova Y. Bone marrow PMN-MDSCs and neutrophils are functionally similar in protection of multiple myeloma from chemotherapy. Cancer Letters. 2016;371:117–124. doi: 10.1016/j.canlet.2015.10.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ries CH, Cannarile MA, Hoves S, Benz J, Wartha K, Runza V, Rey-Giraud F, Pradel LP, Feuerhake F, Klaman I, et al. Targeting tumor-associated macrophages with anti-CSF-1R antibody reveals a strategy for cancer therapy. Cancer Cell. 2014;25:846–859. doi: 10.1016/j.ccr.2014.05.016. [DOI] [PubMed] [Google Scholar]
- Ries CH, Hoves S, Cannarile MA, Ruttinger D. CSF-1/CSF-1R targeting agents in clinical development for cancer therapy. Curr Opin Pharmacol. 2015;23:45–51. doi: 10.1016/j.coph.2015.05.008. [DOI] [PubMed] [Google Scholar]
- Sasse S, Alram M, Muller H, Smardova L, Metzner B, Doehner H, Fischer T, Niederwieser DW, Schmitz N, Schafer-Eckart K, et al. Prognostic relevance of DHAP dose-density in relapsed Hodgkin lymphoma: an analysis of the German Hodgkin-Study Group. Leuk Lymphoma. 2016;57:1067–1073. doi: 10.3109/10428194.2015.1083561. [DOI] [PubMed] [Google Scholar]
- Sionov RV, Assi S, Gershkovitz M, Sagiv JY, Polyansky L, Mishalian I, Fridlender ZG, Granot Z. Isolation and Characterization of Neutrophils with Anti-Tumor Properties. Journal of visualized experiments: JoVE. 2015:e52933. doi: 10.3791/52933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sluijter M, van der Sluis TC, van der Velden PA, Versluis M, West BL, van der Burg SH, van Hall T. Inhibition of CSF-1R supports T-cell mediated melanoma therapy. PLoS One. 2014;9:e104230. doi: 10.1371/journal.pone.0104230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solito S, Marigo I, Pinton L, Damuzzo V, Mandruzzato S, Bronte V. Myeloid-derived suppressor cell heterogeneity in human cancers. Ann NY Acad Sci. 2014;1319:47–65. doi: 10.1111/nyas.12469. [DOI] [PubMed] [Google Scholar]
- Strachan DC, Ruffell B, Oei Y, Bissell MJ, Coussens LM, Pryer N, Daniel D. CSF1R inhibition delays cervical and mammary tumor growth in murine models by attenuating the turnover of tumor-associated macrophages and enhancing infiltration by CD8+ T cells. Oncoimmunology. 2013;2:e26968. doi: 10.4161/onci.26968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szkandera J, Absenger G, Liegl-Atzwanger B, Pichler M, Stotz M, Samonigg H, Glehr M, Zacherl M, Stojakovic T, Gerger A, Leithner A. Elevated preoperative neutrophil/lymphocyte ratio is associated with poor prognosis in soft-tissue sarcoma patients. Br J Cancer. 2013;108:1677–1683. doi: 10.1038/bjc.2013.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talmadge JE, Gabrilovich DI. History of myeloid-derived suppressor cells. Nat Rev Cancer. 2013;13:739–752. doi: 10.1038/nrc3581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tap WD, Wainberg ZA, Anthony SP, Ibrahim PN, Zhang C, Healey JH, Chmielowski B, Staddon AP, Cohn AL, Shapiro GI, et al. Structure-Guided Blockade of CSF1R Kinase in Tenosynovial Giant-Cell Tumor. N Engl J Med. 2015;373:428–437. doi: 10.1056/NEJMoa1411366. [DOI] [PubMed] [Google Scholar]
- Vetsika EK, Koinis F, Gioulbasani M, Aggouraki D, Koutoulaki A, Skalidaki E, Mavroudis D, Georgoulias V, Kotsakis A. A Circulating Subpopulation of Monocytic Myeloid-Derived Suppressor Cells as an Independent Prognostic/Predictive Factor in Untreated Non-Small Lung Cancer Patients. Journal of Immunology Research. 2014;2014:12. doi: 10.1155/2014/659294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang LC, Lo A, Scholler J, Sun J, Majumdar RS, Kapoor V, Antzis M, Cotner CE, Johnson LA, Durham AC, et al. Targeting fibroblast activation protein in tumor stroma with chimeric antigen receptor T cells can inhibit tumor growth and augment host immunity without severe toxicity. Cancer Immunol Res. 2014;2:154–166. doi: 10.1158/2326-6066.CIR-13-0027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber J, Gibney G, Kudchadkar R, Yu B, Cheng P, Martinez AJ, Kroeger J, Richards A, McCormick L, Moberg V, et al. Phase I/II Study of Metastatic Melanoma Patients Treated with Nivolumab Who Had Progressed after Ipilimumab. Cancer Immunol Res. 2016;4:345–353. doi: 10.1158/2326-6066.CIR-15-0193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Escamilla J, Mok S, David J, Priceman S, West B, Bollag G, McBride W, Wu L. CSF1R signaling blockade stanches tumor-infiltrating myeloid cells and improves the efficacy of radiotherapy in prostate cancer. Cancer Res. 2013;73:2782–2794. doi: 10.1158/0008-5472.CAN-12-3981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Lin Y, Shi Y, Li B, Liu W, Yin W, Dang Y, Chu Y, Fan J, He R. FAP Promotes Immunosuppression by Cancer-Associated Fibroblasts in the Tumor Microenvironment via STAT3-CCL2 Signaling. Cancer Res. 2016;76:4124–4135. doi: 10.1158/0008-5472.CAN-15-2973. [DOI] [PubMed] [Google Scholar]
- Zhang S, Ma X, Zhu C, Liu L, Wang G, Yuan X. The Role of Myeloid-Derived Suppressor Cells in Patients with Solid Tumors: A Meta-Analysis. PloS one. 2016a;11:e0164514. doi: 10.1371/journal.pone.0164514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Ma C, Wang M, Hou H, Cui L, Jiang C, Sun J, Qu X. Prognostic significance of immune cells in the tumor microenvironment and peripheral blood of gallbladder carcinoma patients. Clin Transl Oncol. 2016b doi: 10.1007/s12094-016-1553-6. [DOI] [PubMed] [Google Scholar]
- Zhu Y, Knolhoff BL, Meyer MA, Nywening TM, West BL, Luo J, Wang-Gillam A, Goedegebuure SP, Linehan DC, DeNardo DG. CSF1/CSF1R blockade reprograms tumor-infiltrating macrophages and improves response to T-cell checkpoint immunotherapy in pancreatic cancer models. Cancer Res. 2014;74:5057–5069. doi: 10.1158/0008-5472.CAN-13-3723. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
1
2