Subsynaptic spatial organization as a regulator of synaptic strength and plasticity (original) (raw)
. Author manuscript; available in PMC: 2019 Aug 1.
Published in final edited form as: Curr Opin Neurobiol. 2018 Jun 11;51:147–153. doi: 10.1016/j.conb.2018.05.004
Abstract
Synapses differ markedly in their performance, even amongst those on a single neuron. The mechanisms that drive this functional diversification are of great interest because they enable adaptive behaviors and are targets of pathology. Considerable effort has focused on elucidating mechanisms of plasticity that involve changes to presynaptic release probability and the number of postsynaptic receptors. However, recent work is clarifying that nanoscale organization of the proteins within glutamatergic synapses impacts synapse function. Specifically, active zone scaffold proteins form nanoclusters that define sites of vesicle release, and these sites align transsynaptically with clustered postsynaptic receptors. These nanostructural characteristics raise numerous possibilities for how synaptic plasticity could be expressed.
Transsynaptic alignment can control synaptic function
The efficacy of synaptic transmission may rely in part on efficient organization of presynaptic release sites to postsynaptic receptors. While in some synapse types, ultrastructural landmarks such as ribbons or T-bars provide a natural hint for where release occurs, the organization of release sites in brain glutamatergic synapses has been more difficult to measure. New work on this includes increasingly sophisticated physiology and imaging. Release characteristics were examined through patch-clamp at the synapse between cerebellar granule cells and molecular layer interneurons, a prototypic “simple” synapse involving a single contact between one active zone (AZ) and one postsynaptic density (PSD) [1*]. Quantal analysis of release events counted with high temporal precision here revealed that action potentials drove release at multiple discrete and essentially independent sites in the AZ. Consistent with this, a novel imaging approach called pHuse (pHluorin uncovering sites of exocytosis), using vesicle-resident sensors to map sites of single-vesicle fusion events within individual synapses, demonstrated that action potentials evoked vesicle fusion within a smaller area of the presynaptic bouton than spontaneous release in hippocampal terminals [2**]. Critically, trains of stimuli prompt release from repeatedly used sites [3*]. Thus, release probability (Pr) in a single AZ is spatially heterogeneous.
The molecular basis for this map of release likelihood presumably arises from a small set of multidomain “scaffold” proteins that coordinate the positioning of the vesicle, Ca2+ channels, and other release machinery [4, [5]. Amongst these critical scaffold proteins are Rab-interacting molecules (RIMs), Munc13/Unc13, RIM-binding proteins (RBPs), and ELKs family proteins [5]. By super-resolution imaging, these scaffolds form nanoclusters typically of ~60 to 80 nm within the AZ [2**, [6, [7**]. Varied approaches have now been used to deduce that these nanoclusters dictate the spatial distribution of Pr across the AZ surface. Combined pHuse and live-cell, single-molecule imaging of expressed RIM1 found that AZ subregions with high RIM1 density were the preferred sites of vesicle fusion after an action potential [2**]. In the glutamatergic Drosophila neuromuscular junction, (M)Unc13 distribution measured by STED aligned with functional synaptic vesicle release sites as measured by postsynaptic Ca2+ sensor GCaMP [8**]. In particular, the isoform Unc13-A appeared critical for evoked release, as deletion of the isoform resulted in drastic reduction in evoked release stemming from a reduced number of vesicles docked close to Ca2+ channels [9]. In hippocampal cultures, a newly synthesized fluorescent glutamate sensor to measure quantal release characteristics and deduce the number of release sites in individual boutons (~3 to over a dozen), was combined with STORM to measure the distribution of Munc13 in the same terminals [7**]. Remarkably, the number of Munc13 clusters observed per bouton tightly correlated with the number of functionally defined release sites. Interestingly, while modulated by AZ scaffold proteins, Ca2+ channels themselves can be mobile [10] but are frequently clustered [11], and release probability scales not only with the number of channels but the number of channel nanoclusters [12*]. This suggests a model whereby release sites arising from protein clustering may occupy stable positions, but that release likelihood per site may vary as scaffolds and channels move among them.
These observations of AZ organization strikingly parallel nanostructure within the PSD. Both AMPA and NMDA type receptors are subsynaptically nanoclustered, in spatial correlation with PSD-95 [13, [14, [15], which itself is clustered [14, [16]. In fact, PSD nanoclusters are very similar in size and number to AZ protein nanoclusters [2]. Critically, protein clustering on both sides of the synapse creates the potential for alignment or mis-alignment of these functionally critical features. Indeed, in hippocampal neurons both in culture and in acute slices, RIM nanoclusters align transsynaptically with concentrated postsynaptic receptors and scaffolding proteins [2**]. This organization brings to mind a transcellular protein “nanocolumn” that guides release to occur near receptors (Fig. 1). Such alignment is important because the surprisingly small amount of glutamate released from a single vesicle is not likely to activate relatively distant receptors before dispersing by diffusion [5, [14, [17, [18]. Direct experimental support for the idea that spatially confined subsets of receptors are activated by single vesicles remains elusive, but there is substantial indirect evidence [19, [20, [21, [22]. Indeed, given maps of RIM abundance and receptor distribution, the relationship between release location and AMPAR opening probability has been used to model synaptic strength and predict the effect of release misalignment [2**], which can be up to even 50% change in signal amplitude [2**, [14], similar in magnitude to many forms of synaptic plasticity.
Figure 1.
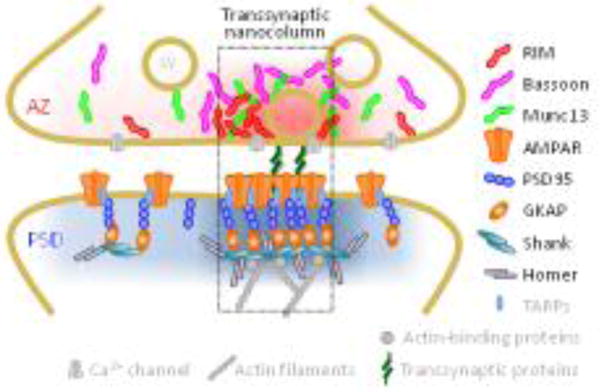
Key synaptic proteins are enriched within nanocolumns. High density nanoclusters of RIM1/2 and (M)Unc13 dictate sites of vesicle fusion following action potentials, which are assembled in alignment with postsynaptic nanoclusters of receptors [2**,8**]. The coupling between the release and receptors through transsynaptic proteins together with the distributions of proteins in grey is hypothetical, while the distributions of color-coded proteins have been confirmed. Adapted from [2**].
Alignment-mediated plasticity arising from postsynaptic reorganization
Sensitivity of receptor activation to alignment with release site suggests many forms of synaptic plasticity (Fig. 2). Classic models of potentiation and depression have established that these canonical forms of plasticity involve alterations in the number of postsynaptic receptors. However, the functional effects of adding receptors, for instance, will depend on their alignment with release sites. In one influential model, synapses contain multiple “modules” spanning the two cells, and under basal conditions, some modules are opposed to release sites but are “silent” because they lack AMPARs [23]. In this model, LTP converts silent modules to become responsive through addition of AMPARs to these regions (Fig. 2). Support for such a modular nature of synaptic organization came from recent work using STED imaging to examine pre- and post-synaptic proteins simultaneously even in live synapses undergoing LTP [24]. Single spines were observed to contain several sometimes widely spaced, distinguishable assemblies of PSD-95, suggestive of modules. These were associated with colabeled presynaptic proteins, and glycine-induced LTP increased the number of these coordinated modules, as suggested in Figure 2h. further work will be required to reveal the nature of these modules and their relation with the nanocolumns seen with STORM. It will also be important to determine whether these modules of PSD-95 contain AMPARs. In fact, the location of receptors newly added during physiological LTP has not (somewhat shockingly) been deduced, and there is not concrete evidence for whether they intermix with pre-existing receptors or occupy distinct sites. An important recent test used an optically regulated protein dimerization system to recruit exogenous AMPARs to the PSD without LTP induction [25**]. Surprisingly, the newly added receptors had no apparent effect on quantal amplitude even though they responded to glutamate uncaging at the synapse. One interpretation of these intriguing results is that the receptors’ position within the PSD was not optimal to sense evoked glutamate release.
Figure 2.
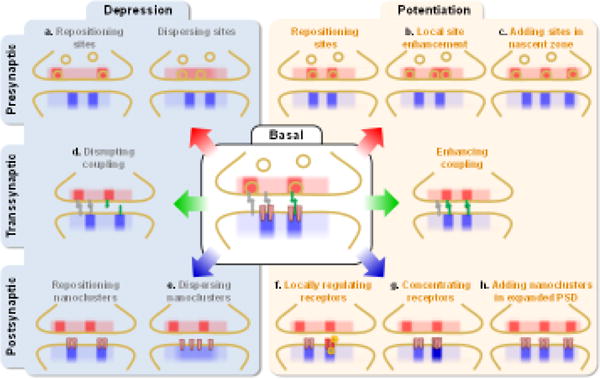
Potential mechanisms of synaptic plasticity mediated by changes in release-receptor alignment. These changes may arise from repositioning or change in the number or position of release sites presynaptically, from altering the number, position, size, or internal density of postsynaptic receptor clusters, or from changing the nature of transsynaptic coupling. Panels with notations depict mechanisms that have support from direct or indirect evidence in the literature.
Posttranslational modification of PSD-resident receptor subsets may alter transmission through the local effects of kinases docked in PSD nanoregions (Fig. 2f), for instance CaMKII on specific NMDARs [26]. Alternatively, regulation of receptor-anchoring PSD proteins may be required to drive receptor exchange in local regions of the synapse. Indeed, multiple mechanisms control PSD-95 abundance, including phosphorylation [27], palmitoylation [13] and direct binding to the actin modulator α-actinin [28]. Because PSD-95 displays such limited intra-PSD mobility [29, [30], spatially restricted modification of the scaffold may be a mechanism for nanoscale remodeling of receptor distribution. Another exciting possibility is the potential for extracellular interactions to control receptors’ ability to be activated. In C elegans, presynaptic secretion of the protein NRAP-1 modifies gating of postsynaptic NMDARs and completely controls their ability to be opened by glutamate (Fig. 2f) [31*]. Whether a related mechanism is at play in vertebrates is unknown, but this could offer a potential mechanism to activate pre-clustered receptors.
Perhaps most intriguingly, reorganization of receptor distribution—regardless of changes in receptor number—could alter synaptic strength (Fig. 2). Simulations indicate that over a broad range of starting geometrical conditions, simply condensing a cluster of existing AMPARs strongly potentiates the AMPAR current [32], and redistribution of NMDARs similarly was predicted to strongly regulate their activation probability [31]. The receptor distribution is likely established and adjusted by diverse mechanisms. Critically, AMPARs can be mobile within the PSD, but the diffusion rate and fraction which is mobile are affected not only by binding of auxiliary subunits [33, [34, [35], but also by synaptic activity [36] and receptor desensitization [37]. Recent work makes clear that the extraordinarily high density of protein within the PSD has profound consequences for receptor mobility. Steric hindrance in the PSD obstructs receptor motion [38*, [39] and may even help lock receptors within dense subdomains [40]. In fact, immediately after chemical LTP induction, PSD-95 is more concentrated within nanoclusters, which may in turn increase the AMPAR density locally within nanocolumns (Fig. 2g) [2**]. Even more unexpectedly, PSD-95 in the synapse can exist in both condensed liquid and aqueous phases, with the transition between these states regulated by density and through binding of the highly abundant GTPase activating protein SynGAP [41**]. It is easy to imagine that mobility of receptors and other proteins may alter within PSD regions of differing phase states. These complex interactions that control receptor exchange and diffusion within the synapse are likely to be particularly important during synaptic plasticity, since reducing receptor mobility (via antibody-based cross-linking) not only magnifies paired-pulse depression [42], but impairs LTP and learning [43*].
Alignment-mediated plasticity arising from presynaptic reorganization
Just as net receptor activation is not completely predicted from only the number of postsynaptic receptors, Pr may not be sufficient per se to determine the functional impact of increasing or decreasing glutamate release. Release position matters, suggesting potential mechanisms of plasticity that arise from alterations to vesicle release sites.
Following the idea of synaptic protein “modules,” release sites may be reorganized by the addition of functional presynaptic units. One mechanism for this may be through “nascent zones,” synaptic areas adjacent to AZs that lack presynaptic vesicles (Fig. 2c) [44]. It has been proposed that with PSD expansion in LTP, nascent zones are converted to functional domains of the AZ through addition of synaptic vesicles [44], though the molecular requirements for such a transition have not been evaluated.
The AZ itself is also a dynamic structure. AZ proteins can undergo vigorous reorganization on a time scale of minutes [45], which may consequently affect release site organization. For instance, following prolonged synaptic silencing, Bassoon was found to be less clustered while overall quantity remained unchanged [46]. This was associated with increased recruitment of presynaptic vesicle fusion machinery including Ca2+ channels [46] and a decreased AZ-PSD distance [47], both presumably facilitating the known homeostatic increase of synaptic transmission. These activity-dependent reorganizations could dynamically modulate the properties of release sites, including not only Pr but also spatial distribution (Fig. 2a). Indeed, release mapping with pHuse showed that high-frequency stimulation reduced the frequency with which release sites were re-used, and shifted their location away from the AZ center [3*]. This redistribution of glutamate release sites would lead to reduced EPSC amplitude due to an increased distance to the receptor densities.
Another mechanism by which the location of release could be triggered to change during plasticity is through regulation of presynaptic Ca2+ channels. This may occur through regulation by AZ scaffold proteins that engage with Ca2+ channels. For example, RBP-null mutants have impaired presynaptic homeostatic plasticity resulting from lack of enhancement in presynaptic Ca2+ influx, looser coupling between Ca2+ channels and vesicles, and the number of releasable vesicles [48]. Alternatively, if mobile Ca2+ channels dynamically regulate effectiveness of individual release sites [10], their mobility may be independently modulated by activity, as suggested by decreased Ca2+ channel mobility after lowering intracellular Ca2+ with chelators [10].
Cleft reorganization
Given these considerations, the organization of the synaptic cleft appears to play a critical role in governing synapse function. Though molecular mechanisms underlying release-receptor alignment are still unknown, the most intriguing hypothesis is that trans-synaptic cell adhesion molecules (CAMs) might organize and modulate these functional modules (Fig. 2). CAMs include a large number of protein families, and some members directly interact with glutamate receptors and scaffolds [49]. Cadherin family members adopt a perisynaptic distribution, and recent EM and super-resolution imaging has revealed nanoclustered organizations of several CAMs including SynCAM [50], neuroligin and LRRTM2 [51], and Neurexin1β [52]. The colocalization of these with vesicle fusion sites or receptors remains to be tested, but CAMs appear poised to play a key role in establishing and maintaining release-receptor alignment [5] and thus controlling synaptic strength. CAMs undergo vigorous activity-dependent remodeling, and may indeed be a critical site of action. An NMDAR activation inducing long-term depression leads to a rapid disassembly of NRXβ1-NLG1 complexes (Fig. 2d) [51], and this may result from proteolytic activity of the extracellular metalloproteases MMP9 and/or ADAM 10 [53, [54] as well as CAMKII dependent phosphorylation [55] of neuroligin. A similar treatment also enlarges postsynaptic SynCAM1 puncta on the cell surface [50]. Further studies are necessary to test whether these reorganizations of CAMs result in dismantling or strengthening of release-receptor alignment, and if so, whether these alignment changes contribute to the alteration in synaptic transmission.
By acting as a signaling bridge, CAMs can also mediate the trans-synaptic coordination during maturation and plasticity of synapses [56]. In C. elegans neuromuscular junction, postsynaptic neurexin1 plays a key role in mediating a retrograde inhibition of presynaptic transmitter release [57**]. Emerging evidence suggests that this kind of retrograde coordination could happen structurally at the nanoscale level. Following chemical LTD induction with NMDA treatment, hippocampal synapses underwent a two-step change of synaptic nanoarchitecture: initially, PSD-95 nanoclusters were disrupted (Fig. 2e), but delayed effects after 30 min recovery included an increase in size of presynaptic RIM nanoclusters that remarkably appeared only in those aligned with postsynaptic PSD-95 and AMPARs (Fig. 2b). Meanwhile there was no change in RIM nanoclusters not aligned with PSD-95, suggesting an alignment-specific process involved in this plasticity [2**].
Conclusions
Together, these observations indicate that glutamatergic synapses are built with a delicate subsynaptic spatial organization of vesicle release sites relative to clustered receptors, and suggest that this architecture may help establish synaptic strength. Many forms of plasticity may thus arise from spatial reorganization of release sites, receptors, or cleft proteins—cooperating with or even instead of changes in release probability or receptor number. Exciting work lies ahead to link particular nanostructural changes to functional changes in synaptic strength, and to discover which of these many potential means of regulation are in fact taken advantage of by cells as they undergo plasticity. A deeper grasp of the diversity of mechanisms, including between synapses and amongst different cell types, will give greater insight to their unique contributions to neural circuits, ultimately enabling a better understanding of both healthy and diseased brain function.
Highlights.
- Synaptic strength depends on nanoscale alignment of release and clustered receptors
- Diverse mechanisms of altered alignment can contribute to synaptic plasticity
- Plasticity thus need not involve changes to release probability or receptor number
Acknowledgments
This work was supported by the Kahlert Foundation and grants to HC (F30 MH105111), AT (NARSAD YIA), and TAB (R01 MH080046 and NS090644).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1*.Malagon G, Miki T, Llano I, Neher E, Marty A. Counting Vesicular Release Events Reveals Binomial Release Statistics at Single Glutamatergic Synapses. The Journal of Neuroscience. 2016;36:4010–4025. doi: 10.1523/JNEUROSCI.4352-15.2016. Quantal analysis of release events at cerebellar granule cell to molecular layer interneuron synapses suggest that action potentials driven release occurs at multiple discrete and essentially independent sites in the AZ. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2**.Tang A-H, Chen H, Li TP, Metzbower SR, MacGillavry HD, Blanpied TA. A trans-synaptic nanocolumn aligns neurotransmitter release to receptors. Nature. 2016;536:210–214. doi: 10.1038/nature19058. Demonstrated AZ protein clusters using STORM, mapped vesicle fusion sites near RIM1 clusters using pHUSE-PALM, characterized transsynaptic alignment of AZ and PSD nanoclusters at baseline and with plasticity in cultured hippocampal neurons. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3*.Maschi D, Klyachko VA. Spatiotemporal Regulation of Synaptic Vesicle Fusion Sites in Central Synapses. Neuron. 2017;94:65–73. e3. doi: 10.1016/j.neuron.2017.03.006. Observed multiple vesicle release clusters in individual boutons in cultured hippocampal neurons, noted that release event distance to AZ center increased with higher frequency stimulation. [DOI] [PubMed] [Google Scholar]
- 4.Sudhof TC. The presynaptic active zone. Neuron. 2012;75:11–25. doi: 10.1016/j.neuron.2012.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Biederer T, Kaeser PS, Blanpied TA. Transcellular Nanoalignment of Synaptic Function. Neuron. 2017;96:680–696. doi: 10.1016/j.neuron.2017.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Grauel MK, Maglione M, Reddy-Alla S, Willmes CG, Brockmann MM, Trimbuch T, Rosenmund T, Pangalos M, Vardar G, Stumpf A, et al. RIM-binding protein 2 regulates release probability by fine-tuning calcium channel localization at murine hippocampal synapses. Proceedings of the National Academy of Sciences. 2016;113:11615–11620. doi: 10.1073/pnas.1605256113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7**.Sakamoto H, Ariyoshi T, Kimpara N, Sugao K, Taiko I, Takikawa K, Asanuma D, Namiki S, Hirose K. Synaptic weight set by Munc13-1 supramolecular assemblies. Nature neuroscience. 2018;21:41. doi: 10.1038/s41593-017-0041-9. Utilized novel glutamate sensor to measure quantal release in cultured hippocampal neurons, and correlated functional release sites to Munc13 clusters. [DOI] [PubMed] [Google Scholar]
- 8**.Reddy-Alla S, Böhme MA, Reynolds E, Beis C, Grasskamp AT, Mampell MM, Maglione M, Jusyte M, Rey U, Babikir H, et al. Stable Positioning of Unc13 Restricts Synaptic Vesicle Fusion to Defined Release Sites to Promote Synchronous Neurotransmission. Neuron. 2017;95:1350–1364. e12. doi: 10.1016/j.neuron.2017.08.016. Showed overlap of release sites by GCAMP and UNC13 distribution by STED in Drosophila NMJ, and deletion of UNC13a reduced evoked release through decreasing the positioning of docked vesicles near Ca channels. [DOI] [PubMed] [Google Scholar]
- 9.Böhme MA, Beis C, Reddy-Alla S, Reynolds E, Mampell MM, Grasskamp AT, Lützkendorf J, Bergeron DD, Driller JH, Babikir H, et al. Active zone scaffolds differentially accumulate Unc13 isoforms to tune Ca 2+ channel–vesicle coupling. Nature neuroscience. 2016;19:1311. doi: 10.1038/nn.4364. [DOI] [PubMed] [Google Scholar]
- 10.Schneider R, Hosy E, Kohl J, Klueva J, Choquet D, Thomas U, Voigt A, Heine M. Mobility of Calcium Channels in the Presynaptic Membrane. Neuron. 2015;86:672–679. doi: 10.1016/j.neuron.2015.03.050. [DOI] [PubMed] [Google Scholar]
- 11.Holderith N, Lorincz A, Katona G, Rózsa B, Kulik A, Watanabe M, Nusser Z. Release probability of hippocampal glutamatergic terminals scales with the size of the active zone. Nature neuroscience. 2012;15:988. doi: 10.1038/nn.3137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12*.Miki T, Kaufmann WA, Malagon G, Gomez L, Tabuchi K, Watanabe M, Shigemoto R, Marty A. Numbers of presynaptic Ca2+ channel clusters match those of functionally defined vesicular docking sites in single central synapses. Proceedings of the National Academy of Sciences. 2017;114:E5246–E5255. doi: 10.1073/pnas.1704470114. In cerebellar granule cell synapses, the number of Ca2+ channel clusters correlates with the number of docked vesicles at different ages. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fukata Y, Dimitrov A, Boncompain G, Vielemeyer O, Perez F, Fukata M. Local palmitoylation cycles define activity-regulated postsynaptic subdomains. The Journal of cell biology. 2013;202:145–161. doi: 10.1083/jcb.201302071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.MacGillavry HD, Song Y, Raghavachari S, Blanpied TA. Nanoscale Scaffolding Domains within the Postsynaptic Density Concentrate Synaptic AMPA Receptors. Neuron. 2013;78:615–622. doi: 10.1016/j.neuron.2013.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nair D, Hosy E, Petersen JD, Constals A, Giannone G, Choquet D, Sibarita J-B. Super-resolution imaging reveals that AMPA receptors inside synapses are dynamically organized in nanodomains regulated by PSD95. The Journal of Neuroscience. 2013;33:13204–13224. doi: 10.1523/JNEUROSCI.2381-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Broadhead MJ, Horrocks MH, Zhu F, Muresan L, Benavides-Piccione R, DeFelipe J, Fricker D, Kopanitsa MV, Duncan RR, Klenerman D, et al. PSD95 nanoclusters are postsynaptic building blocks in hippocampus circuits. Scientific reports. 2016;6:24626. doi: 10.1038/srep24626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Freche D, Pannasch U, Rouach N, Holcman D. Synapse geometry and receptor dynamics modulate synaptic strength. PLoS One. 2011;6:e25122. doi: 10.1371/journal.pone.0025122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Santucci DM, Raghavachari S. The effects of NR2 subunit-dependent NMDA receptor kinetics on synaptic transmission and CaMKII activation. PLoS computational biology. 2008;4:e1000208. doi: 10.1371/journal.pcbi.1000208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu G, Choi S, Tsien RW. Variability of neurotransmitter concentration and nonsaturation of postsynaptic AMPA receptors at synapses in hippocampal cultures and slices. Neuron. 1999;22:395–409. doi: 10.1016/s0896-6273(00)81099-5. [DOI] [PubMed] [Google Scholar]
- 20.McAllister AK, Stevens CF. Nonsaturation of AMPA and NMDA receptors at hippocampal synapses. Proceedings of the National Academy of Sciences. 2000;97:6173–6178. doi: 10.1073/pnas.100126497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Atasoy D, Ertunc M, Moulder KL, Blackwell J, Chung C, Su J, Kavalali ET. Spontaneous and evoked glutamate release activates two populations of NMDA receptors with limited overlap. The Journal of Neuroscience. 2008;28:10151–10166. doi: 10.1523/JNEUROSCI.2432-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sara Y, Bal M, Adachi M, Monteggia LM, Kavalali ET. Use-dependent AMPA receptor block reveals segregation of spontaneous and evoked glutamatergic neurotransmission. The Journal of Neuroscience. 2011;31:5378–5382. doi: 10.1523/JNEUROSCI.5234-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lisman J, Raghavachari S. A unified model of the presynaptic and postsynaptic changes during LTP at CA1 synapses. Sci STKE. 2006;2006:re11–re11. doi: 10.1126/stke.3562006re11. [DOI] [PubMed] [Google Scholar]
- 24*.Hruska M, Henderson N, Le Marchand SJ, Jafri H, Dalva MB. Synaptic nanomodules underlie the organization and plasticity of spine synapses. Nature neuroscience. 2018:1. doi: 10.1038/s41593-018-0138-9. Used live-cell STED to visualize events in both pre and postsynaptic neurons during induction of LTP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25**.Sinnen BL, Bowen AB, Forte JS, Hiester BG, Crosby KC, Gibson ES, Dell’Acqua ML, Kennedy MJ. Optogenetic control of synaptic composition and function. Neuron. 2017;93:646–660.e5. doi: 10.1016/j.neuron.2016.12.037. Optogenetically targeted AMPARs into the PSD and found no change in quantal amplitude in cultured hippocampal neurons. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lu HE, MacGillavry HD, Frost NA, Blanpied TA. Multiple Spatial and Kinetic Subpopulations of CaMKII in Spines and Dendrites as Resolved by Single-Molecule Tracking PALM. The Journal of Neuroscience. 2014;34:7600–7610. doi: 10.1523/JNEUROSCI.4364-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Steiner P, Higley MJ, Xu W, Czervionke BL, Malenka RC, Sabatini BL. Destabilization of the postsynaptic density by PSD-95 serine 73 phosphorylation inhibits spine growth and synaptic plasticity. Neuron. 2008;60:788–802. doi: 10.1016/j.neuron.2008.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28*.Matt L, Kim K, Hergarden AE, Patriarchi T, Malik ZA, Park DK, Chowdhury D, Buonarati OR, Henderson PB, Saraç ÇG, et al. α-Actinin Anchors PSD-95 at Postsynaptic Sites. Neuron. 2018;97:1094–1109.e9. doi: 10.1016/j.neuron.2018.01.036. Identified α-actinin as critical for anchoring PSD-95 at synapses. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Blanpied TA, Kerr JM, Ehlers MD. Structural plasticity with preserved topology in the postsynaptic protein network. Proceedings of the National Academy of Sciences. 2008;105:12587–12592. doi: 10.1073/pnas.0711669105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chazeau A, Giannone G. Organization and dynamics of the actin cytoskeleton during dendritic spine morphological remodeling. Cellular and molecular life sciences. 2016;73:3053–3073. doi: 10.1007/s00018-016-2214-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31*.Lei N, Mellem JE, Brockie PJ, Madsen DM, Maricq AV. NRAP-1 Is a Presynaptically Released NMDA Receptor Auxiliary Protein that Modifies Synaptic Strength. Neuron. 2017;96:1303–1316.e6. doi: 10.1016/j.neuron.2017.11.019. Identified NRAP-1 to be a presynaptically secreted auxiliary protein for postsynaptic NMDARs that modifies receptor gating in C. elegans. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Savtchenko LP, Rusakov DA. Moderate AMPA receptor clustering on the nanoscale can efficiently potentiate synaptic current. Phil Trans R Soc B. 2014;369:20130167. doi: 10.1098/rstb.2013.0167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Opazo P, Sainlos M, Choquet D. Regulation of AMPA receptor surface diffusion by PSD-95 slots. Curr Opin Neurobiol. 2012;22:453–60. doi: 10.1016/j.conb.2011.10.010. [DOI] [PubMed] [Google Scholar]
- 34.Park J, Chavez AE, Mineur YS, Morimoto-Tomita M, Lutzu S, Kim KS, Picciotto MR, Castillo PE, Tomita S. CaMKII Phosphorylation of TARPgamma-8 Is a Mediator of LTP and Learning and Memory. Neuron. 2016;92:75–83. doi: 10.1016/j.neuron.2016.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Klaassen RV, Stroeder J, Coussen F, Hafner AS, Petersen JD, Renancio C, Schmitz LJ, Normand E, Lodder JC, Rotaru DC, et al. Shisa6 traps AMPA receptors at postsynaptic sites and prevents their desensitization during synaptic activity. Nat Commun. 2016;7:10682. doi: 10.1038/ncomms10682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Groc L, Heine M, Cognet L, Brickley K, Stephenson FA, Lounis B, Choquet D. Differential activity-dependent regulation of the lateral mobilities of AMPA and NMDA receptors. Nature neuroscience. 2004;7:695–696. doi: 10.1038/nn1270. [DOI] [PubMed] [Google Scholar]
- 37.Constals A, Penn AC, Compans B, Toulmé E, Phillipat A, Marais S, Retailleau N, Hafner A-S, Coussen F, Hosy E, et al. Glutamate-induced AMPA receptor desensitization increases their mobility and modulates short-term plasticity through unbinding from Stargazin. Neuron. 2015;85:787–803. doi: 10.1016/j.neuron.2015.01.012. [DOI] [PubMed] [Google Scholar]
- 38*.Li TP, Song Y, MacGillavry HD, Blanpied TA, Raghavachari S. Protein Crowding within the Postsynaptic Density Can Impede the Escape of Membrane Proteins. The Journal of Neuroscience. 2016;36:4276–4295. doi: 10.1523/JNEUROSCI.3154-15.2016. Found that AMPAR mobility was affected by both steric hindrance and protein binding in the PSD. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li TP, Blanpied TA. Control of transmembrane protein diffusion within the postsynaptic density assessed by simultaneous single-molecule tracking and localization microscopy. Frontiers in synaptic neuroscience. 2016;8:19. doi: 10.3389/fnsyn.2016.00019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Santamaria F, Gonzalez J, Augustine GJ, Raghavachari S. Quantifying the effects of elastic collisions and non-covalent binding on glutamate receptor trafficking in the post-synaptic density. PLoS Comput Biol. 2010;6:e1000780. doi: 10.1371/journal.pcbi.1000780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41**.Zeng M, Shang Y, Araki Y, Guo T, Huganir RL, Zhang M. Phase transition in postsynaptic densities underlies formation of synaptic complexes and synaptic plasticity. Cell. 2016;166:1163–1175.e12. doi: 10.1016/j.cell.2016.07.008. Observed PSD-95 in both condensed liquid and aqueous phases and identified SynGAP as regulator of phase transition in cultured hippocampal neurons. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Heine M, Groc L, Frischknecht R, Béïque J-C, Lounis B, Rumbaugh G, Huganir RL, Cognet L, Choquet D. Surface mobility of postsynaptic AMPARs tunes synaptic transmission. Science. 2008;320:201–205. doi: 10.1126/science.1152089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43**.Penn A, Zhang C, Georges F, Royer L, Breillat C, Hosy E, Petersen J, Humeau Y, Choquet D. Hippocampal LTP and contextual learning require surface diffusion of AMPA receptors. Nature. 2017;549:384. doi: 10.1038/nature23658. Strongest test to date that AMPAR mobility is critical for expression of LTP, including in vivo. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bell ME, Bourne JN, Chirillo MA, Mendenhall JM, Kuwajima M, Harris KM. Dynamics of nascent and active zone ultrastructure as synapses enlarge during long - term potentiation in mature hippocampus. Journal of Comparative Neurology. 2014;522:3861–3884. doi: 10.1002/cne.23646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Matz J, Gilyan A, Kolar A, McCarvill T, Krueger SR. Rapid structural alterations of the active zone lead to sustained changes in neurotransmitter release. Proceedings of the National Academy of Sciences. 2010;107:8836–8841. doi: 10.1073/pnas.0906087107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Glebov OO, Jackson RE, Winterflood CM, Owen DM, Barker EA, Doherty P, Ewers H, Burrone J. Nanoscale Structural Plasticity of the Active Zone Matrix Modulates Presynaptic Function. Cell Reports. 2017;18:2715–2728. doi: 10.1016/j.celrep.2017.02.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Glebov OO, Cox S, Humphreys L, Burrone J. Neuronal activity controls transsynaptic geometry. Scientific reports. 2016;6:22703. doi: 10.1038/srep22703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Müller M, Genç Ö, Davis GW. RIM-binding protein links synaptic homeostasis to the stabilization and replenishment of high release probability vesicles. Neuron. 2015;85:1056–1069. doi: 10.1016/j.neuron.2015.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Südhof TC. Synaptic Neurexin Complexes: A Molecular Code for the Logic of Neural Circuits. Cell. 2017;171:745–769. doi: 10.1016/j.cell.2017.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.de Arce KP, Schrod N, Metzbower SW, Allgeyer E, Kong GK-W, Tang A-H, Krupp AJ, Stein V, Liu X, Bewersdorf J, et al. Topographic mapping of the synaptic cleft into adhesive nanodomains. Neuron. 2015;88:1165–1172. doi: 10.1016/j.neuron.2015.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chamma I, Letellier M, Butler C, Tessier B, Lim K-H, Gauthereau I, Choquet D, Sibarita J-B, Park S, Sainlos M, et al. Mapping the dynamics and nanoscale organization of synaptic adhesion proteins using monomeric streptavidin. Nature communications. 2016;7:10773. doi: 10.1038/ncomms10773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chamma I, Levet F, Sibarita J-B, Sainlos M, Thoumine O. Nanoscale organization of synaptic adhesion proteins revealed by single-molecule localization microscopy. Neurophotonics. 2016;3:041810. doi: 10.1117/1.NPh.3.4.041810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Peixoto RT, Kunz PA, Kwon H, Mabb AM, Sabatini BL, Philpot BD, Ehlers MD. Transsynaptic signaling by activity-dependent cleavage of neuroligin-1. Neuron. 2012;76:396–409. doi: 10.1016/j.neuron.2012.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Suzuki K, Hayashi Y, Nakahara S, Kumazaki H, Prox J, Horiuchi K, Zeng M, Tanimura S, Nishiyama Y, Osawa S, et al. Activity-dependent proteolytic cleavage of neuroligin-1. Neuron. 2012;76:410–422. doi: 10.1016/j.neuron.2012.10.003. [DOI] [PubMed] [Google Scholar]
- 55.Bemben MA, Shipman SL, Hirai T, Herring BE, Li Y, Badger JD, II, Nicoll RA, Diamond JS, Roche KW. CaMKII phosphorylation of neuroligin-1 regulates excitatory synapses. Nature neuroscience. 2014;17:56. doi: 10.1038/nn.3601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dalva MB, McClelland AC, Kayser MS. Cell adhesion molecules: signalling functions at the synapse. Nature Reviews Neuroscience. 2007;8:206–220. doi: 10.1038/nrn2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57**.Tong X-J, López-Soto EJ, Li L, Liu H, Nedelcu D, Lipscombe D, Hu Z, Kaplan JM. Retrograde Synaptic Inhibition Is Mediated by α-Neurexin Binding to the α2δ Subunits of N-Type Calcium Channels. Neuron. 2017;95:326–340.e5. doi: 10.1016/j.neuron.2017.06.018. Showed that postsynaptic neurexin1 retrogradely inhibits presynaptic release in Drosophila NMJ. [DOI] [PMC free article] [PubMed] [Google Scholar]