Synaptic Strength Modulation after Cortical Trauma: A Role in Epileptogenesis (original) (raw)
Abstract
Traumatic brain injuries are often followed by abnormal hyperexcitability, leading to acute seizures and epilepsy. Previous studies documented the rewiring capacity of neocortical neurons in response to various cortical and subcortical lesions. However, little information is available on the functional consequences of these anatomical changes after cortical trauma and the adaptation of synaptic connectivity to a decreased input produced by chronic deafferentation. In this study, we recorded intracellular (IC) activities of cortical neurons simultaneously with extracellular (EC) unit activities and field potentials of neighboring cells in cat cortex, after a large transection of the white matter underneath the suprasylvian gyrus, in acute and chronic conditions (at 2, 4, and 6 weeks) in ketamine–xylazine-anesthetized cats. Using EC spikes to compute the spike-triggered averages of IC membrane potential, we found an increased connection probability and efficacy between cortical neurons weeks after cortical trauma. Inhibitory interactions showed no significant changes in the traumatized cortex compared with control. The increased synaptic efficacy was accompanied by enhanced input resistance and intrinsic excitability of cortical neurons, as well as by increased duration of silent network periods. Our electrophysiological data revealed functional consequences of previously reported anatomical changes in the injured cortex. We suggest that homeostatic synaptic plasticity compensating the decreased activity in the undercut cortex leads to an uncontrollable cortical hyperexcitability and seizure generation.
Keywords: epilepsy, cortical trauma, synaptic efficacy, intrinsic excitability, homeostatic plasticity, in vivo
Introduction
The CNS reveals a remarkable plasticity, which enables adaptation to changes in incoming volleys while still maintaining activity within functional boundaries (Turrigiano, 1999). Brain injury frequently generates aberrant excitability responsible for the increased frequency of acute seizures (Dinner, 1993; Topolnik et al., 2003b) and chronic epilepsy (Salazar et al., 1985).
Partially isolated neocortex is a well established model of epileptogenesis after cortical trauma (Prince and Tseng, 1993; Hoffman et al., 1994; Jacobs et al., 2000; Topolnik et al., 2003b; Nita et al., 2007). The undercut cortex becomes increasingly hyperexcitable over a few weeks and progressively generates paroxysmal activity, both in vivo (Nita et al., 2006, 2007) and in vitro (Prince and Tseng, 1993; Salin et al., 1995). Several mechanisms may contribute to the development of hyperexcitability after cortical injury, including changes in intrinsic neuronal properties (Esplin et al., 1994; Topolnik et al., 2003a), selective loss of inhibitory synapses (Ribak and Reiffenstein, 1982), increase of both inhibition and excitation (Bush et al., 1999), and axonal sprouting (Salin et al., 1995). The capacity of neocortical neurons to modify their axonal arborization has been documented after various cortical and subcortical lesions (Kuang and Kalil, 1990; McKinney et al., 1997). However, little is known about the functional consequences of these anatomical changes after cortical trauma and the adaptation of synaptic connectivity to a decreased input produced by chronic deafferentation.
Although studies performed in slices or cultures have been crucial in uncovering the role of different substances (Graber and Prince, 1999), ionic channels (Desai et al., 1999), and receptors (Chen et al., 2007) in epilepsy, they cannot recreate the complexity of the whole brain, in which different structures interact to maintain the fine balance between excitation and inhibition. To overcome these limitations, we performed the experiments in vivo, to investigate the gradual changes of synaptic efficacy and intrinsic excitability after cortical undercut. Most previous studies assessing synaptic connectivity after brain trauma used only indirect methods to quantify the changes in synaptic connectivity (Li et al., 2005; Jin et al., 2006), because direct electrophysiological assessment of synaptic properties is difficult with currently available techniques (Li et al., 2005) and was not considered feasible for quantitative analysis of functional connectivity (Jin et al., 2006).
In the present study, we quantified the efficacy of synaptic interactions in vivo, at different time delays after cortical deafferentation, to uncover the synaptic changes responsible for posttraumatic epilepsy. We found a boost in intrinsic excitability and synaptic connectivity several weeks after brain injury, accompanied by an increased efficacy of excitatory connections without significant changes of inhibitory connections, both in acute and chronic conditions. The decreased network activity in the deafferented cortex might have activated homeostatic synaptic plasticity (Turrigiano, 1999; Houweling et al., 2005; Trasande and Ramirez, 2007), leading to increased strength of excitatory synapses to compensate for the reduced inputs to the undercut cortex.
A part of this work has been reported in abstract form (Avramescu and Timofeev, 2006).
Materials and Methods
Animal preparation.
Experiments were performed in 37 ketamine–xylazine-anesthetized cats (10–15 and 2–3 mg/kg, i.m., respectively) of both sexes, divided into three groups: control (n = 13), acute undercut (n = 12), and chronic undercut (n = 12). The animals were paralyzed with gallamine triethiodide (20 mg/kg, i.v.) after the EEG showed typical signs of deep general anesthesia, essentially consisting of sequences of slow oscillation (0.5–1 Hz). Supplementary doses of anesthetics were administered at the slightest change toward an activated EEG pattern. Lidocaine (0.5%) infiltration was used for all pressure points or incision lines. General surgical procedures included: cephalic vein cannulation for systemic liquid delivery (lactated Ringer's solution, 5–10 ml/kg/h) and tracheotomy. The cats were ventilated artificially with the control of end-tidal CO2 at 3.5–3.7% and heart rate (90–100 beats/min). The body temperature was maintained at 37–38°C with a heating pad. The stability of intracellular (IC) recordings was increased by the drainage of the CSF from the cisterna magna, hip suspension, bilateral pneumothorax, and filling the hole made for recordings with a solution of 4% agar.
Cortical deafferentation.
The partial cortical deafferentation was performed by a large transection (13–15 mm posteroanteriorly, 3–4 mm mediolaterally, and 3–4 mm deep) of the white matter under the suprasylvian gyrus. To produce the undercut, a custom-designed “L”-shaped knife was inserted into the posterior part of the suprasylvian gyrus, rotated at a 90° angle, and advanced rostrally, parallel to the surface of the gyrus. Therefore, the white matter below the posterior and middle part of the gyrus was transected while the anterior part was still connected to the underneath structures, hence creating conditions of partial cortical deafferentation.
At the end of experiments, the animals received a lethal dose of sodium pentobarbital (50 mg/kg, i.v.). The brains were removed, sliced into 80 μm sections, and stained with thionine to verify the extension and the position of the undercut (Fig. 1A). All experimental procedures were approved by the committee for animal care of Laval University and were in accordance with the guidelines published in the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Figure 1.
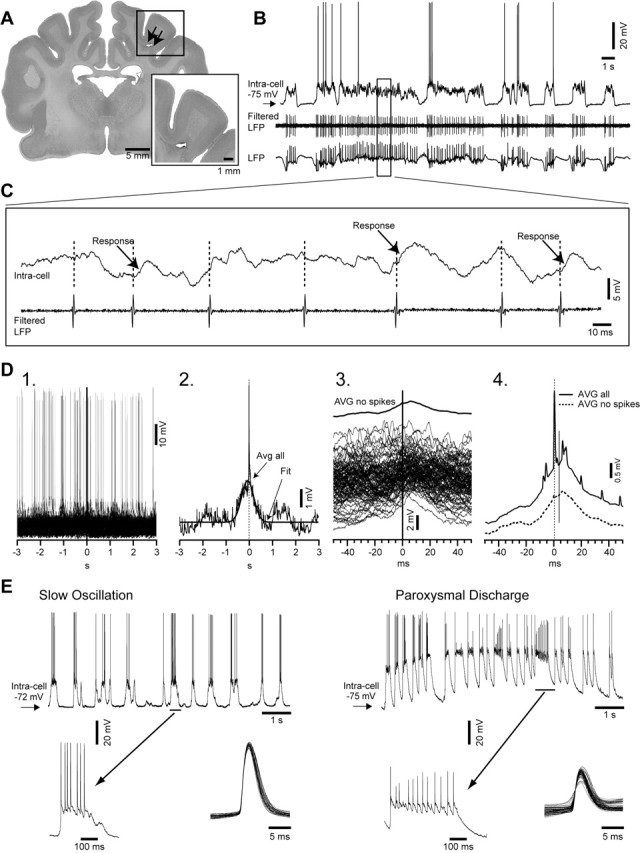
Experimental paradigm and methodology of analysis. A, A frontal section of cat brain (Nissl staining) depicting the large transection of the white matter under the suprasylvian gyrus (expanded in inset). Undercut is indicated by arrows. B, Simultaneous intracellular, local field potential (LFP), and unit activity (filtered LFP), in cortical area 7 left, in a ketamine–xylazine-anesthetized cat. The area designated with a rectangle is expanded in C. C, Intracellular recording (top trace) and filtered local field potential (bottom trace). Different responses related to unit activity are indicated in the recorded neuron. D1, Superimposition of multiple segments (n = 140) triggered by the spikes in the extracellularly recorded neuron. D2, Spike-triggered average (n = 140) of intracellular segments (Avg all) fitted with a Gaussian function (Fit). D3, All segments without action potentials triggered by the extracellular spikes (n = 125), extracted from the intracellular recorded neuron, together with their average (AVG no spikes). D4, The average of all intracellular segments (AVG all) and the average of the segments without action potentials triggered by the presynaptic neuron (AVG no spikes). E, Intracellular recording of the same neuron during slow oscillation period (left) and during paroxysmal discharge (right). Periods indicated by horizontal bars are expanded below. Bottom right insets show superimposition of all action potentials detected during depicted periods of slow oscillation and paroxysmal discharge. Note the smaller amplitude and the higher variability of the action potentials during paroxysmal discharge periods compared with the slow oscillation.
Recordings.
Acute experiments were performed during the same day that the undercut was made, whereas chronic experiments were performed at 2 weeks (n = 4), 4 weeks (n = 4), and 6 weeks (n = 4) after cortical deafferentation, under ketamine–xylazine anesthesia (10–15 and 2–3 mg/kg, i.m.). In control experiments, the recordings were performed in intact cortex.
The experimental approach was similar to the one described by Matsumura et al. (1996). Simultaneous IC and extracellular (EC) activities of neighboring neurons were recorded in the partially deafferented area 7 of the left suprasylvian gyrus (Fig. 1B,C) with glass micropipettes (tip diameter, <0.5 μm). The IC electrode was inserted in a vertical stereotaxic direction, whereas the EC electrode was inserted at an angle of 10–15° from vertical in the parasagittal plane, so that the tips of the electrodes were very close one to each other (<100 μm). The pipettes were filled with potassium acetate (2.5 m; in situ impedance, 35–65 MΩ) for the IC recordings and with sodium chloride (1 m; in situ impedance, 5–10 MΩ) for local field potential and extracellular unit recordings. We carefully verified the morphology of each EC recorded spike, and we only included single-unit recordings of the presynaptic neurons. However, it was possible that in some recordings, the EC spikes corresponded to more than one presynaptic neuron. Each pair of neurons consisted of an EC recorded presynaptic spike and a postsynaptic IC recording.
The intracellular signals were passed through a high-impedance amplifier with an active bridge circuitry (bandwidth DC to 9 kHz). All signals were digitized (20 kHz sampling rate) and stored for off-line analysis.
Twenty animals with deafferented suprasylvian gyrus displayed paroxysmal EEG activities (Fig. 1E). We chose not to include in our analysis this paroxysmal periods for several reasons. First, in the present study, we were interested to quantify the changes in neuronal interactions that precede and favor the development of electrographic seizures. Second, during paroxysmal discharges, the amplitude of the action potentials (29.1 ± 6.2 mV) decreases significantly (p < 0.05, Student's t test), and their variability (SD) increases compared with the slow oscillation (57.2 ± 2.5 mV) periods (Fig. 1E). This may bias the detection of EC spikes, which in this study was based on the morphology of spikes.
Data analysis.
All IC recorded neurons were classified, based on their pattern of response to depolarizing current pulses, into one of four electrophysiological classes of neocortical neurons: regular spiking (RS), intrinsically bursting (IB), fast rhythmic bursting (FRB), or fast spiking (FS) (Connors and Gutnick, 1990; Gray and McCormick, 1996; Steriade et al., 1998). The apparent input resistance of neurons was measured during both active and silent phases of the slow oscillation by averaging cellular responses to five trials of hyperpolarizing current pulses (0.5 nA, 0.1 s) during each phase. To evaluate the firing frequency of RS neurons, we counted the number of spikes and we measured instantaneous firing rate as the reciprocal of the first interspike interval in response to depolarizing current (0.5 nA, 0.75 nA, 1.0 nA, 0.2 s) in control and in undercut cortex. We used the average of the values measured in five consecutive trials from 10 RS neurons with similar membrane potential (−70 ± 5 mV) in each group.
To establish the possible synaptic connections between neurons, the EC recorded units were used to compute spike-triggered averages (STAs) of the membrane potential of IC recorded neurons (Fig. 1). We first averaged all segments (Fig. 1D1,D2,D4), and subsequently, to reliably determine the response of the postsynaptic neuron to the presynaptic trigger, we removed manually all segments that revealed an action potential (AP) during 50 ms before or after the trigger. The average of the remaining segments was identified as “AVG no spikes” (Fig. 1D3,D4). The minimum number of segments to detect a synaptic connection was 100, although most averages were obtained from 100–500 segments that remained after removing the APs.
Based on the polarity and the relative onset times of deflections in “AVG no spikes,” we identified four basic types of interactions between the recorded cells. If the IC membrane potential revealed a depolarization event after the presynaptic AP, we identified it as an EPSP (Fig. 2A), and if the onset of depolarization occurred before the trigger, we classified it as a network excitation (NE) (Fig. 2B). When a hyperpolarizing potential was recorded after the EC spike, we classified it as an IPSP (Fig. 2C), and if the onset of hyperpolarization occurred before the EC spike, we classified it as a network inhibition (NI) (Fig. 2D). If the averaged amplitude of the IC membrane potential during 20 ms before or after the EC revealed no relation to the spike, we considered that the two cells were not interacting (Fig. 2E).
Figure 2.

Different types of responses and possible designs of synaptic topology. A, C, Direct synaptic connections. Panels contain the scheme of a possible synaptic circuit triggering the average EPSP (n = 145) or IPSP (n = 125). The pyramidal-shaped unit depicts the cell that was recorded intracellularly (IC); the round-shaped unit symbolizes the cell recorded extracellularly (EC). Open circles and buttons designate excitatory neurons and synapses, respectively, whereas the black ones designate the inhibitory neurons and synapses. B, D, Synchronous network connections: NE (n = 178) and NI (n = 131). E, Nonresponses (n = 155). F, Methodology used for the quantification of responses. Ampl, Amplitude; Fit, sigmoid fitting.
Each averaged postsynaptic response was fitted with a sigmoid function, and we estimated the beginning of the response at the point representing 10% of the slope. The same averaged responses were used to quantify the amplitude, latency, 0–100% rise time, and duration at half amplitude for the four types of interactions (Fig. 2F).
For all direct synaptic connections (EPSPs and IPSPs), we analyzed each segment manually to distinguish between successful responses and failures. We established a jitter of 0.2 ms for the latency of the responses centered on a time window after the presynaptic trigger during which the responses were expected to occur. We then computed the first derivative of each individual segment included in the STAs, and we considered a “response” those segments that displayed a positive peak (for excitatory connections) or a negative peak (for inhibitory connections) in the differentiated wave, during the established time window (Fig. 3A).
Figure 3.

Examples of responses and failures in direct synaptic connections. A, Top, Superimposition of the first derivative of the individual segments depicted as examples in the bottom panels. Middle, Averages (AVG) of all postsynaptic responses obtained from the pair of recorded neurons. Bottom, Ten individual examples of successful excitatory (left) and inhibitory (right) connection, together with examples of failures recorded in the same postsynaptic neuron. B, Amplitude of excitatory responses (open red circles) and failures (open black circles) plotted against the membrane potential, and histograms of the amplitude of responses (open bars) and background noise (gray bars) in the excitatory connection topology. C, Amplitude of inhibitory responses (open red circles) and failures (open black circles) plotted against the membrane potential, and histograms of the amplitude of responses (open bars) and network noise (gray bars) in the inhibitory connection topology. Noise histograms were fitted using a Gaussian function (plain gray line superimposed on gray histograms). Note the wider distribution to the right of the excitatory responses and of inhibitory responses to the left, compared with the network noise amplitude. The bin of the histogram is 0.2 mV.
To clearly identify the response from the background synaptic noise, we measured from IC traces the amplitude of the synaptic potential in the time window during which the responses were expected to occur, before (to measure the synaptic noise) and after (to measure the amplitude of the response) the EC spike. Thereafter, we computed and superimposed the histograms of amplitude for both synaptic noise (gray bars) and responses and failures (open bars) (Fig. 3B,C). To double-check the presence of responses, we plotted the amplitude of the responses and failures against the membrane potential and observed that for excitatory connections, the responses were positive (open red circles) and the failures were centered on zero (open black circles), being either positive or negative depending on polarity of synaptic noise (Fig. 3B). For the inhibitory connections, the amplitude of responses was negative at a depolarized membrane potential and positive at a more hyperpolarized potential (open red circles), whereas the failures (open black circles) were also centered around the zero line and were either positive or negative (Fig. 3C). Because IPSPs occurring around reversal potential could be mistaken for failures, we calculated failure rates for IPSPs that occurred at least 5 mV above estimated reversal potential for IPSP. We also computed the skewness and kurtosis for the responses (excitatory and inhibitory) and for the noise.
It has been previously shown in vitro, in the undercut model, that GABA-mediated postsynaptic potentials could become depolarizing during high activity periods, because of an impairment of Cl− extrusion (Jin et al., 2005). In the present study, we estimated the reversal potential for all our recordings, and it was always between −60 and −70 mV, whereas the potential for depolarizing GABA events could not be more depolarized than −50 mV. Moreover, neurons in the undercut cortex have a normal resting _E_Cl, and the impairment of Cl− extrusion has been described to convert the hyperpolarizing GABAergic responses into depolarizing ones only during intense neuronal activity, such as during seizures (Jin et al., 2005), but here we only analyzed neuronal interactions during the seizure-free periods. Thus, depolarizing events after presynaptic spike recorded in the current study were EPSPs and not depolarizing IPSPs.
For all direct excitatory connections, we calculated the failure rate as the percentage of all failures from the total number of segments of the STA, and the coefficient of variation (c.v.) was computed for successful responses only.
Statistical significance of comparative data were assessed by performing appropriate statistical tests: Student's t test and Mann–Whitney test. Differences between means were considered significant at p < 0.05. Numerical values are given as means ± SEM except where otherwise specified. In all statistical tests, the results obtained in acute conditions were compared with control and with chronic conditions.
Results
We recorded a total of 135 pairs of neurons in control and 263 in undercut cortex, from which 93 in control and 180 in deafferented cortex displayed an interaction (EPSP, IPSP, NE or NI), whereas in the other EC–IC pairs, the neurons were not connected.
In both control and undercut cortex (acute and chronic), the IC neurons revealed a slow oscillatory pattern (Steriade et al., 1993; Contreras and Steriade, 1995). During depth positivity, the neurons were hyperpolarized and silent, and during depth negativity, they were depolarized (Fig. 1B). The incidence of paroxysmal activities increased after undercut as already reported in previous published in vivo data, both in acute and chronic conditions (Topolnik et al., 2003b; Nita et al., 2006). In acute conditions, 8 of 12 animals displayed paroxysmal EEG patterns consisting of paroxysmal discharges and/or clear-cut electrographic seizures (Fig. 1E), whereas in chronic conditions (2, 4, and 6 weeks after undercut), all animals (n = 12) showed paroxysmal activity.
Synaptic interactions between cortical neurons
We identified four basic types of synaptic interactions between the triggering EC cell and the events in IC recorded neurons, based on the polarity and the relative onset times of deflections in the STAs. As we described in Materials and Methods, a pair of neurons was considered to be connected via a direct excitatory synaptic connection if the STAs revealed an EPSP (Fig. 2A) or a direct inhibitory synaptic connection if the STAs revealed an IPSP (Fig. 2C). We also analyzed the histograms of noise and responses (Fig. 3B,C). The histograms of noise were always near normal distribution (skewness, 0.22 ± 0.17; kurtosis, 0.38 ± 0.08). The skewness for the EPSPs (1.29 ± 0.98) was always higher (p < 0.05, Student's t test) than that for the noise, whereas the skewness for the IPSPs (−0.74 ± 0.51) was smaller (p < 0.05, Student's t test). The kurtosis of the responses (−0.62 ± 0.29 for EPSPs and −1.1 ± 0.32 for IPSPs) was smaller (p < 0.05, Student's t test) than the kurtosis of the noise, suggesting a more flattened distribution of responses. Depolarizing potentials beginning before the presynaptic spike were termed NE (Fig. 2B), and the mechanism that generated them could be a common excitatory synaptic input to both EC and IC cells from the same presynaptic cell(s). Hyperpolarizing potentials beginning before the presynaptic spike, NI (Fig. 2D), were probably produced by a common excitatory synaptic input onto EC and a population of inhibitory interneurons that synapse on IC. There were also several cases in which an EPSP was superimposed on an NE type of response. All other mixed responses, such as an IPSP overlaying an NE, an EPSP over an NI, or an IPSP on top of an NI, were difficult to quantify and therefore were not included in the analysis.
Deafferentation induced changes in synaptic connectivity
The most common type of response in control and after undercut was NE followed by NI, whereas the direct synaptic connections (EPSP and IPSP) were the least frequent (Fig. 4A). The high frequency of NE could reflect the possibility that a spike has a larger chance to be preceded by network or neuronal depolarization. NI responses were less frequent, because they could only be triggered when a local population of inhibitory neurons was able to send common synaptic input to both the EC and the IC cells, and it should also be taken into consideration that there are far less inhibitory neurons compared with excitatory ones.
Figure 4.

Changes of synaptic interactions after cortical deafferentation. A, Variation of the type of interactions: synchronous responses (NE and NI) and direct pure EPSP (EPSP p) and pure IPSP (IPSP p) connections in control (light gray bars) and undercut cortex (dark gray bars). Note that NE was the most frequent type of response both in control and in the undercut cortex. B, Variation of connection probability (only EPSPs and IPSPs were taken into account) at different time delays after cortical injury. Note the increased connectivity in chronic undercut cortex. C, D, The incidence of NE (C) and NI (D) in control and after cortical deafferentation. Ctrl, Control; Ac, acute; W, weeks. n represents the total number of responses recorded in each group. Error bars indicate SEM. *p < 0.05, Mann–Whitney.
We then measured the connection probability as the percentage of both EPSP and IPSP types of responses from the total number of interactions in a group. The probability of connection was similar in control (16.8 ± 2.8%) and in acute undercut (17.63 ± 4.03%) groups and then progressively increased (p < 0.05, Mann–Whitney) to 32.5 ± 7.5% at 2 weeks, 35.64 ± 3.89% at 4 weeks, and 35.5 ± 6.98% at 6 weeks after deafferentation (Fig. 4B). There were no statistical significant changes when analyzing separately the direct excitatory and inhibitory connections.
The higher rate of paired recording in our study compared with connection rate from in vitro studies is probably attributable to the fact that we measured the connectivity in vivo with all the connections intact. In fact, in vitro studies support this assumption, showing that an increased slice thickness only from 400 to 500 μm resulted in more than a threefold increase in the probability of connection (from 1:15 to 7:32) (Thomson et al., 1996).
Because NEs and NIs were the most frequent responses, we followed their dynamics at different time delays after cortical deafferentation. We observed no major changes in their probability of occurrence in acute cortical trauma conditions (77.33 ± 5.19% for NE and 10.01 ± 3.73% for NI) compared with control (77.52 ± 5.49% for NE and 15.98 ± 4.37% for NI). Nevertheless, the incidence of NE and NI at 2 and 4 weeks after deafferentation revealed opposite changes, increasing by 10.9% compared with acute conditions for NE and decreasing by 6.42% for NI at 2 weeks (p < 0.05, Mann–Whitney), whereas at 4 weeks the probability for NE decreased by 13.49% compared with control (p < 0.05, Mann–Whitney), and increased for NI by 15.78% (p < 0.05, Mann–Whitney) (Fig. 4C,D). Therefore, the ratio between NE and NI, which was 4.8 in control, decreased dramatically at 4 weeks, reaching a 2.5 value and then boosted again to 7.2, almost double the control conditions. These data provide support at the cellular level for our previous observation that the number of ictal events showed a plateau in the first 2 weeks, then decreased progressively, being almost absent after ∼4 weeks, and subsequently increased again, remaining constant for a long period of time (Nita et al., 2007).
In conclusion, the probability of connection, which includes both inhibitory and excitatory direct connections, is significantly larger in all the chronic stages of the undercut, whereas NE and NI display variations between the undercut groups. The high frequency of NE at 2 weeks is accompanied by a decreased incidence of NI; on the contrary, at 4 weeks the decreased expression of NE goes together with a higher frequency of NI, and at 6 weeks the NE and NI are balanced. This means that at 2 weeks after the penetrating injury, there is a high incidence of excitation, and at 4 weeks the inhibitory activity prevails; this might explain the decreased frequency of seizures at this time. Thereafter, the incidence of excitation increases again, and the occurrence of seizures remains constant (Nita et al., 2007).
Modulation of synaptic efficacy after penetrating cortical trauma
We identified four types of neurons based on the electrophysiological properties of the intracellular recordings: RS, FRB, IB, and FS. The general distribution expressed as means ± SD was 73.22 ± 9.85% for RS, 11.17 ± 4.03% for FRB, 9.56 ± 4.21% for IB, and 6.05 ± 2.29% for FS neurons. We observed a tendency toward a decreasing number of RS neurons at 4 and 6 weeks, together with an increased percentage of FRB and IB neurons, although it was not statistically significant. This heterogeneity of neurons could have influenced the response of the postsynaptic cell. To test this possibility, we initially analyzed separately for each type of postsynaptic neuron (RS, FRB, IB, and FS) the three parameters used to characterize the responses (amplitude, latency, and rise time) in control conditions. Given that we found no significant differences between the groups, the synaptic efficacy was analyzed together for all recorded neurons.
The amplitude of excitatory connections, both EPSPs and NEs, increased at 2 and 6 weeks after deafferentation compared with acute conditions (p < 0.05, Student's t test), whereas at 4 weeks it was decreased (p < 0.05, Student's t test) (Fig. 5A), showing therefore a similar trend with the incidence of NE. The amplitude of inhibitory connections (IPSP and NI) was not significantly modified after deafferentation (data not shown). However, there was a trend toward a reduction of inhibition at 2 weeks, whereas at 4 weeks after the undercut we observed a tendency toward enhanced inhibition (data not shown). In conclusion, the network excitability shifted toward excitation at 2 weeks after cortical undercut, diminished at 4 weeks, and then increased again at 6 weeks, paralleling the alteration in the number of ictal events reported previously (Nita et al., 2007).
Figure 5.
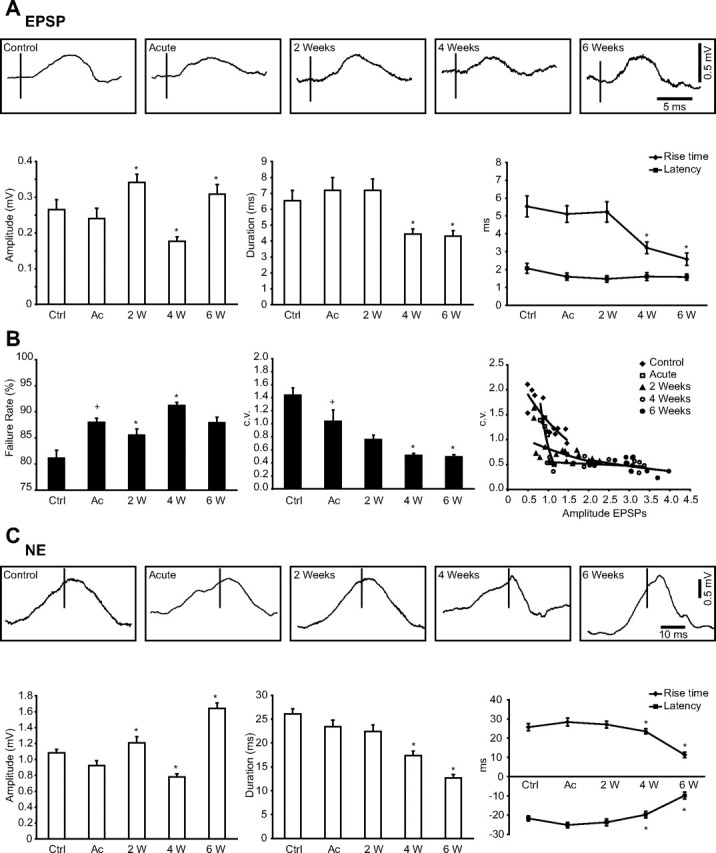
Modulation of synaptic interactions in acute and chronically deafferented cortex. A, Top, Examples of averaged postsynaptic responses for the direct synaptic connection (EPSP) in control and in undercut cortex. Bottom, Variation in amplitude, duration, rise time, and latency of EPSPs in different conditions. B, Variation of the failure rate (left) and the dynamics of the c.v. (middle) in control and after undercut. Right, Plot of the coefficient of variation versus EPSP amplitude in control and in injured cortex. The values were fitted with an exponential function. Note the smallest values of c.v. at 4 and 6 weeks after undercut, supporting the increased synaptic strength. C, Top, Examples of averaged postsynaptic responses for the synchronous excitatory synaptic connections (NE) in control and in undercut cortex. Bottom, Variation in amplitude, duration, rise time, and latency of NEs in different conditions. Ctrl, Control; Ac, acute; W, weeks. Error bars indicate SEM. +p < 0.05, Student's t test (control vs acute); *p < 0.05, Student's t test (acute vs 2, 4, and 6 weeks, respectively, after undercut).
Both the duration and the rise time of EPSPs were significantly reduced 4 and 6 weeks after deafferentation. As expected, the latency was not modified in acute conditions after trauma, or after 2, 4, or 6 weeks, suggesting that axonal velocity and mediator release were similar in all experimental conditions (Fig. 5A, bottom right). Similarly, the duration, rise time, and latency of NEs were reduced at 4 and 6 weeks after cortical deafferentation (Fig. 5C, bottom right). Instead, the duration, rise time, and latency were not significantly modified for IPSPs or NIs (data not shown).
The next step of our study was to identify the mechanism or the mechanisms responsible for the enhanced efficacy of excitatory connections after cortical trauma. The increased amplitude of averaged EPSPs could result from (1) increased release probability, (2) larger number of release sites, (3) increased intrinsic excitability of postsynaptic neurons, or (4) increase in postsynaptic receptor content. The latter mechanism was not tested in the present study.
To test the strength of synaptic connections, we computed the failure rate and the c.v. for each monosynaptic excitatory connection. The failure rate of direct excitatory synaptic connections increased in acute undercut cortex to 85.76 ± 0.78%, compared with control 78.91 ± 1.54% (p < 0.05, Student's t test). Subsequently, it was significantly reduced 2 weeks after trauma (82.32 ± 1.18%) and increased at 4 weeks (88.9 ± 0.61%) compared with acute conditions (p < 0.05, Student's t test) (Fig. 5B, left), explaining therefore the variation of amplitude, which was higher at 2 weeks and the lowest at 4 weeks. However, the failure rate was always higher in the undercut cortex than in control; consequently, supplementary mechanisms should have been responsible for the increased amplitude of averaged excitatory responses in the undercut cortex.
The c.v. of excitatory postsynaptic responses reflects postsynaptic reliability of synaptic transmission (Markram et al., 1997). Similar to our previous observations (Crochet et al., 2005), the c.v. in control conditions was 1.44 ± 0.11, and it decreased to 1.04 ± 0.17 in acute undercut conditions, probably because of injury-induced changes of neuronal interactions. The c.v. continued to decrease slowly even after the initial insult to the brain, was significantly different from acute (p < 0.05, Student's t test) at 4 weeks (0.52 ± 0.03), and appeared to be maintained at a similar value thereafter (Fig. 5B, middle). When investigating the relationship between the c.v. and the amplitude of responses, we found a correlation (Fig. 5B, right) similar to previous studies (Markram et al., 1997). Moreover, a reduced c.v. is associated with the formation of new synapses and appears to be a sign of strong synaptic connection (Berninger et al., 1999).
The apparent discrepancy between the decreased release probability (increased failure rate) and the decreased c.v. likely occurs because the failure rate measures only the efficacy at the presynaptic site, whereas the c.v. of successful responses quantifies postsynaptic efficacy.
Neuronal hyperexcitability after trauma
To assess the intrinsic excitability of neurons in the injured cortex, we computed the neuronal input resistance as well as the firing frequency of neurons in response to depolarizing current pulses.
The input resistance of cortical neurons is higher during silent network states than during active states (Contreras et al., 1996; Steriade et al., 2001; Rudolph et al., 2007), except for layer 2–3 pyramidal neurons from rat somatosensory cortex (Waters and Helmchen, 2006). After cortical trauma, the input resistance of neurons was similar in control (20.3 ± 1.85 MΩ during silent phase and 13.96 ± 1.31 MΩ during active state) and in acute (21.43 ± 1.06 MΩ during silent phase and 14.59 ± 1.14 MΩ during active state) conditions, as reported previously (Topolnik et al., 2003a). However, in chronically injured cortex, the input resistance increased during both silent and active periods of the slow oscillation (p < 0.05, Student's t test), reaching a value of 30.52 ± 3.98 MΩ/20.83 ± 3.74 MΩ at 2 weeks, 40.49 ± 2.37 MΩ/31.44 ± 2.59 MΩ at 4 weeks, and 39.45 ± 2.44 MΩ/26.88 ± 3.14 MΩ 6 weeks after cortical deafferentation (Fig. 6).
Figure 6.

Increased neuronal input resistance in chronically undercut cortex. A, Examples of intracellular recordings of an RS neuron together with the corresponding EEG during intracellularly applied hyperpolarizing current pulses in control (top) and 6 weeks after cortical injury (bottom). B, Averages (Avg) of voltage responses to five trials of hyperpolarizing current (0.5 nA, 0.1 s) during each state of the network (silent and active). The averages correspond to the neurons depicted in A. C, Increased input resistance during both silent (black bars) and active (open bars) phases of the slow oscillation in chronically injured cortex. Ctrl, Control; Ac, acute; W, weeks. Bars represent the mean input resistance from 10 different neurons for each group. Error bars indicate SEM. *p < 0.05, Student's t test.
We further tested the relationship between the firing rate of RS neurons and the intensity of intracellularly applied current pulses in control and in undercut cortex. Our results revealed a significant decrease (p < 0.05, Student's t test) in both number of spikes and instantaneous firing rate after acute trauma to the brain compared with control conditions, for all intensities of current applied (0.5, 0.75, and 1.0 nA). Nevertheless, the number of spikes and instantaneous firing rate increased thereafter at 2, 4, and 6 weeks compared with acute (p < 0.05, Student's t test) (Fig. 7). When we compared the results obtained in chronically injured cortex to the control group, the increased number of spikes and instantaneous firing rate reached statistical significance only at 4 and 6 weeks after the undercut.
Figure 7.

Progressive increase in neuronal excitability after cortical undercut. A, Responses to depolarizing current pulses of RS neurons from control and undercut cortex. The membrane potential is indicated for each example. B, Number of spikes recorded in RS neurons in response to depolarizing current pulses of increasing intensity (0.5 nA in black, 0.75 nA in white, and 1.0 nA in gray). C, Instantaneous firing rate (reciprocal of the first interspike interval) in response to depolarizing current (0.5 nA in black, 0.75 nA in white, and 1.0 nA in gray). Note the decline of both number of spikes and instantaneous firing rate immediately after injury, followed by the progressive increase in excitability at 2, 4, and 6 weeks after trauma. Ctrl, Control; Ac, acute; W, weeks. Error bars indicate SEM. +p < 0.05, Student's t test (control vs acute); *p < 0.05, Student's t test (acute vs 2, 4, and 6 weeks, respectively, after undercut).
In agreement with a previous study (Prince and Tseng, 1993), the firing threshold of neurons in chronic undercut cortex was not different from control (data not shown).
Increased duration of hyperpolarizing periods in the deafferented cortex
The occurrence of seizures during waking state has been related previously to the presence of periods of hyperpolarization during this state of vigilance, after cortical deafferentation (Nita et al., 2007). Therefore, we measured the duration of the hyperpolarizing events at mean membrane potential (Fig. 8) in control and in undercut cortex. To avoid disturbances caused by transient membrane potential fluctuations, we only took into account the hyperpolarizing periods lasting longer than 40 ms, as in a previous study (Volgushev et al., 2006). The duration of hyperpolarizations was significantly increased in acute conditions compared with control (p < 0.05, Student's t test), and it continued to grow progressively at 2, 4, and 6 weeks after the undercut. When we compared the results obtained in chronically injured cortex to the acute group, they were statistically significant at 4 and 6 weeks (p < 0.05, Student's t test); nevertheless, when the comparison was made to the control group, the difference was significant for all three of the chronic groups (2, 4, and 6 weeks) (Fig. 8C).
Figure 8.
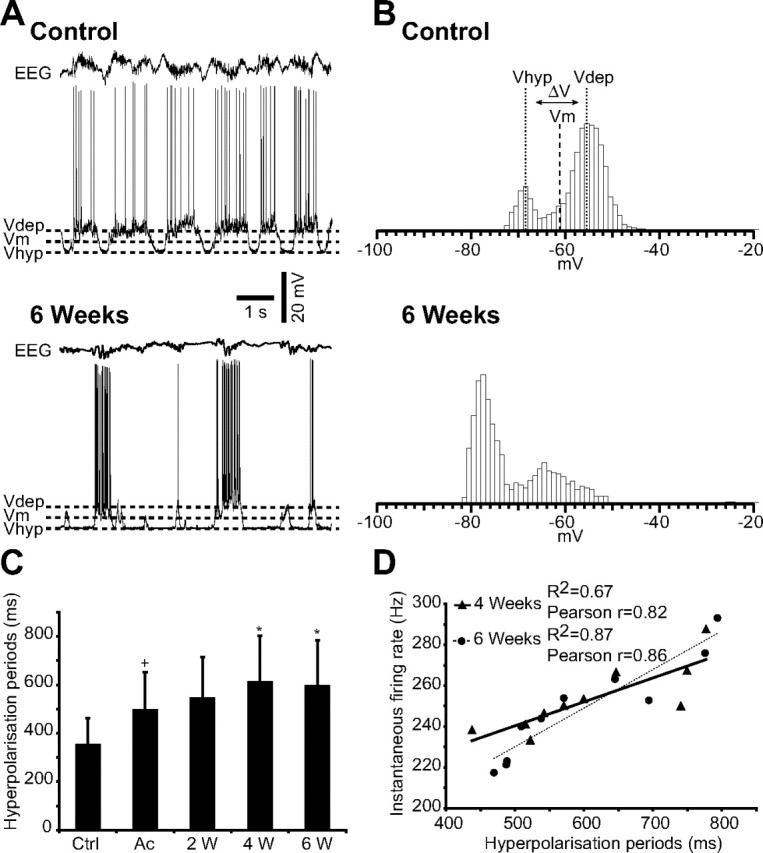
Increased duration of hyperpolarizing periods after cortical trauma. A, Intracellular recordings in control and 6 weeks after undercut, together with the corresponding EEG. Dashed lines indicate the membrane potential of the active (_V_dep) and silent (_V_hyp) phases of the slow oscillation, as well as the mean membrane potential (_V_m). B, Histograms of the cellular membrane potential corresponding to the intracellular recordings in A. The top depicts also how the _V_dep, _V_hyp, and _V_m were calculated. The histograms were computed for 10 s duration, with a bin of 1 mV. C, Mean duration of hyperpolarizing periods in control and after undercut (n = 10 in each group), measured during 10 s for each neuron taken into account. Ctrl, Control; Ac, acute; W, weeks. Error bars indicate SEM. +p < 0.05, Student's t test (control vs acute); *p < 0.05, Student's t test (acute vs 2, 4, and 6 weeks, respectively, after undercut). D, Correlations between the instantaneous firing rate (_y_-axis) and the duration of hyperpolarization periods (_x_-axis) at 4 weeks (triangles, correlation line solid) and at 6 weeks (circles, correlation line dotted) after cortical undercut. The coefficient of correlation _R_2 and the global Pearson coefficient (r) are indicated on the graph.
The histograms of membrane potential computed for all IC recorded neurons after deafferentation revealed always a higher hyperpolarizing peak, in contrast to control neurons, for which the depolarizing peak was higher, reflecting longer periods of hyperpolarization in the undercut versus intact cortex. Furthermore, the histograms of neurons in the undercut cortex were constantly shifted toward a more hyperpolarized membrane potential (Fig. 8B). Similar results were presented formerly only for neurons recorded immediately after cortical trauma, in acute conditions (Topolnik et al., 2003a).
To test the assumption that the increased excitability in chronic undercut cortex is related to the decreased activity in the deafferented gyrus, we made correlations between the neuronal instantaneous firing rates in response to 1.0 nA current pulses and the duration of hyperpolarization in all tested groups. We found a significant correlation (p < 0.01) at 4 and 6 weeks after the deafferentation, characterized at 4 weeks by the equation y = 0.1169_x_ + 181.93, an R_2 = 0.67 and a global Pearson r = 0.82, and at 6 weeks by the equation y = 0.1887_x + 135.78, an _R_2 = 0.87 and a global Pearson r = 0.86 (Fig. 8D).
Discussion
In the present in vivo study, we demonstrated that after undercut, the cortical neuronal network undergoes reorganization, leading to an overall increase in network excitability in chronic conditions. These changes are mediated by an increased connection probability and amplitude, rising phase of EPSPs, decreased c.v., increased input resistance, and increased instantaneous firing rates. There were no significant changes in the efficacy of inhibitory synaptic relations.
The mechanisms accounting for seizure generation after an acute insult to the brain are probably different from those responsible for chronic hyperexcitability and epilepsy. We found an important boost of synaptic interactions, as well as an increased efficacy of excitatory connections, starting with the second week after the brain injury, without any significant changes in acute conditions. These findings support the fact that in humans, early administration of anticonvulsant medication decreases the percentage of early posttraumatic seizures but not that of chronic epilepsy (Temkin et al., 1990, 1999; Chang and Lowenstein, 2003), because the mechanisms of the two are different. Moreover, our study evaluates the progressive transformation of synaptic interactions and intrinsic neuronal properties, therefore advancing the actual knowledge regarding the temporal window in which these synaptic changes occur and consequently the critical period during which antiepileptogenic prophylaxis may be attempted.
Early posttraumatic hyperexcitability (Topolnik et al., 2003a,b) is most likely generated by the dramatic alteration of neuronal milieu induced by the injury rather than by long-term anatomical changes of cortical networks. Previous studies suggest that the major activator of deleterious processes after neuronal injury is the increased Ca2+ influx (Choi, 1988), which might inhibit Na+–K+ pump activity, engendering further accumulation of Ca2+ intracellularly (Fukuda and Prince, 1992) while the extracellular Ca2+ decreases (Morris et al., 1991; Wolf et al., 2001), and this significantly increases the failure of synaptic transmission (Markram et al., 1998; Massimini and Amzica, 2001; Crochet et al., 2005; Seigneur and Timofeev, 2007). Accordingly, we found an increased failure rate of synaptic transmission in the acutely injured cortex compared with control.
Posttraumatic epilepsy arises probably from complex structural changes of neuronal networks (Lowenstein, 1996). The brain restores its activity after injury by extensive reorganization of cortical connectivity, such as axonal sprouting with formation of new synapses (Purpura and Housepian, 1961; Sutula et al., 1989; Salin et al., 1995; Dancause et al., 2005); by selective loss of inhibitory synapses (Ribak and Reiffenstein, 1982); or by increased synaptic and intrinsic neuronal responsiveness (Bush et al., 1999).
We found an increased efficacy of excitatory connections after cortical deafferentation characterized by an increased amplitude and decreased duration and rise time at 2 and 6 weeks after trauma, but also an unexpected decreased amplitude after 4 weeks. This finding could be explained by the simultaneous massive increase in the failure rate of synaptic responses also seen 4 weeks after the injury and is also supported by the increased incidence of NI together with a decreased probability of NE at the same time. This matches very well with previously published data revealing a decreased number of ictal events from day 25 to day 40 after cortical undercut, followed by a subsequent increase of seizures to a stable rate (Nita et al., 2007). Our data follow the course of events also seen in humans, in which acute seizures are viewed as epiphenomena of the underlying brain disorder, with little independent impact on outcome from injury (Bladin et al., 2000), whereas late or remote seizures are thought to be the result of epileptogenesis after chronic changes of neural networks favoring excitation (Herman, 2002). In contrast to patients with early postinjury seizures, those with late-onset seizures are at greater risk of epilepsy (Bladin et al., 2000), probably because they express the already irreversible changes of neuronal network.
We then asked what could generate the increased amplitude of averaged responses at 2 and 6 weeks, respectively, after cortical undercut. We envisaged several factors that could be implicated individually or conjointly, such as modified reliability of synaptic transmission estimated as failure rate of postsynaptic responses, changes of intrinsic neuronal properties, or reorganization of cortical connectivity, with formation of new synapses or revealing of otherwise unapparent excitatory connections. The failure rate was highest at 4 weeks, when the amplitude of responses was dramatically diminished and the number of failures decreased at 2 weeks, compared with acute conditions, which also consisted with increased amplitude of responses. We also observed that although the fluctuations of failure rate in the undercut cortex followed a trend similar to the amplitude of responses, the number of failures after the injury was much higher than in the intact cortex. Therefore, additional factors should have been responsible for the increased amplitude of averaged postsynaptic responses in the undercut cortex.
We found that neuronal input resistance in chronic conditions was increased, confirming previous findings obtained in vitro from chronically deafferented cortical neurons (Prince and Tseng, 1993; Tseng and Prince, 1996). In addition, we observed an increased number of spikes and instantaneous firing rate of neurons located in the traumatized cortex, supporting the hyperactivity of these neurons. Our results are also sustained by in vitro undercut models and by studies done in axotomized corticospinal neurons illustrating a steeper relationship between applied current and adapted spike firing frequency (f–I slope); therefore, for the same inward current, neurons would generate more action potentials (Prince and Tseng, 1993; Tseng and Prince, 1996). We assume that the increased neuronal firing in chronic conditions was a reflection of the increased input resistance, which would make these neurons more responsive to excitatory events (Prince and Tseng, 1993; Jacobs et al., 2000), because we found no differences of action potential threshold, as also reported previously in the in vitro undercut model (Prince and Tseng, 1993). Interestingly, high values of input resistance have also been described in immature neocortical neurons, where they may be a factor that increases vulnerability to seizures (McCormick and Prince, 1987), and in surviving neurons from kainate-lesioned epileptogenic hippocampi (Franck and Schwartzkroin, 1985; Franck et al., 1988).
The potential reorganization of axonal arbor of neocortical neurons has been demonstrated after a variety of injuries (Tsukahara et al., 1975; Villablanca et al., 1988; Kuang and Kalil, 1990). Cortical neuronal sprouting may also occur after direct trauma to the cortex (Maxwell et al., 1990; Fishman and Mattu, 1993) or in human pathological processes such as temporal lobe epilepsy (de Lanerolle et al., 1989; Sutula et al., 1989; Houser et al., 1990). In agreement with in vitro studies (Jin et al., 2006), we illustrate the increased efficacy of synaptic interactions after cortical injury, which could be the consequence of axonal sprouting. A progressively decreasing c.v. of postsynaptic responses in injured cortex also correlated with a higher amplitude of individual responses, which appears to be the hallmark of strong synaptic connections (Berninger et al., 1999). The increased amplitude, together with the decreased duration and rise time of EPSPs after undercut, could also be generated by increased presynaptic synchrony after cortical trauma, which boosts EPSPs and bisynaptic IPSPs. Relatively small reduction in the efficacy of inhibition could lead to enlarged neuronal synchrony and to large expansion of activated cortical territory (Chagnac-Amitai and Connors, 1989; Prince and Tseng, 1993). For example, in the hippocampus, gradual blockade of GABAergic inhibition revealed excitatory connections between previously unconnected neurons (Miles and Wong, 1987).
Our results illustrate a progressive boost of connectivity and efficacy of excitatory connections after chronic cortical deafferentation, which is in agreement with a gradually increased propensity to seizures (Nita et al., 2006). Here, we show physiological consequences of axonal sprouting accompanying posttraumatic epileptogenesis (Salin et al., 1995).
The posttraumatic hyperexcitability of cortical neurons emerges probably from homeostatic synaptic plasticity, a mechanism that works to restore a stable pattern of activity whenever networks are perturbed (Turrigiano, 1999; Abbott and Nelson, 2000; Davis and Bezprozvanny, 2001). However, after prolonged periods of reduced activity, as is the case in the deafferented suprasylvian gyrus, homeostatic synaptic plasticity may increase network excitability in an uncontrollable manner, leading to the development of paroxysmal activity (Houweling et al., 2005). Indeed, long-term activity blockade dramatically enhances cortical excitability and may lead to paroxysmal activity (Murthy et al., 2001; Burrone et al., 2002; Bausch et al., 2006). Here we demonstrate that longer periods of hyperpolarizing states correlate well with the increased firing rate starting 1 month after cortical deafferentation. Previous studies showed the occurrence of hyperpolarization periods during wake and REM sleep, when they do not normally appear, in chronic stages of the undercut (Nita et al., 2006). The same scenario is seen in clinical medical practice after severe head trauma that may produce cortical deafferentation, followed by seizures (Salazar et al., 1985). Early administration of antiepileptic drugs known to enhance inhibition could not decrease the incidence of late epilepsy (Temkin, 2001) after cortical trauma, and furthermore, antiepileptic drugs can exacerbate seizures (Perucca et al., 1998).
A better understanding of the role of homeostatic synaptic plasticity in generating posttraumatic epilepsy is needed, but if this hypothesis proves to be valid, then we can attempt to prevent seizure generation by augmenting cortical activity rather than decreasing it with antiepileptic medication.
Footnotes
This work was supported by Canadian Institutes of Health Research Grants MOP-67175 and MOP-37862 and by Natural Science and Engineering Research Council of Canada Grant 298475. I.T. is a Canadian Institutes of Health Research scholar. S.A. is a Savoy Foundation fellow. We thank P. Giguère for technical assistance and Dr. Dragos A. Nita for critical reading of this manuscript and for useful discussions.
References
- Abbott LF, Nelson SB. Synaptic plasticity: taming the beast. Nat Neurosci. 2000;3(Suppl):1178–1183. doi: 10.1038/81453. [DOI] [PubMed] [Google Scholar]
- Avramescu S, Timofeev I. Synaptic synchrony modulation following cortical injury. Soc Neurosci Abstr. 2006;32:235–26. [Google Scholar]
- Bausch SB, He S, Petrova Y, Wang XM, McNamara JO. Plasticity of both excitatory and inhibitory synapses is associated with seizures induced by removal of chronic blockade of activity in cultured hippocampus. J Neurophysiol. 2006;96:2151–2167. doi: 10.1152/jn.00355.2006. [DOI] [PubMed] [Google Scholar]
- Berninger B, Schinder AF, Poo MM. Synaptic reliability correlates with reduced susceptibility to synaptic potentiation by brain-derived neurotrophic factor. Learn Mem. 1999;6:232–242. [PMC free article] [PubMed] [Google Scholar]
- Bladin CF, Alexandrov AV, Bellavance A, Bornstein N, Chambers B, Cote R, Lebrun L, Pirisi A, Norris JW. Seizures after stroke: a prospective multicenter study. Arch Neurol. 2000;57:1617–1622. doi: 10.1001/archneur.57.11.1617. [DOI] [PubMed] [Google Scholar]
- Burrone J, O'Byrne M, Murthy VN. Multiple forms of synaptic plasticity triggered by selective suppression of activity in individual neurons. Nature. 2002;420:414–418. doi: 10.1038/nature01242. [DOI] [PubMed] [Google Scholar]
- Bush PC, Prince DA, Miller KD. Increased pyramidal excitability and NMDA conductance can explain posttraumatic epileptogenesis without disinhibition: a model. J Neurophysiol. 1999;82:1748–1758. doi: 10.1152/jn.1999.82.4.1748. [DOI] [PubMed] [Google Scholar]
- Chagnac-Amitai Y, Connors BW. Synchronized excitation and inhibition driven by intrinsically bursting neurons in neocortex. J Neurophysiol. 1989;62:1149–1162. doi: 10.1152/jn.1989.62.5.1149. [DOI] [PubMed] [Google Scholar]
- Chang BS, Lowenstein DH. Practice parameter: antiepileptic drug prophylaxis in severe traumatic brain injury: report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology. 2003;60:10–16. doi: 10.1212/01.wnl.0000031432.05543.14. [DOI] [PubMed] [Google Scholar]
- Chen Q, He S, Hu XL, Yu J, Zhou Y, Zheng J, Zhang S, Zhang C, Duan WH, Xiong ZQ. Differential roles of NR2A- and NR2B-containing NMDA receptors in activity-dependent brain-derived neurotrophic factor gene regulation and limbic epileptogenesis. J Neurosci. 2007;27:542–552. doi: 10.1523/JNEUROSCI.3607-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi DW. Glutamate neurotoxicity and diseases of the nervous system. Neuron. 1988;1:623–634. doi: 10.1016/0896-6273(88)90162-6. [DOI] [PubMed] [Google Scholar]
- Connors BW, Gutnick MJ. Intrinsic firing patterns of diverse neocortical neurons. Trends Neurosci. 1990;13:99–104. doi: 10.1016/0166-2236(90)90185-d. [DOI] [PubMed] [Google Scholar]
- Contreras D, Steriade M. Cellular basis of EEG slow rhythms: a study of dynamic corticothalamic relationships. J Neurosci. 1995;15:604–622. doi: 10.1523/JNEUROSCI.15-01-00604.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Contreras D, Timofeev I, Steriade M. Mechanisms of long-lasting hyperpolarizations underlying slow sleep oscillations in cat corticothalamic networks. J Physiol (Lond) 1996;494:251–264. doi: 10.1113/jphysiol.1996.sp021488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crochet S, Chauvette S, Boucetta S, Timofeev I. Modulation of synaptic transmission in neocortex by network activities. Eur J Neurosci. 2005;21:1030–1044. doi: 10.1111/j.1460-9568.2005.03932.x. [DOI] [PubMed] [Google Scholar]
- Dancause N, Barbay S, Frost SB, Plautz EJ, Chen D, Zoubina EV, Stowe AM, Nudo RJ. Extensive cortical rewiring after brain injury. J Neurosci. 2005;25:10167–10179. doi: 10.1523/JNEUROSCI.3256-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis GW, Bezprozvanny I. Maintaining the stability of neural function: a homeostatic hypothesis. Annu Rev Physiol. 2001;63:847–869. doi: 10.1146/annurev.physiol.63.1.847. [DOI] [PubMed] [Google Scholar]
- de Lanerolle NC, Kim JH, Robbins RJ, Spencer DD. Hippocampal interneuron loss and plasticity in human temporal lobe epilepsy. Brain Res. 1989;495:387–395. doi: 10.1016/0006-8993(89)90234-5. [DOI] [PubMed] [Google Scholar]
- Desai NS, Rutherford LC, Turrigiano GG. Plasticity in the intrinsic excitability of cortical pyramidal neurons. Nat Neurosci. 1999;2:515–520. doi: 10.1038/9165. [DOI] [PubMed] [Google Scholar]
- Dinner DS. Posttraumatic epilepsy. In: Wyllie E, editor. The treatment of epilepsy: principles. Philadelphia: Lea and Fibinger; 1993. pp. 654–658. [Google Scholar]
- Esplin MS, Abbott JR, Smart ML, Burroughs AF, Frandsen TC, Litzinger MJ. Voltage-sensitive calcium channel development in epileptic DBA/2J mice suggests altered presynaptic function. Epilepsia. 1994;35:911–914. doi: 10.1111/j.1528-1157.1994.tb02533.x. [DOI] [PubMed] [Google Scholar]
- Fishman PS, Mattu A. Fate of severed cortical projection axons. J Neurotrauma. 1993;10:457–470. doi: 10.1089/neu.1993.10.457. [DOI] [PubMed] [Google Scholar]
- Franck JE, Schwartzkroin PA. Do kainate-lesioned hippocampi become epileptogenic? Brain Res. 1985;329:309–313. doi: 10.1016/0006-8993(85)90540-2. [DOI] [PubMed] [Google Scholar]
- Franck JE, Kunkel DD, Baskin DG, Schwartzkroin PA. Inhibition in kainate-lesioned hyperexcitable hippocampi: physiologic, autoradiographic, and immunocytochemical observations. J Neurosci. 1988;8:1991–2002. doi: 10.1523/JNEUROSCI.08-06-01991.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuda A, Prince DA. Excessive intracellular Ca2+ inhibits glutamate-induced Na(+)-K+ pump activation in rat hippocampal neurons. J Neurophysiol. 1992;68:28–35. doi: 10.1152/jn.1992.68.1.28. [DOI] [PubMed] [Google Scholar]
- Graber KD, Prince DA. Tetrodotoxin prevents posttraumatic epileptogenesis in rats. Ann Neurol. 1999;46:234–242. doi: 10.1002/1531-8249(199908)46:2<234::aid-ana13>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- Gray CM, McCormick DA. Chattering cells: superficial pyramidal neurons contributing to the generation of synchronous oscillations in the visual cortex. Science. 1996;274:109–113. doi: 10.1126/science.274.5284.109. [DOI] [PubMed] [Google Scholar]
- Herman ST. Epilepsy after brain insult: targeting epileptogenesis. Neurology. 2002;59:S21–S26. doi: 10.1212/wnl.59.9_suppl_5.s21. [DOI] [PubMed] [Google Scholar]
- Hoffman SN, Salin PA, Prince DA. Chronic neocortical epileptogenesis in vitro. J Neurophysiol. 1994;71:1762–1773. doi: 10.1152/jn.1994.71.5.1762. [DOI] [PubMed] [Google Scholar]
- Houser CR, Miyashiro JE, Swartz BE, Walsh GO, Rich JR, Delgado-Escueta AV. Altered patterns of dynorphin immunoreactivity suggest mossy fiber reorganization in human hippocampal epilepsy. J Neurosci. 1990;10:267–282. doi: 10.1523/JNEUROSCI.10-01-00267.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houweling AR, Bazhenov M, Timofeev I, Steriade M, Sejnowski TJ. Homeostatic synaptic plasticity can explain post-traumatic epileptogenesis in chronically isolated neocortex. Cereb Cortex. 2005;15:834–845. doi: 10.1093/cercor/bhh184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs KM, Graber KD, Kharazia VN, Parada I, Prince DA. Postlesional epilepsy: the ultimate brain plasticity. Epilepsia. 2000;41(Suppl 6):S153–S161. doi: 10.1111/j.1528-1157.2000.tb01574.x. [DOI] [PubMed] [Google Scholar]
- Jin X, Huguenard JR, Prince DA. Impaired Cl− extrusion in layer V pyramidal neurons of chronically injured epileptogenic neocortex. J Neurophysiol. 2005;93:2117–2126. doi: 10.1152/jn.00728.2004. [DOI] [PubMed] [Google Scholar]
- Jin X, Prince DA, Huguenard JR. Enhanced excitatory synaptic connectivity in layer v pyramidal neurons of chronically injured epileptogenic neocortex in rats. J Neurosci. 2006;26:4891–4900. doi: 10.1523/JNEUROSCI.4361-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuang RZ, Kalil K. Specificity of corticospinal axon arbors sprouting into denervated contralateral spinal cord. J Comp Neurol. 1990;302:461–472. doi: 10.1002/cne.903020304. [DOI] [PubMed] [Google Scholar]
- Li H, Bandrowski AE, Prince DA. Cortical injury affects short-term plasticity of evoked excitatory synaptic currents. J Neurophysiol. 2005;93:146–156. doi: 10.1152/jn.00665.2004. [DOI] [PubMed] [Google Scholar]
- Lowenstein DH. Recent advances related to basic mechanisms of epileptogenesis. Epilepsy Res. 1996;11(Suppl):45–60. [PubMed] [Google Scholar]
- Markram H, Lubke J, Frotscher M, Sakmann B. Regulation of synaptic efficacy by coincidence of postsynaptic APs and EPSPs. Science. 1997;275:213–215. doi: 10.1126/science.275.5297.213. [DOI] [PubMed] [Google Scholar]
- Markram H, Wang Y, Tsodyks M. Differential signaling via the same axon of neocortical pyramidal neurons. Proc Natl Acad Sci USA. 1998;95:5323–5328. doi: 10.1073/pnas.95.9.5323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massimini M, Amzica F. Extracellular calcium fluctuations and intracellular potentials in the cortex during the slow sleep oscillation. J Neurophysiol. 2001;85:1346–1350. doi: 10.1152/jn.2001.85.3.1346. [DOI] [PubMed] [Google Scholar]
- Matsumura M, Chen D, Sawaguchi T, Kubota K, Fetz EE. Synaptic interactions between primate precentral cortex neurons revealed by spike-triggered averaging of intracellular membrane potentials in vivo. J Neurosci. 1996;16:7757–7767. doi: 10.1523/JNEUROSCI.16-23-07757.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maxwell WL, Follows R, Ashhurst DE, Berry M. The response of the cerebral hemisphere of the rat to injury. I. The mature rat. Philos Trans R Soc Lond B Biol Sci. 1990;328:479–500. doi: 10.1098/rstb.1990.0121. [DOI] [PubMed] [Google Scholar]
- McCormick DA, Prince DA. Post-natal development of electrophysiological properties of rat cerebral cortical pyramidal neurones. J Physiol (Lond) 1987;393:743–762. doi: 10.1113/jphysiol.1987.sp016851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinney RA, Debanne D, Gahwiler BH, Thompson SM. Lesion-induced axonal sprouting and hyperexcitability in the hippocampus in vitro: implications for the genesis of posttraumatic epilepsy. Nat Med. 1997;3:990–996. doi: 10.1038/nm0997-990. [DOI] [PubMed] [Google Scholar]
- Miles R, Wong RK. Inhibitory control of local excitatory circuits in the guinea-pig hippocampus. J Physiol (Lond) 1987;388:611–629. doi: 10.1113/jphysiol.1987.sp016634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris ME, Leblond J, Agopyan N, Krnjevic K. Temperature dependence of extracellular ionic changes evoked by anoxia in hippocampal slices. J Neurophysiol. 1991;65:157–167. doi: 10.1152/jn.1991.65.2.157. [DOI] [PubMed] [Google Scholar]
- Murthy VN, Schikorski T, Stevens CF, Zhu Y. Inactivity produces increases in neurotransmitter release and synapse size. Neuron. 2001;32:673–682. doi: 10.1016/s0896-6273(01)00500-1. [DOI] [PubMed] [Google Scholar]
- Nita DA, Cisse Y, Timofeev I, Steriade M. Increased propensity to seizures after chronic cortical deafferentation in vivo. J Neurophysiol. 2006;95:902–913. doi: 10.1152/jn.00742.2005. [DOI] [PubMed] [Google Scholar]
- Nita DA, Cisse Y, Timofeev I, Steriade M. Waking-sleep modulation of paroxysmal activities induced by partial cortical deafferentation. Cereb Cortex. 2007;17:272–283. doi: 10.1093/cercor/bhj145. [DOI] [PubMed] [Google Scholar]
- Perucca E, Gram L, Avanzini G, Dulac O. Antiepileptic drugs as a cause of worsening seizures. Epilepsia. 1998;39:5–17. doi: 10.1111/j.1528-1157.1998.tb01268.x. [DOI] [PubMed] [Google Scholar]
- Prince DA, Tseng GF. Epileptogenesis in chronically injured cortex: in vitro studies. J Neurophysiol. 1993;69:1276–1291. doi: 10.1152/jn.1993.69.4.1276. [DOI] [PubMed] [Google Scholar]
- Purpura DP, Housepian EM. Morphological and physiological properties of chronically isolated immature neocortex. Exp Neurol. 1961;4:377–401. doi: 10.1016/0014-4886(61)90025-5. [DOI] [PubMed] [Google Scholar]
- Ribak CE, Reiffenstein RJ. Selective inhibitory synapse loss in chronic cortical slabs: a morphological basis for epileptic susceptibility. Can J Physiol Pharmacol. 1982;60:864–870. doi: 10.1139/y82-122. [DOI] [PubMed] [Google Scholar]
- Rudolph M, Pospischil M, Timofeev I, Destexhe A. Inhibition determines membrane potential dynamics and controls action potential generation in awake and sleeping cat cortex. J Neurosci. 2007;27:5280–5290. doi: 10.1523/JNEUROSCI.4652-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salazar AM, Jabbari B, Vance SC, Grafman J, Amin D, Dillon JD. Epilepsy after penetrating head injury. I. Clinical correlates: a report of the Vietnam Head Injury Study. Neurology. 1985;35:1406–1414. doi: 10.1212/wnl.35.10.1406. [DOI] [PubMed] [Google Scholar]
- Salin P, Tseng GF, Hoffman S, Parada I, Prince DA. Axonal sprouting in layer V pyramidal neurons of chronically injured cerebral cortex. J Neurosci. 1995;15:8234–8245. doi: 10.1523/JNEUROSCI.15-12-08234.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seigneur J, Timofeev I. Extracellular calcium depletion associated with low-threshold calcium spikes controls presynaptic mediator release in thalamus. Soc Neurosci Abstr. 2007;33:827–4. [Google Scholar]
- Steriade M, Nuñez A, Amzica F. A novel slow (<1 Hz) oscillation of neocortical neurons in vivo: depolarizing and hyperpolarizing components. J Neurosci. 1993;13:3252–3265. doi: 10.1523/JNEUROSCI.13-08-03252.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steriade M, Timofeev I, Durmuller N, Grenier F. Dynamic properties of corticothalamic neurons and local cortical interneurons generating fast rhythmic (30–40 Hz) spike bursts. J Neurophysiol. 1998;79:483–490. doi: 10.1152/jn.1998.79.1.483. [DOI] [PubMed] [Google Scholar]
- Steriade M, Timofeev I, Grenier F. Natural waking and sleep states: a view from inside neocortical neurons. J Neurophysiol. 2001;85:1969–1985. doi: 10.1152/jn.2001.85.5.1969. [DOI] [PubMed] [Google Scholar]
- Sutula T, Cascino G, Cavazos J, Parada I, Ramirez L. Mossy fiber synaptic reorganization in the epileptic human temporal lobe. Ann Neurol. 1989;26:321–330. doi: 10.1002/ana.410260303. [DOI] [PubMed] [Google Scholar]
- Temkin NR. Antiepileptogenesis and seizure prevention trials with antiepileptic drugs: meta-analysis of controlled trials. Epilepsia. 2001;42:515–524. doi: 10.1046/j.1528-1157.2001.28900.x. [DOI] [PubMed] [Google Scholar]
- Temkin NR, Dikmen SS, Wilensky AJ, Keihm J, Chabal S, Winn HR. A randomized, double-blind study of phenytoin for the prevention of post-traumatic seizures. N Engl J Med. 1990;323:497–502. doi: 10.1056/NEJM199008233230801. [DOI] [PubMed] [Google Scholar]
- Temkin NR, Dikmen SS, Anderson GD, Wilensky AJ, Holmes MD, Cohen W, Newell DW, Nelson P, Awan A, Winn HR. Valproate therapy for prevention of posttraumatic seizures: a randomized trial. J Neurosurg. 1999;91:593–600. doi: 10.3171/jns.1999.91.4.0593. [DOI] [PubMed] [Google Scholar]
- Thomson AM, West DC, Hahn J, Deuchars J. Single axon IPSPs elicited in pyramidal cells by three classes of interneurones in slices of rat neocortex. J Physiol (Lond) 1996;496:81–102. doi: 10.1113/jphysiol.1996.sp021667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topolnik L, Steriade M, Timofeev I. Hyperexcitability of intact neurons underlies acute development of trauma-related electrographic seizures in cats in vivo. Eur J Neurosci. 2003a;18:486–496. doi: 10.1046/j.1460-9568.2003.02742.x. [DOI] [PubMed] [Google Scholar]
- Topolnik L, Steriade M, Timofeev I. Partial cortical deafferentation promotes development of paroxysmal activity. Cereb Cortex. 2003b;13:883–893. doi: 10.1093/cercor/13.8.883. [DOI] [PubMed] [Google Scholar]
- Trasande CA, Ramirez JM. Activity deprivation leads to seizures in hippocampal slice cultures: is epilepsy the consequence of homeostatic plasticity? J Clin Neurophysiol. 2007;24:154–164. doi: 10.1097/WNP.0b013e318033787f. [DOI] [PubMed] [Google Scholar]
- Tseng GF, Prince DA. Structural and functional alterations in rat corticospinal neurons after axotomy. J Neurophysiol. 1996;75:248–267. doi: 10.1152/jn.1996.75.1.248. [DOI] [PubMed] [Google Scholar]
- Tsukahara N, Hultborn H, Murakami F, Fujito Y. Electrophysiological study of formation of new synapses and collateral sprouting in red nucleus neurons after partial denervation. J Neurophysiol. 1975;38:1359–1372. doi: 10.1152/jn.1975.38.6.1359. [DOI] [PubMed] [Google Scholar]
- Turrigiano GG. Homeostatic plasticity in neuronal networks: the more things change, the more they stay the same. Trends Neurosci. 1999;22:221–227. doi: 10.1016/s0166-2236(98)01341-1. [DOI] [PubMed] [Google Scholar]
- Villablanca JR, Fomez-Pinilla F, Sonnier BJ, Hovda DA. Bilateral pericruciate cortical innervation of the red nucleus in cats with adult or neonatal cerebral hemispherectomy. Brain Res. 1988;453:17–31. doi: 10.1016/0006-8993(88)90139-4. [DOI] [PubMed] [Google Scholar]
- Volgushev M, Chauvette S, Mukovski M, Timofeev I. Precise long-range synchronization of activity and silence in neocortical neurons during slow-wave oscillations [corrected] J Neurosci. 2006;26:5665–5672. doi: 10.1523/JNEUROSCI.0279-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waters J, Helmchen F. Background synaptic activity is sparse in neocortex. J Neurosci. 2006;26:8267–8277. doi: 10.1523/JNEUROSCI.2152-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf JA, Stys PK, Lusardi T, Meaney D, Smith DH. Traumatic axonal injury induces calcium influx modulated by tetrodotoxin-sensitive sodium channels. J Neurosci. 2001;21:1923–1930. doi: 10.1523/JNEUROSCI.21-06-01923.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]