Disease-Modifying Pathways in Neurodegeneration (original) (raw)
Identifying effective therapies for adult-onset neurodegenerative diseases has been a major challenge. For example, although the gene that causes Huntington's disease (HD) has been identified, its protein product appears to lack receptor or enzymatic activity that could be disrupted with therapeutic small molecules (Beal and Ferrante, 2004). For more common diseases, such as Alzheimer's disease (AD), Parkinson's disease (PD), and amyotrophic lateral sclerosis (ALS), the underlying causes remain mostly unknown. In a minority of cases, these disorders have been linked to genetic mutations; however, the affected proteins also appear to be poor pharmacological targets. Because of these difficulties, indirect treatment strategies are appealing. Putative disease-modifying genes have been identified for some disorders (Djousse et al., 2004; Wexler et al., 2004). These genes, especially those in which loss-of-function (LOF) suppresses disease, could encode proteins that are more attractive therapeutic targets. For example, in HD, variation in the age of onset can be substantial. If the pathways responsible for the variation could be pharmacologically targeted, symptoms might be delayed for decades.
Here we summarize recent work on disease-modifying pathways in neurodegeneration that was presented as a symposium at the 36th Annual Meeting of the Society for Neuroscience. We describe general strategies for identifying disease-modifying pathways and review examples in which these strategies have provided promising insights. First, we describe the use of survival analysis to “look” at neurons. Second, we describe genetic experiments in yeast that identified a potential disease-modifying pathway for HD present only in microglia. Third, we describe a series of experiments in Caenorhabditis elegans in which genes that control aging modify protein misfolding disease in worms. Finally, we describe the role of autophagy in neurodegenerative diseases.
Our understanding of disease-modifying pathways in neurodegenerative disease reflects decades of careful pathological analyses. Such studies provided beautiful descriptions of the subpopulations of affected neurons for each disorder, the characteristic gross and microscopic pathological changes, and a timeline for the changes.
As valuable as pathological studies of human disease have been, their findings can be misinterpreted. First, it is tempting to infer stepwise disease mechanisms from the time course of pathological changes, as one might infer mechanisms of development by studying different stages of embryogenesis. However, such an approach could be misleading (Fig. 1). With embryogenesis, it is reasonable to assume that intermediate stages are steps along a well established path that lead to a properly developed organism. In contrast, pathological changes could be part of a single pathogenic pathway or they could well represent components of different pathways that emanate from the initiating insult. Moreover, some “pathological” changes may actually reflect beneficial responses. Indeed, synaptic plasticity, which is so critical for development and learning and memory, is induced in the CNS in response to injury (Emery et al., 2003). To make matters even more complex, disease-modifying pathways may also include homeostatic responses evolved to mitigate pathogenic perturbations (Nadeau, 2003).
Figure 1.

Neurodegenerative diseases display phenotypes that can be beneficial, pathogenic, or incidental. Whether an observed change is pathogenic or beneficial is a common question that has enormous therapeutic importance. The top depicts the time course over which clinical hallmarks appear based on standard pathology studies. However, some changes may actually be beneficial (bottom, 4) or inconsequential (bottom, 2), rather than pathogenic. Activation of homeostatic pathways may mitigate pathogenic perturbations, and promoting these beneficial coping responses could be effective therapies. The timing of the events can further confound recognition of the true disease pathways. If pathogenic processes elicit nearly simultaneous coping responses, they will be highly correlated with each other and with the course of disease. New strategies will enable us to identify disease-modifying pathways and may identify new therapeutic targets for these devastating diseases.
Whether a change is pathogenic or beneficial is a common question that has enormous therapeutic importance. Most therapies focus on inhibiting pathogenic processes (Thakur et al., 2004; Zhang et al., 2005), but promoting beneficial coping responses could be equally effective (Bodner et al., 2006). Unfortunately, the nature of most neurodegenerative diseases makes this determination extremely difficult with conventional approaches, which depend on inferring underlying relationships from “snap shots” in time. Although these diseases are devastating, many progress slowly over years or decades, possibly indicating that the initial trigger is only moderately toxic, that effective coping responses are triggered early and in parallel with disease, or both. If pathogenic processes elicit nearly simultaneous coping responses, they will be highly correlated with each other and with the course of disease.
Survival analysis: a new way to look at neurons
To overcome the limitations of conventional approaches to disentangle pathogenic, incidental, and coping responses, we developed a new strategy for the study of neurodegenerative disease. We reasoned that, if we could observe the same neuron repeatedly, as neurodegeneration unfolded, we could link the appearance of particular changes within that neuron to its ultimate fate. Moreover, if we could quickly collect similar information for millions of neurons, we could identify natural variation in the levels or timing of the changes and any associated changes in future fate and establish predictive relationships. In turn, this information would help us categorize these changes qualitatively as pathogenic, incidental, or beneficial and quantitatively as major or minor.
The practical embodiment of this strategy has two components. First, we developed an automated microscope and software that can acquire highly focused images of cultured primary neurons and return to precisely the same neuron or field of neurons, even after the culture plate has been removed from and returned to the microscope stage (Arrasate and Finkbeiner, 2005). Information can be collected from up to 1 million neurons in as little as 10 min, and individual cells can be tracked as often and for as long as we want. For our HD studies, we use primary striatal neurons expressing mutant huntingtin (htt) and typically observe them once daily for ∼2 weeks.
Second, we applied survival analysis to quantify relationships between particular changes within neurons and some future fate (Arrasate and Finkbeiner, 2005). Although it is a well established statistical tool for quantifying relationships between two events separated by time, survival analysis is not limited to the study of survival per se. It is particularly well suited to quantifying neurodegeneration, in which the precise timing of meaningful events (e.g., cell death) appears to be stochastic and affects a relatively small subpopulation of cells (Clarke et al., 2000). The cumulative nature of the analysis makes it much more sensitive (∼102- to 103-fold) and less variable than approaches that depend on average responses measured at particular time points (Arrasate and Finkbeiner, 2005).
Our approach follows the fate of every cell from the beginning of the monitoring period until the end. As a result, it is essentially insensitive to a common problem with conventional approaches, observer bias: that the duration of an event affects the likelihood of observing it.
In neurodegeneration, short-lived changes may be the most critical steps and the most difficult to observe. Cells exhibiting these changes die more quickly and are less available to study. Conversely, homeostatic changes tend to prolong the lifetimes of affected cells, making these changes easier to observe. These beneficial changes could themselves become so closely associated with disease progression that they are mistaken for pathogenic factors.
As one example, the abnormal deposition of mutant htt protein in intracellular structures called inclusion bodies is a pathological hallmark of HD (Davies et al., 1997). The pathogenic role of inclusion bodies has been controversial, in part because conventional time course data from animal and cell models of the disease were insufficient to determine whether those structures were pathological, beneficial, or incidental (Sisodia, 1998; Orr, 2004). To address this controversy, we applied automated microscopy and survival analysis to the study of striatal neurons expressing normal or disease-associated versions of an N-terminal htt fragment. Surprisingly, neurons that formed inclusion bodies lived longer than those that did not, and we were able to show that the level of more diffuse forms of htt predicted whether and when neurodegeneration would occur (Arrasate et al., 2004).
The ability of survival analysis to uncover predictive relationships that are not susceptible to observer bias is one of the major reasons why this approach, unlike conventional correlations, can address causation per se. Indeed, gross correlations made with conventional approaches can be resolved further with survival analysis into changes that are associated with an increase, a decrease, or no change in the likelihood of reaching an end point within a defined interval. Factors associated with accelerated neurodegeneration are likely to be pathogenic, those associated with decelerated neurodegeneration are likely to be coping responses, and those associated with no significant relationship to the rate of neurodegeneration are likely to be incidental.
In addition to resolving qualitative relationships, survival analysis can quantify the magnitude of the change in risk associated with a factor. This is especially helpful because many biological processes may go awry during neurodegeneration, but only some may be relevant to patient's symptoms and therefore important therapeutic targets. Quantitative measures of the risk of neurodegeneration associated with identifiable factors provide a way to rank them in order of importance.
Finally, survival analysis and related statistical approaches make it possible to build quantitative models that incorporate each factor shown to bear a predictive relationship (including factors that promote or inhibit a fate) to the outcome of interest. This makes it more feasible to satisfy a critical requirement for establishing cause-and-effect relationships: to simultaneously measure (or exclude) the contributions of other potential mechanisms to a putative cause-and-effect relationship. This type of model building can reveal the extent to which neurodegeneration can be predicted, and therefore fully understood, or whether important stochastic or deterministic contributions remain to be elucidated (Herndon et al., 2002).
From yeast to microglia: the role of the kynurenine pathway in HD
In recent years, many laboratories have undertaken genetic screens in model organisms (including yeast, flies, and worms) to identify and dissect disease-modifying pathways for neurodegenerative diseases. These studies, and many others based on candidate gene approaches, have laid the foundation for rigorous analyses of the relevance and causality of diverse pathways by genetic and pharmacological approaches in animal models and will provide tools for future studies in more physiologically relevant models of disease. Understanding disease-modifying pathways at a mechanistic level will likely yield novel drug targets and candidate therapeutics for the treatment of neurodegenerative disorders.
We recently used the model organism Saccharomyces cerevisiae to identify, on a genome-wide scale, loss-of-function enhancers (Willingham et al., 2003) and suppressors (Giorgini et al., 2005) of mutant htt toxicity. In the LOF enhancer screen, we identified mutations in 52 yeast genes that enhanced mutant htt toxicity, and the majority of these genes are involved in stress-activated signaling and protein folding. In the suppressor screen, we identified mutations in 28 yeast genes that suppressed mutant htt toxicity that are known or predicted to play roles in vesicle transport, vacuolar degradation, transcription, and prion-like aggregation. Among the most potent suppressors was BNA4 (biosynthesis of nicotinic acid protein 4), which encodes the yeast homolog of kynurenine 3-monnooxygenase (KMO) that catalyzes the hydroxylation of kynurenine in the kynurenine pathway (KP) of tryptophan degradation (Fig. 2). Accumulating evidence has implicated the KP, which in the brain is expressed predominantly in microglia and astrocytes, as an important factor in HD (Schwarcz and Pellicciari, 2002). Many of the distinct neuropathological features in HD neostriatum can be duplicated in experimental animals by an intrastriatal injection of the excitotoxin quinolinic acid (QUIN) (Schwarcz and Kohler, 1983; Beal et al., 1986; Davies and Roberts, 1988). These findings led to the suggestion that QUIN, a selective NMDA receptor agonist endogenous to the mammalian brain, might be causally involved in HD. QUIN is a metabolite of the KP (Fig. 2). This pathway contains two additional neuroactive products, the neurotoxic QUIN precursor 3-hydroxykynurenine (3-HK) and the neuroinhibitory and neuroprotective agent kynurenic acid (KYNA), which is formed in a side arm of the metabolic cascade. KYNA and 3-HK may also be linked to the pathophysiology of HD: the former because of its ability to preferentially prevent QUIN-induced neurodegeneration by antagonizing NMDA receptors, and the latter because it produces free radicals and potentiates QUIN neurotoxicity. Enzymes of the KP are expressed constitutively in human microglia and astrocytes but not in neurons (Heyes et al., 1996; Guillemin et al., 1999, 2001). In human cells, KMO, the biosynthetic enzyme of 3-HK, is expressed exclusively in microglia, whereas the enzymatic transamination of kynurenine to KYNA occurs mainly in astrocytes. Neostriatal and neocortical levels of 3-HK and QUIN are significantly increased in early- grade HD brains (Guidetti et al., 2004) and in three transgenic mouse models of HD (Guidetti et al., 2006). Together, these results suggest that enhanced flux through the KP to generate 3-HK and QUIN in microglia might contribute to neuronal degeneration in the early phases of HD.
Figure 2.
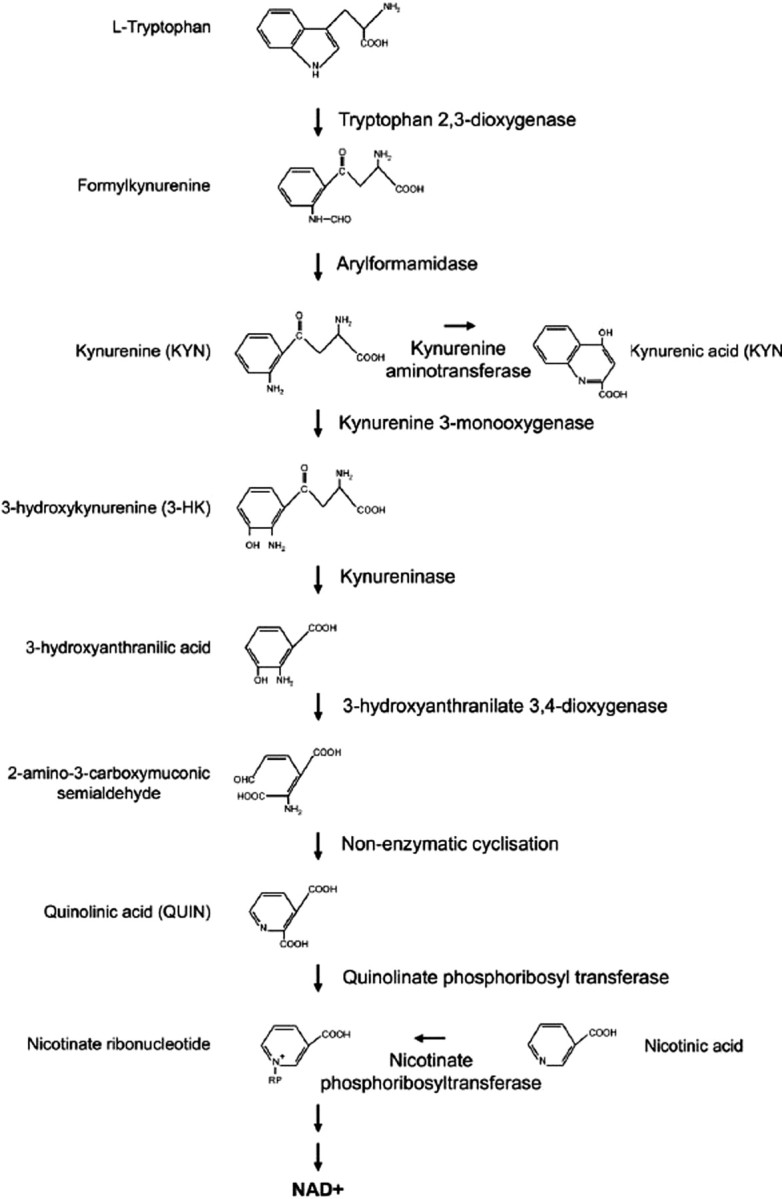
The kynurenine pathway of tryptophan degradation. This metabolic pathway is abnormal in HD patients and transgenic mouse models of HD. Toxic metabolites in this pathway generated by microglia may contribute to neurodegeneration in HD by causing excitotoxicity and/or generation of reactive oxygen species.
As in mouse models of HD and in HD patients, a control yeast strain expressing a mutant htt fragment (Htt103Q) had significantly greater increases in 3-HK and QUIN than the same strain expressing a wild-type (WT) htt fragment (Htt25Q) (Giorgini et al., 2005). Genetic or pharmacological inhibition of Bna4 prevented these increases and correlated with decreased levels of reactive oxygen species (ROS) and Htt103Q-mediated toxicity. Moreover, a mutation in the last enzyme upstream of QUIN (3-hydroxyanthranilate 3,4-dioxygenase or _bna1_Δ) partially blocked Htt103Q toxicity, whereas mutations in enzymes in the side arms of the pathway (kynurenine aminotransferase or _aro9_Δ, and nicotinate phosphoribosyltransferase or _npt1_Δ) enhanced 3-HK and QUIN levels and Htt103Q toxicity (Giorgini et al., 2005). Ro 61-8048 (3,4-dimethoxy-_N_-[4-(nitrophenyl)thiazol-2-yl]benzenesulfonamide), a small molecule orally bioavailable inhibitor of KMO (Rover et al., 1997), prevented mutant htt-induced increases in 3-HK and partially suppressed toxicity and ROS generation in yeast (Giorgini et al., 2005). More importantly, in a small pilot study, Ro 61-8048 improved multiple outcome measures in the R6/2 transgenic mouse model of HD (P. J. Muchowski, L. Menalled, and D. S. Howland, unpublished observations). These promising results, currently being confirmed, suggest that blocking the KP in microglia in vivo may provide therapeutic benefit in HD.
Our studies in yeast elucidated the molecular basis for mutant htt-induced elevation of the KP and demonstrated that this induction occurs at the level of transcription and is dependent on histone deacetylase (HDAC) activity in microglia (F. Giorgini, T. Möller, J. L. Wacker, S. Hong, L. L. Tsai, C. S. Cheah, R. Schwarcz, P. Guidetti, and P. J. Muchowski, unpublished observation). Mutations in the yeast Rpd3 HDAC complex (conserved in mammals) suppressed mutant htt toxicity by causing repression of KP gene transcription. Rpd3 is a target for HDAC inhibitors, which have shown promising results in preclinical studies in HD mice (Ferrante et al., 2003; Hockly et al., 2003; Gardian et al., 2005) and are in a clinical trial for HD. HDAC-dependent regulation of the KP was also observed in a mouse model of HD, in which treatment with an HDAC inhibitor blocked activation of the KP in microglia. HDAC inhibitors are thought to exert beneficial effects in HD mice by increasing the transcription of multiple target genes in neurons that are repressed attributable to expression of mutant htt. Interestingly, our results demonstrate that HDAC inhibitors can modulate mutant htt-induced transcriptional perturbations not only by increasing transcription in neurons but also by repressing transcription of the KP in microglia.
Two recent studies provide compelling evidence that microglia contribute to neurodegeneration in mouse models of multiple sclerosis and ALS. In the first study, transgenic mice were constructed to express herpes simplex thymidine kinase under the control of a promoter specific for macrophages and microglia (Heppner et al., 2005). Treatment of these mice (called CD11b-HSVTK mice) with ganciclovir in vivo kills all cells of the macrophage/monocyte lineage, including microglia. However, because systemic ganciclovir administration to CD11b-HSVTK mice causes hematopoietic toxicity, bone marrow chimeras were generated in which WT bone marrow was transferred to CD11b-HSVTK mice. In bone marrow chimeras, ganciclovir blocked microglial activation and repressed the development of experimental autoimmune encephalomyelitis, a model for multiple sclerosis (Heppner et al., 2005). In the second study, Boillee et al. (2006) evaluated the effects of conditional expression of a superoxide dismutase (SOD) mutant in neurons and macrophages/microglia in a mouse model of ALS. Astonishingly, simply decreasing expression of the SOD mutant in a fraction of macrophages/microglia resulted in much slower disease progression than when the SOD mutant was expressed in these cells at normal levels.
Although these two studies suggest that microglia exert properties detrimental to neurons that may contribute to their ultimate demise, these cells also provide many protective functions in the normal brain and possibly in disease states (Fig. 3). As just one example, microglia modulate the size and abundance of amyloid plaques in transgenic models of AD (Simard et al., 2006). Microglia may initially serve a protective role in neurodegenerative diseases but, ultimately, may acquire toxic properties (Wyss-Coray and Mucke, 2002). Surprisingly, in at least some diseases (such as HD and ALS), these toxic properties may be directly attributable to expression of mutant proteins in these cells.
Figure 3.

The yin and yang of microglia in neurodegeneration. Under normal conditions, microglia provide trophic support to surrounding neurons. However, in certain disease states, they can become dysfunctional and secrete toxic molecules that may contribute to the demise of surrounding neurons. Such a scenario might be caused by accumulation of misfolded protein directly in microglia, as may occur in HD and ALS.
The most surprising result from our yeast genetic screen was the identification of a potential disease-modifying pathway exclusive to microglia, which were not previously thought to be involved in HD pathogenesis. These results underscore the value of unbiased genetic approaches in model organisms to identify disease-modifying pathways.
Genes that control aging modify protein misfolding disease in worms
Neurodegenerative diseases are associated with the expression of misfolded proteins, which, over time, form oligomers and accumulate as aggregates that can disrupt cellular protein-folding homeostasis and lead to cellular dysfunction and death. To investigate the relationship between the stress of misfolded proteins, cellular toxicity, and aging, we developed models for expression of disease-associated misfolded proteins in C. elegans. Analysis of the dynamics of polyQ aggregate formation in neurons and muscle cells over the lifespan of individual animals has identified genes controlling aging and the heat shock response as determinants of aggregation toxicity.
As a model system, C. elegans has unique characteristics with extensive genetic and molecular tools and a completely characterized cell lineage. Adult animals contain 959 somatic cells, of which 302 are neurons and provide opportunities to examine cell-type specificity, developmental processes, and aging. We have C. elegans models that express polyQ expansion proteins and other aggregation-prone proteins in different cells with pan-neuronal or neuron-specific promoters or by similar regulatory strategies for body-wall muscle, intestine, and hypodermal cells.
As one example of these models, we created transgenic animals that express polyQ repeats of different lengths (from Q0 to Q86) fused to the yellow fluorescent protein (YFP) under the control of a body-wall muscle cell or neuronal promoter. Expression of polyQ proteins below a threshold of mid-Q30s results in diffuse soluble polyQ protein, whereas transgenic animals expressing proteins of Q40 or longer formed discrete aggregate structures (Fig. 4).
Figure 4.

PolyQ length-dependent aggregation in C. elegans. Length-dependent aggregation of polyQ–YFP fusion proteins in C. elegans. Epifluorescence micrographs of 3- to 4-d-old C. elegans expressing different lengths of polyQ–YFP (Q0, Q19, Q29, Q33, Q35, Q40, Q44, Q64, and Q82). Scale bar, 0.1 mm. [Adapted from Morley et al. (2002).].
Expression of expanded polyQ proteins in muscle and neuronal cells is cytotoxic and results in a dramatic loss of movement (Fig. 5). Neuronal Q86 aggregates appear in early larval stages and persist throughout the lifespan, suggesting that length-dependent changes in aggregation state are independent of neuronal subtype. However, at the lower threshold lengths of Q35 and Q40, neuronal cell bodies and processes are clearly delineated, indicating a soluble protein state, yet behavioral deficits were observed. Behavioral toxicity in these animals resulted from neuron-specific aggregation that was detected in fluorescence recovery after photobleaching (FRAP) experiments. In the ventral cord neurons (VNCs), Q40::YFP exists in both soluble and immobile states, whereas in neurons in head or tail ganglia, only soluble Q40 was observed. To further establish that neuronal Q40 can form multiple biophysical states, we used fluorescence resonance energy transfer (FRET) to show that Q40-containing proteins can exist as multiple populations within the VNC. In animals expressing short or long polyQ proteins, htt was clearly either soluble or aggregated, respectively. The complementary techniques of FRET and FRAP reveal that intermediate-length Q40 proteins exhibit polymorphic subcellular distributions, solubilities, and intermolecular interactions in the neurons of individual animals (Brignull et al., 2006).
Figure 5.
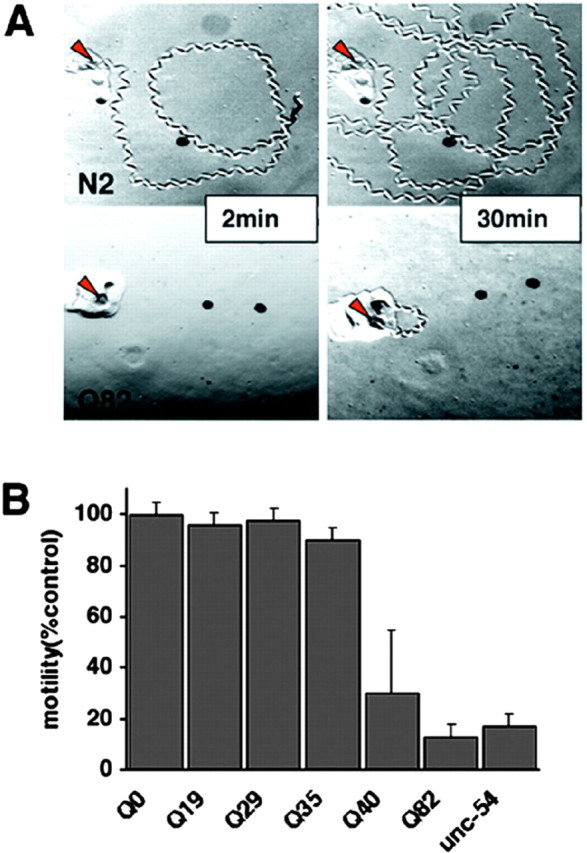
Expression of polyQ expansions in C. elegans muscle results in a motility defect that directly corresponds to aggregate formation. A, Time-lapse micrographs illustrating tracks left by 5-d-old wild-type (N2) and Q82 animals 2 and 30 min after being placed at the position marked by the red arrow. B, Quantitation of motility index for 4- to 5-d-old Q0, Q19, Q29, Q35, Q40, Q82, and unc-54(r293) animals. Data are mean ± SD for at least 50 animals of each type as a percentage of N2 motility. [Adapted from Morley et al. (2002).].
C. elegans expressing different polyQ proteins in body-wall muscle cells exhibit a striking polyQ length-dependent aggregation phenotype (Fig. 4). Relative to Q40 and Q82 animals that rapidly accumulate aggregates and a corresponding loss of motility, intermediate Q33 and Q35 animals show an initial lag and then gradual accumulation of aggregates to a much lower extent than for Q40 or Q82. Up to 3 d of age, only animals expressing Q40 or greater displayed aggregates. At 4–5 d, the threshold shifts, and aggregates are detected in Q33 and Q35 animals. For animals expressing Q29, aggregation is not observed until 8–9 d of age. These results establish that the threshold for polyQ aggregation is dynamic and reflects a balance of different factors, including repeat length and age-dependent changes in the protein-folding environment (Morley et al., 2002) (Fig. 6).
Figure 6.

Influence of aging on polyQ aggregation and toxicity. A, Accumulation of aggregates in Q82 (○), Q40 (●), Q35 (□), Q33 (■), Q29 (▵), and Q0 (▴) during aging. Data are mean ± SEM. Twenty-four animals of each type are represented at day 1. Cohort sizes decreased as animals died during the experiment, but each data point represents at least five animals. B, Motility index as a function of age for the same cohorts of animals described in A. Data are mean ± SD as a percentage of age-matched Q0 animals. C–E, Epifluorescence micrographs of the head of an individual Q35 animal at 4 (C), 7 (D), and 10 (E) days of age, illustrating age-dependent accumulation of aggregates. Arrowheads indicate positions of the same aggregates on different days. In E, the animal is rotated slightly relative to its position in D. [Adapted from Morley et al. (2002).].
Does this dynamic behavior result from the intrinsic properties of the polyQ motif, or do changes over time reflect the influence of aging-related alterations in the cell? The idea that the molecular determinants of longevity might influence polyQ-mediated toxicity is supported by observations that the time before pathology develops (e.g., days in C. elegans, weeks in Drosophila, months in mice, and years in humans) correlates approximately with the lifespan of the organism. We demonstrated a role for lifespan as a modifier of protein misfolding by expressing Q82 on the background of the age-1(hx546) (aging alteration protein 1) mutation or age-1 RNA interference. AGE-1 encodes phosphoinositide-3 kinase, which functions in an insulin-like signaling (ILS) pathway. Mutations in this gene extend lifespan by 1.5- to 2-fold. Q82 on the age-1(hx546) background (Q82;age-1) exhibited reduced aggregate formation in embryos relative to Q82 in the WT background. Q82 aggregate formation was 30–50% lower during larval stages (1–2 d old) in age-1 animals than in wild-type animals and remained significantly lower through 4–5 d of age (Morley et al., 2002). Parallel motility assays also demonstrated a delay in the onset of the motility defect, consistent with slower aggregate accumulation in Q82;age-1 animals. In WT animals, the kinase activity of AGE-1 is required in a signaling cascade that results in constitutive repression of the forkhead transcription factor DAF-16 (abnormal dauer formation protein 16), leading to normal lifespan. Derepression of DAF-16 in age-1 animals results in extended lifespan, and daf-16 mutations suppress the longevity phenotype. To examine whether the age-1 effects on longevity and polyQ aggregation and toxicity are mediated through similar regulatory pathways, we showed that Q82;age-1;daf-16 animals exhibited aggregation and motility phenotypes similar to Q82 expressed on a wild-type background. Thus, the dual effects of age-1 on longevity and polyQ-mediated toxicity share a common genetic pathway (Morley et al., 2002).
Our demonstration that a mutation conferring longevity also delays polyQ aggregation and toxicity suggests an important link between the genetic regulation of lifespan and aging-associated protein misfolding disease. The molecular link between these pathways is regulated, in part, by factors that detect and respond to misfolded proteins, namely heat shock transcription factor (HSF) and molecular chaperones/heat shock proteins. For example, inhibition of HSF-1 function leads to decreased lifespan and an accelerated aging phenotype in C. elegans (Hsu et al., 2003; Morley and Morimoto, 2004). Conversely, overexpression of HSF-1 in C. elegans extends lifespan (Hsu et al., 2003; Morley and Morimoto, 2004). Additionally, inactivation of daf-16, hsf-1, or the genes encoding small heat shock proteins in C. elegans accelerated the aggregation of polyQ expansion proteins, supporting the idea that ILS could coordinately influence aging and protein aggregation through the action of DAF-16, HSF-1, and molecular chaperones.
Our studies in C. elegans led us to propose that expression of polyQ proteins causes cellular dysfunction by a generalized destabilization of the cellular protein-folding environment. To test the hypothesis, we recently took a genetic approach. Specifically, we used diverse and unrelated C. elegans temperature-sensitive (ts) mutations as folding sensors to examine whether their functionality at the permissive condition is affected by expression of aggregation-prone polyQ expansions (Gidalevitz et al., 2006). Because many ts mutant proteins are highly dependent on the cellular-folding environment, they are highly sensitive indicators of a disruption in protein homeostasis.
Animals expressing a ts mutant of the C. elegans homolog of a muscle paramyosin [UNC-15 (uncoordinated protein 15)] were crossed to C. elegans polyQ strains, and the phenotypes were examined at both permissive and restrictive temperatures. The paramyosin ts animals alone do not display overt phenotypes at the permissive temperature. However, at the restrictive temperature, the ts mutation disrupts thick filament formation and leads to embryonic and larval lethality phenotypes and slow movement in adults. In animals expressing only Q40 in muscle cells, neither lethality nor paralysis was observed. In contrast, >40% of embryos coexpressing Q40 and paramyosin ts failed to hatch or move at the permissive temperature. This effect was polyQ length dependent, because coexpression of smaller polyQ expansions with paramyosin(ts) decreased penetrance of ts phenotypes. Examination of muscle structure in paramyosin(ts)+Q40 embryos at the permissive temperature revealed a disrupted pattern of actin staining, similar to the pattern in paramyosin(ts) embryos at the restrictive temperature and absent at the permissive temperature or in WT animals. Thus, in muscle cells, expression of an aggregation-prone polyQ protein is sufficient to cause a paramyosin ts mutation to exhibit its mutant phenotype at the permissive temperature.
To understand the nature of this interaction, we examined the cellular localization of paramyosin(ts) protein when it was coexpressed with Q40. The paramyosin ts mutation affects protein interactions that, at restrictive temperature, result in mislocalization into foci. When paramyosin(ts) protein is coexpressed with Q40, it mislocalizes into foci at the permissive temperature, distinct from Q40 aggregates. Thus, expression of Q40 uncovers the protein-folding defect in paramyosin(ts) mutant.
We subsequently demonstrated that the effect of polyQ expansions on folding homeostasis extends to other muscle cell proteins (e.g., myosin, perlecan) and neuronal cell proteins (e.g., dynamin-1 and ras). Animals expressing a ts mutation in the neuronal protein dynamin-1 or ras become paralyzed at the restrictive temperature but have normal motility at the lower, permissive temperature. With the coexpression of pan-neuronal Q40, dynamin(ts) and ras(ts), animals display severe neuronal impairment at the permissive temperature. This effect was polyQ length dependent because no unusual phenotypes were observed in animals coexpressing the non-aggregating Q19. Thus, expression of polyQ expansions phenocopies ts mutations in both muscle and neuronal cells at permissive temperature, and this genetic interaction reflects the propensity of polyQ proteins to aggregate. Thus, the chronic expression of an aggregation-prone polyQ protein interferes with the function of multiple structurally and functionally unrelated proteins.
Aggregation-prone proteins may exert their destabilizing effects by placing stress on the folding capacity of the cell. If the levels of polyQ affect the folding of the ts protein, does the misfolding of the ts protein, in turn, intensify misfolding of polyQ? To address this, we quantified mQ40 aggregation in ts backgrounds and found that aggregation was enhanced dramatically. In contrast, mutations that cause a loss-of-function rather than a ts phenotype did not enhance aggregation. From a genetic perspective, ts mutations in proteins unrelated to cellular folding or clearance pathways behaved as modifiers of polyQ aggregation.
These observations suggest that a positive feedback mechanism exists to enhance the disruption of cellular-folding homeostasis. In one interpretation of these results, the protein-folding capacity of the cell, integrated at a systems level, is a reflection of expressed protein polymorphisms and random mutations, which in themselves do not lead to disease because of the balance achieved by folding and clearance mechanisms (Gidalevitz et al., 2006). However, these proteins may misfold and in turn contribute to the progressive disruption of the folding environment when this balance becomes overwhelmed (e.g., by the expression of an aggregation-prone protein in conformational diseases).
Involvement of autophagy in neurodegeneration
Although molecular chaperones are the front line of defense to combat the accumulation of misfolded proteins, they can be overwhelmed by high levels of misfolded proteins or when cellular conditions are not favorable for refolding/repair. In these cases, a second set of intracellular surveillance mechanisms, the proteolytic systems, removes altered proteins from cells. Two major proteolytic systems participate in clearing abnormal proteins in cells. The proteasome system has been extensively investigated by many laboratories and will not be reviewed here. Our laboratory has focused on understanding the role of lysosome-mediated protein degradation (i.e., autophagy) and its possible involvement in the pathogenesis of neurodegenerative disorders.
In mammalian cells, three different types of autophagy have been described: macroautophagy, microautophagy, and chaperone-mediated autophagy (CMA) (Fig. 7, left) (Cuervo et al., 2005; Yorimitsu and Klionsky, 2005). In macroautophagy and microautophagy, complete regions of the cytosol, including soluble proteins and organelles, are delivered at once into lysosomes. In CMA, only soluble proteins translocate one-by-one across the lysosomal membrane. In most tissues, microautophagy is considered to be the constitutive mechanism for degrading intracellular components in lysosomes and to be responsible for continuous turnover of long-lived proteins and organelles. Although some basal level of macroautophagy and CMA occurs, these two pathways are maximally activated in response to stress. Starvation and exogenous aggressors, including pathogens (i.e., virus and bacteria) and physical insults (i.e., irradiation), activate macroautophagy. CMA is maximally activated in response to prolonged starvation, mild oxidative stress, and chemical agents that selectively alter particular intracellular proteins (Massey et al., 2006a). Activation of CMA in these two last conditions seems aimed at the removal of the altered proteins without affecting the unaltered proteins nearby.
Figure 7.

Autophagy and neurodegenerative disorders. Left, Mammalian cells use three types of autophagy. Microautophagy and macroautophagy involve the sequestration of complete cytosolic regions directly by the lysosomes or in an intermediate compartment, the autophagic vacuole, which is then delivered to lysosomes. In chaperone-mediated autophagy, single soluble proteins are recognized by a cytosolic chaperone and a receptor at the lysosomal membrane that mediates their translocation across the membrane into the lysosomal lumen. Right, Misfolded or altered proteins are selectively degraded by the ubiquitin/proteasome system or by chaperone-mediated autophagy (STAGE I). However, when these altered proteins organize in toxic multimeric complexes, they often alter the proteolytic activity of these two pathways. Upregulation of macroautophagy can compensate for this deficit (STAGE II). Aggravating factors, such as oxidative stress and aging, can precipitate the failure of macroautophagy with the consequent detrimental effect on cell functioning, often resulting in cellular death (STAGE III).
The selective nature of CMA makes it and the ubiquitin/proteasome system the ideal candidates for removing misfolded proteins when refolding is not possible (Fig. 7, right, STAGE I). In fact, numerous studies support the participation of the ubiquitin/proteasome system in the removal of proteins known to be altered in common neurodegenerative disorders, such as α-synuclein in PD and mutant htt in HD (Rubinsztein et al., 2005). Likewise, oxidized and misfolded CMA substrates are more readily degraded through this autophagic pathway (Massey et al., 2006b).
However, proteins with altered conformation tend to organize into toxic multimeric complexes that can no longer be degraded through these pathways and that, in addition, often have a negative effect on the activity of these proteolytic systems. Fragments of mutant htt and mutant forms of α-synuclein block the proteasome (Rubinsztein et al., 2005). By a still unknown mechanism, blockage of this major protease activates macroautophagy, which would remove the cytosolic aggregates that the altered proteins often form (Iwata et al., 2005). We showed that mutant forms of α-synuclein block CMA (Cuervo et al., 2004). Wild-type α-synuclein bears a CMA-targeting motif in its sequence and so can be degraded through this selective lysosomal pathway. Pathogenic forms of α-synuclein are recognized by a cytosolic chaperone and targeted to the lysosomal membrane, in which they interact with a lysosomal receptor. Unlike the wild-type protein, mutant α-synucleins do not translocate into the lysosomal lumen. Instead, they remain tightly bound to the lysosomal membrane, blocking the translocation of other CMA substrates through this pathway (Cuervo et al., 2004). Our initial studies were limited by the fact that, although α-synuclein is found in the protein aggregates in all forms of PD, pathogenic mutant forms of α-synuclein are only found in <10% of patients with PD. We recently found that particular modifications of the wild-type protein, namely those induced after exposure to oxidized forms of dopamine, mimic the effects of mutant forms of α-synuclein on CMA. This finding is of particular relevance because dopaminergic neurons are a preferred target of this disease, and the neurons of patients contain higher levels of oxidized dopamine in their cytosol. Thus, pathogenic α-synuclein mutants and modifications on the wild-type protein alter their degradation via CMA, but, most importantly, their accumulation at the lysosomal membrane results in severe blockage of CMA. Blockage of CMA in cultured cells results in compensatory activation of macroautophagy, which preserves normal levels of protein turnover and proper removal of aggregate proteins (Massey et al., 2006b). However, macroautophagy cannot completely replace CMA. CMA-deficient cells are more vulnerable to stress and rapidly undergo apoptosis after exposure to oxidants, pro-oxidants, and ultraviolet light (Massey et al., 2006b).
Although some level of constitutive macroautophagic activity in neurons is essential to maintain proper neuronal homeostasis (Hara et al., 2006; Komatsu et al., 2006), this pathway can be maximally activated in these protein conformational disorders in response to both blockage of the proteasome and blockage of CMA (Fig. 7, right, STAGE II) (Iwata et al., 2005; Massey et al., 2006b). The presence of aggregate proteins also seems to be enough to activate macroautophagy in different systems, because this is the only mechanism able to degrade these protein complexes. Although macroautophagy is usually nonselective, a certain degree of selectivity has been proposed in the removal of aggregates by this pathway. Thus, under these circumstances, macroautophagy seems to occur preferentially in the peri-centriolar area in which aggregate proteins are delivered by microtubules. This way, the ratio of aggregate to soluble cytosolic protein trapped by the autophagosomes is unbalanced toward the removal of the aggregates, with minimal incorporation of other cytosolic components. The critical role of macroautophagy in the removal of aggregate proteins has been further supported by recent studies in mice in which macroautophagy has been selectively ablated in neurons. Lack of macroautophagy in these animals resulted in accumulation of aggregate proteins in neurons and rapid neurodegeneration, even in the absence of a prone-to-aggregate protein (Hara et al., 2006; Komatsu et al., 2006).
Activation of macroautophagy maintains normal rates of protein degradation, proper organelle turnover, and facilitates the removal of the aggregate proteins. However, depending solely on this proteolytic system leaves affected cells particularly vulnerable. Any added stressor, such as oxidative stress, common in these disorders, would increase the macroautophagic load. With the decrease with age in the activity of the different proteolytic systems (Cuervo et al., 2005), including macroautophagy, this could precipitate the failure of this system and lead to the accumulation of undegraded products inside cells. We described a defect in the ability of lysosomes to take up proteins via CMA, and other studies have shown inefficient clearing up of already formed autophagic vacuoles in different tissues in old animals (Cuervo et al., 2005). The persistence of the engulfed material in these vacuoles often favors nonspecific intermolecular cross-linking, decreasing the susceptibility of cargo to proteases (Cuervo et al., 2005). The inability to properly remove aggregate proteins and other altered components, along with the accumulation of autophagic vacuoles, is likely to contribute to cellular failure in these advanced states (Fig. 7, right, STAGE III).
The removal of aggregated proteins by macroautophagy has stimulated growing interest in activators of macroautophagy as a possible therapeutic to clear aggregates in neurodegenerative disorders. In fact, this approach has been used to remove α-synuclein aggregates in culture cells and of mutant htt both in culture cells and mouse models (Rubinsztein et al., 2005). In this last case, aggregate clearance by upregulation of macroautophagy improves neural symptomathology. Although there was an initial concern about how long macroautophagy could be efficiently upregulated and about the consequences that this accelerated turnover of intracellular components could have for the cells, the proposed selectivity in the removal of aggregates, along with the beneficial effects observed in the animal models, now supports the validity of this approach. Future efforts should, however, focus on the identification of new activators of macroautophagy. So far, the most efficient way of upregulating macroautophagy is by blocking mammalian target of rapamycin (mTOR), one of the major intracellular kinases, also involved in the regulation of other critical intracellular processes. The first mTOR-independent mechanism for activating macroautophagy was described recently, although its suitability for the treatment of these disorders untested. Moreover, future interventions should consider acting at earlier stages, for example preventing the blockage of the ubiquiting/proteasome system and of CMA. A better understanding of the cross talk among different proteolytic systems, and of the mechanisms that regulate the upregulation of one of these systems in response to failure of another, should also provide novel ways to promote the clearance of the toxic protein products that accumulate in different neurodegenerative disorders.
Summary
The identification and characterization of putative disease-modifying pathways in neurodegenerative disorders has enormous potential for discovery of novel therapeutic agents that target these pathways. In studies of these putative pathways, it is critical to rigorously evaluate causality to the disease process. Findings in model organisms require validation in more physiologically relevant models but have the benefit of identifying targets amenable to modulation by small molecule therapeutics, possibly including drugs already in clinical use. Although many disease-modifying pathways have been reported, few have been validated in vivo in animal models of disease. Nonetheless, given the powerful genetic tools currently available, including a growing list of mouse mutants, the future looks promising and one can expect to see the number of clinical trials based on interventions that target disease-modifying pathways to continue growing.
Footnotes
This work was supported by grants from the National Institute of Neurological Disease and Stroke (S.F., P.J.M.), the National Institute of General Medical Science (R.I.M.), the National Institute for Aging (S.F., A.M.C., R.I.M.), the High Q Foundation (S.F., P.J.M.), the Hereditary Disease Foundation Lieberman Award (S.F.), the Huntington's Disease Society of America (A.M.C.) Coalition for the Cure (R.I.M.), and the Therapeutics Initiative Award (S.F.), the Amyloid Lateral Sclerosis Association (R.I.M.), the National Center for Research Resources (S.F.), the Taube Family Foundation Program in Huntington's Disease Research (S.F., P.J.M.), the Daniel F. and Ada L. Rice Foundation (R.I.M.), the J. David Gladstone Institutes (S.F., P.J.M.), and the Ellison Medical Foundation (A.M.C.). We thank many members of our laboratories past and present who contributed to this work both intellectually and technically. We thank Stephen Ordway and Gary Howard for editorial assistance.
References
- Arrasate M, Finkbeiner S. Automated microscope system for determining factors that predict neuronal fate. Proc Natl Acad Sci USA. 2005;102:3840–3845. doi: 10.1073/pnas.0409777102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arrasate M, Mitra S, Schweitzer ES, Segal MR, Finkbeiner S. Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature. 2004;431:805–810. doi: 10.1038/nature02998. [DOI] [PubMed] [Google Scholar]
- Beal MF, Ferrante RJ. Experimental therapeutics in transgenic mouse models of Huntington's disease. Nat Rev Neurosci. 2004;5:373–384. doi: 10.1038/nrn1386. [DOI] [PubMed] [Google Scholar]
- Beal MF, Kowall NW, Ellison DW, Mazurek MF, Swartz KJ, Martin JB. Replication of the neurochemical characteristics of Huntington's disease by quinolinic acid. Nature. 1986;321:168–171. doi: 10.1038/321168a0. [DOI] [PubMed] [Google Scholar]
- Bodner RA, Outeiro TF, Altmann S, Maxwell MM, Cho SH, Hyman BT, McLean PJ, Young AB, Housman DE, Kazantsev AG. Pharmacological promotion of inclusion formation: a therapeutic approach for Huntington's and Parkinson's disease. Proc Natl Acad Sci USA. 2006;103:4246–4251. doi: 10.1073/pnas.0511256103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boillee S, Yamanaka K, Lobsiger CS, Copeland NG, Jenkins NA, Kassiotis G, Kollias G, Cleveland DW. Onset and progression in inherited ALS determined by motor neurons and microglia. Science. 2006;312:1389–1392. doi: 10.1126/science.1123511. [DOI] [PubMed] [Google Scholar]
- Brignull HR, Moore FE, Tang SJ, Morimoto RI. Polyglutamine proteins at the pathogenic threshold display neuron-specific aggregation in a pan-neuronal Caenorhabditis elegans model. J Neurosci. 2006;26:7597–7606. doi: 10.1523/JNEUROSCI.0990-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke G, Collins RA, Leavitt BR, Andrews DF, Hayden MR, Lumsden CJ, McInnes RR. A one-hit model of cell death in inherited neuronal degenerations. Nature. 2000;406:195–199. doi: 10.1038/35018098. [DOI] [PubMed] [Google Scholar]
- Cuervo AM, Stefanis L, Fredenburg R, Lansbury PTJ, Sulzer D. Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science. 2004;305:1292–1295. doi: 10.1126/science.1101738. [DOI] [PubMed] [Google Scholar]
- Cuervo AM, Bergamini E, Brunk UT, Droge W, Ffrench M, Terman A. Autophagy and aging: the importance of maintaining “clean” cells. Autophagy. 2005;1:131–140. doi: 10.4161/auto.1.3.2017. [DOI] [PubMed] [Google Scholar]
- Davies SW, Roberts PJ. Model of Huntington's disease. Science. 1988;241:474–475. doi: 10.1126/science.2969147. [DOI] [PubMed] [Google Scholar]
- Davies SW, Turmaine M, Cozens BA, DiFiglia M, Sharp AH, Ross CA, Scherzinger E, Wanker EE, Mangiarini L, Bates GP. Formation of neuronal intranuclear inclusions underlies the neurological dysfunction in mice transgenic for the HD mutation. Cell. 1997;90:537–548. doi: 10.1016/s0092-8674(00)80513-9. [DOI] [PubMed] [Google Scholar]
- Djousse L, Knowlton B, Hayden MR, Almqvist EW, Brinkman RR, Ross CA, Margolis RL, Rosenblatt A, Durr A, Dode C, Morrison PJ, Novelletto A, Frontali M, Trent RJ, McCusker E, Gomez-Tortosa E, Mayo Cabrero D, Jones R, Zanko A, Nance M. Evidence for a modifier of onset age in Huntington disease linked to the HD gene in 4p16. Neurogenetics. 2004;5:109–114. doi: 10.1007/s10048-004-0175-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emery DL, Royo NC, Fishcher I, Saatman KE, McIntosh TK. Plasticity following injury to the adult central nervous system: is recapitulation of a developmental state worth promoting. J Neurotrauma. 2003;20:1271–1292. doi: 10.1089/089771503322686085. [DOI] [PubMed] [Google Scholar]
- Ferrante RJ, Kubilus JK, Lee J, Ryu H, Beesen A, Zucker B, Smith K, Kowall NW, Ratan RR, Luthi-Carter R, Hersch SM. Histone deacetylase inhibition by sodium butyrate chemotherapy ameliorates the neurodegenerative phenotype in Huntington's disease mice. J Neurosci. 2003;23:9418–9427. doi: 10.1523/JNEUROSCI.23-28-09418.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardian G, Browne SE, Choi DK, Klivenyi P, Gregorio J, Kubilus JK, Ryu H, Langley B, Ratan RR, Ferrante RJ, Beal MF. Neuroprotective effects of phenylbutyrate in the N171–82Q transgenic mouse model of Huntington's disease. J Biol Chem. 2005;280:556–563. doi: 10.1074/jbc.M410210200. [DOI] [PubMed] [Google Scholar]
- Gidalevitz T, Ben-Zvi A, Ho KH, Brignull HR, Morimoto RI. Progressive disruption of cellular protein folding in models of polyglutamine diseases. Science. 2006;311:1471–1474. doi: 10.1126/science.1124514. [DOI] [PubMed] [Google Scholar]
- Giorgini F, Guidetti P, Nguyen Q, Bennett SC, Muchowski PJ. A genomic screen in yeast implicates kynurenine 3-monooxygenase as a therapeutic target for Huntington disease. Nat Genet. 2005;37:526–531. doi: 10.1038/ng1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guidetti P, Luthi-Carter RE, Augood SJ, Schwarcz R. Neostriatal and cortical quinolinate levels are increased in early grade Huntington's disease. Neurobiol Dis. 2004;17:455–461. doi: 10.1016/j.nbd.2004.07.006. [DOI] [PubMed] [Google Scholar]
- Guidetti P, Bates GP, Hayden MR, Leavitt BR, MacDonald ME, Wheeler VC, Woodman B, Schwarcz R. Elevated brain 3-hydroxykynurenine and quinolinic acid in mouse models of Huntington's disease. Neurobiol Dis. 2006;23:190–197. doi: 10.1016/j.nbd.2006.02.011. [DOI] [PubMed] [Google Scholar]
- Guillemin GJ, Kerr SJ, Smythe GA, Armati PJ, Brew BJ. Kynurenine pathway metabolism in human astrocytes. Adv Exp Med Biol. 1999;467:125–131. doi: 10.1007/978-1-4615-4709-9_18. [DOI] [PubMed] [Google Scholar]
- Guillemin GJ, Kerr SJ, Smythe GA, Smith DG, Kapoor V, Armati PJ, Croitoru J, Brew BJ. Kynurenine pathway metabolism in human astrocytes: a paradox for neuronal protection. J Neurochem. 2001;78:842–853. doi: 10.1046/j.1471-4159.2001.00498.x. [DOI] [PubMed] [Google Scholar]
- Hara T, Nakamura K, Matsui M, Nakahara Y, Suzuki-Migishima R, Yokoyama M, Saito I, Okano H. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature. 2006;441:885–889. doi: 10.1038/nature04724. [DOI] [PubMed] [Google Scholar]
- Heppner FL, Greter M, Marino D, Falsig J, Raivich G, Hovelmeyer N, Waisman A, Rulicke T, Prinz M, Priller J, Becher B, Aguzzi A. Experimental autoimmune encephalomyelitis repressed by microglial paralysis. Nat Med. 2005;11:146–152. doi: 10.1038/nm1177. [DOI] [PubMed] [Google Scholar]
- Herndon LA, Schmeissner PJ, Dudaronek JM, Brown PA, Listner KM, Sakano Y, Paupard MC, Hall DH, Driscoll M. Stochastic and genetic factors influence tissue-specific decline in ageing C. elegans. Nature. 2002;419:808–814. doi: 10.1038/nature01135. [DOI] [PubMed] [Google Scholar]
- Heyes MP, Achim CL, Wiley CA, Major EO, Saito K, Markey SP. Human microglia convert l-tryptophan into the neurotoxin quinolinic acid. Biochem J. 1996;320:595–597. doi: 10.1042/bj3200595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hockly E, Richon VM, Woodman B, Smith DL, Zhou X, Rosa E, Sathasivam K, Ghazi-Noori S, Mahal A, Lowden PA, Steffan JS, Marsh JL, Thompson LM, Lewis CM, Marks PA, Bates GP. Suberoylanilide hydroxamic acid, a histone deacetylase inhibitor, ameliorates motor deficits in a mouse model of Huntington's disease. Proc Natl Acad Sci USA. 2003;100:2041–2046. doi: 10.1073/pnas.0437870100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu AL, Murphy CT, Kenyon C. Regulation of aging and age-related disease by DAF-16 and heat-shock factor. Science. 2003;300:1142–1145. doi: 10.1126/science.1083701. [DOI] [PubMed] [Google Scholar]
- Iwata A, Riley BE, Johnston JA, Kopito RR. HDAC6 and microtubules are required for autophagic degradation of aggregated huntingtin. J Biol Chem. 2005;280:40282–40292. doi: 10.1074/jbc.M508786200. [DOI] [PubMed] [Google Scholar]
- Komatsu M, Waguri S, Chiba T, Murata S, Iwata J, Tanida I, Ueno T, Koike M, Uchiyama Y, Kominami E. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature. 2006;441:880–884. doi: 10.1038/nature04723. [DOI] [PubMed] [Google Scholar]
- Massey AC, Zhang C, Cuervo A. Chaperone-mediated autophagy in aging and disease. Curr Top Dev Biol. 2006a;73:205–235. doi: 10.1016/S0070-2153(05)73007-6. [DOI] [PubMed] [Google Scholar]
- Massey AC, Kaushik S, Sovak G, Kiffin R, Cuervo AM. Consequences of the selective blockage of chaperone-mediated autophagy. Proc Natl Acad Sci USA. 2006b;103:5805–5810. doi: 10.1073/pnas.0507436103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morley JF, Morimoto RI. Regulation of longevity in Caenorhabditis elegans by heat shock factor and molecular chaperones. Mol Biol Cell. 2004;15:657–664. doi: 10.1091/mbc.E03-07-0532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morley JF, Brignull HR, Weyers JJ, Morimoto RI. The threshold for polyglutamine-expansion protein aggregation and cellular toxicity is dynamic and influenced by aging in Caenorhabditis elegans. Proc Natl Acad Sci USA. 2002;99:10417–10422. doi: 10.1073/pnas.152161099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadeau JH. Modifier genes and protective alleles in humans and mice. Curr Opin Genet Dev. 2003;13:290–295. doi: 10.1016/s0959-437x(03)00061-3. [DOI] [PubMed] [Google Scholar]
- Orr HT. Neuron protection agency. Nature. 2004;431:747–748. doi: 10.1038/431747a. [DOI] [PubMed] [Google Scholar]
- Rover S, Cesura AM, Huguenin P, Kettler R, Szente A. Synthesis and biochemical evaluation of N-(4-phenylthiazol-2-yl)benzenesulfonamides as high-affinity inhibitors of kynurenine 3-hydroxylase. J Med Chem. 1997;40:4378–4385. doi: 10.1021/jm970467t. [DOI] [PubMed] [Google Scholar]
- Rubinsztein D, DiFiglia M, Heintz N, Nixon RA, Qin Z-H, Ravikumar B, Stefanis L, Tolkovsky AM. Autophagy and its possible roles in nervous system diseases, damage and repair. Autophagy. 2005;1:11–22. doi: 10.4161/auto.1.1.1513. [DOI] [PubMed] [Google Scholar]
- Schwarcz R, Kohler C. Differential vulnerability of central neurons of the rat to quinolinic acid. Neurosci Lett. 1983;38:85–90. doi: 10.1016/0304-3940(83)90115-5. [DOI] [PubMed] [Google Scholar]
- Schwarcz R, Pellicciari R. Manipulation of brain kynurenines: glial targets, neuronal effects, and clinical opportunities. J Pharmacol Exp Ther. 2002;303:1–10. doi: 10.1124/jpet.102.034439. [DOI] [PubMed] [Google Scholar]
- Simard AR, Soulet D, Gowing G, Julien JP, Rivest S. Bone marrow-derived microglia play a critical role in restricting senile plaque formation in Alzheimer's disease. Neuron. 2006;49:489–502. doi: 10.1016/j.neuron.2006.01.022. [DOI] [PubMed] [Google Scholar]
- Sisodia SS. Nuclear inclusions in glutamine repeat disorders: are they pernicious, coincidental, or beneficial? Cell. 1998;95:1–4. doi: 10.1016/s0092-8674(00)81743-2. [DOI] [PubMed] [Google Scholar]
- Thakur AK, Yang W, Wetzel R. Inhibition of polyglutamine aggregate cytotoxicity by a structure-based elongation inhibitor. FASEB J. 2004;18:923–925. doi: 10.1096/fj.03-1238fje. [DOI] [PubMed] [Google Scholar]
- Wexler NS, Lorimer J, Porter J, Gomez F, Moskowitz C, Shackell E, Marder K, Penchaszadeh G, Roberts SA, Gayan J, Brocklebank D, Cherny SS, Cardon LR, Gray J, Dlouhy SR, Wiktorski S, Hodes ME, Conneally PM, Penney JB, Gusella J. Venezuelan kindreds reveal that genetic and environmental factors modulate Huntington's disease age of onset. Proc Natl Acad Sci USA. 2004;101:3498–3503. doi: 10.1073/pnas.0308679101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willingham S, Outeiro TF, DeVit MJ, Lindquist SL, Muchowski PJ. Yeast genes that enhance the toxicity of a mutant huntingtin fragment or alpha-synuclein. Science. 2003;302:1769–1772. doi: 10.1126/science.1090389. [DOI] [PubMed] [Google Scholar]
- Wyss-Coray T, Mucke L. Inflammation in neurodegenerative disease: a double-edged sword. Neuron. 2002;35:419–432. doi: 10.1016/s0896-6273(02)00794-8. [DOI] [PubMed] [Google Scholar]
- Yorimitsu T, Klionsky DJ. Autophagy: molecular machinery for self-eating. Cell Death Differ. 2005;12:1542–1552. doi: 10.1038/sj.cdd.4401765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Smith DL, Meriin AB, Engemann S, Russel DE, Roark M, Washington SL, Maxwell MM, Marsh JL, Thompson LM, Wanker EE, Young AB, Housman DE, Bates GP, Sherman MY, Kazantsev AG. A potent small molecule inhibits polyglutamine aggregation in Huntington's disease neurons and suppresses neurodegeneration in vivo. Proc Natl Acad Sci USA. 2005;102:892–897. doi: 10.1073/pnas.0408936102. [DOI] [PMC free article] [PubMed] [Google Scholar]