A Mechanism for Ca2+/Calmodulin-Dependent Protein Kinase II Clustering at Synaptic and Nonsynaptic Sites Based on Self-Association (original) (raw)
Abstract
The activity of Ca2+/calmodulin-dependent protein kinase II (CaMKII) plays an integral role in regulating synaptic development and plasticity. We designed a live-cell-imaging approach to monitor an activity-dependent clustering of green fluorescent protein (GFP)-CaMKII holoenzymes, termed self-association, a process that we hypothesize contributes to the translocation of CaMKII to synaptic and nonsynaptic sites in activated neurons. We show that GFP-CaMKII self-association in human embryonic kidney 293 (HEK293) cells requires a catalytic domain and multimeric structure, requires Ca2+ stimulation and a functional Ca2+/CaM-binding domain, is regulated by cellular pH and Thr286 autophosphorylation, and has variable rates of dissociation depending on Ca2+ levels. Furthermore, we show that the same rules that govern CaMKII self-association in HEK293 cells apply for extrasynaptic and postsynaptic translocation of GFP-CaMKII in hippocampal neurons. Our data support a novel mechanism for targeting CaMKII to postsynaptic sites after neuronal activation. As such, CaMKII may form a scaffold that, in combination with other synaptic proteins, recruits and localizes additional proteins to the postsynaptic density. We discuss the potential function of CaMKII self-association as a tag of synaptic activity.
Keywords: protein translocation, autophosphorylation, postsynaptic density, aggregation, calmodulin, synaptic tag
Introduction
Proposed mechanisms of long-term synaptic plasticity in the mammalian CNS have evolved to overcome a series of difficult engineering issues. For example, a single hippocampal neuron contains >10,000 excitatory synapses that are each isolated computational units that can be strengthened or weakened independently. Plasticity lasting more than a few hours is thought to involve upregulation of nuclear gene expression and subsequent protein translation in the soma and, as such, introduces a particularly unique problem. How can synapse-specific plasticity be maintained in the face of nuclear changes that should impact synapses cell wide? This could be achieved through synaptic “tags” (Frey and Morris, 1998; Martin, 2002; Martin and Kosik, 2002), which are laid down at specific synapses to somehow capture the necessary components from the cell wide supply.
Despite the importance of the concept of synaptic tagging, no specific molecule that underlies the process has been identified. Martin and Kosik (2002) proposed criteria that the tag should be spatially restricted, time-limited and reversible, and should interact with the cell-wide events that produce long-term synapse-specific strengthening, such as long-term potentiation (LTP). Additionally, Frey and Morris (1998) hypothesized that “an economical arrangement would be one in which the biochemical cascade responsible for early LTP included the phosphorylation of a protein that, in addition to its immediate effects on synaptic transmission, was also responsible for sequestering proteins later” (Frey and Morris, 1998).
Ca2+/calmodulin-dependent protein kinase II (CaMKII) has well-documented functions in the induction of LTP (Lisman et al., 2002) and possesses structural and regulatory features that fulfill the criteria for a synaptic tag. First, CaMKII is a multisubunit complex (Kolodziej et al., 2000; Hoelz et al., 2003; Gaertner et al., 2004) with the catalytic domains extending out from the core, making them available to interact simultaneously with and phosphorylate multiple substrates and anchoring proteins, including those associated with the postsynaptic density (PSD) (Bayer and Schulman, 2001; Soderling et al., 2001; Colbran, 2004). Second, the enzyme can rapidly translocate to postsynaptic regions after NMDA receptor activation and has variable rates of dissociation from individual synapses (Shen and Meyer, 1999; Shen et al., 2000; Gleason et al., 2003; Otmakhov et al., 2004). Third, electron microscopy (EM) studies of isolated PSDs revealed that CaMKII was not uniformly distributed and tended to form tower-like structures of variable height that extended away from the synaptic membrane (Peterson et al., 2003). Finally, CaMKII holoenzymes were shown to be capable of associating with one another (or self-associate) in response to Ca2+ stimulation (Hudmon et al., 1996, 2001), revealing that the enzyme has an inherent capacity to form higher-order complexes. Self-associated holoenzymes may in fact produce the tower-like PSD-associated CaMKII. As such, CaMKII self-association could be a mechanism to anchor the kinase at specific synapses and be involved with other associated postsynaptic molecules as a tag.
However, the process of CaMKII self-association, as demonstrated in vitro, has not been described in cells. In the present study, an imaging approach was developed to demonstrate and dissect the mechanism behind green fluorescent protein (GFP)-CaMKII self-association in living cells. Using this approach, we also show that the recruitment of activated CaMKII to synaptic sites is mechanistically consistent with GFP-CaMKII binding to other CaMKII holoenzymes in the PSD. The unique multimeric structure of the enzyme would thus provide a geometrically constrained scaffold onto which other synaptic proteins could be assembled, making CaMKII self-association an ideal synaptic tagging mechanism.
Materials and Methods
GFP-CaMKII constructs. In this study, we used three different GFP-CaMKII constructs, which were all made independently from Clontech (Mountain View, CA) enhanced GFP (eGFP) vectors. The first (gift from Tobias Meyer, Stanford University, Stanford, CA) contained the linker (LRSRAQASNSAVDGTAGPGSSAR) and was constructed by digesting the full-length rat cDNA encoding the α subunit of CaMKII from SRα with _Bam_HI/_Xba_I and placing the insert into the _Bam_HI/_Xba_I sites of eGFP-C1. The second contained the linker (RTQISSSSFEFPMAN) and was constructed by digesting the full-length rat cDNA encoding α-CaMKII from pGBT-α (Kolb et al., 1998) with _Eco_RI/_Sal_I and placing the insert into the _Eco_RI/_Sal_I sites of eGFP-C2. These two constructs were used in preliminary experiments, but because eGFP tends to dimerize, we made a third construct, termed mGFP-CaMKII, in which a mutation (A207K) in the eGFP was introduced to render GFP monomeric (Zacharias et al., 2002). We chose to use this construct for all of our remaining experiments to avoid GFP-dependent aggregation. For simplicity, we refer only to GFP-CaMKII throughout this study. The following various mutant forms of CaMKII were described previously: 286Ala (Meyer et al., 1992), 286Asp (Waldmann et al., 1990), 205Lys (Bayer et al., 2001), 302Arg (Shen and Meyer, 1999), 1-326 (Hanson et al., 1994), 315-478 (Kolb et al., 1998), 305-306/Ala-Ala (Hanson and Schulman, 1992), and 305-306/Asp-Asp (Rich and Schulman, 1998) except for 286-287/Asp-Asp and 286Asp-305-306Ala-Ala, which were generated by mutagenesis and verified by sequence analysis. The various mutant forms of the kinase were subcloned into the wild-type (WT) GFP-CaMKII backbone using a _PflM_I restriction insert.
Time-lapse imaging of transfected human embryonic kidney 293 cells. Human embryonic kidney 293 (HEK293) cells grown to 70-80% confluence in DMEM growth media supplemented with 10% fetal bovine serum and l-glutamine (Invitrogen, San Diego, CA) were transfected using Lipofectamine 2000 (Invitrogen) as described by the manufacturer. On the following day, transfected cells were plated onto poly-d-lysine-coated Aclar (22 C grade; ∼120 μm thickness; Honeywell, Morristown, NJ) coverslips (13 mm in diameter) in 24-well tissue culture plates (Falcon, Franklin Lakes, NJ), and imaging experiments were performed 6-16 h later. Imaging solutions, made fresh, were either HBSS (plus 25 mm HEPES, pH 7.4, with 2 mm CaCl2) or a nigericin-based buffer, which was composed of 20 mm HEPES, 140 mm KCl, 2 mm CaCl2, 5 mm NaCl, 1 mg/ml glucose, at pH 7.5 or 6.5. Low Ca2+ (∼150 nm) buffer was made using 15 mm EGTA and 1 mm CaCl2, pH 6.5, whereas 0 Ca2+ buffer was made by replacing CaCl2 with 5 mm BAPTA. Working solutions of nigericin were 10 μg/ml (1:1000 dilution of a methanol stock) and were 10 μm for ionomycin (1:1000 dilution of a methanol stock). Unless otherwise stated, the standard Ca2+/ionomycin-pH drop protocol was as follows: (1) to perfuse the coverslips with nigericin solution at pH 7.5 without ionomycin (Time-0 image), (2) to switch to the same solution with ionomycin/CaCl2 for 2 min, and (3) to switch to a nigericin solution at pH 6.5 with ionomycin/CaCl2. The cells were perfused (0.5-1 ml/min) using a custom-made stop flow multivalve perfusion system in a low-volume custom-made perfusion chamber constructed by Robert Schneevies (Department of Neurobiology, Stanford University, Stanford, CA). For time-lapse series, 12-bit images were taken with a CoolSnap-FX CCD (Roper Scientific, Duluth, GA) through a 40× oil immersion [1.4 numerical aperture (NA)] objective mounted on a Nikon (Tokyo, Japan) Diaphot microscope, controlled by Meta-Morph software (Universal Imaging Corporation, West Chester, PA).
Immunocytochemistry of HEK293 cells. For immunostaining experiments, HEK293 cells were transfected as described above with GFP-αCaMKII or with pCDNA 3.1 expression vectors containing αCaMKII. After stimulation (2 min at pH 7.5 followed by 3 min at pH 6.5), the cells were immediately fixed in 4% paraformaldehyde (0.1 m phosphate buffer, pH 7.4) for 10 min and washed in PBS three times. For labeling with mouse monoclonal anti-αCaMKII antibody (CBα2; 1:5000), cells were permeabilized in methanol at -20°C for 10 min, rehydrated in PBS, and incubated for 30 min in blocking solution (PBS, 2% normal goat serum). For labeling with rabbit anti-phospho-Thr286-CaMKII (1:1250; Promega, Madison, WI), cells were permeabilized for 30 min with 0.02% Triton X-100 at room temperature in blocking solution. Primary antibodies were incubated for 2 h at room temperature in blocking solution. After washes, secondary antibodies (goat anti-mouse or anti-rabbit Alexa 594, 1:1000; Molecular Probes, Eugene, OR) were incubated in blocking solution for 45 min at room temperature. Coverslips were mounted in Prolong Gold Antifade mounting media (Molecular Probes).
Thin-section immunogold labeling and electron microscopy. HEK293 cells, transfected or not transfected with GFP-CaMKII, were treated with ionomycin/nigericin (±Ca2+) as described above and were then fixed immediately in 4% paraformaldehyde. The coverslips were then immersed in 30% sucrose, freeze-thawed over liquid nitrogen, rinsed in phosphate buffer (PB) and incubated with CBα2 (1:10,000). This was followed by washes in PBS and incubation with goat anti-mouse 1 nm gold-conjugated antibody (1:200; Aurion, Wageningen, The Netherlands). Both antibody incubations were for 8 h at 4°C, which was followed by silver intensification (SE-EM; Aurion, Conches, France). Cells were then treated with 1% osmium-tetroxide in 0.1 m PB for 30 min, dehydrated in ethanol and propylene-oxide, and embedded in Durcupan ACM (Fluka, Buchs, Switzerland). During dehydration, the cells were stained with 1% uranyl acetate (Sigma-Aldrich, Deisenhofen, Germany) in 70% ethanol for 25 min. Selected immunoreactive profiles were reembedded for ultrathin (60 nm) sectioning using a Leica (Nussloch, Germany) Ultracut S. Electron micrographs were obtained using a Philips Tecnai 12 microscope equipped with MegaView II digital camera.
Rat hippocampal cultures. Rat hippocampi were dissected out of postnatal day 1 (P1) to P3 rats, and cells were dissociated enzymatically and mechanically (trituration through Pasteur pipette). The Papain solution (12 U/ml; Worthington, Freehold, NJ) contained Ca2+/Mg2+-free HBSS, 0.42 mg/ml cysteine (Sigma), 250U/ml DNase 1 (type IV; Sigma), 25 mm NaHCO3, penicillin (50 U/ml)/streptomycin (50 μg/ml), 1 mm sodium pyruvate, and 1 mg/ml glucose (Invitrogen). After dissociation, the cells were washed and centrifuged in Neurobasal medium containing BSA (2 mg/ml and 20 mg/ml, respectively), Pen/Strep, glucose, pyruvate, and DNase1 (as above) and then plated on poly-d-lysine-coated Aclar coverslips at high density (∼2000 cells/mm2). Growth media (1 ml/well) consisted of Neurobasal and B27, supplemented with penicillin/streptomycin (50 U/ml; 50 μg/ml), and 0.5 mm l-glutamax (Invitrogen). Cytosine β-d-arabinofuranoside (5 μm; Sigma) was added 24 h after plating to reduce the number of non-neuronal cells. After 4 d in culture and twice each week thereon, half of the growth medium was replaced with medium without Ara-C.
Transfection, stimulation, and immunocytochemistry of neurons. Neurons [14-18 d in vitro (DIV)] were transfected with GFP-CaMKII WT and mutants as described above, except that the DNA and Lipofectamine 2000 reagents were applied for 5 h only and then the coverslips were transferred to fresh growth media supplemented with 100 μm AP-5 (Calbiochem, La Jolla, CA). The following day, the cells were washed in HBSS (plus 10 mm HEPES, pH 7.4, 1.2 mm CaCl2, 1 mm MgCl2) and then stimulated with the same HBSS solution containing glutamate/glycine (100 μm/10 μm) for 1 or 5 min. Neurons were immediately fixed in 4% paraformaldehyde (0.1 m PB, pH 7.4, 4% sucrose) for 10 min, washed in PBS three times, and incubated for 30 min in blocking solution with 0.05% Triton X-100. Cells were then incubated overnight at 4°C with mouse monoclonal anti-PSD95 antibody (1:250 Ab IC9; Affinity BioReagents, Golden, CO) in blocking solution. Secondary antibody (goat anti-mouse Alexa 546; 1:1000) application and coverslip mounting were done as described above. For the assessment of postsynaptic translocation of GFP-CaMKII in neurons, independent cultures were fixed before and immediately after a 1 or 5 min glutamate/glycine treatment and then immunostained for PSD95.
Confocal imaging and quantification of GFP-CaMKII clustering. To quantify the degree of self-association of different CaMKII mutants, HEK293 cells were fixed immediately after the Ca2+/pH drop protocol as described above. Twelve-bit images (1024 × 1024) from randomly selected cells (blind of the condition) were acquired on a Zeiss (Thornwood, NY) LSM510 META-Axioskop FS2 Plus confocal system, using a 63× oil immersion objective (1.4 NA), scanning a 0.5-μm-thick optical slice through the center of each cell. GFP was excited with an Argon laser line at 488 nm and detected through a long-pass filter (LP505 nm). The number of clusters per optical slice was computed by a morphometric analysis using Meta-Morph. A cluster was detected as follows: (1) a contiguous group of pixels corresponding to an area ranging from 0.1 to 1.0 μm2 that (2) were at least twofold brighter than the average fluorescence of all of the pixels in the cell, and (3) that had a shape factor (4πA/P2; A, area; P, perimeter) >0.5. The clustering factor was expressed as the total brightness of all of the detected clusters divided by the total fluorescence of the cell optical slice.
For the Thr286 phospho-CaMKII labeling, GFP and Alexa 594 were excited with a 488 and a 543 nm laser line, respectively, and the emitted signal was filtered through a P505-530 nm and a LP560 filter, respectively.
To quantify the degree of GFP-CaMKII translocation in neurons, we used the following two approaches. First, we scanned randomly selected dendrites (z-stack of three 0.5-μm-thick optical slices) or cell bodies (one optical slice through the center) of fluorescent neurons (blind of the condition). For postsynaptic translocation, regions of interests (ROIs) were drawn on synaptic (PSD95 positive) and adjacent extrasynaptic regions (see Fig. 9_A_). After applying a threshold on the images of selected dendrites and subtracting background, we scored the average pixel intensities of [(synaptic ROI) - (extrasynaptic ROI)]/total dendrite intensity. For extrasynaptic translocation, we used the same morphometric analysis, as described for the HEK293 cells, on one optical slice (0.7 μm) from the center of the cell body. The number of pixels in the detected clusters corresponded to a range of 0.04-0.32 μm2. Second, we sampled by visual observation under the microscope a large number of transfected neurons (n > 100) and applied a semiquantitative score of GFP-CaMKII translocation (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). Both methods of quantification yielded similar conclusions.
Figure 9.
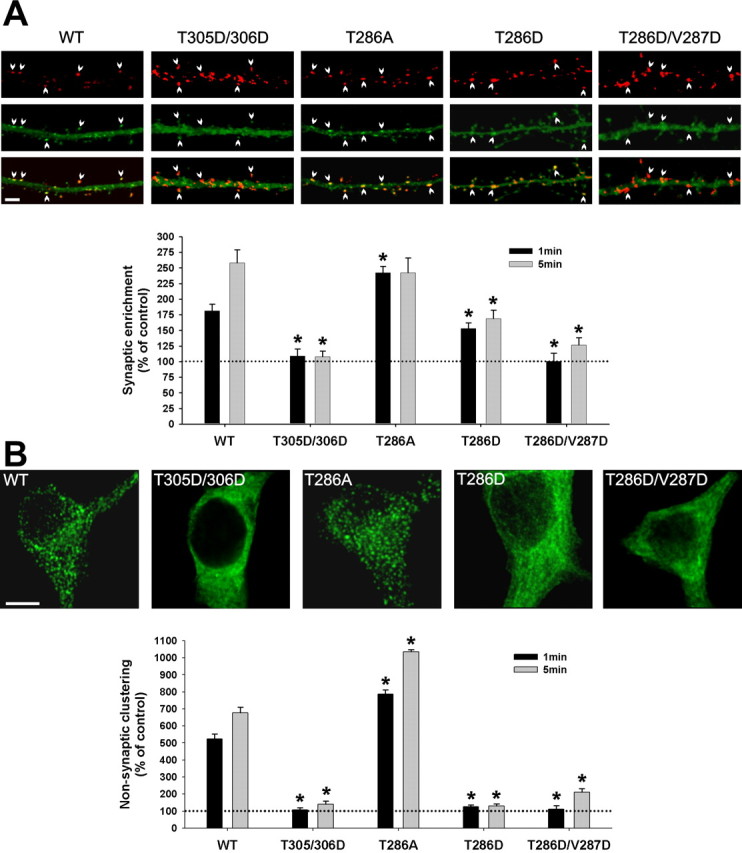
Role of CaMKII self-association in its activity-dependent translocation in hippocampal neurons. Hippocampal neurons (14 DIV) transfected with WT or mutated GFP-α-CaMKII were stimulated with glutamate/glycine (100 μm/10 μm) for 1 or 5 min and immediately fixed. A, Postsynaptic translocation of GFP-WT-CaMKII and mutants. The top panels show representative confocal images of dendrites from neurons expressing different GFP-CaMKII constructs (green; middle), which were treated for 1 min and immunostained for PSD95 (red; top). Arrowheads point to synapses. Scale bar, 3 μm. The histogram shows mean (±SEM; n = 40 synapses per construct) synaptic fluorescence intensities over the total intensity of the corresponding dendrite, relative to nonstimulated control (see Materials and Methods). B, Nonsynaptic translocation of GFP-WT-CaMKII and mutants. The top panels show confocal optical slices (0.7 μm) through the middle of neuronal cell bodies of stimulated (5 min) neurons. Scale bar, 5 μm. The histogram shows the mean (±SEM; n = 10 neurons per construct) degree of GFP-CaMKII clustering in stimulated neurons relative to nonstimulated controls (see Materials and Methods). Asterisks in A and B indicate a statistical difference (p > 0.001; one-way ANOVA followed by a Dunnett's test; p > 0.001) compared with WT for each treatment (1 or 5 min). To sample from a larger set of neurons (n > 100), we also designed a semiquantitative analysis, which yielded similar conclusions (see supplemental Fig. 2, available at www.jneurosci.org as supplemental material).
Statistics. A one-way ANOVA was conducted to assess overall group differences (F test; p < 0.001), with a subsequent Dunnett's test to identify specific pairwise differences (p < 0.001) between the means. Analyses were conducted using SPSS version 10.1.3 (SPSS, Chicago, IL). Additional statistical analyses were performed and described in supplemental Figure 2 (available at www.jneurosci.org as supplemental material).
Results
Glutamate receptor stimulation induces CaMKII translocation to synaptic and nonsynaptic sites
Endogenous CaMKII or transfected GFP-CaMKII were shown to translocate to synaptic sites in cultured hippocampal neurons stimulated by glutamate/glycine (Shen and Meyer, 1999; Shen et al., 2000; Dosemeci et al., 2001; Gleason et al., 2003). We confirmed this observation with GFP-CaMKII in hippocampal neurons but observed that the enzyme also translocated to nonsynaptic sites (Fig. 1). In unstimulated neurons, GFP-CaMKII was distributed throughout the soma and neurites (Fig. 1_A_) and exhibited a largely diffuse pattern of fluorescence. GFP-CaMKII did not significantly colocalize with PSD95, a marker of the postsynaptic side of synapses in dendrites and the soma (Fig. 1_A_, middle, right). Stimulation with glutamate/glycine led to a different pattern of fluorescence (Fig. 1_B_). Discrete puncta of GFP-CaMKII fluorescence appeared at synaptic (PSD95-positive) sites (Fig. 1_B_, middle), suggesting that the enzyme had been recruited to synapses (Shen and Meyer, 1999; Shen et al., 2000). However, in these same neurons, we also observed discrete puncta of GFP-CaMKII in the soma, which did not overlap with PSD95-immunoreactive sites (Fig. 1_B_, right). The right panels in Figure 1 represent a 1-μm-thick optical slice through the center of the cell body, indicating that the somatic puncta were mostly intracellular. The appearance of these nonsynaptic GFP-CaMKII puncta in neurons has not been described previously. We hypothesized that the rapid relocation of GFP-CaMKII into synaptic and nonsynaptic intracellular clusters, after glutamate/glycine stimulation, involves a CaMKII self-association process (Hudmon et al., 1996, 2001). To investigate whether the formation of GFP-CaMKII clusters in cells can be explained by the self-association of GFP-CaMKII holoenzymes, we first used transfected HEK293 cells, which are essentially devoid of synaptic proteins.
Figure 1.

Activity-dependent translocation of GFP-CaMKII to synaptic and nonsynaptic sites in neurons. Hippocampal neurons (18 DIV) transfected with GFP-CaMKII (green) were stimulated 24 h after transfection with glutamate/glycine (100 μm/10 μm) for 5 min. Neurons were immediately fixed, immunostained for PSD95 (red), and imaged with confocal microscopy. A, GFP-CaMKII fluorescence is primarily diffuse in the soma (right) and dendrites (middle) of nonstimulated neurons, with occasional colocalization with PSD95 (synaptic sites) immunostaining. B, Glutamate/glycine stimulation caused a translocation of GFP-CaMKII into fluorescent puncta in both the soma and dendrites. GFP-CaMKII fluorescence in the distal dendrites (middle) appears localized primarily to PSD95-positive sites (synapses), whereas CaMKII fluorescence in the soma and initial segments of the dendrite do not (right). Images of the entire neuron (left) and cropped dendrites (middle) represent maximum projections from z-stacks, whereas those of cell bodies (right) represent a single optical slice (1.0 μm) from their center. Scale bars: (in B) left, 25 μm; (in A) middle, 3 μm; (in A) right, 5 μm.
Ca2+-dependent translocation of GFP-CaMKII in HEK293 cells
Treatment of GFP-CaMKII transfected cells with ionomycin in the presence of external Ca2+ resulted in the formation of discrete GFP-CaMKII fluorescence puncta distributed throughout the cytosol (Fig. 2_A_). This effect is reminiscent of the GFP-CaMKII redistribution observed in glutamate/glycine-stimulated neurons (Fig. 1). Before stimulation, GFP-CaMKII exhibited a relatively uniform non-nuclear distribution in these cells as reported previously (Bayer et al., 2001) (Fig. 2_A_). Hence, Ca2+ elevation can induce GFP-CaMKII to form fluorescent puncta in the absence of other synaptic proteins. Previous studies showed that in HEK293 cells transfected with NMDA receptor subunit NR2B, ionomycin treatment caused the translocation of the fluorescence into puncta (Bayer et al., 2001). For comparison, we repeated this experiment and observed the appearance of elongated puncta (Fig. 2_B_, arrows), which formed an annular-like pattern of fluorescence that was morphologically distinct from that seen in HEK293 cells transfected with the GFP-CaMKII alone (Fig. 2_B_). This is consistent with GFP-CaMKII translocating to the perinuclear endoplasmic reticulum, where overexpressed NR2B is concentrated in the absence of NR1 (Bayer et al., 2001; Fukaya et al., 2003).
Figure 2.

Ionomycin-induced GFP-CaMKII translocation in HEK293 cells. GFP-CaMKII was transiently transfected into HEK293 cells in the absence (A) or presence (B) of the NR2B subunit of the NMDA receptor. A, During ionomycin stimulation in HBSS (25 mm HEPES, pH 7.4, 2 mm CaCl2), the initially diffuse fluorescence became progressively punctate throughout the cytosol during 5 min of stimulation. Arrowheads in the right panel point to the spherical puncta. B, In contrast, in cells cotransfected with NR2B, the GFP-CaMKII fluorescence translocated to perinuclear endoplasmic reticulum regions, in which overexpressed NR2B subunits accumulate. Arrowheads in the right panel point to the elongated (rather than spherical) puncta. Scale bars, (in A) 6 μm. These images are representative of 8-12 experiments. To fully appreciate the difference in the two kinds of puncta observed, it is useful to look at the complete sequences of images available as movies (supplemental Fig. 2-movie1.mov and Fig. 2-movie2.mov, available at www.jneurosci.org as supplemental material).
We also noted that the CaMKII puncta formed in the presence and absence of NR2B differed in their time course of appearance. In cotransfected (GFP-CaMKII plus NR2B) HEK293 cells, puncta appeared within 2-3 min of ionomycin exposure. In the absence of NR2B, GFP-CaMKII puncta first appeared at ∼5 min of ionomycin stimulation. Also, once formed, the NR2B-bound GFP-CaMKII clusters appeared static, whereas those formed in the absence of NR2B were moving randomly within the cytoplasm. These features of CaMKII translocation can be best compared by looking at the complete time-lapse sequences provided as movies (supplemental Fig. 2-movie1.mov and Fig. 2-movie2.mov, available at www.jneurosci.org as supplemental material). The random-like movement of CaMKII puncta formed in the absence of the NR2B subunit suggests that they were not anchored to any stable intracellular structure. The more static appearance of the puncta formed in the presence of NR2B is consistent with the binding of the enzyme to NR2B embedded into cellular membranes.
pH influences the rate of CaMKII cluster formation in HEK293 cells
During the ionomycin treatment and preceding the formation of puncta in the HEK293 cells transfected with GFP-CaMKII, we observed that the fluorescence intensity decreased noticeably (20-30%). Because GFP fluorescence is sensitive to pH, it is likely that the reduction in GFP fluorescence was attributable to an ionomycin/Ca2+-induced acidification of the cell cytoplasm as reported previously (Patterson et al., 1997; Llopis et al., 1998). A pH dependence to the formation of GFP-CaMKII puncta would be congruent with a self-association mechanism underlying this translocation (Hudmon et al., 1996, 2001).
To address the regulatory role of pH in the formation of these CaMKII fluorescent puncta, we used the monovalent ionophore nigericin to control the intracellular pH (pHi) (Thomas et al., 1979). We compared the extent of ionomycin-induced clustering of GFP-CaMKII at pH 7.5 versus pH 6.5. The lower pH value was chosen, in part, because brief NMDA receptor stimulation was shown to cause pHi to drop to 6.5 in cultured neurons (Hartley and Dubinsky, 1993; Irwin et al., 1994), and a pH level of 6.5 enhances CaMKII self-association in vitro (Hudmon et al., 1996, 2001). At pHi 7.5, the fluorescent signal remained diffuse and homogenous after ionomycin/Ca2+ treatment for ≥5 min (Fig. 3 A). The pH clamping by nigericin eliminated the drop in the cell fluorescence intensity, which was observed above, after ionomycin stimulation. Reequilibrating these same cells in low Ca2+ and then lowering the pH to 6.5 also had no effect on GFP-CaMKII distribution (Fig. 3, compare left panels in A and B) but did cause a reduction in the cell fluorescence intensity (Fig. 3_B_, compare left and right panels). In contrast, lowering the pH to 6.5 in the presence of ionomycin/Ca2+ was rapidly (within 20 s) associated with the appearance of punctate GFP-CaMKII fluorescence throughout the cytosol (Fig. 3_C_). A time-lapse movie (supplemental Fig. 3-movie3.mov, available at www.jneurosci.org as supplemental material) illustrates the evolution of CaMKII self-association after Ca2+ elevation at pH 6.5. The formation of CaMKII clusters is detectable within the first 20-40 s of the pH drop. Cells transfected with GFP alone never exhibited puncta following the same pH and Ca2+ treatment (data not shown).
Figure 3.
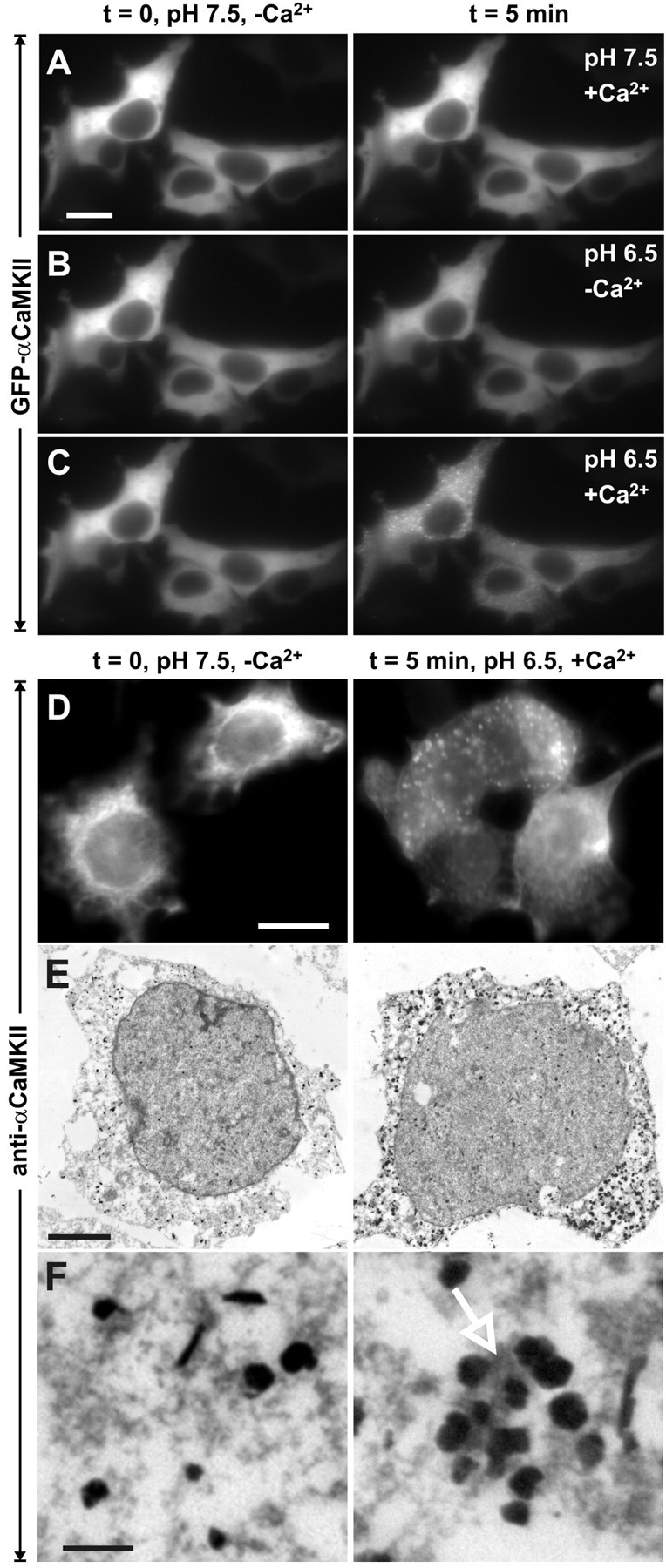
Ionomycin-induced GFP-CaMKII translocation in HEK293 cells is regulated by pH. GFP-CaMKII fluorescence was imaged following sequential ionomycin treatments in a nigericin-based extracellular buffer designed to clamp pHi at 7.5 or 6.5 as indicated. Ionomycin/Ca2+ treatment induced translocation of GFP-CaMKII at pH 6.5, as in C, but ionomycin treatment at pH 7.5 in the presence of Ca2+ (A) or at pH 6.5 in the absence of Ca2+ (B) did not induce translocation. In B and C (left), the cells were reequilibrated in nigericin/low-Ca2+/pH 7.5 buffer for 5 min before the solution changes shown in the right panels. D, Non-GFP-tagged CaMKII also translocates into clusters as revealed by immunocytochemistry for α CaMKII (right). Scale bars, (in A, D) 6 μm. E, F, Electron microscopic images of GFP-CaMKII clusters detected with anti-αCaMKII and immunogold labeling. In stimulated cells (right), groups of gold particles surround electron-dense material (arrow), whereas the gold particles are dispersed in nonstimulated cells (left) and are not associated with electron-dense material. Scale bars: E, 3 μm; F, 250 nm.
To assess whether the GFP tag on CaMKII contributed to the formation of these puncta, we transfected HEK293 cells with non-GFP-tagged αCaMKII and exposed them to the same low pH/high Ca2+ stimulation protocol. We immediately fixed the cells and immunostained them with a monoclonal antibody (CBα2) against CaMKII. Untagged CaMKII also formed discrete puncta indistinguishable from the GFP-CaMKII clusters after stimulation (Fig. 3_D_).
CaMKII clusters at the ultrastructural level
Although nonsynaptic CaMKII clusters had not been reported in stimulated neurons with fluorescent imaging, they have been observed with EM (Dosemeci et al., 2000; Tao-Cheng et al., 2001, 2002, 2005). We examined whether the GFP-CaMKII clusters formed in HEK293 cells could be detected at the ultrastructural level with a monoclonal antibody specific for αCaMKII and immunogold labeling. Figure 3, E (low magnification) and F (high magnification), shows electron micrographs of examples of electron-dense material heavily labeled with immunogold particles in HEK293 cells exposed to the lowered pH/high-Ca2+ stimulation (right panels). Little or no immunogold labeling was evident in nontransfected cells or when the primary antibody was omitted (data not shown). In nonstimulated cells or in cells stimulated without Ca2+, gold particles were more evenly distributed and generally not associated with dense material (Fig. 3_E,F_, left panels). This electron-dense immunolabeling appears analogous to the clusters of CaMKII that Dosemeci and colleagues have reported at the ultrastructural level in neurons after ischemic or brief NMDA receptor stimulation (Dosemeci et al., 2000; Tao-Cheng et al., 2001, 2002, 2005).
Reversibility of CaMKII cluster formation
To test whether the Ca2+ and pH-dependent translocation of CaMKII can be reversed, we induced CaMKII cluster formation using the standard Ca2+/ionomycin-pH drop protocol (Fig. 4, sequence a to c) (see Materials and Methods) and then removed extracellular Ca2+ with the Ca2+ chelator BAPTA and/or raised the pHi back to 7.5. Within ∼2 min of BAPTA perfusion, still at pHi 6.5, the punctate fluorescence disappeared (Fig. 4_A_) (supplemental Fig. 4-movie4.mov, available at www.jneurosci.org as supplemental material), indicating that CaMKII clustering can be reversed by removing extracellular Ca2+ and thus is not caused by an irreversible aggregation of denatured kinase. Raising the pHi to 7.5 in absence of extracellular Ca2+ led to a similar reversal of CaMKII clusters (data not shown). However, raising the pHi to 7.5 in the continuous presence of Ca2+ did not reverse the clusters by 30 min (Fig. 4_B_), indicating that although their formation is initially pH regulated, elevated Ca2+ is both necessary and sufficient to maintain CaMKII in the clustered state. We next examined whether the kinase would fully dissociate under Ca2+ concentration that reflected basal intracellular levels by washing the cells with low (∼150 nm) Ca2+-containing solution. Under these conditions, we observed a partial dissociation by 30 min; at that time point, ∼50% of the clusters formed in each cell (n = 11) were still present. These results indicate that the dissociation rate of GFP-CaMKII puncta is sensitive to Ca2+ levels and suggests that this clustered state can be relatively long-lasting in basal Ca2+ levels.
Figure 4.

Reversibility of GFP-CaMKII translocation in HEK293 cells. GFP-CaMKII translocation was induced using our standard Ca2+/ionomycin-pH drop protocol: a, prewash with nigericin-based extracellular solution, pH 7.5, without ionomycin (Time-0 image); b, switch to the same solution with ionomycin/CaCl2 for 2 min; c, switch to the same solution but at pH 6.5. A, To assess reversibility, ionomycin/Ca2+ was replaced with 5 mm BAPTA after punctate fluorescence peaked (240 s). Under these conditions, clusters disappeared within 2-5 min (20 experiments). B, Reversing the pH back to 7.5, after CaMKII translocation in the presence of Ca2+/ionomycin, produced little change in the punctate appearance of GFP-CaMKII (10 experiments), indicating that the reversibility is primarily dependent on the removal of Ca2+. Note that the fluorescence intensity rose slightly between Bc and Bd, likely a result of the pH sensitivity of GFP emission. Scale bar, (in Ba) 8 μm. a-e on the timeline correspond to a-e on the images. The complete sequence of images from A is available as a supplemental movie (supplemental Fig. 4-movie4.mov, available at www.jneurosci.org as supplemental material).
The reversibility of the clustering with the high/zero (BAPTA) Ca2+-shift protocol afforded the opportunity to assess whether CaMKII puncta form in the same subcellular location during each high-Ca2+ cycle. By repeating cycles of zero Ca2+/pH 7.5 and high-Ca2+/pH 6.5, we observed that clusters did not repeatedly form in the same location (supplemental Fig. 1, available at www.jneurosci.org as supplemental material), suggesting that CaMKII clusters are produced at random sites throughout the cytosol, rather than at spatially fixed subcellular sites. Thus, this activity-dependent clustering of GFP-CaMKII in HEK293 cells requires Ca2+, is regulated by pH, and does not appear to be associated with any subcellular structure, consistent with the self-association of CaMKII observed in vitro (Hudmon et al., 1996, 2001).
Structural features of CaMKII required for self-association
CaMKII is a multimer, with each holoenzyme composed of 12 catalytic/autoregulatory domains and potential sites of interaction with targeting proteins. If the clusters that were forming in HEK293 cells and neurons were attributable to CaMKII holoenzymes interacting with one another through multivalent interactions, then producing monomeric forms of the kinase should prevent cluster formation. As illustrated in a linear domain map of a single subunit of CaMKII, the catalytic domain is contiguous with an autoregulatory domain, composed of a pseudosubstrate-type sequence and a CaM-binding domain (Fig. 5_A_). In addition, each subunit also possesses a C-terminal association domain responsible for holoenzyme assembly. We fused GFP to the N terminus of a truncated catalytic domain composed of amino acids 1-326, lacking the association domain necessary for holoenzyme assembly. This GFP-1-326-CaMKII was diffusely distributed throughout the cytoplasm and the nucleus of transfected HEK293 cells (Fig. 5_B_, left) as described previously (Shen and Meyer, 1998). However, the monomeric GFP-CaMKII was resistant to the formation of puncta with the low-pH/high-Ca2+ treatment (Figs. 5_B_, right, 7_B_). These data indicate that the multimeric structure of CaMKII is necessary for the Ca2+-dependent cluster formation of GFP-CaMKII in HEK293 cells.
Figure 5.

Self-association requires both catalytic and association domains. A, Schematic diagram of αCaMKII (1-478), with the catalytic (1-326) and association domains (315-478) illustrated. Cells were imaged using the standard Ca2+/ionomycin-pH drop protocol (Fig. 4_a-c_), with example frames shown for 0 min (left) and 5 min (right). B, Cells transfected with GFP-1-326-CaMKII (catalytic/autoregulatory domain). C, Cells transfected with GFP-315-478-CaMKII (association domain). Note the absence of fluorescent clusters. Scale bar, (in B) 10 μm.
Figure 7.
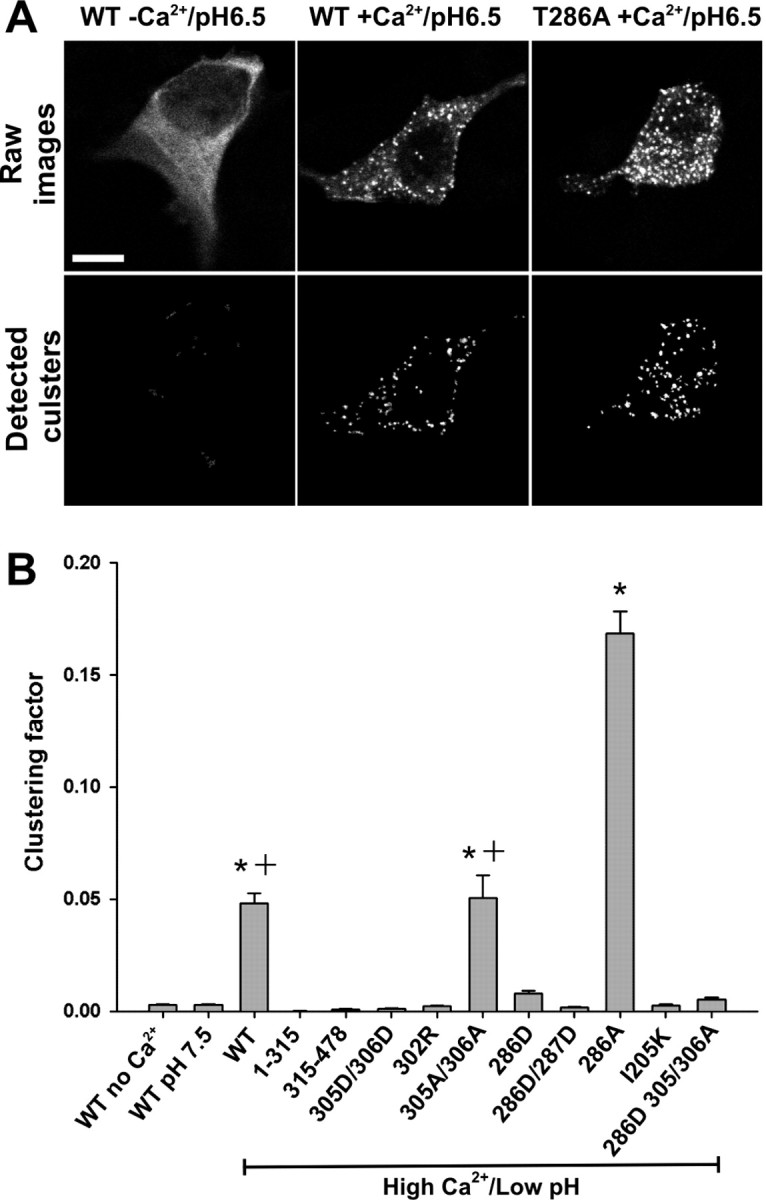
Quantification of self-association capacity by CaMKII mutants. A, Examples of confocal images (0.5-μm-thick optical slice; top) from HEK293 cells fixed after stimulation as described in Figure 4. Scale bar, 10 μm. The bottom panels show the clusters detected by our morphometric analysis (see Materials and Methods). B, Quantification of the clustering from A (n = 40 for each construct). Clustering factor, Total fluorescence inside the clusters/total fluorescence inside the cell. The asterisk indicates one-way ANOVA (F test; p < 0.001), followed by a Dunnett's test (p < 0.001) compared with control (stimulation without Ca2+). The plus sign indicates a significant difference (p < 0.001) from every condition.
We also tested whether the oligomeric association domain lacking the catalytic domain was capable of forming clusters. Previous studies indicated that the construct containing amino acids 315-478 assembled into a dodecameric core of CaMKII (Kolodziej et al., 2000). The GFP-fusion protein of CaMKII that contains the C-terminal association domain (amino acids 315-478) diffused throughout the cytosol of transfected HEK293 cells but was excluded from the nucleus (Fig. 5_C_, left), consistent with the multimeric assembly of this construct (Shen and Meyer, 1998). As observed for the monomeric catalytic domain (Fig. 5_B_), the multimer of association domains was also resistant to cluster formation when HEK293 cells were stimulated with low-pH/high-Ca2+ (Figs. 5_C_, right, 7_B_). Together, these data suggest that the binding site for the self-association reaction likely resides in the catalytic/autoregulatory portion of CaMKII and that the multivalent architecture of the holoenzyme is essential for the Ca2+-dependent cluster formation.
CaM binding is necessary for CaMKII self-association
Self-association in vitro was demonstrated previously to require Ca2+/CaM (Hudmon et al., 2001), and the GFP-CaMKII cluster formation revealed here (Figs. 3, 4) shows a strong dependence on elevated Ca2+. We thus tested whether CaM binding to the enzyme was required in GFP-CaMKII cluster formation. A single-point mutation, Ala302Arg, within the CaM-binding domain was shown previously to both ablate CaM binding (Mukherji and Soderling, 1994) and disrupt the activity-dependent translocation of CaMKII to synaptic sites in neurons (Shen and Meyer, 1999). This mutant form of CaMKII did not form clusters in HEK293 cells subjected to the low-pH/high-Ca2+ treatment (Figs. 6_A_, 7_B_). We observed similar results with another form of CaMKII that possesses a nonfunctional CaM-binding domain (Thr305Asp/Thr306Asp) (Elgersma et al., 2002) (Fig. 7_B_). This was in contrast to GFP-CaMKII harboring the Thr305Ala/Thr306Ala mutations, a construct that retains the ability to bind CaM (Hanson and Schulman, 1992), which formed clusters to the same extent as for WT-CaMKII (Fig. 7_B_). These results demonstrate that a functional CaM-binding domain is an essential component to Ca2+-induced clusters of CaMKII holoenzymes and also support the notion that this process reflects CaMKII self-association, a Ca2+/CaM-dependent reaction (Hudmon et al., 2001). One potential mechanism that can be inferred from these results is that the regions important for this reaction are not exposed or available on the inactive kinase. Because Ca2+/CaM binding disrupts the intrasubunit interaction between the catalytic and the autoinhibitory regions of the enzyme, thereby enabling kinase activity, we hypothesized that these same regions become available to interact with other exposed binding sites on CaM-bound subunits of neighboring holoenzymes to generate the self-associated state.
Figure 6.

Self-association requires CaM binding, involves interactions between the autoregulatory and catalytic domains, and is regulated by Thr286 autophosphorylation. Cells were stimulated as described in Figure 4_a-c_, with example frames shown for 0 min (top) and 5 min (bottom) for Ala302Arg (A), Ile205Lys (B), Thr286Asp (C), Thr286Ala (D), and Thr286Val287/Asp286Asp287 (E). The arrow in C indicates a rare example of an HEK293 cell exhibiting clusters with Thr286Asp mutant. Scale bar, (in A) 10 μm
Determinants within the catalytic domain important for CaMKII self-association
We first tested whether the catalytic domain contains specific binding sites necessary for CaMKII holoenzymes to interact with one another after cell stimulation. We assayed the self-association potential of a CaMKII construct containing a point mutation in the anchoring protein-binding site in the catalytic domain (Ile205Lys), shown previously to prevent activity-dependent GFP-CaMKII binding to NR2B (Bayer et al., 2001). This GFP-Ile205Lys-CaMKII mutant did not undergo self-association in HEK293 cells subjected to the low-pH/high-Ca2+ treatment (Figs. 6_B_, 7_B_). These data lend additional support that the catalytic domain of CaMKII constitutes one of the sites of interaction that lead to self-association.
Role of Thr286 autophosphorylation in CaMKII self-association
A potential binding interaction that could underlie CaMKII self-association would involve the autoinhibitory region surrounding Thr286 on one subunit with the catalytic regions of another subunit from a separate holoenzyme. In the inactive state of the kinase, the catalytic domain, including the anchoring protein-binding site around Ile205, interacts with the autoinhibitory region surrounding the Thr286 residue of the same subunit. Ca2+/CaM binds to the autoinhibitory domain and disrupts this interaction. Autophosphorylation of Thr286 in the autoinhibitory domain also disrupts this interaction by preventing reassociation of this domain with the catalytic domain (for review, see Hudmon and Schulman, 2002). The catalytic/autoinhibitory interaction hypothesis can be directly tested by mutating the Thr286 to mimic autophosphorylation (Thr286Asp), as we would predict that it would prevent self-association, by inhibiting the autoinhibitory and catalytic regions from interacting (Waldmann et al., 1990). Figures 6_C_ and 7_B_ show that mutating Thr286 to Asp strongly reduced self-association in our HEK293 cell translocation assay. In contrast, the GFP-Thr286Ala-CaMKII mutant exhibited more self-association than WT-CaMKII (Figs. 6_D_, 7), suggesting that activation of WT-CaMKII was associated with sufficient Thr286 autophosphorylation to reduce the extent of self-association.
A possible mechanism underlying the reduction of self-association by Thr286Asp mutant is the autonomous phosphorylation of the neighboring Thr305/306, which would inhibit CaM binding, as was mimicked by the Thr305/306Asp mutant (Fig. 7_B_). To test this hypothesis, we assessed self-association of triple mutant Thr286Asp-Thr305/306Ala. This mutant did not form more clusters than the Thr286Asp mutant (Fig. 7_B_), suggesting that Thr305/306 phosphorylation was not responsible for the reduced self-association by GFP-Thr286Asp-CaMKII.
These results favor the hypothesis that the negative charge of phosphorylated Thr286 (or Thr286Asp) has reduced interactions with the catalytic domains of other subunits involved in self-association, in a similar way that this phosphorylation allows for autonomous activity of the kinase. To further support this hypothesis, we made the region surrounding Thr286 more negatively charged by adding another mutation (Thr286Asp/Val287Asp), which exhibited even more autonomous activity in an in vitro kinase reaction (data not shown), consistent with a stronger repulsion of the autoinhibitory domain. This mutant was even more resistant to cluster formation following the low-pH/high-Ca2+ treatment (Figs. 6_E_, 7_B_), strengthening the notion that Thr286 autophosphorylation of CaMKII is a negative regulator of self-association, presumably via an analogous mechanism to the one underlying the generation of autonomous activity.
Thr286 autophosphorylation of self-associated CaMKII
The finding that Thr286Asp mutant exhibits poor self-association would seem to argue that autophosphorylation at Thr286 is incompatible with CaMKII self-association. However, our observation that GFP-Thr286Ala-CaMKII exhibited more self-association compared with GFP-WT-CaMKII (Fig. 7_B_) is consistent with the notion that the WT kinase did undergo at least partial autophosphorylation in stimulated HEK293 cells. To test this more directly, we immunostained GFP-CaMKII-transfected HEK293 cells with a phospho-Thr286-CaMKII-specific antibody. Figure 8_A_ shows that clusters of GFP-WT-CaMKII are immunoreactive to this antibody, whereas GFP-WT-CaMKII in unstimulated cells (Fig. 8_B_) or GFP-Thr286Ala-CaMKII from stimulated cells are not (Fig. 8_C_). These results suggest that the multimeric nature of CaMKII allows for both self-association and Thr286 autophosphorylation reactions to follow Ca2+/CaM activation.
Figure 8.
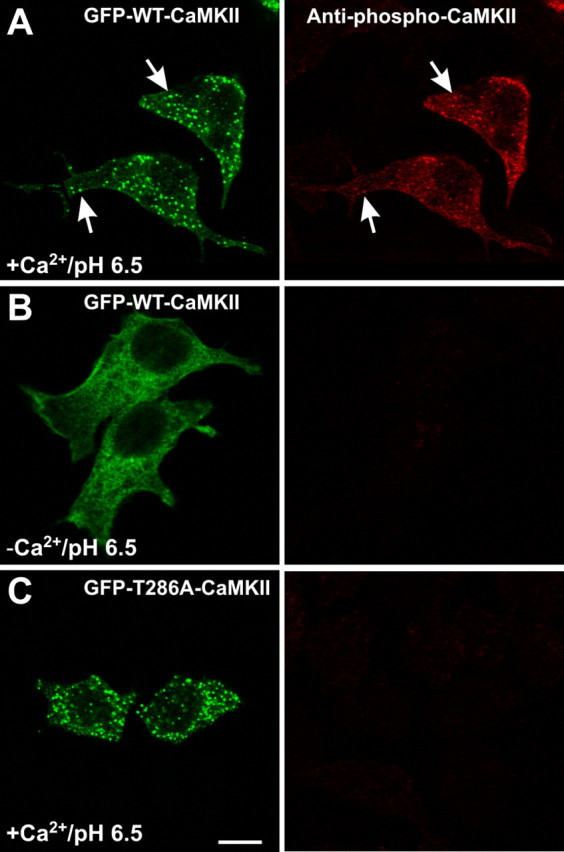
Autophosphorylation of CaMKII during self-association. HEK293 cells, stimulated as in Figure 4_a-c_, were fixed and immunostained for phospho-Thr286-CaMKII. A, GFP-WT-CaMKII clusters are autophosphorylated at Thr286 (arrows). B, In the absence of Ca2+, GFP-WT-CaMKII is not autophosphorylated at Thr286. C, GFP-Thr286A-CaMKII clusters are not immunoreactive to the anti-phospho-Thr286 antibody, showing its specificity. Scale bar, (in C) 10 μm.
CaMKII translocation in neurons
CaMKII self-association in HEK293 cells requires the oligomeric structure and Ca2+/CaM binding and is regulated by Thr286 autophosphorylation, as revealed by using GFP-CaMKII mutants. If CaMKII self-association is contributing to either postsynaptic or nonsynaptic activity-dependent translocation in hippocampal neurons, we would expect that the rules governing self-association in HEK293 cell should apply in neurons. These features of CaMKII self-association were tested by quantifying the degree of postsynaptic (Fig. 9_A_) and nonsynaptic translocation (Fig. 9_B_) of GFP-CaMKII mutants after glutamate/glycine stimulation in dissociated neurons. In nonstimulated neurons, all of the CaMKII constructs transfected produced a uniformly distributed fluorescence as seen in Figure 1_A_. Figure 9 shows that a mutant form of CaMKII that cannot bind CaM (Thr305Asp/306Asp) does not exhibit any detectable translocation anywhere inside the stimulated neurons, consistent with the requirement of Ca2+/CaM binding for CaMKII translocation in HEK293 cells (Figs. 6_A_, 7_B_) and as shown previously for CaMKII postsynaptic translocation in neurons (Shen et al., 1998). We also observed that the monomeric GFP-CaMKII (Fig. 5_B_) did not translocate after neuronal stimulation (data not shown). Mutants designed to mimic autophosphorylation (Thr286Asp and Thr286Asp/Val287Asp) exhibited, respectively, little to almost no detectable translocation, either inside the cell body or postsynaptically (Fig. 9) (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). These results suggest that CaMKII self-association contributes to its postsynaptic and nonsynaptic translocation in neurons. Although NMDA receptor stimulation causes CaMKII autophosphorylation postsynaptically, it is always submaximal (Molloy and Kennedy, 1991; Ocorr and Schulman, 1991; Fukunaga et al., 1993). Thus, as we have seen for self-association in HEK293 cells, submaximal autophosphorylation is compatible with CaMKII accumulation postsynaptically. We would then predict that the probability or speed of postsynaptic translocation should be affected by the fraction of subunits autophosphorylated. Indeed, we observed that the GFP-Thr286Ala-CaMKII mutant translocated postsynaptically slightly more rapidly than the WT enzyme (Fig. 9_A_) (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). Although GFP-Thr286Ala-CaMKII translocated postsynaptically to a higher degree by 1 min compared to WT-CaMKII, by 5 min stimulation, both forms accumulated to a similar level at synapses.
Discussion
By examining GFP-CaMKII dynamic translocation in HEK293 cells, we provide strong evidence that CaMKII can undergo a regulated and reversible form of self-association in the cytosol. The rules governing self-association of multimeric GFP-CaMKII in HEK293 cells are as follows: (1) correlated with the appearance of CaMKII immunoreactive structures detectable at the ultrastructural level, (2) directly and reversibly linked to increased [Ca2+]i, (3) regulated by pHi, (4) dependent on Ca2+/CaM binding, and (5) regulated by autophosphorylation of Thr286. Based on our living cell study and previous in vitro data (Hudmon et al., 1996, 2001), a working model for CaMKII self-association is illustrated in Figure 10. In the inactive state, the autoregulatory domain of CaMKII makes contacts with the catalytic domain to inhibit enzyme activity. The binding site for the autoregulatory domain is represented by two regions within the catalytic domain, the targeting site (denoted as T-Site), and the substrate-binding site (denoted as S-Site). The Ile205 residue within the catalytic domain is part of the targeting site, which was previously shown to interact with the NR2B-subunit (Bayer et al., 2001) and proposed to interact with the region around Thr286 of the autoregulatory domain (Yang and Schulman, 1999). We targeted the T-site for self-association, because our previous work had demonstrated that a peptide designed after that region of the autoregulatory domain blocked self-association (Hudmon et al., 2001). Ca2+/CaM binding exposes the catalytic domain as well as the targeting site, allowing self-association to occur through binding of this site to an autoregulatory domain on another subunit of a different holoenzyme. In addition, we show that self-association is regulated by Thr286 autophosphorylation and pHi.
Figure 10.

Working model for CaMKII self-association. Top, Left, Illustration of one catalytic domain, the location of key amino acids (yellow circles), and the intramolecular contacts formed between the autoregulatory and the catalytic cleft in an inactive subunit of a CaMKII holoenzyme. Top, Right, The first step in the self-association reaction, as well as enzyme activation, is Ca2+/CaM binding, which releases the autoregulatory domain to expose the catalytic domain, the substrate site, and presumably the targeting site. Middle, Left, Holoenzyme with a mix of inactive (white), Ca2+-CaM activated (green), and Thr286 autophosphorylated (red) subunits. Middle, Self-association involves binding between activated subunits from two separate holoenzymes, under the regulation by intracellular pH, which is hypothesized to affect displacement of the autoinhibitory domain from the catalytic pocket (middle, right). Bottom panels (circles 1-4) represent hypothesized interactions that activated subunits undergo, depending on the various possible combinations of Thr286 phosphorylation (red circles). 1, 2, Autophosphorylated Thr286 cannot interact with the targeting site, preventing association. 3, 4, Thr286 region of activated subunits (but not phosphorylated) can interact with the targeting site of another subunit, whether (3) or not (4) the latter is phosphorylated. Those potential interactions are exemplified in the cluster of CaMKII (see 1-4 in the middle).
Self-association in situ and in vitro (Hudmon et al., 1996, 2001) is enhanced at acidic pH, possibly because protons (H+) stabilize the binding of subunits between holoenzymes or expose a binding site or conformation that permits activated holoenzymes to self-associate. Previous data support the latter mechanism, because autoregulatory interactions with the catalytic domain have been shown to be altered via the protonation state of His282 (Smith et al., 1992). This site is adjacent to Thr286 and could function as a pH-sensitive switch to regulate the exposure of the targeting site or the presentation of the autoregulatory domain itself.
We used a pH shift protocol from 7.5 to 6.5 that produced robust self-association, allowing us to study the molecular mechanisms of this process. However, the pH dependence of CaMKII self-association in vitro is in fact graded between pH 6.5 and 7.5 (Hudmon et al., 1996, 2001). The pH sensitivity of self-association may have biological relevance, because alterations in the pHi of neurons have been associated with physiological and pathological signaling. Sustained elevation in [Ca2+]i, after cerebral ischemia is also associated with intracellular acidification (Silver and Erecinska, 1992) and the appearance of nonsynaptic clusters of CaMKII (Dosemeci et al., 2000; Tao-Cheng et al., 2001, 2002), most likely related to the nonsynaptic GFP-CaMKII clusters observed here. In addition, ischemic-like conditions also promote the accumulation of CaMKII to the PSD (Suzuki et al., 1994; Aronowski and Grotta, 1996; Dosemeci et al., 2001; Tao-Cheng et al., 2002). Changes in pHi with physiological activity in neurons are not as robust as with pathological perturbations (e.g., ischemia); however, intracellular acidification is associated with Ca2+ signaling in neurons (Chesler and Kaila, 1992; Hartley and Dubinsky, 1993; Irwin et al., 1994; Koch and Barish, 1994). These changes are well within the range of where self-association is observed (Hudmon et al., 1996, 2001), particularly within neuronal microdomains and synapses (Schwiening and Willoughby, 2002; Willoughby and Schwiening, 2002).
The regulation of self-association by Thr286 autophosphorylation may result from a mechanism analogous to the generation of autonomous activity. The negative charges associated with phosphorylated Thr286 are thought to prevent the autoregulatory domain from reacquiring stable intramolecular contacts with the catalytic domain after CaM dissociation, thus permitting catalytic activity in the absence of Ca2+/CaM (Hudmon and Schulman, 2002). In a similar model, we hypothesize that the Thr286 phosphorylated autoregulatory domain is not able to participate in self-association, because it cannot establish contacts with the catalytic domain of a subunit from another holoenzyme. However, as shown in Figure 10, the catalytic site of a Thr286-phosphorylated subunit could presumably still associate with the autoregulatory domain of an activated, nonphosphorylated subunit. If so, we might expect the latter interaction to be prolonged by autophosphorylation-dependent CaM trapping (Meyer et al., 1992). This hypothesis is supported by experiments in which conditions that reduce dephosphorylation of postsynaptically targeted (Shen et al., 2000) or intracellularly clustered (Tao-Cheng et al., 2005) CaMKII lead to a slower detranslocation of the enzyme. A submaximal Thr286 phosphorylation associated with self-association (Fig. 8) is consistent with several studies performed in tissue or dissociated cells, where autonomous activity, a readout of Thr286 autophosphorylation, was typically <25% (Molloy and Kennedy, 1991; Ocorr and Schulman, 1991; Fukunaga et al., 1993).
The rules that govern CaMKII self-association in HEK293 cells apply for the activity-dependent translocation in neurons shown here and previously (Rongo and Kaplan, 1999; Shen and Meyer, 1999; Shen et al., 2000; Dosemeci et al., 2001). Indeed, both are initiated by an increase in [Ca2+]i, require a functional CaM-binding domain and an oligomeric structure, are reduced by Thr286 mutation to Asp, and are reversible. Autophosphorylation at Thr286 does occur during self-association, which may prolong the lifetime of the kinase postsynaptically (Shen and Meyer, 1999). The small amount of GFP-Thr286Asp-CaMKII that accumulated postsynaptically (Fig. 9_A_) (Rongo and Kaplan, 1999) may reflect predominantly NMDA receptor binding (Strack and Colbran, 1998; Strack et al., 2000; Bayer et al., 2001; Leonard et al., 2002). Thus, during NMDA receptor stimulation, Ca2+-CaM-activated CaMKII may bind to PSD partners, such as the NMDA receptor, as well as other postsynaptic CaMKII holoenzymes, which could themselves be already bound to PSD partners.
The lifetime of CaMKII clusters postsynaptically, after synaptic stimulation, may be important for long-term synaptic plasticity. Under chelated levels of Ca2+, they disappeared within minutes (Shen and Meyer, 1999; Shen et al., 2000), as do the GFP-CaMKII clusters in HEK293 cells (Fig. 4). However, under homeostatic level of synaptic transmission, with occasional Ca2+ spikes at the synapse, we might expect that a significant fraction of CaMKII holoenzymes would remain self-associated postsynaptically. Indeed, we found that the dissociation rate of CaMKII clusters was linked to the intracellular Ca2+, and that in low Ca2+ (∼150 nm), it was considerably prolonged. Interestingly, after chemical LTP induction in hippocampal slices, Otmakhov et al. (2004) reported persistent postsynaptic localization of GFP-CaMKII.
What function could an activity-dependent polymerization of CaMKII serve at synaptic and nonsynaptic sites? The unique multimeric structure of CaMKII and the local concentrating effect of self-association could serve to organize or stabilize the interaction of CaMKII with other proteins in the PSD, regulate substrate phosphorylation by subcellular and/or steric restrictions, and affect the structure of the spine (Matsuzaki et al., 2004). Indeed, CaMKII phosphorylates or anchors to several proteins within the PSD that are involved in synaptic signaling (e.g., NMDA and AMPA receptors and SynGAP) and synaptic structure (e.g., densin-180, α-actinin, actin, PSD95) (Bayer and Schulman, 2001; Colbran, 2004). The potential role of nonsynaptic CaMKII self-association is less clear, but one function, as previously postulated by Dosemeci and colleagues, may be to limit or restrict Ca2+/CaM signaling during excessive or pathological Ca2+ signaling (Dosemeci et al., 2000; Tao-Cheng et al., 2001, 2002).
During development, the incorporation of the α-isoform of CaMKII into the PSD correlates with one of the most active periods of synapse formation (Kelly and Cotman, 1981; Scholz et al., 1988; Wu and Cline, 1998). The unique three-dimensional structure of CaMKII, with catalytic domains extending outward from a central core, may provide the kinase with a spatially organized set of binding sites that could play a scaffolding role in the organization of the PSD during development (Lisman et al., 2002) (supplemental Fig. 3, available at www.jneurosci.org as supplemental material). Finally, as local mRNA translation of α-CaMKII is central to synaptic plasticity (Steward, 2002; Steward and Worley, 2002), self-association would be an effective targeting mechanism to localize and concentrate this newly synthesized CaMKII in dendrites specifically to active synapses.
Our findings suggest that CaMKII self-association possesses many of the criteria postulated by Martin and Kosik (2002) for a molecular tag of synaptic activity. Indeed, the process of self-association ensures a relatively immobile pool of CaMKII in the context of intracellular diffusion providing a spatially restricted marker. Furthermore, self-association is reversible, but the time course of reversibility is influenced by the local level of Ca2+ or the degree of Thr286 phosphorylation. Finally, CaMKII is characterized as a multifunctional protein kinase attributable to the wide variety of substrates that it phosphorylates and thus is appropriately poised to interact with a multitude of signaling processes at synapses. We hypothesize that self-associated CaMKII, possibly intertwined with other molecules at the PSD or at the junction between the dendrite and the spine neck, could mark that particular spine for both short- and long-term synaptic modification. Future studies will be necessary to dissect the role of self-association in synaptic physiology and pathology.
Footnotes
This work was supported by the National Institutes of Health (H.S., M.N.W.), the Canadian Institute for Health Research (CIHR) (P.D.K., A.S.), and the National Alliance for Research on Schizophrenia and Disorders (P.D.K.). P.D.K. received a Career Award from the Burroughs Wellcome Fund and is now a Scholar of the Fond de la Recherche en Santé du Québec. E.L. is supported by a studentship from CIHR and the K. M Hunter Foundation. A.H. was a Fellow of the American Heart Society. We thank Dr. Michael Bradshaw, Jenny Tsui, Karen Hudmon, and members of the De Koninck laboratory for helpful discussions and comments on this manuscript. The GFP-CaMKII (Ala302Arg) and the wild-type GFP-CaMKII were generous gifts from Dr. Tobias Meyer (Stanford University, Stanford, CA). We thank Francine Nault and Salma Behna (Laval University) for excellent technical assistance with neuronal and HEK293 cell cultures, immunocytochemistry, recombinant DNA, and mutagenesis. We also thank Philippe Lemieux for preparing samples for electron microscopy.
Correspondence should be addressed to either of the following: Dr. Neal Waxham, Department of Neurobiology and Anatomy, University of Texas Health Science Center at Houston, Room 7.254, 6431 Fannin, Houston, TX 77030, E-mail: m.n.waxham@uth.tmc.edu; or Dr. Paul De Koninck, Centre de Recherche Université Laval Robert-Giffard, Beauport, Québec, Canada G1J 2G3, E-mail: paul.dekoninck@crulrg.ulaval.ca.
A. Hudmon's present address: Department of Neurology, Yale University, New Haven, CT 06516.
H. Schulman's present address: SurroMed, Menlo Park, CA 94025.
Copyright © 2005 Society for Neuroscience 0270-6474/05/256971-13$15.00/0
*
M.N.W. and P.D.K. are collaborative principal investigators and contributed equally to this work.
References
- Aronowski J, Grotta JC (1996) Ca2+/calmodulin-dependent protein kinase II in postsynaptic densities after reversible cerebral ischemia in rats. Brain Res 709: 103-110. [DOI] [PubMed] [Google Scholar]
- Bayer KU, Schulman H (2001) Regulation of signal transduction by protein targeting: the case for CaMKII. Biochem Biophys Res Commun 289: 917-923. [DOI] [PubMed] [Google Scholar]
- Bayer KU, De Koninck P, Leonard AS, Hell JW, Schulman H (2001) Interaction with the NMDA receptor locks CaMKII in an active conformation. Nature 411: 801-805. [DOI] [PubMed] [Google Scholar]
- Chesler M, Kaila K (1992) Modulation of pH by neuronal activity. Trends Neurosci 15: 396-402. [DOI] [PubMed] [Google Scholar]
- Colbran R (2004) Targeting of calcium/calmodulin-dependent protein kinase II. Biochem J 15: 1-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dosemeci A, Reese TS, Petersen J, Tao-Cheng JH (2000) A novel particulate form of Ca2+/CaMKII-dependent protein kinase II in neurons. J Neurosci 20: 3076-3084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dosemeci A, Tao-Cheng JH, Vinade L, Winters CA, Pozzo-Miller L, Reese TS (2001) Glutamate-induced transient modification of the postsynaptic density. Proc Natl Acad Sci USA 98: 10428-10432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elgersma Y, Fedorov NB, Ikonen S, Choi ES, Elgersma M, Carvalho OM, Giese KP, Silva AJ (2002) Inhibitory autophosphorylation of CaMKII controls PSD association, plasticity, and learning. Neuron 36: 493-505. [DOI] [PubMed] [Google Scholar]
- Frey U, Morris RG (1998) Synaptic tagging: implications for late maintenance of hippocampal long-term potentiation. Trends Neurosci 21: 181-188. [DOI] [PubMed] [Google Scholar]
- Fukaya M, Kato A, Lovett C, Tonegawa S, Watanabe M (2003) Retention of NMDA receptor NR2 subunits in the lumen of endoplasmic reticulum in targeted NR1 knockout mice. Proc Natl Acad Sci USA 100: 4855-4860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukunaga K, Stoppini L, Miyamoto E, Muller D (1993) Long-term potentiation is associated with an increased activity of Ca2+/calmodulin-dependent protein kinase II. J Biol Chem 268: 7863-7867. [PubMed] [Google Scholar]
- Gaertner TR, Kolodziej SJ, Wang D, Kobayashi R, Koomen JM, Stoops JK, Waxham MN (2004) Comparative analyses of the three-dimensional structures and enzymatic properties of alpha, beta, gamma and delta isoforms of Ca2+-calmodulin-dependent protein kinase II. J Biol Chem 279: 12484-12494. [DOI] [PubMed] [Google Scholar]
- Gleason MR, Higashijima S, Dallman J, Liu K, Mandel G, Fetcho JR (2003) Translocation of CaM kinase II to synaptic sites in vivo. Nat Neurosci 6: 217-218. [DOI] [PubMed] [Google Scholar]
- Hanson PI, Schulman H (1992) Inhibitory autophosphorylation of multifunctional Ca2+/calmodulin-dependent protein kinase analyzed by site-directed mutagenesis. J Biol Chem 267: 17216-17224. [PubMed] [Google Scholar]
- Hanson PI, Meyer T, Stryer L, Schulman H (1994) Dual role of calmodulin in autophosphorylation of multifunctional CaM kinase may underlie decoding of calcium signals. Neuron 12: 943-956. [DOI] [PubMed] [Google Scholar]
- Hartley Z, Dubinsky JM (1993) Changes in intracellular pH associated with glutamate excitotoxicity. J Neurosci 13: 4690-4699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoelz A, Nairn AC, Kuriyan J (2003) Crystal structure of a tetradecameric assembly of the association domain of Ca2+/calmodulin-dependent kinase II. Mol Cell 11: 1241-1251. [DOI] [PubMed] [Google Scholar]
- Hudmon A, Schulman H (2002) Structure-function of the multifunctional Ca2+/calmodulin-dependent protein kinase II. Biochem J 364: 593-611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudmon A, Aronowski J, Kolb SJ, Waxham MN (1996) Inactivation and self-association of Ca2+/calmodulin-dependent protein kinase II during autophosphorylation. J Biol Chem 271: 8800-8808. [DOI] [PubMed] [Google Scholar]
- Hudmon A, Kim SA, Kolb SJ, Stoops JK, Waxham MN (2001) Light scattering and transmission electron microscopy studies reveal a mechanism for calcium/calmodulin-dependent protein kinase II self-association. J Neurochem 76: 1364-1375. [DOI] [PubMed] [Google Scholar]
- Irwin RP, Lin SZ, Long RT, Paul SM (1994) _N_-methyl-d-aspartate induces a rapid, reversible, and calcium-dependent intracellular acidosis in cultured fetal rat hippocampal neurons. J Neurosci 14: 1352-1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly PT, Cotman CW (1981) Developmental changes in morphology and molecular composition of isolated synaptic junctional structures. Brain Res 206: 251-271. [DOI] [PubMed] [Google Scholar]
- Koch RA, Barish ME (1994) Perturbation of intracellular calcium and hydrogen ion regulation in cultured mouse hippocampal neurons by reduction of the sodium ion concentration gradient. J Neurosci 14: 2585-2593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolb SJ, Hudmon A, Ginsberg TR, Waxham MN (1998) Identification of domains essential for the assembly of calcium/calmodulin-dependent protein kinase II holoenzymes. J Biol Chem 273: 31555-31564. [DOI] [PubMed] [Google Scholar]
- Kolodziej SJ, Hudmon A, Waxham MN, Stoops JK (2000) Three-dimensional reconstructions of calcium/calmodulin-dependent (CaM) kinase IIalpha and truncated CaM kinase IIalpha reveal a unique organization for its structural core and functional domains. J Biol Chem 275: 14354-14359. [DOI] [PubMed] [Google Scholar]
- Leonard AS, Bayer KU, Merrill MA, Lim IA, Shea MA, Schulman H, Hell JW (2002) Regulation of calcium/calmodulin-dependent protein kinase II docking to _N-_methyl-d-aspartate receptors by calcium/calmodulin and alpha-actinin. J Biol Chem 277: 48441-48448. [DOI] [PubMed] [Google Scholar]
- Lisman J, Schulman H, Cline H (2002) The molecular basis of CaMKII function in synaptic and behavioural memory. Nat Rev Neurosci 3: 175-190. [DOI] [PubMed] [Google Scholar]
- Llopis J, McCaffery JM, Miyawaki A, Farquhar MG, Tsien RY (1998) Measurement of cytosolic, mitochondrial, and Golgi pH in single living cells with green fluorescent proteins. Proc Natl Acad Sci USA 95: 6803-6808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin KC (2002) Synaptic tagging during synapse-specific long-term facilitation of Aplysia sensory-motor neurons. Neurobiol Learn Mem 78: 489-497. [DOI] [PubMed] [Google Scholar]
- Martin KC, Kosik KS (2002) Synaptic tagging-who's it? Nat Rev Neurosci 3: 813-820. [DOI] [PubMed] [Google Scholar]
- Matsuzaki M, Honkura N, Ellis-Davies GC, Kasai H (2004) Structural basis of long-term potentiation in single dendritic spines. Nature 429: 761-766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer T, Hanson PI, Stryer L, Schulman H (1992) Calmodulin trapping by calcium-calmodulin-dependent protein kinase. Science 256: 1199-1201. [DOI] [PubMed] [Google Scholar]
- Molloy SS, Kennedy MB (1991) Autophosphorylation of type II Ca2+/calmodulin-dependent protein kinase in cultures of postnatal rat hippocampal slices. Proc Natl Acad Sci USA 88: 4756-4760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherji S, Soderling TR (1994) Regulation of Ca2+/calmodulin-dependent protein kinase II by inter- and intrasubunit-catalyzed autophosphorylations. J Biol Chem 269: 13744-13747. [PubMed] [Google Scholar]
- Ocorr KA, Schulman H (1991) Activation of multifunctional Ca2+/calmodulin-dependent kinase in intact hippocampal slices. Neuron 6: 907-914. [DOI] [PubMed] [Google Scholar]
- Otmakhov N, Tao-Cheng JH, Carpenter S, Asrican B, Dosemeci A, Reese TS, Lisman J (2004) Persistent accumulation of calcium/calmodulin-dependent protein kinase II in dendritic spines after induction of NMDA receptor-dependent chemical long-term potentiation. J Neurosci 24: 9324-9331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson GH, Knobel SM, Sharif WD, Kain SR, Piston DW (1997) Use of the green fluorescent protein and its mutants in quantitative fluorescence microscopy. Biophys J 73: 2782-2790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson JD, Chen X, Vinade L, Dosemeci A, Lisman JE, Reese TR (2003) Distribution of postsynaptic density (PSD)-95 and Ca2+/calmodulin-dependent protein kinase II at the PSD. J Neurosci 23: 11270-11278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rich RC, Schulman H (1998) Substrate-directed function of calmodulin in autophosphorylation of Ca2+/calmodulin-dependent protein kinase II. J Biol Chem 273: 28424-28429. [DOI] [PubMed] [Google Scholar]
- Rongo C, Kaplan JM (1999) CaMKII regulates the density of central glutamatergic synapses in vivo. Nature 402: 195-199. [DOI] [PubMed] [Google Scholar]
- Scholz WK, Baitinger C, Schulman H, Kelly PT (1988) Developmental changes in Ca2+/calmodulin-dependent protein kinase II in cultures of hippocampal pyramidal neurons and astrocytes. J Neurosci 8: 1039-1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwiening CJ, Willoughby D (2002) Depolarization-induced pH microdomains and their relationship to calcium transients in isolated snail neurones. J Physiol (Lond) 538: 371-382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen K, Meyer T (1998) In vivo and in vitro characterization of the sequence requirement for oligomer formation of Ca2+/calmodulin-dependent protein kinase IIalpha. J Neurochem 70: 96-104. [DOI] [PubMed] [Google Scholar]
- Shen K, Meyer T (1999) Dynamic control of CaMKII translocation and localization in hippocampal neurons by NMDA receptor stimulation. Science 284: 162-167. [DOI] [PubMed] [Google Scholar]
- Shen K, Teruel MN, Subramanian K, Meyer T (1998) CaMKIIb functions as an F-actin targeting module that localizes CaMKIIa/b heterooligomers to dendritic spines. Neuron 21: 593-606. [DOI] [PubMed] [Google Scholar]
- Shen K, Teruel MN, Connor JH, Shenolikar S, Meyer T (2000) Molecular memory by reversible translocation of calcium/calmodulin-dependent protein kinase II. Nat Neurosci 3: 881-886. [DOI] [PubMed] [Google Scholar]
- Silver IA, Erecinska M (1992) Ion homeostasis in rat brain in vivo: intra- and extracellular [Ca2+] and [H+] in the hippocampus during recovery from short-term, transient ischemia. J Cereb Blood Flow Metab 12: 759-772. [DOI] [PubMed] [Google Scholar]
- Smith MK, Colbran RJ, Brickey DA, Soderling TR (1992) Functional determinants in the autoinhibitory domain of calcium/calmodulin-dependent protein kinase II. Role of His282 and multiple basic residues. J Biol Chem 267: 1761-1768. [PubMed] [Google Scholar]
- Soderling TR, Chang B, Brickey D (2001) Cellular signaling through multifunctional Ca2+/calmodulin-dependent protein kinase II. J Biol Chem 276: 3719-3722. [DOI] [PubMed] [Google Scholar]
- Steward O (2002) mRNA at synapses, synaptic plasticity, and memory consolidation. Neuron 36: 338-340. [DOI] [PubMed] [Google Scholar]
- Steward O, Worley P (2002) Local synthesis of proteins at synaptic sites on dendrites: role in synaptic plasticity and memory consolidation? Neurobiol Learn Mem 78: 508-527. [DOI] [PubMed] [Google Scholar]
- Strack S, Colbran RJ (1998) Autophosphorylation-dependent targeting of calcium/calmodulin-dependent protein kinase II by the NR2B subunit of the _N-_methyl-d-aspartate receptor. J Biol Chem 273: 20689-20692. [DOI] [PubMed] [Google Scholar]
- Strack S, McNeill RB, Colbran RJ (2000) Mechanism and regulation of calcium/calmodulin-dependent protein kinase II targeting to the NR2B subunit of the _N-_methyl-d-aspartate receptor. J Biol Chem 275: 23798-23806. [DOI] [PubMed] [Google Scholar]
- Suzuki T, Okumura-Noji K, Tanaka R, Tada T (1994) Rapid translocation of cytosolic Ca2+/calmoduiln-dependent protein kinase II into postsynaptic density after decapitation. J Neurochem 63: 1529-1537. [DOI] [PubMed] [Google Scholar]
- Tao-Cheng JH, Vinade L, Smith C, Winters CA, Ward R, Brightman MW, Reese TS, Dosemeci A (2001) Sustained elevation of calcium induces Ca2+/calmodulin-dependent protein kinase II clusters in hippocampal neurons. Neuroscience 106: 69-78. [DOI] [PubMed] [Google Scholar]
- Tao-Cheng JH, Vinade L, Pozzo-Miller LD, Reese TS, Dosemeci A (2002) Calcium/calmodulin-dependent protein kinase II clusters in adult rat hippocampal slices. Neuroscience 115: 435-440. [DOI] [PubMed] [Google Scholar]
- Tao-Cheng JH, Vinade L, Winters CA, Reese TS, Dosemeci A (2005) Inhibition of phosphatase activity facilitates the formation and maintenance of NMDA-induced calcium/calmodulin-dependent protein kinase II clusters in hippocampal neurons. Neuroscience 130: 651-656. [DOI] [PubMed] [Google Scholar]
- Thomas JA, Buchsbaum RN, Zimniak A, Racker E (1979) Intracellular pH measurements in Ehrlich ascites tumor cells utilizing spectroscopic probes generated in situ. Biochemistry 18: 2210-2218. [DOI] [PubMed] [Google Scholar]
- Waldmann R, Hanson PI, Schulman H (1990) Multifunctional Ca2+/calmodulin-dependent protein kinase made Ca2+-independent for functional studies. Biochemistry 29: 1679-1684. [DOI] [PubMed] [Google Scholar]
- Willoughby D, Schwiening CJ (2002) Electrically evoked dendritic pH transients in rat cerebellar Purkinje cells. J Physiol (Lond) 544: 487-499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu GY, Cline HT (1998) Stabilization of dendritic arbor structure in vivo by CaMKII. Science 279: 222-226. [DOI] [PubMed] [Google Scholar]
- Yang E, Schulman H (1999) Structural examination of autoregulation of multifunctional calcium/calmodulin-dependent protein kinase II. J Biol Chem 274: 26135-26140. [DOI] [PubMed] [Google Scholar]
- Zacharias DA, Violin JD, Newton AC, Tsien RY (2002) Partitioning of lipid-modified monomeric GFPs into membrane microdomains of live cells. Science 296: 913-916. [DOI] [PubMed] [Google Scholar]