Synchronous Neuronal Activity Is a Signal for Axonal Sprouting after Cortical Lesions in the Adult (original) (raw)
Abstract
The ability of the adult brain to form new connections in areas denervated by a lesion (axonal sprouting) is more widespread than previously thought, but mechanisms remain unknown. We have previously demonstrated an unexpected, robust axonal sprouting of contralateral corticostriatal neurons into the denervated striatum after ischemic cortical lesions. We now take advantage of marked differences in the degree of axonal sprouting from contralateral homotypic cortex after two types of cortical lesions to define the role of neuronal activity in this response. Thermal–ischemic lesions (TCL) of sensorimotor cortex, which induce axonal sprouting, produced two sequential patterns of low-frequency, synchronized neuronal activity that are not seen after similarly sized aspiration lesions, which do not induce axonal sprouting. An early rhythm of synchronous neuronal activity occurred in perilesion cortex on day 1 after lesion, with a frequency range of 0.2–2 Hz. A later pattern of activity occurred on days 2 and 3 after lesion, with a frequency range of 0.1–0.4 Hz. This second rhythm synchronized neuronal activity across widespread areas, including the cortical areas that contain the cell bodies of the sprouting axons. TTX was used to block this patterned neuronal activity and determine whether axonal sprouting was prevented. Chronic TTX infusion into the lesion site blocked the synchronous neuronal activity after TCL as well as axonal sprouting. Thus, both after different types of lesions and in the blockade experiments axonal sprouting was strongly correlated with synchronous neuronal activity, suggesting a role for this activity in anatomical reorganization after brain lesion in the adult.
Keywords: cerebral ischemia, tetrodotoxin, striatum, repair, neuroplasticity, regeneration, activity, EEG
Axonal sprouting is emerging as an important component of the CNS response to injury and may be critical for functional plasticity and recovery. Indeed, axonal sprouting has long been thought restricted to the immature brain or to discrete, highly plastic regions in the adult, notably the hippocampus. However, axonal sprouting also occurs in the cortex after lesions (Napieralski et al., 1996; Carmichael et al., 2001). Remarkably, the cell bodies of the neurons involved are often located at a considerable distance from the lesion (Napieralski et al., 1996). It is unclear what signals, originating either at the lesion site or in the denervated area, participate in the sprouting response of distant neurons. Electrophysiological activity is a prime candidate for such a signal. Recent evidence suggests that specific patterns of activity could be involved not only in synaptic refinement (Feller, 1999), but also in axonal growth during development (Catalano and Shatz, 1998; Dantzker and Callaway, 1998). Therefore, we investigated the role of in vivo electrophysiological activity in a model of long distance axonal sprouting in the adult brain.
This research has been limited by the difficulty in distinguishing between the degenerative effects of a lesion and signals specifically associated with sprouting in most experimental models in which sprouting occurs. In a model system that addresses this question, we have shown that ischemic lesions of the sensorimotor cortex induced by thermocoagulation of pial blood vessels in adult rats are followed by a massive sprouting of axons from the contralateral homotypic cortex into the denervated striatum (Napieralski et al., 1996; Uryu et al., 2001). In contrast, ablation of the exact same cortical area by acute aspiration does not induce a sprouting response. In this model, axonal sprouting can first be detected by alterations in growth cone markers at 7 d after lesion (M. H. Shomer and M. F. Chesselet, unpublished observations) and at 16 d by ultrastructural study (Uryu et al., 2001) and, in the striatum, is limited to the region of the dorsolateral striatum denervated by the lesion (Napieralski et al., 1996). Postlesion axonal sprouting closely overlaps with an area containing activated astrocytes, as detected by an increase in glial fibrillary acidic protein immunostaining (Szele et al., 1995). This experimental model has the advantages that axonal sprouting occurs from cortical sites in the hemisphere opposite the ischemic lesion and that both aspiration and ischemic lesions share similar degrees of tissue damage and axotomy. Thus, this cortical lesion model provides the ability to eliminate lesion effects unrelated to sprouting by comparing two similarly sized lesions that differ primarily in induced axonal sprouting.
In the present study, we have used this model to examine the role of neuronal activity as a signal for axonal sprouting from cortex contralateral to the lesion into striatum ipsilateral to the lesion in the adult. Neuronal activity was recorded for 7 d in perilesion and in contralateral cortex in adult rats with either a thermocoagulatory (TCL) or aspiration (ASP) lesion of the sensorimotor cortex. Only the sprouting-inducing thermocoagulatory lesions were followed by a characteristic pattern of activity, first in the perilesion cortex and secondarily on the side opposite to the lesion. To confirm that this transient network of neuronal activity induced by the thermocoagulatory lesion was necessary for axonal sprouting, tetrodotoxin (TTX) was infused chronically into the perilesion cortex. This treatment blocked the lesion-induced activity and abolished axonal spouting from contralateral cortex into the denervated striatum.
MATERIALS AND METHODS
Physiological preparation. Animal procedures were conducted within National Institutes of Health and University of California Los Angeles Department of Laboratory Animal Medicine guidelines. Adult Sprague Dawley rats (250–400 gm, 15 males) were anesthetized with Equithesin (1.0 ml/300gm, i.p.) and immobilized in a stereotaxic frame. Skull screw electrodes for ground and indifferent channels were implanted over both cerebellar hemispheres. Four insulated tungsten microelectrodes (0.04 mm diameter, 0.2–0.5 MΩ impedance at 1 kHz) were placed through small burr holes in cortical sites that would become, after the cortical lesion (see below): (1) perilesion cortex [relative to bregma: anteroposterior (AP) −7 mm, mediolateral (ML) 2 mm; Paxinos and Watson, 1997], (2) contralateral parietal cortex (AP −4, ML 3), (3) contralateral frontal cortex (AP 0, ML 4), and (4) contralateral occipital cortex (AP −7, ML 3) (see Fig. 1D). In four animals (two with TCL and two with ASP), sintered Ag–AgCl DC electrodes (A-M Systems, Carlsborg, WA) were also implanted in perilesion cortex, contralateral parietal cortex, and ipsilateral cerebellum. All electrode leads were immobilized in a dental acrylic headset. After a 1 d recovery, the animals were reanesthetized, and a craniotomy was cut above frontal and parietal cortex in the left hemisphere and either ASP (n = 5) or TCL (n = 5) produced as previously described (Szele et al., 1995). The bone defect was sealed with sterilized plastic wrap and dental cement. In five physiology sham animals skull screw and microelectrodes were placed, but no lesion was given.
Fig. 1.

Axonal sprouting after focal cortical lesions. Coronal sections through rat brain with BDA tracer injection (arrowheads) in right frontal cortex and sham (A) and ASP (B) or TCL (D) in left frontoparietal cortex. After TCL (D) there is a substantial increase in axonal labeling in contralateral striatum and in cortex medial to the lesion site (arrows). Scale bar: B, 500 μm.C, Quantification of corticostriatal projections by lesion condition. Measurements are taken from three levels in each animal (r, rostral; m, middle;c, caudal). Asterisks denote significance of p < 0.01 (Tukey–Kramer _post hoc_test, n = 3; see Results for ANOVA values).E, Electrode placement for in vivo_physiological recording. Vertical line represents the level of coronal sections in A, B, and_D.
Lesions. Two types of cortical lesions were produced as previously described (Szele et al., 1995). A craniotomy was cut above frontal and parietal cortex. In TCL, pial blood vessels were thermocoagulated with a heater probe, leaving the dura intact. For ASP, the dura was opened and the cortex gently aspirated with a fine pipette to a point just above the subcortical white matter (Uryu et al., 2001). Sham animals received a craniotomy alone.
Recording. Spontaneous cortical activity was recorded before and after the lesions in awake, unrestrained animals in 1 hr sessions each day for 7 d after the lesion. Thus, each animal served as its own control. AC signals were amplified, filtered, and digitized (six channel simultaneously recording at 2.5 kHz; Axoscope 7.0; Axon Instruments, Foster City, CA). DC activity was recorded in reference to the cerebellar skull screw electrode and with a cerebellar DC electrode as indifferent (Grass DC amplifier; 3 Hz low-pass filter). Baseline DC electrode drift was not significant until 5 d after the lesion, at which time DC recording was terminated. Cortical depolarization shifts or spreading depressions were recorded in DC mode. In addition, AC recordings were analyzed during each recording session for the characteristic transient depression produced in this mode by spreading depressions (Hossmann, 1996).
TTX physiological experiments. Seven day infusing osmotic minipumps (Alza 1007D; Alza, Palo Alto, CA) filled with TTX (Calbiochem, San Diego, CA) dissolved in sterile citrate buffer (Reiter et al., 1986) were primed at 37oC in sterile saline overnight. Operative procedures were as above. Tungsten microelectrodes were implanted in perilesion cortex and contralateral parietal cortex, skull electrodes were placed over both cerebellar hemispheres, and TCL was produced. Minipumps were placed in the interscapular area and specially constructed cannulas (Kasamatsu and Schmidt, 1997) implanted into the lesion bed 1.0 mm deep to the dural surface. In preliminary experiments, a single minipump implanted into the middle of the lesion was inadequate to block action potentials in both the rostral and caudal peri-infarct cortex. Two minipumps were thus implanted: 3 mm from the rostral margin of the lesion and 3 mm from the caudal margin of the lesion. Minipump cannulas were fixed with dental cement. Spontaneous cortical activity from the two AC channels was filtered, amplified, and digitized.
Data processing. Five minute epochs of cortical activity from each electrode at each recording session were subjected to Fast Fourier Transform (Clampfit 8.0; Axon Instruments). These power spectra were then averaged across animals in the same lesion group to give the average power of the cortical activity for each brain region, for each condition (ASP, TCL, sham, TCL plus vehicle, TCL plus TTX) at each time point before and after each lesion. These values were compared across lesion conditions using factorial ANOVA and a Tukey–Kramer post hoc test (Statview 1.2; SAS Institute Inc., Carey, NC).
Multiunit activity. Multiunit activity was obtained offline by high-pass filtering each recording session at 300 Hz (Colder et al., 1996). Three minute epochs of multiunit action potential activity simultaneously recorded from the four AC electrodes in the first electrophysiology experiments or two AC electrodes in the TTX experiments were passed through a window discriminator set to twice background (Origin 6.0; Microcal Software, Northampton, MA). In individual animals, multiunit cross-correlations were computed (Sears and Stagg, 1976) using a 2 msec bin width and a 200 msec sampling window (Stranger; Biographics, Inc., Winston-Salem, NC). Cross-correlation bin counts were converted to firing rate. Statistical significance was tested using Poisson statistics. Confidence limits of 99% for an independent firing relationship were calculated based on the average firing frequency (Abeles, 1982; Stranger; Biographics, Inc.). Additionally, cross-correlation bin counts were averaged for all animals within each experimental group to generate average firing rate histograms per cortical area per time point after lesion. These were statistically compared using the same cross-correlation function as above. Differences in spontaneous multiunit activity between TCL plus vehicle and TCL plus TTX were tested with an unpaired Student's_t_ test (Statview 1.2; SAS Institute Inc.).
Anatomical tracing and histology. In the electrophysiology experiments, 7 d after the lesion or 8 d after electrode implantation in sham, animals were anesthetized and perfused with buffered saline and 4% paraformaldehyde. Brains were post-fixed, cryoprotected, frozen-sectioned at 50 μm, and stained with cresyl violet. Sections were analyzed for lesion size, including involvement of subcortical white matter and corpus callosum. Two sets of animals received biotinylated dextran amine (BDA) injections to directly demonstrate axonal sprouting: a set of ASP, TCL, and craniotomy (sham) controls (n = 3 for each group) and a set of TCL plus TTX and TCL plus vehicle animals (n = 5 for each group). Twenty-eight days after the lesion, animals received four, 160 nl injections into cortical layer V at AP 1.2, ML 3.O (Paxinos and Watson, 1997) of a 10% solution of BDA (Molecular Probes, Eugene, OR) as described previously (Carmichael et al., 2001). Seven days later, the animals were perfused as above, and 50 μm tissue sections were processed for BDA visualization (Carmichael et al., 2001). BDA is a bidirectional label (Reiner et al., 2000), but we quantified the surface area of labeled axonal profiles in the anterograde-only corticostriatal projection (Heimer et al., 1995). Digital photomicrographs of the dorsolateral striatum ipsilateral and contralateral to the cortical injection at three coronal levels within the region of the contralateral corticostriatal projection (Napieralski et al., 1996; AP 1.45, AP 0.8, AP –0.7; Paxinos and Watson, 1997) were given a gray scale threshold to include only labeled axons. The surface area of labeled axons was measured (NIH Image 1.6) in three separate regions within the projection zone in each section and averaged to give a mean surface area per section of corticostriatal labeling for both sides of the striatum. The contralateral surface area was then divided by that in the ipsilateral striatum to give a contralateral–ipsilateral ratio as a measure of axonal sprouting (see Fig. 1D). The contralateral–ipsilateral labeling ratios were compared by condition using factorial ANOVA and a Tukey–Kramer post hoc test (Statview 1.2; SAS Institute Inc.). In these same groups of animals, a separate series of 50 μm sections was stained for Nissl. The surface area of the lesion was measured in five sections through the lesion site (SPOT 3.2.4 software; Diagnostic Instruments, Inc., Sterling Heights, MI), and the volume of the lesion was computed by multiplying each surface area by the distance between sections. The lesion size of TCL plus TTX and TCL plus vehicle was compared with the Mann–Whitney U test, and the correlation of contralateral–ipsilateral labeling to lesion size was tested using linear regression analysis (Statview 1.2; SAS Institute Inc.).
Fluorojade-B histochemistry was used to measure neuronal degeneration 1 d after TCL plus TTX and TCL plus vehicle as described (Schmued and Hopkins, 2000). A separate group of TCL plus vehicle and TCL plus TTX (n = 5 for each group) was prepared as above. At 1 d after lesion, the animals were anesthetized, perfused, and the brains were frozen and sectioned at 25 μm. Sections were processed with fluorojade B (Schmued and Hopkins, 2000), labeled cells were counted at 400× in three sections through the lesion at the AP levels described for axonal quantification, and the effect of TCL plus TTX versus TCL plus vehicle tested statistically (Mann–Whitney U test; Statview 1.2; SAS Institute Inc.).
RESULTS
Different types of cortical lesions elicit distinct axonal sprouting responses
Axonal sprouting after cortical lesions was first quantified after two types of cortical lesions known to produce markedly different degrees of postlesion axonal sprouting in adult rats (Szele et al., 1995; Napieralski et al., 1996). TCLs were produced by coagulating pial blood vessels in frontoparietal cortex. These lesion produced a total cell loss in all cortical layers down to the subcortical white matter over a period of 3–5 d (Szele et al., 1995) (Fig.1D). In another group of rats, the same cortical region was acutely removed by aspiration with a glass pipette (ASP), taking care to leave the corpus callosum intact as well (Uryu et al., 2001) (Fig. 1B). Sham animals received only a craniotomy (Fig. 1A).
To demonstrate the pattern of axonal connections from cortex contralateral to the lesion, small injections of the axonal tracer BDA were placed into frontal cortex contralateral to the lesion 28 d after surgery. Each injection was exactly matched in volume and stereotaxic position. The resultant axonal labeling showed that the normally small, crossed corticostriatal pathway (Fig.1A) had sprouted into the denervated striatum on the side of the lesion after TCL (Fig. 1D) but not ASP (Fig. 1B), as we have described previously (Napieralski et al., 1996). There is also an increase in axonal labeling in the medial perilesion cortex after TCL that is not seen in sham animals or after ASP (Fig. 1).
Crossed corticostriatal projections were quantified by measuring the surface area of the axonal projection in three coronal sections through the territory of this projection. The ratio of the contralateral to ipsilateral projections was calculated to account for possible differences in labeling caused by small variations in the diffusion distance of the tracer immediately surrounding the injection site from animal to animal. ASP did not change the ratio of contralateral versus ipsilateral labeling compared with sham. In contrast, this ratio was increased twofold to threefold after TCL (Fig. 1C). This difference is statistically significant in TCL versus ASP (p < 0.01 at all levels; Tukey–Kramer_post hoc_ test; F(1,4) = 44.61 for rostral, F(1,4) = 210.28 for mid, F(1,4) = 18.32 for caudal) and_p_ < 0.01 TCL versus sham (F(1,4) = 49.90 for rostral,F(1,4) = 279.84 for mid,F(1,4) = 15.33 for caudal). There was no significant difference between ASP and sham (Fig. 1C). Thus, although ASP and TCL removed similar regions of cortex, only TCL produced axonal sprouting in perilesion cortex and striatum. Because this sprouting occurs from a distant, connected cortical site (Fig.1A), we hypothesized that the trigger for axonal sprouting may be an electrophysiological signal in the connections between the damaged cortex and contralateral, homotypic cortex.
Lesions that produce axonal sprouting trigger rhythmic neuronal activity in perilesion cortex
We recorded spontaneous cortical activity for 7 d after TCL, ASP, and in sham animals, the time frame of axonal sprouting in other models of neuronal plasticity in adults (Steward et al., 1988) (Fig.1E). The pattern of spontaneous cortical activity was initially similar among sham, ASP, and TCL (Fig.2A). However, 1 d after the induction of TCL, activity in perilesion cortex consisted entirely of high-voltage slow waves (Fig. 2A). These were biphasic positive–negative waves with a frequency centered at 1 Hz. Each wave ranged from 200 to 700 μV in amplitude and 200–500 msec in duration (Fig. 2B). This activity pattern was maximal at day 1 and absent by day 5 after the lesion (Fig.2A). In contrast, spontaneous cortical activity after ASP lesions remained low voltage and desynchronized as in sham cases throughout the 7 d recording period (Fig. 2A). Multiunit recording during the slow waves induced by TCL showed action potential activity synchronized on the negative phase of the wave (Fig.2B). Thus, slow waves are induced by TCL in perilesion cortex early and transiently after the lesion and synchronize action potential activity.
Fig. 2.

Spontaneous cortical activity in perilesion cortex. A, Spontaneous cortical activity in single animals over the recording period. Each trace is from a single animal before and after lesion, or from equivalent days after electrode implantation in sham animals. B, Coincident action potential and slow wave activity after TCL. Slow wave activity and multiunit action potential activity taken from the same electrode in perilesion cortex on day 1 after TCL.
To quantify this activity pattern, power spectra were computed for each animal in each condition for each time point after the lesion. Power spectral analysis in sham and ASP animals showed no change in the pattern of spontaneous electrical activity in perilesion cortex over the 7 d recording period (Fig. 3). In contrast, animals with a TCL had a substantial increase in power spectra in the 0.2–2 Hz frequency range on days 1–3 after the lesion in the perilesion cortex (Fig. 3C), the same frequency of the slow waves in this region (Fig. 2A). The mean of all power spectra for each lesion group in perilesion cortex was significantly different in a frequency range from 0.226 Hz (F(1,8) = 18.03) to 2.2 Hz (F(1,8) = 28.15) on day 1 after the TCL compared with ASP and for TCL compared with sham conditions (0.226 Hz, F(1,8) = 2.69–2.2 Hz; F1,8 = 31.03) (n = 5 for each group; p < 0.05) (Fig. 3E), but not at other time points.
Fig. 3.
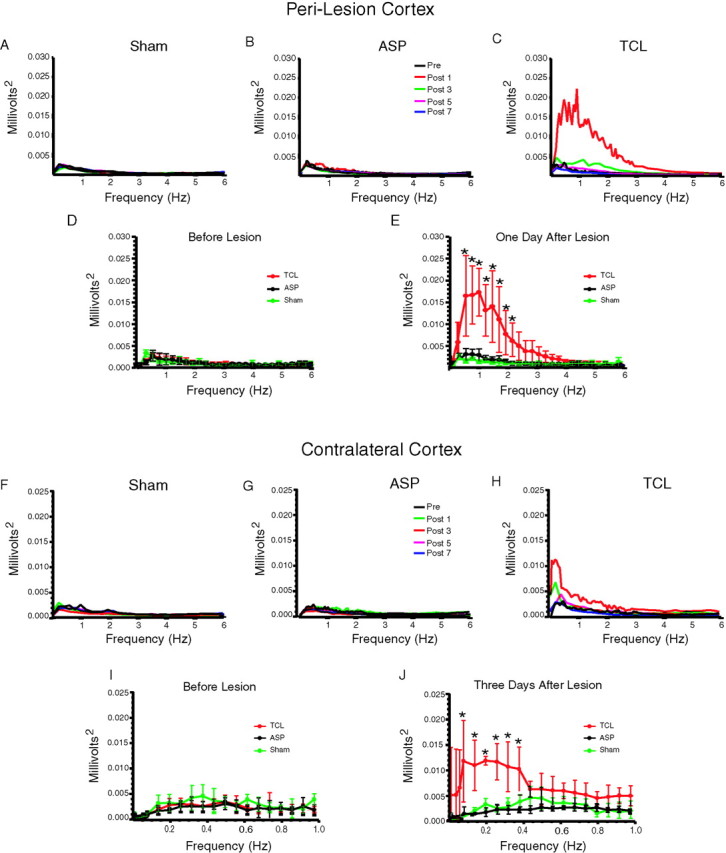
Quantitative analysis of spontaneous cortical activity in perilesion cortex. A–E, Pattern of spontaneous cortical activity in perilesion cortex. The top panels show averaged power spectra in all animals in each group for each time point: A, sham; B, ASP;C, TCL groups. D, E, Quantitative analysis of spontaneous cortical activity in perilesion cortex before in D and 1 d after in E ASP (black) and TCL (red), or at 1 and 2 d after electrode implantation in sham (green) animals. In all cases, graphs are scaled to match that in_E_. Asterisks denote values significant at_p_ < 0.05 for TCL versus ASP and sham (Tukey–Kramer post hoc test, n = 5; see Results for ANOVA values). F–J, Pattern of spontaneous cortical activity in contralateral cortex. Same conventions as in A–E. The top panels show averaged power spectra for all animals in each group for each time point:F, sham; G, ASP; H, TCL groups. I, J, Quantitative analysis of cortical activity on day 3 in contralateral cortex. Asterisks denote values significant at p < 0.05 for TCL versus ASP and sham (Tukey–Kramer post hoc test,n = 5; see Results for ANOVA values).
Lesions that produce axonal sprouting trigger rhythmic neuronal activity in contralateral cortical areas
Synchronous spontaneous activity was also detected with in vivo recordings in the contralateral cortex after TCL, but not after ASP or sham. Large, very low-frequency waves first occurred in cortex contralateral to the lesion on day 1 and became maximal by day 3 after TCL (Fig. 4A). Within single animals, these slow waves occurred with an irregular range of frequencies, at one wave per 2.5–12.5 sec (Fig.4A,B). These waves did not correspond to seizure activity, sleep, or body movement. Each wave consists of a complex of 0.8–1 sec duration with an initial positive component, followed by a longer negative phase (Fig. 4B,C). Smaller waves at 4–6 Hz or 12–16 Hz were superimposed on the negative phase (Fig.4B,C). Quantitative analysis of this pattern of spontaneous cortical activity using power spectra showed that most of the overall amplitude of the spontaneous cortical activity after TCL occurred between 0.1 and 1 Hz on days 1 and 3 after the lesion (Fig.3H). The mean of all power spectra at each frequency for each lesion group was significantly different at day 3 in a range from 0.113 Hz (F(1,8) = 4.29) to 0.396 Hz (F(1,8) = 13.98) in TCL compared with ASP, and in TCL compared with sham (0.113 Hz,F(1,8) = 4.73–0.396 Hz;F(1,8) = 10.23) (n = 5 for each group; p < 0.05) (Fig. 3J). There were no differences between ASP and sham at this (Fig.3J) or other time points (Fig.3F,G,I).
Fig. 4.

Slow wave and multiunit activity in contralateral cortex after TCL. A, Periodic slow wave activity in cortex contralateral to TCL on day 2 after the lesion. The_top_ and bottom traces in each panel were simultaneously recorded from frontal and parietal cortex contralateral to TCL. B, Periodic slow waves on day 3 after TCL in contralateral cortex. Traces are from a different animal than in A and show the morphology of the individual waves. C, D, Simultaneous slow wave and action potential activity in cortex contralateral to TCL on day 3. C and_D_ are taken from recordings from frontal and parietal cortex contralateral to TCL in different animals. _E,_Coincident multiunit action potential activity in two regions of cortex contralateral to TCL on day 3 in the same animal.
As with the earlier perilesion slow waves on day 1 after TCL, multiunit neuronal discharges were synchronized on the negative phase of the slow waves in cortex contralateral to TCL on days 2 and 3 (Fig. 4D). Multiunit activity appears synchronized across distant cortical sites after TCL during this slow wave activity (Fig. 4E). Indeed, cross-correlation analysis showed that action potential activity after TCL was synchronized across widespread cortical areas contralateral and ipsilateral to the lesion site in individual animals during slow wave activity on day 3 after TCL (Fig. 5A). Furthermore, a cross-correlation of the average of the multiunit activity for all TCL animals at each cortical site from sampling windows taken during low-frequency slow waves showed significant correlation in the action potential activity on days 2 and 3 after TCL (Fig. 5B). This correlation represents coincident neuronal activity in cortical areas separated by 3.5 mm (parietal to occipital), 5 mm (frontal to parietal), and across hemispheres for a 2 d period after TCL in awake, behaving animals. It was no longer observed by day 5, after slow waves have subsided (Fig. 5A,B).
Fig. 5.
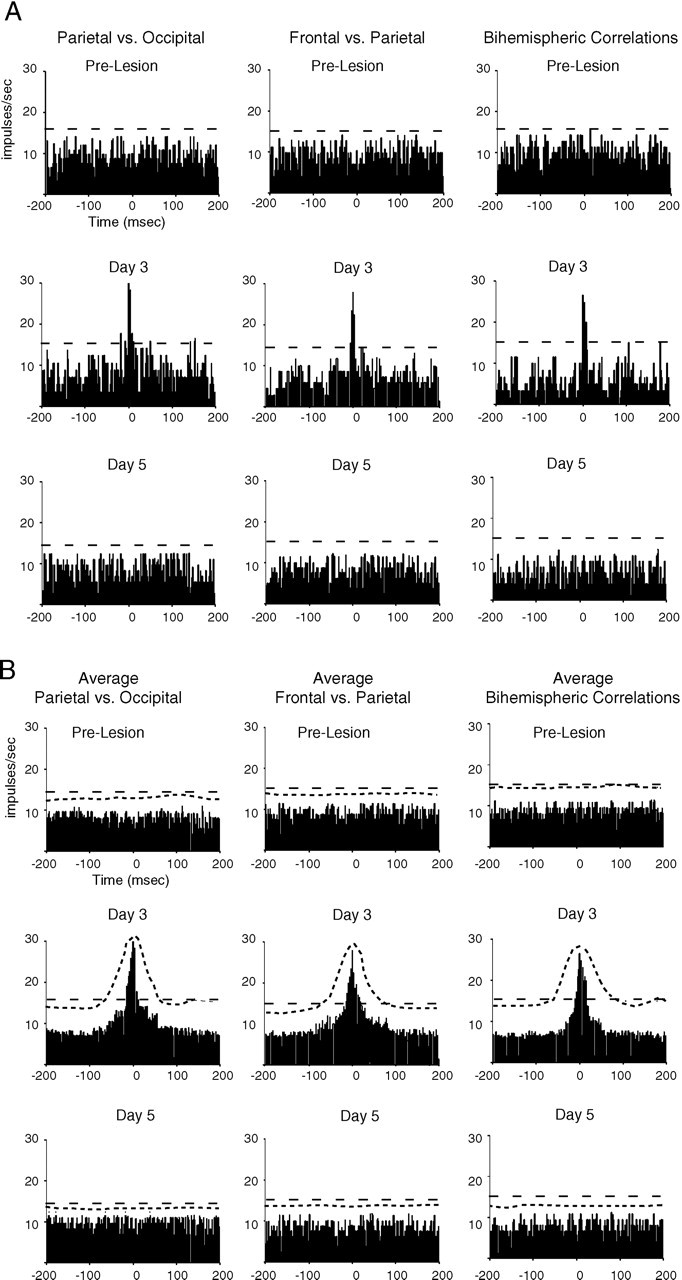
Cross-correlation of multiunit activity after TCL.A, Correlograms show multiunit activity from one recording site compared with that occurring at a reference site, before and on days 3 and 5 after TCL. Above each column of correlograms the listed site on the left is the reference site. The dashed line in each graph shows the 99% confidence level for a correlation greater than chance. These were computed from one animal. B, Average correlograms for all animals before and on days 3 and 5 after TCL (_n_= 5). The ordinate shows the average firing frequency in each 2 msec bin. The more closely spaced _dashed line_shows the upper limit of the SD for each 2 msec bin. The more broadly spaced dashed line shows the 99% confidence level for a correlation grater than chance.
Spreading depressions
Focal cortical lesions are known to trigger spreading depressions, which could also serve as a signal for postlesion neuronal plasticity (Hossmann, 1996). We measured spreading depressions during each recording session. Spreading depressions occurred solely ipsilateral to the lesion immediately after lesion induction. These were infrequent, and their number did not differ between TCL (mean of 1.6 ± 0.89 events/hr) and ASP lesions (mean of 1.3 ± 0.45 events/hr). Each spreading depression lasted from 0.6 to 1.3 min. These features are consistent with the characteristics of spreading depressions after other types of brain lesions (Hossmann, 1996).
TTX infusion into perilesion cortex blocks postlesion synchronous activity
The previous experiments showed that a sequential pattern of synchronous neuronal activity arises first in perilesion cortex and then in contralateral cortex only in the type of lesion (TCL) that produces axonal sprouting. To determine whether this activity was directly causative in the axonal sprouting response, we used activity blockade to block the synchronous activity pattern and test whether this in fact blocked axonal sprouting after TCL. Synchronous activity could either be blocked in the perilesion cortex where it originates or in cortex contralateral to the lesion, the site of the cell bodies of the sprouting axons. However, sustained drug delivery into cortex contralateral to the lesion produces a secondary lesion at the delivery site, either with minipumps (Jablonska et al., 1993) or with Elvax-polymer slow release (our unpublished observations). TTX infused into the lesion to block action potential activity in perilesion cortex circumvents this problem: it is infused directly into a lesion and thus produces no additional damage of adjacent brain (see below for quantification). Also, the diffusion distance of TTX at this site is limited by the physical barrier of the corpus callosum and the ischemia-induced reduction in water diffusion (see below). Furthermore, TTX has not been shown to produce general effects on normal neuronal function or on neuronal lesions, outside of its blockade of voltage-dependent sodium channels (Reiter et al., 1986; Olson and Meyer, 1994; Catalano and Shatz, 1998).
In pilot experiments, increasing concentrations of TTX were tested, beginning with the concentrations used in earlier studies in developing animals (Reiter et al., 1986). Five and 10 μm TTX only produced a partial activity blockade in perilesion cortex. These concentrations were not used. Fifteen micromolar TTX infused into the lesion bed blocked most action potentials in the perilesion cortex of the awake, behaving animal but had no effect on multiunit firing in cortex contralateral to the lesion (Fig.6A,B). The difference in firing rate between TTX and vehicle-treated animals in perilesion cortex is statistically significant (unpaired t test: day 1, df = 8, t = −7.28, p < 0.001; day 3, df = 8, t = −6.2, p = 0.003). However, 15 μm TTX had no effect on the baseline frequency of action potentials in contralateral cortex (Fig.6B,C). Thus, the direct effect of TTX infusion, which is blockade of voltage-gated sodium channels and as a result action potential generation, is seen only adjacent to the lesion, not in cortex contralateral to the lesion.
Fig. 6.

TTX blocks action potential activity in perilesion cortex and low-frequency slow waves in both hemispheres. A, B, The top trace in each panel shows slow waves, and the bottom trace shows multiunit activity, simultaneously recorded from the same electrode during either TTX or vehicle infusion. Traces are from different vehicle- and TTX-treated animals. C, Power spectral analysis of effect of TTX infusion on slow wave activity in perilesion cortex. Plots show average and SD of all animals in TCL alone, TCL plus vehicle, and TCL plus TTX groups. Asterisks denote_p_ < 0.05; TCL plus TTX versus TCL alone and versus TCL plus vehicle (Tukey–Kramer post hoc test,n = 5 for each group). D, Power spectral analysis of effect of TTX infusion on slow wave activity in cortex contralateral to the lesion. Asterisks denote_p_ < 0.05; TCL plus TTX versus TCL alone and versus TCL plus vehicle (Tukey–Kramer post hoc test,n = 5 for each group). _Pound sign_denotes p < 0.05; TCL plus TTX versus TCL alone (Tukey–Kramer post hoc test, n = 5 for each group). E, Effect of TTX on action potential activity. n = 5 for TTX and vehicle groups.Asterisks denote p < 0.001; TTX versus vehicle on day 1; p < 0.003; TTX versus vehicle on day 3 (unpaired Student's t test,n = 5, see Results). F, TTX infusion blocks the increase in the surface area of axonal labeling in contralateral striatum after TCL. Same conventions as in Figure1C. n = 4 TCL plus TTX and TCL plus vehicle, n = 5 for TCL and n = 3 for sham. Asterisks denote p < 0.01 for vehicle and TCL versus sham and TTX (Tukey–Kramer post hoc test).
By blocking action potential activity in perilesion cortex, TTX blocked the synchronous pattern of slow waves in perilesion cortex after TCL (Fig. 6A). This effect was quantified through power spectral analysis of the activity pattern in perilesion cortex in TCL plus TTX, TCL plus vehicle, and TCL alone. In perilesion cortex, TCL plus TTX produced a pattern of cortical activity that was significantly different from TCL alone and TCL plus vehicle from 0.46 Hz (F(1,8) = 16.61 for TCL plus TTX vs TCL; F(1,8) = 27.69 for TCL plus TTX vs TCL plus vehicle) to 1.38 Hz (F(1,8) = 17.40 for TCL plus TTX vs TCL; F(1,8) = 12.55 for TCL plus TTX vs TCL plus vehicle) (p < 0.05;n = 5 for each group) (Fig.7D). This corresponds to a blockade of the lower frequencies in the synchronous activity triggered by TCL in perilesion cortex (Fig. 3D).
Fig. 7.
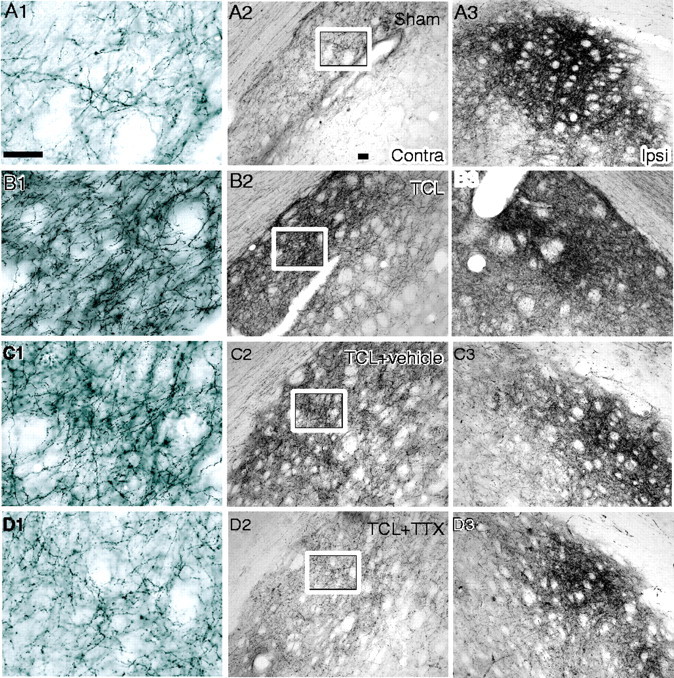
Blockade of rhythmic slow wave activity prevents axonal sprouting after TCL. BDA was injected into cortex contralateral to TCL 28 d after the lesion. A, Corticostriatal projections in sham-operated animal. B, Corticostriatal projections in TCL. C, Corticostriatal projections in TCL plus vehicle. D, Corticostriatal projections in TCL plus TTX. The middle column contains photomicrographs from striatum contralateral to the lesion. The right column contains photomicrographs from striatum ipsilateral to the lesion. The left column contains high-powered photomicrographs from the area within the box in the_middle column_. Scale bars: A1 (applies to_left column_), 50 μm; A2 (applies to_middle_ and right columns), 100 μm.
As a consequence of the blockade of action potential and slow wave activity in perilesion cortex, the pattern of slow wave activity in contralateral cortex after TCL did not develop. TCL plus TTX did not produce a pattern of slow wave activity significantly different from control animals in contralateral cortex (Tukey–Kramer post hoc test, for frequencies 0, 0.23. 0.46, 0.69, 0.92, 1.16, 1.40 Hz; F(5,24) = 1.84, 1,47, 1.10, 0.60, 1.58, 1.71, and 2.10, respectively) (compare Fig. 3F and6E). In fact, TCL plus TTX produced a pattern of activity that was significantly different from TCL alone and TCL plus vehicle at a range of 0.231 (F(1,8) = 17.49 for TCL plus TTX vs TCL; F(1,8)= 35.62 for TCL plus TTX vs TCL plus vehicle) to 0.92 Hz (F(1,8) = 23.79 for TCL plus TTX vs TCL; F(1,8) = 5.33 for TCL plus TTX vs TCL plus vehicle) (n = 5 for each group;p < 0.05) (Fig. 6E). In the previous studies the frequency range of spontaneous cortical activity that differed between TCL and sham/ASP in contralateral cortex was 0.113–0.396 (Fig. 3J). TCL plus TTX produced significantly less power in the contralateral cortex when compared with TCL alone up to a frequency of 1.16 Hz (F(1,8) = 11.58; p < 0.05) (Fig. 6E). TTX infusion into perilesion cortex thus blocks the development of the local perilesion pattern of synchronous activity and the later, more distributed pattern of synchronous activity after TCL in the area of origin of the sprouting axons.
TTX infusion does not influence lesion size after TCL
To compare axonal sprouting after TCL plus TTX as compared with TCL plus vehicle, it is important to establish that TTX did not alter the size of the lesion. Lesion size was measured at 35 d after the lesion, the end point of the tracing experiments (see below). Because of the technical constraints created by the need to secure the delivery cannula to the skull, lesion sizes were usually smaller and more variable in these experiments with both vehicle and TTX delivery than when no minipump–cannula setup was put in place. Nevertheless, lesion size was not significantly different in TCL plus TTX, 8.99 ± 4.36 mm3, compared with TCL plus vehicle, 9.13 ± 3.01 mm3 (Mann–Whitney_U_ test; z = −0.289; p = 0.77;n = 4 for each group). Furthermore, a linear regression analysis of axonal sprouting ratio (see below) and lesion size showed there was no relationship between lesion size and degree of axonal sprouting among animals within TCL plus TTX and TCL plus vehicle groups (TCL plus TTX: _r_2 = −0.44,p = 0.79; TCL plus vehicle,_r_2 = −0.16, _p_= 0.52).
The effect of TCL plus TTX on spontaneous cortical activity was present at 1 d after lesion. To evaluate any effect of TTX on the degree of neuronal degeneration at this time point, we compared the effect of TCL plus TTX versus TCL plus vehicle 1 d after the lesion by measuring the number of degenerating neurons quantitatively in the two groups using fluorojade B histochemistry (Schmued and Hopkins, 2000;Zuch et al., 2000), a marker of both necrotic and apoptotic cells (Zuch et al., 2000). There was no difference in the number of degenerating neurons in the sampled brain region between TCL plus TTX, 261 ± 72 cells, and TCL plus vehicle, 257 ± 85 cells (Mann–Whitney_U_ test; z = −0.104; p = 0.92;n = 5 for each group).
Blockade of synchronous neuronal activity after cortical lesions blocks axonal sprouting
To assay the effects of blocking the TCL-induced synchronous cortical activity on axonal sprouting, neuroanatomical tract tracing was performed in rats with and without TTX infusion after TCL. BDA was injected into frontal cortex contralateral to TCL plus TTX and TCL plus vehicle 28 d after the lesion was produced, and these results were compared with data previously obtained in rats with TCL alone, ASP, and in sham. Cortical projections are heavily labeled in the dorsolateral striatum ipsilateral to the injection site in all experimental groups (Fig. 7A3–D3). A smaller contralateral corticostriatal projection is present in sham animals (Fig. 7A1,A2). Axonal sprouting after TCL has produced a substantial increase in the labeled axons projecting to the contralateral striatum (Fig. 7B1,B2) that is not altered by vehicle infusion (Fig.7C1,C2). TTX infusion blocks this increase in axonal labeling in contralateral striatum (Fig.7D1,D2). Quantitative analysis of the ratio of the surface area of the corticostriatal projection confirmed these observations (Fig. 6F). There was no statistical difference in the pattern of striatal labeling between TCL plus vehicle and TCL alone. Comparison of sham, TCL, TCL plus vehicle, and TCL plus TTX showed that the pattern of axonal labeling in TCL plus TTX was significantly different from TCL plus vehicle (rostral section,F(1,6) = 51.64; mid section_F_(1,6) = 108.39; caudal section_F_(1,6) = 56.29) and from TCL alone (rostral section, F(1,5) = 70.35; mid section F(1,5) = 178.75; caudal section F(1,5) = 15.12) (p < 0.01, Tukey–Kramer _post hoc_test; n = 4 for TTX groups; n = 3 for TCL alone) but not from sham. Thus, blockade of the synchronous neuronal activity induced by TCL blocks axonal sprouting after TCL.
DISCUSSION
Axonal sprouting from neurons in cortex contralateral to the lesion site was induced by an ischemic–thermal lesion, but not by a similarly sized aspiration lesion of cortex. This lesion-induced sprouting response is remarkable in the adult brain: it dramatically expands long distance projections from the cortex of one hemisphere into the striatum of the other. The similar features of tissue damage and axotomy shared by TCL and ASP allowed us to separate alterations common to both lesions from those directly related to axonal sprouting, which are unique to the TCL. We have found that only those cortical lesions that induce axonal sprouting produced a transient network of low-frequency synchronized neuronal activity and that blockade of this activity prevented axonal sprouting. This lesion-induced activity is strikingly different from seizure activity in the hippocampus. Indeed in the hippocampus cellular activity is characterized by repeated afterdischarges: seconds to minutes of sustained synaptic activity, action potential discharges, and behavioral seizures (Racine, 1972;Traub and Jefferys, 1994).
Rhythmic neuronal activity after cortical lesions
TCL induced two types of synchronous activity. The early pattern, with a frequency range of 0.2–2.2 Hz in perilesion cortex 1 d after the lesion, resembles polymorphic delta waves (Gloor et al., 1977; Sharbrough, 1999). In experimental animals, this activity pattern was thought to arise from injury involving subcortical structures (Gloor et al., 1977). However, these older studies did not record longer than 6 hr after lesion. We have found that this activity pattern develops 1 d after the lesion. Polymorphic delta waves have long been recognized in the clinical electrophysiology literature (Sharbrough, 1999), but their significance has remained unknown. Our results suggest that this EEG pattern may be part of a lesion-induced signal for anatomical reorganization within adult brain.
A second, slower and more distributed rhythm became maximal 3 d after the lesion. This activity pattern crossed functional cortical areas, occurring over much of the contralateral hemisphere and in bilateral occipital areas. The distribution and synchrony of the slow waves and neuronal bursting in this rhythm suggest that it involves a network of cortical and subcortical structures (Steriade, 1998). The timing of synchronous activity first in perilesion cortex then in cortex contralateral to the lesion suggests a progression from sites adjacent to the damage to those in distant, connected areas. The fact that blockade of neuronal activity in perilesion cortex prevents later rhythmic activity in contralateral cortex also suggests a progression of rhythmic activity from the lesion site to connected areas. The underlying mechanism of these two types of patterned activity remains unclear. Our data do not allow a determination of whether perilesional or contralateral activity patterns, or both, are important for axonal sprouting. However the data clearly show that this distributed pattern of cortical activity develops only after the progressive destruction associated with ischemic–thermal injury and not the immediate cortical injury that occurs after aspiration.
We have previously shown that these two types of lesions do not induce differential changes in the expression levels of many molecules that could directly influence sprouting, including chondroitan sulfate proteoglycan, laminin, tenascin, β fibroblast growth factor, glial fibrillary acidic protein, or the polysialylated from of the neural cell adhesion molecule (Szele et al., 1995). However, ASP but not TCL does induce a marked increase in the mRNA encoding NogoA, a myelin-associated inhibitory protein, both in the surrounding cortex and underlying corpus callosum (Shomer and Chesselet, unpublished observations).
One major difference between these two lesion types pertains to timing. Unlike the acute injury of cortical aspiration, ischemic cortical injury produces a progression of tissue damage over days (Salin and Chesselet, 1992; Szele et al., 1995; Wei et al., 1998) that may account for the different physiological responses of cortex to these two lesions. Baseline neuronal firing frequency increases as early as 1 d after ischemic lesions (Schiene et al., 1996), the time point of rhythmic multiunit firing in the perilesion cortex in the present study. In vitro experiments have shown that ischemic injury results in prolonged EPSPs to afferent stimulation, diminished IPSPs and paired pulse inhibition (Neumann-Haeflin et al., 1995;Mittmann et al., 1998), and a facilitated induction of LTP (Hagemann et al., 1998) days to weeks after the lesion. In the present_in vivo_ study a lesion-specific state of repetitive, synchronous neuronal activity in cortical networks was necessary for axonal sprouting in the days during which the lesion is still evolving (Salin and Chesselet, 1992; Szele et al., 1995). Thus, although equivalent amounts of cortex may be lost after brain lesions, the mechanisms and timing of actual damage contain important variables in both the physiological and anatomical processes of neuroplasticity.
TTX blocked the development of synchronous neuronal activity after TCL. The direct diffusion distance of TTX is difficult to determine in this model. TTX currently cannot be produced in a radiolabeled or biologically tagged form, and electrophysiologically establishing the area of TTX influence would require cannula–head set removal and serial microelectrode penetrations, causing secondary lesioning of the TCL site and adjacent cortex. Nevertheless, four lines of evidence suggest that the TTX effect on axonal sprouting was attributable to a specific blockade of the synchronous activity signal, rather than to a general effect of TTX in more distant brain regions. First, there was no direct effect of TTX on the contralateral cortex, the site of the cell bodies of the sprouting axons. Second, a concentration of TTX just sufficient to block local action potential activity in peri-infarct cortex was used. Third, in developing brain similar concentrations of TTX have a limited diffusion distance (Reiter et al. 1986) that is likely to be substantially smaller in adult brain (Lehmenkühler et al., 1993). Fourth, TTX was infused into an ischemic lesion, a site of substantially reduced extracellular diffusion (Li et al., 1999; Liu et al., 2001). Therefore, our data indicate that ipsilateral action potential blockade prevents ipsilateral and contralateral synchronous activity after TCL.
Effect of synchronous neuronal activity on axonal sprouting
Lesion-induced axonal sprouting was prevented when synchronous activity was blocked by TTX. TTX is unlikely to have had an effect on the uptake and transport of BDA, because BDA was injected into the cortex contralateral to the infusion site and, furthermore, anterograde tracers with similar uptake and transport properties to BDA (Reiner et al., 2000) are unaffected by TTX activity blockade (Olson and Meyer, 1994). Moreover, a diffuse effect of TTX in the striatum deep to the infusion site or beyond the perilesion cortex might be expected to increase axonal sprouting (Catalona and Shatz, 1998; Cohen-Cory, 1999), whereas the exact opposite effect is reported here. Therefore, the data show a very strong association between the patterns of neuronal activity induced by TCL and the sprouting of crossed corticostriatal projections into the denervated striatum. It will be interesting to impose low-frequency synchronous activity similar to that observed after TCL to the cortex of animals with ASP lesions to further establish the role of patterned neuronal activity in inducing axonal sprouting.
In our model, statistical differences in the pattern of cortical activity between TCL, sham, and ASP were limited to the first 3 d after the lesion, and thus we propose that early synchronous neuronal activity is an initial trigger for axonal sprouting. In other cortical injury models, dendritic sprouting occurs early after the lesion, in the sensorimotor cortex contralateral to electrolytic forelimb cortex lesions, but is then partially pruned back to control levels after this period (Jones and Schallert, 1994). This late pruning phase can be prevented by MK-801 administration, suggesting that neuronal activity or glutamate receptors are involved in delayed neuronal reorganization after cortical lesions (Kozlowski and Schallert, 1998). In contrast, in our model axonal sprouting in the denervated striatum is maintained for at least 1 month after the lesion. Therefore, the transient nature of the electrophysiological activity we have observed may be critical for the induction and maintenance of axonal sprouting after ischemic brain lesions.
Although the type of activity we have observed has never, to our knowledge, been associated with axonal sprouting in the adult brain, synchronous cellular activity is widely present in many CNS regions during the development of axonal projections (Feller, 1999). However, the prevalent hypothesis concerning the role of cellular activity during development has been that it primarily plays a role in the refinement of already formed connections (Tessier-Lavigne and Goodman, 1996; McCormick, 1999). Recent studies have begun to challenge this idea by showing that activity also may play a role in axonal pathfinding between neuronal groups (Catalano and Shatz, 1998; Dantzker and Callaway, 1998; Goldberg et al., 2002). Similarly, there is emerging evidence from in vitro growth cone guidance studies that cellular activity could alter cues for axonal pathfinding (Ming et al., 2001). Although the synchronous neuronal activity associated with the development of neuronal connections has a very low frequency in subcortical projections, developing cortical circuits exhibit spontaneous rhythmic activity at similar frequencies to that reported in the present study (Weliky and Katz, 1999). Thus, the sprouting of new connections in the adult brain after a lesion is associated with electrical rhythms that could play a role in a similar process in the developing animal.
Although our data indicate that axonal sprouting does not occur in the absence of the slow rhythmic waves induced by TCL, they do not distinguish between a permissive or inductive role for this activity on axonal growth. Patterned cellular activity could play a permissive role by altering the normally inhibitory adult cellular environment. In support of this, short bursts of low-frequency neuronal activity have been shown to alter the inhibitory effect of myelin and netrin-1 into attractive growth cone cues for developing neurons (Ming et al., 2001). ASP does induce a small number of growth cones in the region immediately adjacent to the corpus callosum. These do not invade the denervated dorsolateral striatum and hence do not lead to the substantial axonal sprouting response seen after TCL (Uryu et al., 2001). This suggests that the small, localized growth cone response seen after ASP is restricted by an inability to overcome an environment that is inhibitory to axonal growth in the absence of the synchronous cellular activity signal. An alternative but nonexclusive possibility is that the rhythmic slow wave activity produces a pattern of gene expression in the contralateral cortex that induces axonal sprouting.
In conclusion, this study shows that axonal sprouting in long distance connections after cortical insults in the mature brain is associated with the same type of periodic synchronized neuronal activity known to be an important organizing force in the formation of new connections in the developing brain. These findings provide new insights into how the brain reorganizes as a result of injury and may provide important clues for the improvement of recovery from stroke and other CNS insults. They also suggest that consideration should be given to the effects of future neuroprotective therapies on patterns of neuronal activity in addition to their direct effects on neurodegeneration.
Footnotes
This work was supported by a Howard Hughes Medical Institute Postdoctoral Research Fellowship for Physicians (S.T.C.) and National Institutes of Health Grant NS 29230. We thank Drs. Charles Wilson and Anatol Bragin for technical assistance.
Correspondence should be addressed to Dr. S. Thomas Carmichael, Department of Neurology, University of California Los Angeles School of Medicine, 710 Westwood Plaza, Los Angeles, CA 90095. E-mail:scarmichael@mednet.ucla.edu.
REFERENCES
- 1.Abeles M. Quantification, smoothing, and confidence limits for single-units' histograms. J Neurosci Methods. 1982;5:317–325. doi: 10.1016/0165-0270(82)90002-4. [DOI] [PubMed] [Google Scholar]
- 2.Carmichael ST, Wei L, Rovainen CM, Woolsey TA. New patterns of intra-cortical projections after focal cortical stroke. Neurobiol Dis. 2001;8:910–922. doi: 10.1006/nbdi.2001.0425. [DOI] [PubMed] [Google Scholar]
- 3.Catalona SM, Shatz CJ. Activity-dependent cortical target selection by thalamic axons. Science. 1998;281:559–562. doi: 10.1126/science.281.5376.559. [DOI] [PubMed] [Google Scholar]
- 4.Cohen-Cory S. BDNF modulates, but does not mediate, activity-dependent branching and remodeling of optic axon arbors in vivo. J Neurosci. 1999;19:9996–10003. doi: 10.1523/JNEUROSCI.19-22-09996.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Colder BW, Wilson CL, Frysinger RC, Chao LC, Harper RM, Engel J., Jr Neuronal synchrony in relation to burst discharges in epileptic human temporal lobes. J Neurophysiol. 1996;75:2496–2508. doi: 10.1152/jn.1996.75.6.2496. [DOI] [PubMed] [Google Scholar]
- 6.Dantzker JL, Callaway EM. The development of local, layer-specific visual cortical axons in the absence of extrinsic influences and intrinsic activity. J Neurosci. 1998;18:4145–4154. doi: 10.1523/JNEUROSCI.18-11-04145.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Feller MB. Spontaneous correlated activity in developing neural circuits. Neuron. 1999;22:653–656. doi: 10.1016/s0896-6273(00)80724-2. [DOI] [PubMed] [Google Scholar]
- 8.Gloor P, Ball G, Schaul N. Brain lesions that produce delta waves in the EEG. Neurology. 1977;27:326–333. doi: 10.1212/wnl.27.4.326. [DOI] [PubMed] [Google Scholar]
- 9.Goldberg JL, Espinosa JS, Xu Y, Davidson N, Kovacs GTA, Barres BA. Retinal ganglion cells do not extend axons by default: promotion by neurotrophic signaling and electrical activity. Neuron. 2002;33:689–702. doi: 10.1016/s0896-6273(02)00602-5. [DOI] [PubMed] [Google Scholar]
- 10.Hagemann G, Redecker C, Neumann-Haeflin, Freund HJ, Witte OW. Increased long-term potentiation in the surround of experimentally induced focal cortical infarction. Ann Neurol. 1998;44:255–258. doi: 10.1002/ana.410440217. [DOI] [PubMed] [Google Scholar]
- 11.Heimer L, Zahn DS, Alheid GF. Basal ganglia. In: Paxinos G, editor. The rat nervous system. Ed 2. Academic; San Diego: 1995. pp. 579–628. [Google Scholar]
- 12.Hossmann KA. Periinfarct depolarizations. Cerebrovasc Brain Metab Rev. 1996;8:195–208. [PubMed] [Google Scholar]
- 13.Jablonska B, Gierdalski M, Kublik A, Skangiel-Kramska J, Kossut M. Effects of implantation of Alzet 1007D osmotic minipumps upon 2-deoxyglucose uptake in the cerebral cortex of mice. Acta Neurobiol Exp. 1993;53:577–580. [PubMed] [Google Scholar]
- 14.Jones TA, Schallert T. Use-dependent growth of pyramidal neurons after neocortical damage. J Neurosci. 1994;14:2140–2152. doi: 10.1523/JNEUROSCI.14-04-02140.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kasamatsu T, Schmidt EK. Continuous and direct infusion of drug solutions in the brain of awake animals: implementation, strengths and pitfalls. Brain Res Protocols. 1997;1:57–69. doi: 10.1016/s1385-299x(96)00008-6. [DOI] [PubMed] [Google Scholar]
- 16.Kozlowski DA, Schallert T. Relationship between dendritic pruning and behavioral recovery following sensorimotor cortex lesions. Behav Brain Res. 1998;97:89–98. doi: 10.1016/s0166-4328(98)00030-8. [DOI] [PubMed] [Google Scholar]
- 17.Lehmenkühler A, Syková E, Svoboda J, Zilles K, Nicholson C. Extracellular space parameters in the rat neocortex and subcortical white matter during postnatal development determined by diffusion analysis. Neuroscience. 1993;55:339–351. doi: 10.1016/0306-4522(93)90503-8. [DOI] [PubMed] [Google Scholar]
- 18.Li TQ, Chen ZG, Hindmarsh T. Diffusion-weighted MR imaging of acute cerebral ischemia. Acta Radiol. 1999;39:460–473. doi: 10.1080/02841859809172209. [DOI] [PubMed] [Google Scholar]
- 19.Liu KF, Li F, Tatlisumak T, Garcia JH, Sotak CH, Fisher M, Fenstermacher JD. Regional variations in the apparent diffusion coefficient and the intracellular distribution of water in rat brain during acute focal ischemia. Stroke. 2001;32:1897–1905. doi: 10.1161/01.str.32.8.1897. [DOI] [PubMed] [Google Scholar]
- 20.McCormick DA. Spontaneous activity: signal or noise. Science. 1999;285:541–543. doi: 10.1126/science.285.5427.541. [DOI] [PubMed] [Google Scholar]
- 21.Ming G, Henley J, Tessier-Lavigne M, Song H, Poo M. Electrical activity modulates growth cone guidance by diffusible factors. Neurons. 2001;29:441–452. doi: 10.1016/s0896-6273(01)00217-3. [DOI] [PubMed] [Google Scholar]
- 22.Mittmann T, Qü M, Zilles K, Luhmann HJ. Long-term cellular dysfunction after focal cerebral ischemia: in vitro analyses. Neuroscience. 1998;85:15–27. doi: 10.1016/s0306-4522(97)00638-6. [DOI] [PubMed] [Google Scholar]
- 23.Napieralski JA, Butler AK, Chesselet M-F. Anatomical and functional evidence for lesion-specific sprouting of corticostriatal input in the adult rat. J Comp Neurol. 1996;373:484–497. doi: 10.1002/(SICI)1096-9861(19960930)373:4<484::AID-CNE2>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 24.Neumann-Haeflin T, Hagemann G, Witte OW. Cellular correlates of neuronal hyperexcitability in the vicinity of photochemically induced cortical infarcts in the rat in vitro. Neurosci Lett. 1995;193:101–104. doi: 10.1016/0304-3940(95)11677-o. [DOI] [PubMed] [Google Scholar]
- 25.Olson MD, Meyer RL. Activity-dependent and retinotopic refinement in a low-density retinotectal projection in the goldfish: evidence favoring synaptic cooperation over competition. J Neurosci. 1994;14:208–218. doi: 10.1523/JNEUROSCI.14-01-00208.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Paxinos P, Watson C. The rat brain in stereotaxic coordinates, Ed 3. Academic; San Diego: 1997. [DOI] [PubMed] [Google Scholar]
- 27.Racine RJ. Modification of seizure activity by electrical stimulation: II. Motor seizure. Electroencalogr Clin Neurophysiol. 1972;32:281–294. doi: 10.1016/0013-4694(72)90177-0. [DOI] [PubMed] [Google Scholar]
- 28.Reiner A, Veenman CL, Medina L, Jiao Y, Del Mar N, Honig MG. Pathway tracing using biotinylated dextran amines. J Neurosci Methods. 2000;103:23–37. doi: 10.1016/s0165-0270(00)00293-4. [DOI] [PubMed] [Google Scholar]
- 29.Reiter HO, Waitzman DM, Stryker MP. Cortical activity blockade prevents ocular dominance plasticity in the kitten visual cortex. Exp Brain Res. 1986;65:182–188. doi: 10.1007/BF00243841. [DOI] [PubMed] [Google Scholar]
- 30.Salin P, Chesselet M-F. Paradoxical increase in striatal neuropeptide gene expression following ischemic lesions of the cerebral cortex. Proc Natl Acad Sci USA. 1992;89:9954–9958. doi: 10.1073/pnas.89.20.9954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schiene K, Breuhl C, Zilles K, Qü M, Hagemann G, Kraemer M, Witte OW. Neuronal hyperexcitability and reduction of GABAA-receptor expression in the surround of cerebral photothrombosis. J Cereb Blood Flow Metab. 1996;1:906–914. doi: 10.1097/00004647-199609000-00014. [DOI] [PubMed] [Google Scholar]
- 32.Schmued LC, Hopkins KJ. Fluoro-jade B: a high affinity fluorescent marker for the localization of neuronal degeneration. Brain Res. 2000;874:123–130. doi: 10.1016/s0006-8993(00)02513-0. [DOI] [PubMed] [Google Scholar]
- 33.Sears TA, Stagg D. Short-term synchronization of the intercostal motoneurone activity. J Physiol (Lond) 1976;263:357–381. doi: 10.1113/jphysiol.1976.sp011635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sharbrough FW. Nonspecific abnormal EEG patterns. In: Niedermeyer F, Lopes Da Silva F, editors. Electroencephalography. Basic principles, clinical applications and related fields. Ed 4. Williams and Wilkins; Baltimore: 1999. pp. 215–234. [Google Scholar]
- 35.Steriade M. Corticothalamic networks, oscillations and plasticity. Adv Neurol. 1998;77:105–134. [PubMed] [Google Scholar]
- 36.Steward O, Vinsant SL, Davis L. The process of reinnervation in the dentate gyrus of adult rats: an ultrastructural study of changes in presynaptic terminals as a result of sprouting. J Comp Neurol. 1988;267:203–210. doi: 10.1002/cne.902670205. [DOI] [PubMed] [Google Scholar]
- 37.Szele FG, Alexander C, Chesselet M-F. Expression of molecules associated with neuronal plasticity after aspiration and thermocoagulatory lesions of the cerebral cortex in adult rats. J Neurosci. 1995;15:4429–4448. doi: 10.1523/JNEUROSCI.15-06-04429.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tessier-Lavigne M, Goodman CS. The molecular biology of axon guidance. Science. 1996;274:1123–1133. doi: 10.1126/science.274.5290.1123. [DOI] [PubMed] [Google Scholar]
- 39.Traub RD, Jefferys JG. Are there unifying principles underlying the generation of epileptic afterdischarges in vitro? Prog Brain Res. 1994;102:383–394. doi: 10.1016/S0079-6123(08)60554-3. [DOI] [PubMed] [Google Scholar]
- 40.Uryu K, MacKenzie L, Chesselet M-F. Ultrastructural evidence for differential axonal sprouting in the striatum after thermocoagulatory and aspiration lesions of the cerebral cortex in adult rats. Neuroscience. 2001;105:307–316. doi: 10.1016/s0306-4522(01)00203-2. [DOI] [PubMed] [Google Scholar]
- 41.Wei L, Craven K, Erinjeri J, Liang GE, Bereckzi D, Rovainen CM, Woolsey TA. Local cerebral blood flow during the first hour following acute ligation of multiple arterioles in rat whisker barrel cortex. Neurobiol Dis. 1998;5:142–158. doi: 10.1006/nbdi.1998.0199. [DOI] [PubMed] [Google Scholar]
- 42.Weliky M, Katz LC. Correlational structure of spontaneous neuronal activity in the developing lateral geniculate nucleus in vivo. Science. 1999;285:599–604. doi: 10.1126/science.285.5427.599. [DOI] [PubMed] [Google Scholar]
- 43.Zuch CL, Nordstroem VK, Briedrick LA, Hoernig GR, Granholm A-C, Bickford PC. Time course of degenerative alterations in nigral dopaminergic neurons following a 6-hydroxydopamine lesion. J Comp Neurol. 2000;427:440–454. doi: 10.1002/1096-9861(20001120)427:3<440::aid-cne10>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]