Potassium-Coupled Chloride Cotransport Controls Intracellular Chloride in Rat Neocortical Pyramidal Neurons (original) (raw)
Abstract
Chloride (Cl−) homeostasis is critical for many cell functions including cell signaling and volume regulation. The action of GABA at GABAA receptors is primarily determined by the concentration of intracellular Cl−. Developmental regulation of intracellular Cl− results in a depolarizing response to GABA in immature neocortical neurons and a hyperpolarizing or shunting response in mature neocortical neurons. One protein that participates in Cl− homeostasis is the neuron-specific K+–Cl− cotransporter (KCC2). Thermodynamic considerations predict that in the physiological ranges of intracellular Cl− and extracellular K+ concentrations, KCC2 can act to either extrude or accumulate Cl−. To test this hypothesis, we examined KCC2 function in pyramidal cells from rat neocortical slices in mature (18–28 d postnatal) and immature (3–6 d postnatal) rats. Intracellular Cl− concentration was estimated from the reversal potential of whole-cell currents evoked by local application of exogenous GABA. Both increasing and decreasing the extracellular K+ concentration resulted in a concomitant change in intracellular Cl−concentration in neurons from mature rats. KCC2 inhibition by furosemide caused a change in the intracellular Cl−concentration that depended on the concentration of pipette Cl−; in recordings with low pipette Cl−, furosemide lowered intracellular Cl−, whereas in recordings with elevated pipette Cl−, furosemide raised intracellular Cl−. In neurons from neonatal rats, manipulation of extracellular K+ had no effect on intracellular Cl− concentration, consistent with the minimal KCC2 mRNA levels observed in neocortical neurons from immature animals. These data demonstrate a physiologically relevant and developmentally regulated role for KCC2 in Cl− homeostasis via both Cl− extrusion and accumulation.
Keywords: potassium chloride cotransporter, GABA receptors, KCC2, development, chloride homeostasis, rt-PCR
The principal inhibitory neurotransmitter in the neocortex is GABA. Early studies_in vivo_ demonstrated that fast IPSPs and responses to iontophoretically applied GABA have similar reversal potentials and ionic sensitivities (Krnjevic and Schwartz, 1967; Dreifuss et al., 1969). Subsequent studies in vitro showed that both GABA responses and IPSPs reverse near the expected chloride (Cl−) equilibrium potential and are blocked by bicuculline, suggesting mediation by GABAA receptors (GABAARs) (Weiss and Hablitz, 1984; Howe et al., 1987). The main permeant ion of GABAA receptor channel complexes is Cl−, although permeability to bicarbonate ions has been demonstrated (Bormann et al., 1987; Kaila et al., 1993). The membrane potential and the transmembrane gradients of permeant ions determine ionic flux through the GABAA receptor. In most mature neurons, the resting potential is close to the Cl− equilibrium potential, and activation of GABAARs results in shunting inhibition (Andersen et al., 1980); i.e., activation of GABAA receptors with minimal net ionic flux results in decreased excitability. If the neuron is depolarized relative to the Cl− equilibrium potential, GABAA receptor activation results in an inward flux of Cl− that hyperpolarizes the neuron. Immature neocortical neurons have an elevated [Cl−]i, and GABAAR activation results in a depolarizing response to GABA (Owens et al., 1996).
Electroneutral cotransport of Cl− ions plays a critical role in [Cl−]ihomeostasis (see Alvarez-Leefmans, 1990; Kaila et al., 1993). One mechanism for accumulating Cl− is the Na+–K+–2 Cl− cotransporter (NKCC) (Kakazu et al., 1999). The primary Cl− extrusion mechanism is K+–Cl−cotransport (Alvarez-Leefmans, 1990). A neuron-specific form of the ubiquitous K+-coupled Cl− cotransporter (KCC2) has been characterized recently (Payne, 1997; Rivera et al., 1999; Williams et al., 1999). The expression of this transporter increases with development and is believed to support the developmental changes in GABAAR-mediated signaling (Lu et al., 1999;Rivera et al., 1999).
The direction of K+–Cl−cotransport is determined by the transmembrane K+ and Cl−gradients. Under conditions of low [Cl−]i and elevated [K+]o, thermodynamic considerations suggest that KCC2 could operate in reverse to accumulate Cl− (Payne, 1997; Jarolimek et al., 1999; see also Kakazu et al., 2000). A role for KCC2 in maintaining [K+]o, i.e., that KCC2 could lower [K+]o by cotransport of K+ and Cl− ions into neurons, has also been proposed (Payne, 1997). A consequence of such a mechanism would be the accumulation of [Cl−]i, a phenomenon consistent with activity-dependent decreases in GABAergic inhibition (Thompson and Gähwiler, 1989a; Ling and Benardo, 1995).
In the present study, we tested the hypothesis that developmental changes in the expression of KCC2 result in the coupling of [Cl−]i and [K+]o via the activity of the furosemide-sensitive K+-coupled Cl− cotransporter. Our results demonstrate that manipulations of [K+]o and furosemide altered [Cl−]i in a manner consistent with either accumulation or extrusion of Cl− via a K+-coupled Cl− cotransport mechanism. [K+]omanipulations in immature neurons had no effect on [Cl−]i, consistent with the low expression of KCC2 mRNA detected in cytoplasm harvested from these cells. Expression of KCC2 results in lowered [Cl−]i and translates physiological changes in [K+]o to marked changes in [Cl−]i.
MATERIALS AND METHODS
Brain slice preparation, maintenance, and electrophysiological recording. Animals were housed and handled according to approved guidelines. Brain slices were prepared from postnatal day 3 (P3) to P6 and P18 to P28 animals. Rats were anesthetized with ketamine (100 mg/kg) before decapitation. The brain was rapidly removed and submerged in oxygenated (95% O2/5% CO2) ice-cold saline with no added calcium [containing (in mm ):125 NaCl, 3.5 KCl, 26 NaHCO3, 10 d-glucose, and 4 MgCl2]. Coronal sections (300 μm) containing somatosensory cortex were cut with a Vibratome. Slices were stored in saline consisting of (in mm ): 125 NaCl, 5 KCl, 26 NaHCO3, 10 d-glucose, 2.5 CaCl2, and 1.3 MgCl2, bubbled with 95% O2/5% CO2.
Whole-cell voltage-clamp recordings were made from visually identified neocortical pyramidal cells in layer II/III. Cells were identified by their distance from the pial surface, pyramidal shape, and the presence of a prominent apical dendrite. Recordings were made at 30°C and at a holding potential of −70 mV. Pipettes were pulled from 1.5 mm glass capillaries (KG-33; Garner Glass Company). They had resistances of 2–4 MΩ when filled with the intracellular solution. Series resistance (Rs) was carefully monitored throughout each experiment by the use of a small hyperpolarizing voltage step applied before each acquisition (see Fig. 1A, inset). The peak of the capacitative transient was used to estimate series resistance (equal to the instantaneous current divided by the voltage step: Rs =_I_peak/Vstep). Only recordings with stable Rs (<20 MΩ with <5 MΩ change in 20 min) were used in the analysis.
Fig. 1.
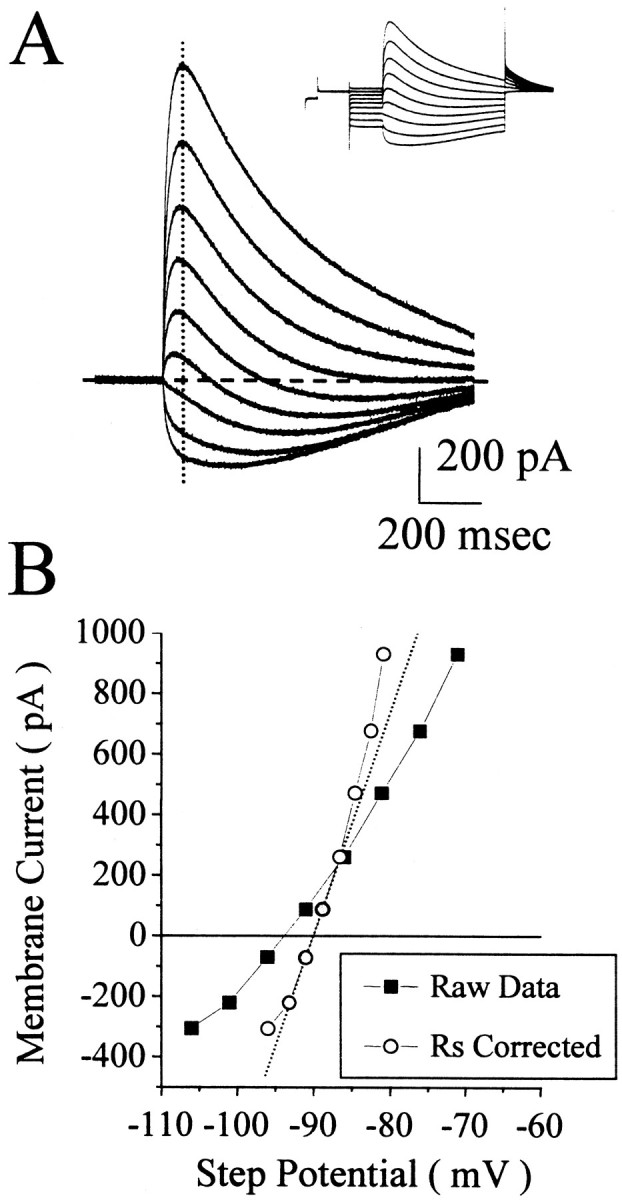
The reversal potential of the GABA-evoked current measured from the amplitudes of the responses at different step potentials. A, The baseline current at each step potential was subtracted from the raw traces (shown in the inset). B, Then the amplitude of the current at the vertical dotted line in _A_was plotted as a function of step potential. _Squares_illustrate the amplitudes as a function of the command potential.Circles represent current amplitude versus membrane potential corrected for the series resistance (Rs) error. A linear fit (dotted line) to the current as a function of the _Rs_-corrected step potential was obtained from the three consecutive data points closest to zero current (gray circles). This recording was obtained from a P18–P28 neuron with 1 mm pipette Cl−.
The internal solutions contained (in mm): 135 K-gluconate, 5 EGTA, 10 HEPES, 2 MgATP, 0.2 NaGTP, and 0.2 CaCl2. KCl was substituted for K-gluconate to arrive at the desired Cl− concentration. The pH was adjusted to 7.3 with 1 mm KOH and HCl such that the final added Cl− concentration equaled 1, 20, or 40 mm and the final [K+]i was always 150 mm. Sucrose was added to achieve a final osmolarity of 300 mOsm. Liquid junction potentials for all solutions were measured, and all voltages reported are corrected values (Neher, 1992).
During recording, slices were continuously perfused with the storage saline listed above with the addition of 0.5 μm TTX (Calbiochem) to block Na+-dependent action potentials, 2–5 mm kynurenic acid (Tocris) to block ionotropic glutamate receptors, and 10 μm SCH50911 (Research Biochemicals, Natick, MA) to antagonize GABAB receptors. Extracellular K+ was varied by making K+-free saline and adding 1, 3.5, or 10 mm KCl. Saline containing 1 mm furosemide was sonicated for 30 min and then oxygenated for at least 30 min before bath application. All drugs (except GABA) were bath applied, and each cell served as its own control. Bath temperature was maintained at 30°C by the use of an in-line heating unit (Warner Instruments). Whole-cell and excised patch recordings (Sakmann and Neher, 1995) were made with an Axopatch-1B amplifier. No series resistance compensation was used. Recordings were digitized at 5–10 kHz by the use of a Digidata 1200 data acquisition system and Clampex software (Axon Instruments). Data analysis was performed with custom scripts written for Origin Pro (Microcal Software). All averages are reported as the mean ± SEM. A Student's t test was used to determine significance (p < 0.05).
Pressure application of GABA. Under direct visual guidance, GABA (250 μm or 1 mm) was pressure applied to the soma of the recorded neuron or to excised patches. Pipettes for pressure applications were fabricated in the same manner as patch electrodes described above. GABA was applied in a solution consisting of (in mm): 125 NaCl, 3.5 KCl, 20 HEPES, and 10 glucose; pH is 7.3 with NaOH. Pressure applications were controlled by the use of a Picospritzer II (General Valve, Fairfield, NJ). Pulses of 5–15 msec were delivered at 3–12 psi. These settings were kept constant during recording. Application of the pressure pipette solution without GABA did not evoke a detectable current (n = 3).
Reverse transcription-PCR. The methods used for determination of mRNA in single neurons have been described (Poth et al., 1997; Devay et al., 1999). In brief, cytoplasm and pipette solution (∼6 μl) were reverse transcribed in a 20 μl reaction (1 hr at 37°C) containing 1 mm each dNTP, 100 pmol of 18-mer polyT, 20 U of RNase inhibitor, and 40 U of AMV reverse transcriptase. Sets of specific primers were constructed such that the annealed products crossed at least one intron/exon boundary, excluding the possibility of amplification of genomic DNA. PCR reactions (50 μl) contained aliquots of the reverse transcriptase (rt) product, 1 mm dNTPs, 2.5 mm MgCl2, 10 pmol of each forward and reverse primer, and 5 U of _Taq_polymerase. The PCR cycling parameters were as follows: 5 cycles of 94°C for 1 min, 55°C for 1 min, and 72°C for 2 min followed by 35 cycles of 94°C for 1 min, 65°C for 1 min, and 72°C for 2 min. The reaction products were then spun through CentriSep columns to remove excess primers and subjected to reamplification (one or two additional amplification steps) by the use of 35 cycles of 94°C for 1 min, 65°C for 1 min, and 72°C for 2 min. Final reaction products were purified by phenol/chloroform extraction. The PCR products were subjected to analysis on 2% agarose gels. rt-PCR was performed without knowledge of the age of the animal or the contents of each centrifuge tube (control extracellular solution or intracellular harvest).
The primers for the experiments were as follows: actin (GenBank accession number L08165) sense, ATCTTTCTTGGGTATGGA, and antisense, ACATCTGCTGGAAGGTGG; KCC2 (GenBank accession number AJ011033) sense, GCAGAGAGTACGATGGCAGG, and antisense, CGTGCCAAGGATGTACATAGC.
_Intracellular chloride calculation._[Cl−]i was calculated from the reversal potential of GABA-evoked currents. Cells were voltage-clamped to −70 mV and stepped to various test potentials. The series resistance (estimated from the peak transient during a 10 mV test pulse given before each trial) and the amplitude of the current (before baseline correction, see Fig. 1A,inset) 100–200 msec from the time of the pressure application were used to calculate the voltage error caused by uncorrected series resistance errors. The baseline current (taken 20 msec before the time of the pressure pulse) was subtracted from the absolute current amplitude (see Fig. 1A, baseline-corrected traces). These values were plotted as a function of the series resistance-corrected membrane potential (see Fig. 1B). The three consecutive data points whose sum was closest to zero current were selected for a linear fit. The_x_-axis intercept (y = 0) of this fit was verified by eye and referred to throughout this paper as the reversal potential (Vrev). [Cl−]i was calculated from the reversal potential by the use of a derivation from the Nernst equation:
[Cl−]in=[Cl−]outexp(−VrevF/RT).
[Cl−]o was set to the extracellular Cl−concentration, corrected for activity (Robinson and Stokes, 1959) (e.g., in 3.5 mm[K+]o, 139.6 mm × 76% = 106.1 mm). Averaged changes in reversal potential and [Cl−]i reflect the mean of the differences from the control condition (3.5 mm[K+]o) for each cell; thus, each cell served as its own control.
The role of bicarbonate and other anions. Bicarbonate is known to permeate GABAA receptors (Bormann et al., 1987). We did not account for a bicarbonate component of the GABAA current because our results suggest that bicarbonate does not contribute significantly to the whole-cell currents under our recording conditions. When 1 mmCl− was included in the pipette, the reversal potential of GABAA-mediated currents was depolarized relative to the reversal potential obtained in excised patches (see Fig. 2A). This result is consistent with a bicarbonate efflux caused by 20 mmintracellular bicarbonate and a permeability ratio of 1:5 (HCO3−/Cl−). However, lowering [K+]o and cotransport antagonism with furosemide both lowered the reversal potential to values similar to those obtained in excised patches (see Results). Because these types of manipulations are not expected to affect intracellular bicarbonate, changes in Vrevare attributed to alterations in Cl− and K+–Cl−cotransport.
Fig. 2.
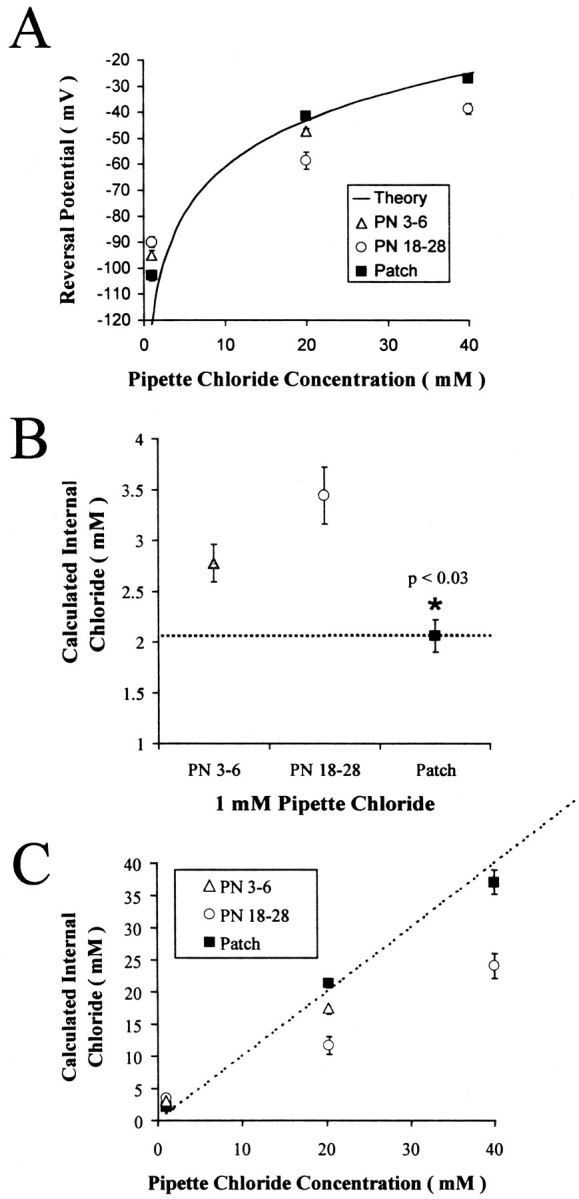
Reversal potentials for GABAA currents recorded with different pipette Cl− concentrations support Cl− accumulation and extrusion in neocortical pyramidal cells. A, Reversal potentials obtained at three pipette Cl− concentrations in postnatal days 3–6 (PN 3–6), PN 18–28, and outside-out patches are plotted as a function of pipette Cl− concentration. For comparison, the theoretical relationship between [Cl−]i and reversal potential (assuming 103.4 mm[Cl−]o) is shown as a_solid line_. Reversal potentials obtained from outside-out patches were closest to theoretical. The reversal potentials from PN 3–6 and PN 18–28_were significantly different from outside-out patches at all pipette Cl− concentrations (p < 0.03). B, Mean calculated [Cl−]i was significantly lower in outside patches (∗) (p < 0.03) and 1.1 mm higher than the added KCl concentration (1 mm). The differences in whole-cell [Cl−]i from the excised patch values suggest a Cl− accumulation mechanism in both neonatal and PN 18–28 neurons. C, Calculated [Cl−]i is plotted as a function of pipette Cl− concentration. As in_A both PN 3–6 and PN 18–28 neurons had significantly lower calculated [Cl−]i compared with that in excised patches, suggesting the action of a Cl− extrusion mechanism.
Another source of error was the purity of the compound providing the primary anion in the internal solution. We obtained potassium-d-gluconate, “puriss” grade, from Fluka. Other sources of the potassium salts of isethionate (Eastman Kodak) and methylsulfate (J. T. Baker Chemical Company) resulted in reversal potential measurements indicative of 10–15 mm internal Cl− when no Cl− had been added to the pipette solution. This suggests that these compounds were contaminated with significant amounts of chloride (or another substance permeable through GABAA receptors). It has also been reported that gluconate can pass through GABAA channels (Barker and Harrison, 1988). Our results with excised patches demonstrated an effective concentration of pipette Cl− of 2.06 ± 0.16 mm when only 1 mm KCl was added. Assuming that this difference from added Cl− is caused solely by gluconate permeability, the relative permeability of gluconate to Cl− is ∼0.008.
RESULTS
Disparity between pipette chloride and calculated intracellular chloride
We assessed the intracellular Cl−concentration of neocortical pyramidal neurons by measuring the reversal potential of GABAA receptor-mediated currents. Responses to pressure application of 250 μmGABA are shown in Figure1A. The pipette Cl− concentration was 1 mm in this recording. The _inset_illustrates the currents evoked by the voltage step protocol and local pressure application of GABA. The currents are shown superimposed on a faster time base after zeroing the baseline currents. GABA response amplitude was measured 100–200 msec after the pressure pulse, at the time indicated by the vertical dotted line. Response amplitude is plotted as a function of membrane potential in Figure1B (squares). After correction for the series resistance error (circles), the reversal potential was calculated from a linear fit to the three consecutive data points closest to zero current (dotted line). This indicated a Vrev of −89.9 mV, >30 mV depolarized from the expected value of −121 mV calculated for 1 mm[Cl−]i.
The observed discrepancy in Vrev implies the existence of a homeostatic mechanism that regulates [Cl−]i. To test this hypothesis and to determine whether such a mechanism is developmentally regulated, measurements of Vrevwere made in P3–P6 and P18–P28 animals at various pipette Cl− concentrations. Reversal potentials determined for each of these experimental conditions, at a [K+]o of 3.5 mm, are shown in Figure2A. The mean reversal potentials for the three pipette chloride concentrations are shown for recordings from P18 to P28 (white circles) and P3 to P6 (gray triangles) neurons. This plot also shows the theoretical relationship between [Cl−]i and the reversal potential (solid line). In whole-cell recordings with 1 mm pipette chloride, the reversal potential in both P3–P6 and P18–P28 neurons was always more depolarized than the value predicted from the Nernst equation. This implies the existence of a Cl−accumulation mechanism in both groups. At higher pipette chloride concentrations (20 and 40 mm) the measured reversal potential was consistently below the predicted reversal potential, consistent with a Cl−extrusion mechanism.
We also estimated reversal potentials in excised patches, reasoning that the large volume of the pipette solution should minimize the effects of Cl− homeostatic mechanisms residing in the small patch of membrane in the pipette tip. The reversal potentials from excised patches were closer to values calculated from the expected pipette [Cl−] than to those obtained in whole-cell recordings, as shown in Figure 2A(black squares). Also, the reversal potentials in excised patches were insensitive to manipulations that effected KCC2 function in whole-cell recordings (changes in [K+]o and furosemide; data not shown). These data support the existence of homeostatic [Cl−]i regulation involving a Cl− accumulation mechanism at low pipette Cl− concentrations and a Cl− extrusion mechanism when pipette Cl− is elevated.
Figure 2B plots the calculated [Cl−]i for each group when the pipette contained 1 mmCl−. In P18–P28 neurons, [Cl−]i was 3.73 ± 0.35 mm (n = 11), whereas it was significantly lower in P3–P6 cells (2.82 ± 0.16 mm;p < 0.04; _n_= 5). Recordings from excised patches indicated a pipette Cl− concentration of 2.10 ± 0.16 mm (n = 4). Estimates of [Cl−]i in P3–P6 and P18–P28 neurons were significantly greater than that in excised patches (p < 0.03). If we assume that the reversal potential of currents in excised patches with 1 mm pipette Cl−reflects the actual pipette Cl−concentration, these results suggest that in 3.5 mm[K+]o and with low pipette Cl−, P3–P6 and P18–P28 neurons can accumulate 0.7–1.6 mmCl−.
Figure 2C plots the calculated [Cl−]i as a function of pipette chloride concentration. In 3.5 mm[K+]o and elevated pipette Cl−, the reversal potentials of whole-cell currents evoked by GABA were consistently lower than the expected values calculated from the Nernst equation. They were also lower than the values obtained in outside-out patches. With 20 mm pipette Cl−, the calculated [Cl−]iin P3–P6 neurons was 17.55 ± 0.71 mm(n = 5), whereas in P18–P28 neurons, [Cl−]i was significantly lower (11.9 ± 1.1 mm;p < 0.002; n = 11). [Cl−]i determined in excised patches was significantly higher than that in either P3–P6 or P18–P28 neurons (21.69 ± 0.72 mm;n = 7; p < 0.005). Assuming that the reversal potential in excised patches reflects the actual pipette Cl− concentration, our results imply that the neuronal Cl− homeostatic mechanisms can extrude 4–9 mmCl− in 3.5 mm[K+]o. These data support a prominent role for Cl−extrusion in determining [Cl−]i in these neurons.
Alterations in [K+]o affect [Cl−]i
If K+-coupled Cl− cotransport is involved in the accumulation and extrusion of Cl−demonstrated above, manipulations of [K+]o should change the reversal potential of the GABA-evoked currents. Lowering [K+]o should lower the Cl− set point and thus enhance extrusion and/or retard accumulation. Raising [K+]o should have the opposite effect by raising the Cl−set point via impairment of extrusion and/or enhancement of accumulation.
Figure 3 illustrates the effect of decreasing [K+]oon whole-cell responses to GABA. The recordings were obtained from a P18–P28 neuron by the use of 1 mmCl− in the recording pipette. GABA responses in 3.5 and 1 mm[K+]o are shown in Figure 3A. At a holding potential of −85 mV, the GABA-evoked current changes from inward to outward when the [K+]o was changed from 3.5 to 1 mm. Lowering [K+]o consistently shifted the reversal potential in the negative direction, indicating a decrease in [Cl−]i. In whole-cell recordings from P18 to P28 neurons with 1 mm pipette Cl−, the mean reversal potential (−100.1 ± 1.3 mV; n = 5) and calculated [Cl−]i (2.27 ± 0.11 mm) measured in 1 mm[K+]o were not significantly different from the mean values obtained in outside-out patches (−102.8 ± 2.0 mV and 2.06 ± 0.16 mm, respectively; p > 0.3). This difference was significant (p < 0.002) in 3.5 and 10 mm[K+]o. The change in reversal potential occurred slowly and was reversible, as shown in Figure 3B.
Fig. 3.
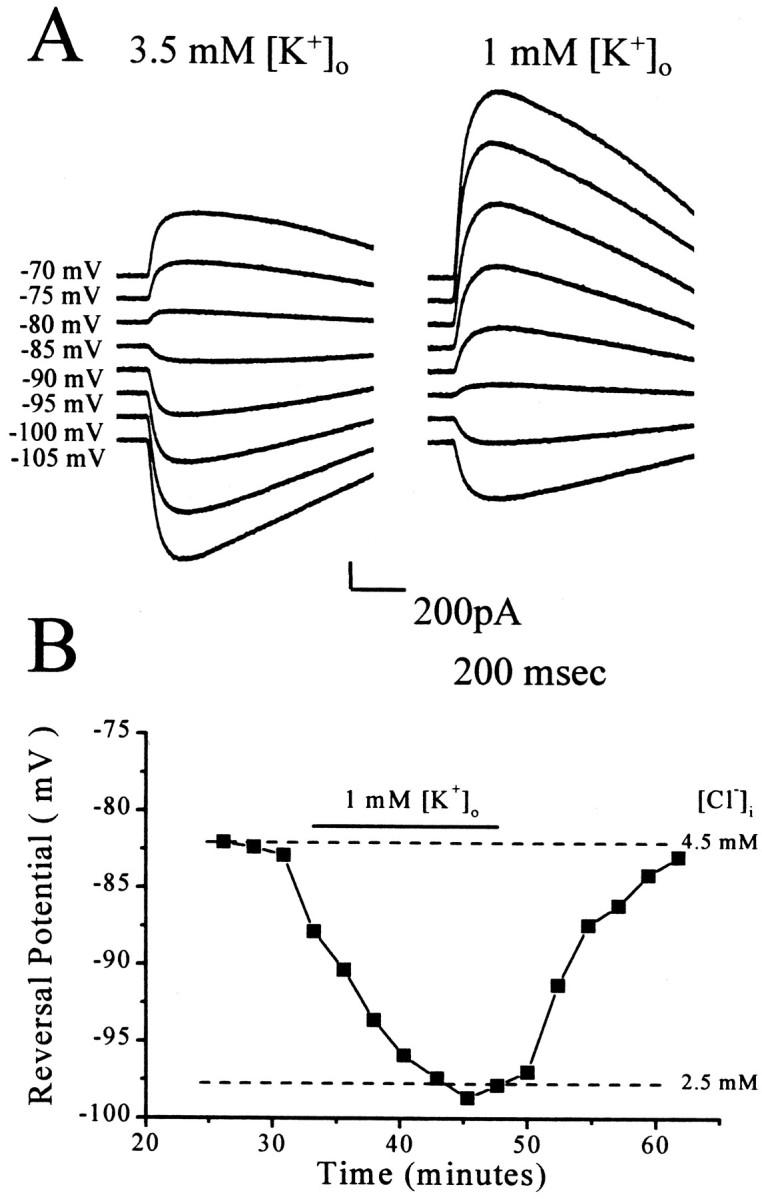
Lowering extracellular potassium lowers intracellular chloride. A, GABA (250 μm) was applied at 20 sec intervals to the soma during voltage steps to the indicated potentials. The pipette solution contained 1 mmadded Cl−. Relative to the currents evoked by GABA application in 3.5 mm[K+]o, a 15 mV drop in the reversal potential is apparent. B, A time course plot of the change in reversal potential is shown.
In previous studies, raising extracellular potassium inhibited Cl− extrusion (Thompson et al., 1988a,b;Thompson and Gähwiler, 1989b). Payne (1997) estimated the apparent affinity (_K_m) of KCC2 for [K+]o to be ∼5 mm. This led to the hypothesis that KCC2 could operate in reverse in the presence of elevated [K+]o and accumulate both K+ and Cl−. As shown in Figure4, raising [K+]o reversibly shifted the reversal potential in the depolarizing direction. In these experiments, the cell was initially maintained in 3.5 mm[K+]o. The bath concentration of K+ was then raised to 10 mm, resulting in a >15 mV depolarization in the reversal potential. This indicates a corresponding 3.1 mm increase in [Cl−]i. With low pipette Cl−, raising [K+]o increased [Cl−]i by increasing the net influx of Cl− via K+–Cl−cotransport.
Fig. 4.

Potassium and furosemide alter intracellular chloride. A 10 min application of 10 mm[K+]o reversibly depolarized the reversal potential and raised the [Cl−]i by 3.1 mm. After washout, bath application of 1 mm furosemide hyperpolarized the reversal potential and lowered [Cl−]i by 1.9 mm. In this figure, 0 min represents the first current–voltage measurement taken ∼5 min after whole-cell mode was achieved. Furo., Furosemide.
Furosemide is a loop diuretic that blocks chloride cotransport. Application of furosemide in the presence of 10 mm[K+]o shifted the reversal potential back toward initial levels (Fig. 4). Lowering [K+]o to 3.5 mm in the presence of furosemide made the reversal potential more negative (Fig. 4), approaching levels observed with excised patches. This effect was reversible. In furosemide and 1 mm pipette Cl−, both the reversal potential (−93.5 ± 6.8 mV; n = 4, 4 animals) and the calculated [Cl−]i (3.19 ± 0.72 mm) were not significantly different from the same parameters recorded in outside-out patches (p > 0.2). This effect is similar to the lowering of [Cl−]i observed in 1 mm[K+]o. The combination of these results suggests that the Cl− accumulation mechanism responsible for the deviations from low pipette Cl−concentrations is a K+-coupled Cl− cotransporter.
To test the hypothesis that K+-coupled Cl− extrusion lowers [Cl−]i in recordings with elevated pipette Cl−, we examined the effects of manipulating [K+]o and blocking transport with furosemide. Figure5A summarizes the effects of manipulating [K+]oand furosemide application on reversal potentials in P18–P28 neurons. The changes in [Cl−]i are shown in Figure 5B. When recordings were made with 1 mm pipette Cl−, lowering [K+]o to 1 mm produced a −12.7 ± 2.3 mV (n = 5) change in reversal potential, indicating a 1.54 ± 0.41 mm decrease in [Cl−]i. A similar change in both parameters was observed with 20 mmpipette Cl−. Likewise, the mean changes in reversal potential and calculated [Cl−]i when [K+]o was raised to 10 mm were similar in both 1 and 20 mm pipette Cl−groups. The effect of furosemide depended on the pipette Cl− concentration. In 1 mm pipette Cl−, furosemide shifted the reversal potential to more negative values and lowered calculated [Cl−]i. In the 20 mm pipette Cl−group, furosemide produced a positive shift in the reversal and raised the calculated [Cl−]i.
Fig. 5.
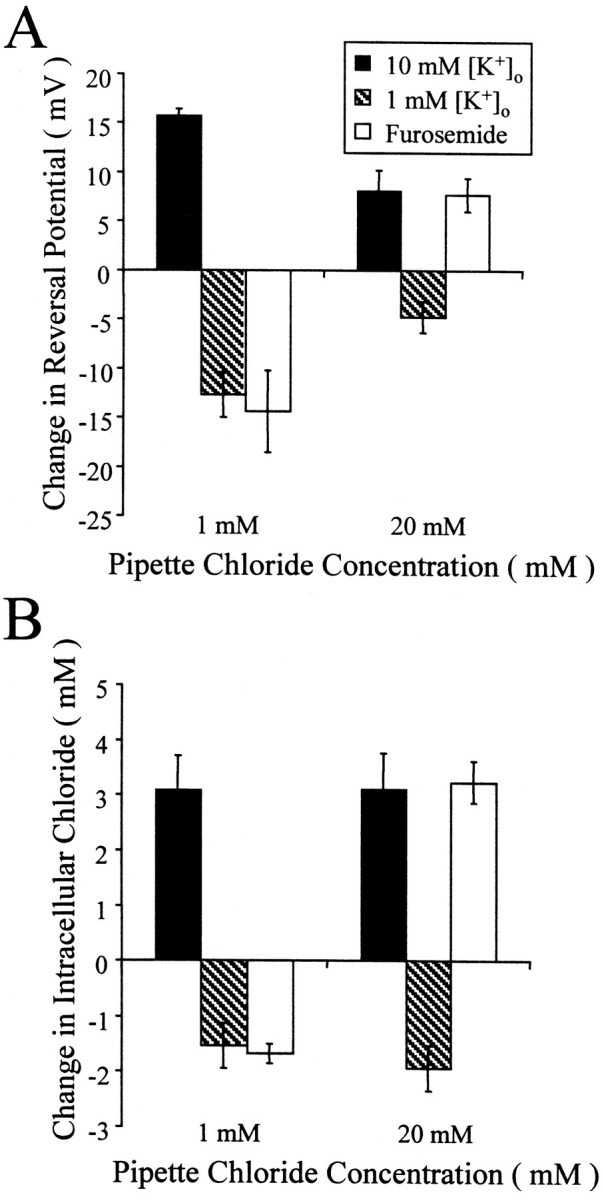
The action of furosemide depends on pipette chloride. A, Changes in reversal potential were smaller in 20 mm pipette Cl− but resulted in similar changes in calculated [Cl−]i. B, Furosemide lowered the reversal potential and [Cl−]i when pipette Cl− was low and raised both the reversal potential and [Cl−]i when pipette Cl− was high. The magnitude of the effect of furosemide was not significantly different from that of the effect of lowering [K+]o in 1 mmpipette Cl− or raising [K+]o in 20 mm pipette Cl−.
The dependency of the action of furosemide on pipette Cl− supports the hypothesis that Cl− homeostatic mechanisms can operate either to accumulate or to extrude Cl−. In 10 mm[K+]o the reversal potential and calculated [Cl−]i were still significantly lower than those obtained with outside-out patches (p < 0.002; n = 5; 20 mm[Cl−]pipette;n = 3; 40 mm[Cl−]pipette). This suggests that although K+–Cl−cotransport plays an important role in maintaining [Cl−]i, other mechanisms may also contribute to Cl−extrusion.
Developmental regulation of KCC2 function and expression
Previous studies have indicated that KCC2 expression is developmentally regulated (Lu et al., 1999; Rivera et al., 1999). We tested the hypothesis that the reversal potential of Cl− currents in neurons lacking KCC2 would be relatively insensitive to changes in [K+]o. Figure6A shows the lack of effect of changes in [K+]o on the Cl− reversal potential in a P3 animal. The reversal potential immediately after establishment of whole-cell recording with 20 mm pipette Cl− reached a value near −49 mV. Only minimal changes in the reversal potential were subsequently observed. In P3–P6 neurons, neither raising [K+]o to 10 mm (n = 3) nor lowering [K+]o to 1 mm (n = 3) had any significant effect on the reversal potential for GABA responses (Fig.6B), consistent with a lack of expression of KCC2. Comparison of the reversal potentials in 3.5 mm[K+]o between P3–P6 and P18–P28 neurons provides further support for reduced function of KCC2 in neonates. As shown above in Figure2A, the reversal potential and [Cl−]i in P3–P6 neurons were significantly different from that in P18–P28 cells, although both groups were also significantly different from the measurements made in outside-out patches (p < 0.03). Under conditions that support Cl−accumulation by KCC2 (low pipette Cl−), the reversal potential and [Cl−]i in the P3–P6 group were significantly lower than that in P18–P28 neurons (p < 0.05). Conversely, with elevated pipette Cl−, these values were significantly higher in P3–P6 than in the older neurons (p < 0.002). These results suggest that KCC2 does not substantially contribute to Cl− homeostasis in P3–P6 neocortical neurons.
Fig. 6.

Single-cell rt-PCR of neurons from neocortical slices reveals developmentally regulated expression of KCC2 RNA.A, Manipulations of [K+]o had no effect in this example recording from a P3 neuron with 20 mm pipette Cl−. B, The mean changes in reversal potential and calculated [Cl−]i were significantly less than those observed in P18–P28 neurons. Data from both 1 and 20 mm pipette Cl− were pooled. C, Single neurons were analyzed from neocortical slices of P3 or P25 rats. rt-PCR, as described in Materials and Methods, was used to amplify a specific KCC2 fragment from pyramidal neurons or interneurons. Control lanes(C) refer to rt-PCR performed on samples taken by aspirating extracellularly within the slice. D, Cell contents from several of the cells in C were subjected to rt-PCR amplification of both KCC2 and actin.
The physiological data presented above are consistent with a developmental role of KCC2 in chloride homeostasis and suggest the hypothesis that KCC2 expression levels will be reduced in P3 neurons compared with P25 neurons. To test this hypothesis, we examined KCC2 mRNA levels in P3 and P25 neurons using single-cell rt-PCR procedures (Fig. 6C). KCC2 mRNA was detected in all P25 neurons including pyramidal cells and interneurons. However, only one of four P3 neurons showed detectable levels of KCC2 mRNA. The presence of KCC2 in all P25 cells was unlikely to be caused by contamination during the rt-PCR procedure because control samples in which the cell cytoplasm was not harvested failed to reveal KCC2 mRNA expression. To ensure that the lack of KCC2 mRNA expression in P3 neurons was not caused by a failure to detect all mRNAs during the rt-PCR process, we examined, in the same sample, the expression of actin mRNA (Fig.6D). Actin mRNA was detected in all P3 and P25 cells. The actin controls also allowed for the semiquantitative analysis of KCC2 levels in the one P3 neuron expressing KCC2 mRNA. The amount of KCC2 mRNA in the P3 neuron (relative to actin mRNA measured in the same cell) was 14% of that in P25 neurons. These data show that the developmental changes in K+-coupled and furosemide-sensitive Cl− accumulation and extrusion are correlated with the expression of KCC2 mRNA and reinforce the role of KCC2 in the developmental shift to lowered [Cl−]i.
Functional consequences of thermodynamic driving forces for cotransporters
The results presented above support the role of KCC2 in both accumulation and extrusion of Cl− under physiological conditions. To determine whether this action of KCC2 is consistent with the chemical forces acting on the transporter, we used a theoretical model to predict the thermodynamic driving forces as a function of [Cl−]i and [K+]o. The direction of transport of an electroneutral cotransporter is governed by the chemical potentials of the ions transported [derived from Stein (1990), Eq. 2.3]:
U=RT∑j=1n mjln[Xj]in[Xj]out,
where n is the number of different ion species transported (KCC2, n = 2; NKCC, n = 3),mj is the number of molecules transported for each species (for NKCC, _m_Cl = 2;m = 1 otherwise), [_Xj_]in and [_Xj_]out are the concentrations of ion species j inside and outside the cell, and RT is the product of the gas constant and absolute temperature. The thermodynamic driving force (U) is plotted in Figure 7 for two chloride cotransporter subtypes: NKCC and KCC2. The direction of transport in NKCC is relatively insensitive to changes in [Cl−]i or [K+]o over the range of physiological concentrations of the two ions. However, the direction of KCC2 transport is sensitive to small changes in [Cl−]i and [K+]o near physiological levels. Below the expected [Cl−]i for mature pyramidal cells [∼10 mm (R. A. DeFazio and S. Keros, unpublished observations)], KCC2 is predicted to accumulate intracellular chloride when [K+]o is >1 mm (Fig. 7, gray shaded region).
Fig. 7.

The thermodynamic driving force determines the direction of electroneutral Cl− cotransport. The_x_-axis represents the intracellular Cl− concentration. Along the _y_-axis,positive values of the driving force represent Cl− extrusion, whereas negative values indicate Cl− accumulation. The sum of the chemical potentials of Na+, K+, and 2 Cl− inside and outside the cell as a function of intracellular Cl−concentration is represented by the dashed line for 3.5 mm [K+]o. The solid lines represent the driving force for the cotransport of both K+ and Cl− (thus KCC2) at different [K+]o. The gray shaded region represents the range of ion concentrations that give rise to intracellular K–Cl accumulation; thus at elevated [K+]o and low [Cl−]i, thermodynamic calculations predict that the KCC2 activity will raise intracellular Cl−.
DISCUSSION
Our results demonstrate the existence of a powerful homeostatic mechanism that maintains intracellular Cl− concentration. Despite whole-cell dialysis with the low Cl− pipette solution, K+–Cl−cotransport activity raised the calculated intracellular Cl− concentration by 0.7–1.6 mm. This is a net change in Cl− concentration; in light of the continuous dialysis of the cell with the low Cl− pipette solution, it is likely that the pumps were accumulating substantially more than ∼1 mmchloride. Likewise despite elevated pipette Cl− concentrations, reversal potentials indicated intracellular Cl−concentrations well below those calculated from excised patches. This implies that the pumps were extruding at least several millimolar Cl−. It is unlikely that NKCC, the pump traditionally associated with Cl−accumulation, plays a role under these recording conditions because both lowering extracellular potassium (affecting only K+–Cl−cotransport) and cotransport antagonism with furosemide (affecting both NKCC and K+–Cl−cotransport) lowered intracellular Cl− to levels similar to those obtained in excised patches. This surprising result suggests that K+–Cl−cotransport function alone is sufficient for the homeostatic maintenance of intracellular Cl−; i.e., under these recording conditions, K+–Cl−cotransport is both the primary extrusion and accumulation mechanism in mature neocortical neurons.
Bicarbonate flux has been proposed to be a major contributor to GABAA receptor function (Kaila et al., 1993;Staley and Proctor, 1999). Our initial finding that the reversal potential with low pipette Cl− was much more depolarized than those obtained in excised patches could be interpreted to reflect a strong bicarbonate flux (see Materials and Methods). However, when Cl− accumulation via K+–Cl−cotransport was reduced by lowering extracellular potassium or by cotransport antagonism with furosemide, the reversal potentials were no longer significantly different from those of excised patches, which lacked a bicarbonate component. These manipulations of extracellular potassium and cotransport antagonism are not expected to modify intracellular or extracellular bicarbonate. These results support the contention that the depolarized reversal observed with low pipette Cl− was caused by Cl− accumulation via K+–Cl−cotransport and not by an unmasked bicarbonate efflux. Bicarbonate can be an important component of GABA responses (Kaila et al., 1993, 1997;Staley and Proctor, 1999); however, under the present recording conditions bicarbonate did not appear to play a major role in determining the response to GABA. A lack of a bicarbonate contribution to depolarizing GABA responses in hippocampal pyramidal cells has also been reported (Grover et al., 1993).
Differential expression of the transport proteins involved in Cl− homeostasis has been proposed to explain differences in resting Cl−concentrations between cell types (Rohrbough and Spitzer, 1996; Ulrich and Huguenard, 1997) and during development (Owens et al., 1996; Kakazu et al., 1999; Rivera et al., 1999). The absence of K+-coupled Cl− cotransport in our recordings from neonatal neurons correlated well with the reduced expression of KCC2 mRNA detected in single-cell harvests. Although other extrusion mechanisms must exist in neonatal neurons, the absence of a potassium-coupled mechanism is consistent with the elevated intracellular Cl− concentration reported in neonatal neurons. In addition, because of a role for neuronal K+–Cl−cotransport in the maintenance of extracellular potassium (Payne, 1997), the lack of KCC2 expression in neonatal neurons may explain differences in extracellular potassium regulation observed during development (Hablitz and Heinemann, 1989).
The presence of a powerful potassium-coupled Cl− cotransport mechanism has important functional implications. Our results suggest that K+–Cl−cotransport alone is sufficient for homeostatic maintenance of intracellular Cl−. The Cl− set point (the concentration at which the pump exhibits no net flux) is coupled to [K+]o. If intracellular Cl− exceeds the set point, K+–Cl−cotransport can extrude Cl−. If Cl− goes below the set point, K+–Cl−cotransport can accumulate Cl−. We have demonstrated the capacity of these pumps to maintain intracellular Cl− concentrations despite whole-cell dialysis via a patch pipette. Such a Cl−load or sink is substantially less than the physiological Cl− load or sink because of GABAA receptor-mediated Cl− flux, thus demonstrating reserve capacity, an important aspect of numerous physiological systems. It is clear that other Cl− homeostatic mechanisms are present in the neuron. For example, the Cl− reversal potential is still quite hyperpolarized in neurons from P3 to P6 animals, suggesting that although immature cells have reduced KCC2 mRNA levels some other mechanism is lowering intracellular Cl−.
Payne (1997) proposed that KCC2 could contribute to extracellular potassium homeostasis. Although the relative contribution of neurons to the spatial buffering of extracellular potassium remains to be determined, one of the primary consequences of a rise in extracellular potassium would be elevation of the intracellular Cl− concentration. In our experiments, raising extracellular potassium from 3.5 to 10 mm increased intracellular Cl− ∼3 mm(with either 1 or 20 mm pipette Cl−). A 3 mm increase in intracellular Cl− from a resting Cl− concentration of 10 mmwould raise the Cl− reversal potential from −61 to −54 mV. Under these conditions, activation of GABAA receptors could depolarize the cell to −54 mV, much closer to the threshold for action potential generation in neocortical neurons. Large increases in extracellular potassium have been described during seizure-like activity both in vivo(Lux et al., 1974; Xiong and Stringer, 1999) and in vitro(Benninger et al., 1980; Swann et al., 1986; Hablitz and Heinemann, 1989). It is possible that elevations in neuronal Cl− could contribute to the difficulty in controlling prolonged seizures.
The net effect on excitability of a K+-dependent accumulation of intracellular Cl− is not clear. A rise in extracellular potassium has multiple effects on neurons; in addition to increased intracellular Cl−, input resistance decreases and the membrane potential depolarizes. In the present study at a holding potential of −70 mV, raising extracellular potassium from 3.5 to 10 mm decreased the input resistance by 41 ± 10 MΩ and induced a depolarizing current of 388 ± 122 pA. To a considerable degree the most prominent effect of an acute increase in extracellular potassium is membrane depolarization such as that described by Kaila et al. (1997). They demonstrated an ∼20 mV depolarization because of a transient increase in extracellular potassium of up to 7.4 mm in response to a high-frequency stimulus train. Such a depolarization is sufficient to place the membrane potential above the elevated Cl−reversal potential and result in a hyperpolarizing response to synaptic GABA. Neuronal buffering of extracellular potassium probably does not have pathological consequences for neuronal function because the elevated intracellular Cl− concentration is balanced by a decrease in input resistance and membrane depolarization.
Recent studies suggest a strong neuronal contribution to buffering of [K+]o (Ransom et al., 2000; Xiong and Stringer, 2000). It is difficult to infer a direct role of KCC2 in these studies because the manipulations that would affect the cotransporter also altered the spontaneous activity that gave rise to the potassium transient. Lowering extracellular Cl−, furosemide, and ouabain all raised the ceiling of the extracellular potassium transient (Xiong and Stringer, 2000). Although Xiong and Stringer conclude a role for the neuronal Na+–K+ATPase, it is clear that breakdown of the potassium gradient because of inhibition of the Na+–K+ATPase could also inhibit K+-coupled Cl− cotransport. KCC2-mediated Cl− accumulation in elevated [K+]o is consistent with an enhancement of the capacity of the homeostatic mechanisms that regulate extracellular K+. In addition to its major role in neuronal Cl− homeostasis, KCC2 expression may result in a more rapid return to normal levels of [K+]o and/or a lower maximal level that [K+]o can attain.
In summary, we have shown that developmental changes in the expression of KCC2 result in the coupling of [Cl−]i and [K+]o via the activity of the furosemide-sensitive K+-coupled Cl− cotransporter. [K+]omanipulations in immature neurons had no effect on [Cl−]i, consistent with the low expression of KCC2 mRNA detected in cytoplasm harvested from these cells. Expression of KCC2 results in lowered [Cl−]i and translates physiological changes in [K+]o to marked changes in [Cl−]i. These data demonstrate a physiologically relevant and developmentally regulated role for KCC2 in Cl−homeostasis and neuronal excitability.
Footnotes
This work was funded by the American Epilepsy Society with support from the Milken Family Foundation (R.A.D.) and National Institute of Neurological Disorders and Stroke Grant NS-22373 (J.J.H.). We wish to thank Alison Margolies for excellent technical assistance.
Correspondence should be addressed to Dr. John J. Hablitz at the above address. E-mail: hablitz@nrc.uab.edu.
REFERENCES
- 1.Alvarez-Leefmans FJ. Intracellular Cl− regulation and synaptic inhibition in vertebrate and invertebrate neurons. In: Alvarez-Leefmans FJ, Russel JM, editors. Chloride channels and carriers in nerve, muscle, and glial cells. Plenum; New York: 1990. pp. 109–158. [Google Scholar]
- 2.Andersen P, Dingledine R, Gjerstad L, Langmoen IA, Laursen AM. Two different responses of hippocampal pyramidal cells to application of gamma-aminobutyric acid. J Physiol (Lond) 1980;305:279–296. doi: 10.1113/jphysiol.1980.sp013363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barker JL, Harrison NL. Outward rectification of inhibitory postsynaptic currents in cultured rat hippocampal neurones. J Physiol (Lond) 1988;403:41–55. doi: 10.1113/jphysiol.1988.sp017237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Benninger C, Kadis J, Prince DA. Extracellular calcium and potassium changes in hippocampal slices. Brain Res. 1980;187:165–182. doi: 10.1016/0006-8993(80)90502-8. [DOI] [PubMed] [Google Scholar]
- 5.Bormann J, Hamill OP, Sakmann B. Mechanism of anion permeation through channels gated by glycine and gamma-aminobutyric acid in mouse cultured spinal neurones. J Physiol (Lond) 1987;385:243–286. doi: 10.1113/jphysiol.1987.sp016493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Devay P, McGehee DS, Yu CR, Role LW. Target-specific control of nicotinic receptor expression at developing interneuronal synapses in chick. Nat Neurosci. 1999;2:528–534. doi: 10.1038/9183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dreifuss JJ, Kelly JS, Krnjevic K. Cortical inhibition and gamma-aminobutyric acid. Exp Brain Res. 1969;9:137–154. doi: 10.1007/BF00238327. [DOI] [PubMed] [Google Scholar]
- 8.Grover LM, Lambert NA, Schwartzkroin PA, Teyler TJ. Role of HCO3− ions in depolarizing GABAA receptor-mediated responses in pyramidal cells of rat hippocampus. J Neurophysiol. 1993;69:1541–1555. doi: 10.1152/jn.1993.69.5.1541. [DOI] [PubMed] [Google Scholar]
- 9.Hablitz JJ, Heinemann U. Alterations in the microenvironment during spreading depression associated with epileptiform activity in the immature neocortex. Dev Brain Res. 1989;46:243–252. doi: 10.1016/0165-3806(89)90288-5. [DOI] [PubMed] [Google Scholar]
- 10.Howe JR, Sutor B, Zieglgansberger W. Characteristics of long-duration inhibitory postsynaptic potentials in rat neocortical neurons in vitro. Cell Mol Neurobiol. 1987;7:1–18. doi: 10.1007/BF00734986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jarolimek W, Lewen A, Misgeld U. A furosemide-sensitive K+-Cl− cotransporter counteracts intracellular Cl− accumulation and depletion in cultured rat midbrain neurons. J Neurosci. 1999;19:4695–4704. doi: 10.1523/JNEUROSCI.19-12-04695.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kaila K, Voipio J, Paalasmaa P, Pasternack M, Deisz RA. The role of bicarbonate in GABAA receptor-mediated IPSPs of rat neocortical neurones. J Physiol (Lond) 1993;464:273–289. doi: 10.1113/jphysiol.1993.sp019634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kaila K, Lamsa K, Smirnov S, Taira T, Voipio J. Long-lasting GABA-mediated depolarization evoked by high-frequency stimulation in pyramidal neurons of rat hippocampal slice is attributable to a network-driven, bicarbonate-dependent K+ transient. J Neurosci. 1997;17:7662–7672. doi: 10.1523/JNEUROSCI.17-20-07662.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kakazu Y, Akaike N, Komiyama S, Nabekura J. Regulation of intracellular chloride by cotransporters in developing lateral superior olive neurons. J Neurosci. 1999;19:2843–2851. doi: 10.1523/JNEUROSCI.19-08-02843.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kakazu Y, Uchida S, Nakagawa T, Akaike N, Nabekura J. Reversibility and cation selectivity of the K+-Cl− cotransport in rat central neurons. J Neurophysiol. 2000;84:281–288. doi: 10.1152/jn.2000.84.1.281. [DOI] [PubMed] [Google Scholar]
- 16.Krnjevic K, Schwartz S. The action of gamma-aminobutyric acid on cortical neurones. Exp Brain Res. 1967;3:320–336. doi: 10.1007/BF00237558. [DOI] [PubMed] [Google Scholar]
- 17.Ling DS, Benardo LS. Activity-dependent depression of monosynaptic fast IPSCs in hippocampus: contributions from reductions in chloride driving force and conductance. Brain Res. 1995;670:142–146. doi: 10.1016/0006-8993(94)01298-v. [DOI] [PubMed] [Google Scholar]
- 18.Lu J, Karadsheh M, Delpire E. Developmental regulation of the neuronal-specific isoform of the K-Cl cotransporter in postnatal rat brains. J Neurobiol. 1999;39:558–568. [PubMed] [Google Scholar]
- 19.Lux HD, Heinemann U, Dietzel I. Ionic changes and alterations in the size of the extracellular space during epileptic activity. In: Delgado-Escueta AV, Ward AA Jr, Woodbury DM, Porter RJ, editors. Advances in neurology, Vol 44. Raven; New York: 1974. pp. 619–639. [PubMed] [Google Scholar]
- 20.Neher E. Correction for liquid junction potentials in patch clamp experiments. Methods Enzymol. 1992;207:123–131. doi: 10.1016/0076-6879(92)07008-c. [DOI] [PubMed] [Google Scholar]
- 21.Owens DF, Boyce LH, Davis MB, Kriegstein AR. Excitatory GABA responses in embryonic and neonatal cortical slices demonstrated by gramicidin perforated-patch recordings and calcium imaging. J Neurosci. 1996;16:6414–6423. doi: 10.1523/JNEUROSCI.16-20-06414.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Payne JA. Functional characterization of the neuronal-specific K+-Cl− cotransporter: implications for [K+]o regulation. Am J Physiol. 1997;273:C1516–C1525. doi: 10.1152/ajpcell.1997.273.5.C1516. [DOI] [PubMed] [Google Scholar]
- 23.Poth K, Nutter TJ, Cuevas J, Parker MJ, Adams DJ, Luetje CW. Heterogeneity of nicotinic receptor class and subunit mRNA expression among individual parasympathetic neurons from rat intracardiac ganglia. J Neurosci. 1997;17:586–596. doi: 10.1523/JNEUROSCI.17-02-00586.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ransom CB, Ransom BR, Sontheimer H. Activity-dependent extracellular K+ accumulation in rat optic nerve: the role of glial and axonal Na+ pumps. J Physiol (Lond) 2000;522:427–442. doi: 10.1111/j.1469-7793.2000.00427.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rivera C, Voipio J, Payne JA, Ruusuvuori E, Lahtinen H, Lamsa K, Pirvola U, Saarma M, Kaila K. The K+/Cl− co-transporter KCC2 renders GABA hyperpolarizing during neuronal maturation. Nature. 1999;397:251–255. doi: 10.1038/16697. [DOI] [PubMed] [Google Scholar]
- 26.Robinson RA, Stokes RH. Electrolyte solutions. Butterworths; London: 1959. [Google Scholar]
- 27.Rohrbough J, Spitzer NC. Regulation of intracellular Cl− levels by Na+-dependent Cl− cotransport distinguishes depolarizing from hyperpolarizing GABAA receptor-mediated responses in spinal neurons. J Neurosci. 1996;16:82–91. doi: 10.1523/JNEUROSCI.16-01-00082.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sakmann B, Neher E. Single-channel recording. Plenum; New York: 1995. [Google Scholar]
- 29.Staley KJ, Proctor WR. Modulation of mammalian dendritic GABAA receptor function by the kinetics of Cl− and HCO3− transport. J Physiol (Lond) 1999;519:693–712. doi: 10.1111/j.1469-7793.1999.0693n.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stein WD. Channels, carriers, and pumps: an introduction to membrane transport. Academic; San Diego: 1990. [Google Scholar]
- 31.Swann JW, Smith KL, Brady RJ. Extracellular K+ accumulation during penicillin-induced epileptogenesis in the CA3 region of immature rat hippocampus. Brain Res. 1986;395:243–255. doi: 10.1016/s0006-8993(86)80203-7. [DOI] [PubMed] [Google Scholar]
- 32.Thompson SM, Gähwiler BH. Activity-dependent disinhibition. I. Repetitive stimulation reduces IPSP driving force and conductance in the hippocampus in vitro. J Neurophysiol. 1989a;61:501–511. doi: 10.1152/jn.1989.61.3.501. [DOI] [PubMed] [Google Scholar]
- 33.Thompson SM, Gähwiler BH. Activity-dependent disinhibition. II. Effects of extracellular potassium, furosemide, and membrane potential on ECl− in hippocampal CA3 neurons. J Neurophysiol. 1989b;61:512–523. doi: 10.1152/jn.1989.61.3.512. [DOI] [PubMed] [Google Scholar]
- 34.Thompson SM, Deisz RA, Prince DA. Outward chloride/cation co-transport in mammalian cortical neurons. Neurosci Lett. 1988a;89:49–54. doi: 10.1016/0304-3940(88)90479-x. [DOI] [PubMed] [Google Scholar]
- 35.Thompson SM, Deisz RA, Prince DA. Relative contributions of passive equilibrium and active transport to the distribution of chloride in mammalian cortical neurons. J Neurophysiol. 1988b;60:105–124. doi: 10.1152/jn.1988.60.1.105. [DOI] [PubMed] [Google Scholar]
- 36.Ulrich D, Huguenard JR. Nucleus-specific chloride homeostasis in rat thalamus. J Neurosci. 1997;17:2348–2354. doi: 10.1523/JNEUROSCI.17-07-02348.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Weiss DS, Hablitz JJ. Interaction of penicillin and pentobarbital with inhibitory synaptic mechanisms in neocortex. Cell Mol Neurobiol. 1984;4:301–317. doi: 10.1007/BF00733594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Williams JR, Sharp JW, Kumari VG, Wilson M, Payne JA. The neuron-specific K-Cl cotransporter, KCC2. Antibody development and initial characterization of the protein. J Biol Chem. 1999;274:12656–12664. doi: 10.1074/jbc.274.18.12656. [DOI] [PubMed] [Google Scholar]
- 39.Xiong Z, Stringer J. Sodium pump activity, not glial spatial buffering, clears potassium after epileptiform activity induced in the dentate gyrus. J Neurophysiol. 2000;83:1443–1451. doi: 10.1152/jn.2000.83.3.1443. [DOI] [PubMed] [Google Scholar]
- 40.Xiong ZQ, Stringer JL. Astrocytic regulation of the recovery of extracellular potassium after seizures in vivo. Eur J Neurosci. 1999;11:1677–1684. doi: 10.1046/j.1460-9568.1999.00587.x. [DOI] [PubMed] [Google Scholar]