Genomic characterization of the 2019 novel human-pathogenic coronavirus isolated from a patient with atypical pneumonia after visiting Wuhan (original) (raw)
ABSTRACT
A mysterious outbreak of atypical pneumonia in late 2019 was traced to a seafood wholesale market in Wuhan of China. Within a few weeks, a novel coronavirus tentatively named as 2019 novel coronavirus (2019-nCoV) was announced by the World Health Organization. We performed bioinformatics analysis on a virus genome from a patient with 2019-nCoV infection and compared it with other related coronavirus genomes. Overall, the genome of 2019-nCoV has 89% nucleotide identity with bat SARS-like-CoVZXC21 and 82% with that of human SARS-CoV. The phylogenetic trees of their orf1a/b, Spike, Envelope, Membrane and Nucleoprotein also clustered closely with those of the bat, civet and human SARS coronaviruses. However, the external subdomain of Spike’s receptor binding domain of 2019-nCoV shares only 40% amino acid identity with other SARS-related coronaviruses. Remarkably, its orf3b encodes a completely novel short protein. Furthermore, its new orf8 likely encodes a secreted protein with an alpha-helix, following with a beta-sheet(s) containing six strands. Learning from the roles of civet in SARS and camel in MERS, hunting for the animal source of 2019-nCoV and its more ancestral virus would be important for understanding the origin and evolution of this novel lineage B betacoronavirus. These findings provide the basis for starting further studies on the pathogenesis, and optimizing the design of diagnostic, antiviral and vaccination strategies for this emerging infection.
KEYWORDS: Coronavirus, Wuhan, SARS, emerging, genome, respiratory, virus, bioinformatics
Introduction
Coronaviruses (CoVs) are enveloped, positive-sense, single-stranded RNA viruses that belong to the subfamily Coronavirinae, family Coronavirdiae, order Nidovirales. There are four genera of CoVs, namely, Alphacoronavirus (αCoV), Betacoronavirus (βCoV), Deltacoronavirus (δCoV), and Gammacoronavirus (γCoV) [1]. Evolutionary analyses have shown that bats and rodents are the gene sources of most αCoVs and βCoVs, while avian species are the gene sources of most δCoVs and γCoVs. CoVs have repeatedly crossed species barriers and some have emerged as important human pathogens. The best-known examples include severe acute respiratory syndrome CoV (SARS-CoV) which emerged in China in 2002–2003 to cause a large-scale epidemic with about 8000 infections and 800 deaths, and Middle East respiratory syndrome CoV (MERS-CoV) which has caused a persistent epidemic in the Arabian Peninsula since 2012 [2,3]. In both of these epidemics, these viruses have likely originated from bats and then jumped into another amplification mammalian host [the Himalayan palm civet (Paguma larvata) for SARS-CoV and the dromedary camel (Camelus dromedarius) for MERS-CoV] before crossing species barriers to infect humans.
Prior to December 2019, 6 CoVs were known to infect human, including 2 αCoV (HCoV-229E and HKU-NL63) and 4 βCoV (HCoV-OC43 [lineage A], HCoV-HKU1 [lineage A], SARS-CoV [lineage B] and MERS-CoV [lineage C]). The βCoV lineage A HCoV-OC43 and HCoV-HKU1 usually cause self-limiting upper respiratory infections in immunocompetent hosts and occasionally lower respiratory tract infections in immunocompromised hosts and elderly [4]. In contrast, SARS-CoV (lineage B βCoV) and MERS-CoV (lineage C βCoV) may cause severe lower respiratory tract infection with acute respiratory distress syndrome and extrapulmonary manifestations, such as diarrhea, lymphopenia, deranged liver and renal function tests, and multiorgan dysfunction syndrome, among both immunocompetent and immunocompromised hosts with mortality rates of ∼10% and ∼35%, respectively [5,6]. On 31 December 2019, the World Health Organization (WHO) was informed of cases of pneumonia of unknown cause in Wuhan City, Hubei Province, China [7]. Subsequent virological testing showed that a novel CoV was detected in these patients. As of 16 January 2020, 43 patients have been diagnosed to have infection with this novel CoV, including two exported cases of mild pneumonia in Thailand and Japan [8,9]. The earliest date of symptom onset was 1 December 2019 [10]. The symptomatology of these patients included fever, malaise, dry cough, and dyspnea. Among 41 patients admitted to a designated hospital in Wuhan, 13 (32%) required intensive care and 6 (15%) died. All 41 patients had pneumonia with abnormal findings on chest computerized tomography scans [10].
We recently reported a familial cluster of 2019-nCoV infection in a Shenzhen family with travel history to Wuhan [11]. In the present study, we analyzed a 2019-nCoV complete genome from a patient in this familial cluster and compared it with the genomes of related β CoVs to provide insights into the potential source and control strategies.
Materials and methods
Viral sequences
The complete genome sequence of 2019-nCoV HKU-SZ-005b was available at GenBank (accession no. MN975262) (Table 1). The representative complete genomes of other related βCoVs strains collected from human or mammals were included for comparative analysis. These included strains collected from human, bats, and Himalayan palm civet between 2003 and 2018, with one 229E coronavirus strain as the outgroup.
Table 1. List of coronaviruses used in this study.
| Accession number | Name displayed on the tree | Name of full-length genome | Year |
|---|---|---|---|
| AY274119 | Human SARS-CoV Tor2 2003 | SARS-related coronavirus isolate Tor2 | 2003 |
| AY278488 | Human SARS-CoV BJ01 2003 | SARS coronavirus BJ01 | 2003 |
| AY278491 | SARS coronavirus HKU-39849 2003 | SARS coronavirus HKU-39849 2003 | 2003 |
| AY390556 | Human SARS-CoV GZ02 2003 | SARS coronavirus GZ02 | 2003 |
| AY391777 | Human CoV OC43 2003 | Human coronavirus OC43 | 2003 |
| AY515512 | Paguma SARS CoV HC/SZ/61/03 2003 | SARS coronavirus HC/SZ/61/03 (paguma SARS) | 2018 |
| EF065513 | Bat CoV HKU9-1 2006 | Bat coronavirus HKU9-1 | 2006 |
| FJ588686 | Bat SL-CoV Rs672 2006 | Bat SARS CoV Rs672/2006 | 2006 |
| KC881005 | Bat SL-CoV RsSHC014 2013 | Bat SARS-like coronavirus RsSHC014 | 2013 |
| KC881006 | Bat SL-CoV Rs3367 2013 | Bat SARS-like coronavirus Rs3367 | 2013 |
| KY417146 | Bat SL-CoV Rs4231 2016 | Bat SARS-like coronavirus isolate Rs4231 | 2016 |
| KY417149 | Bat SL-CoV Rs4255 2016 | Bat SARS-like coronavirus isolate Rs4255 | 2016 |
| MG772933 | Bat SL-CoV ZC45 2018 | Bat SARS-like coronavirus isolate bat-SL-CoVZC45 | 2018 |
| MG772934 | Bat SL-CoV ZXC21 2018 | Bat SARS-like coronavirus isolate bat-SL-CoVZXC21 | 2018 |
| MK211377 | Bat CoV YN2018C 2018 | Coronavirus BtRs-BetaCoV/YN2018C | 2018 |
| MK211378 | Bat CoV YN2018D 2018 | Coronavirus BtRs-BetaCoV/YN2018Da | 2018 |
| MN975262 | HKU-SZ-005b | Human 2019-nCoV HKU-SZ-005b | 2020 |
| NC002645 | Human CoV 229E 2000 | Human coronavirus 229E | 2000 |
| NC006577 | Human CoV HKU1 2004 | Human coronavirus HKU1 | 2004 |
| NC009019 | Bat CoV HKU4-1 2006 | Bat coronavirus HKU4-1 | 2006 |
| NC009020 | Bat CoV HKU5-1 2006 | Bat coronavirus HKU5-1 | 2006 |
| NC014470 | Bat SARS-related CoV BM48-31 2009 | Bat coronavirus BM48-31/BGR/2008 | 2008 |
| NC019843 | Human MERS-CoV 2012 | Middle East respiratory syndrome coronavirus | 2012 |
Genome characterization and phylogenetic analysis
Phylogenetic tree construction by the neighbour joining method was performed using MEGA X software, with bootstrap values being calculated from 1000 trees [12]. The percentage of replicate trees in which the associated taxa clustered together in the bootstrap test (1000 replicates) was shown next to the branches [13]. The tree was drawn to scale, with branch lengths in the same units as those of the evolutionary distances used to infer the phylogenetic tree. The evolutionary distances were computed using the Poisson correction method and were in the units of the number of amino acid substitutions per site [14]. All ambiguous positions were removed for each sequence pair (pairwise deletion option). Evolutionary analyses were conducted in MEGA X [15]. Multiple alignment was performed using CLUSTAL 2.1 and further visualized using BOXSHADE 3.21. Structural analysis of orf8 was performed using PSI-blast-based secondary structure PREDiction (PSIPRED) [16]. For the prediction of protein secondary structure including beta sheet, alpha helix, and coil, initial amino acid sequences were input and analysed using neural networking and its own algorithm. Predicted structures were visualized and highlighted on the BOXSHADE alignment. Prediction of transmembrane domains was performed using the TMHMM 2.0 server (http://www.cbs.dtu.dk/services/TMHMM/). Secondary structure prediction in the 5′-untranslated region (UTR) and 3′-UTR was performed using the RNAfold WebServer (http://rna.tbi.univie.ac.at/cgi-bin/RNAWebSuite/RNAfold.cgi) with minimum free energy (MFE) and partition function in Fold algorithms and basic options. The human SARS-CoV 5′- and 3′- UTR were used as references to adjust the prediction results.
Results and discussion
Genome organization
The single-stranded RNA genome of the 2019-nCoV was 29891 nucleotides in size, encoding 9860 amino acids. The G + C content was 38%. Similar to other βCoVs, the 2019-nCoV genome contains two flanking untranslated regions (UTRs) and a single long open reading frame encoding a polyprotein. The 2019-nCoV genome is arranged in the order of 5′-replicase (orf1/ab)-structural proteins [Spike (S)-Envelope (E)-Membrane (M)-Nucleocapsid (N)]−3′ and lacks the hemagglutinin-esterase gene which is characteristically found in lineage A β-CoVs (Figure 1).
Figure 1.
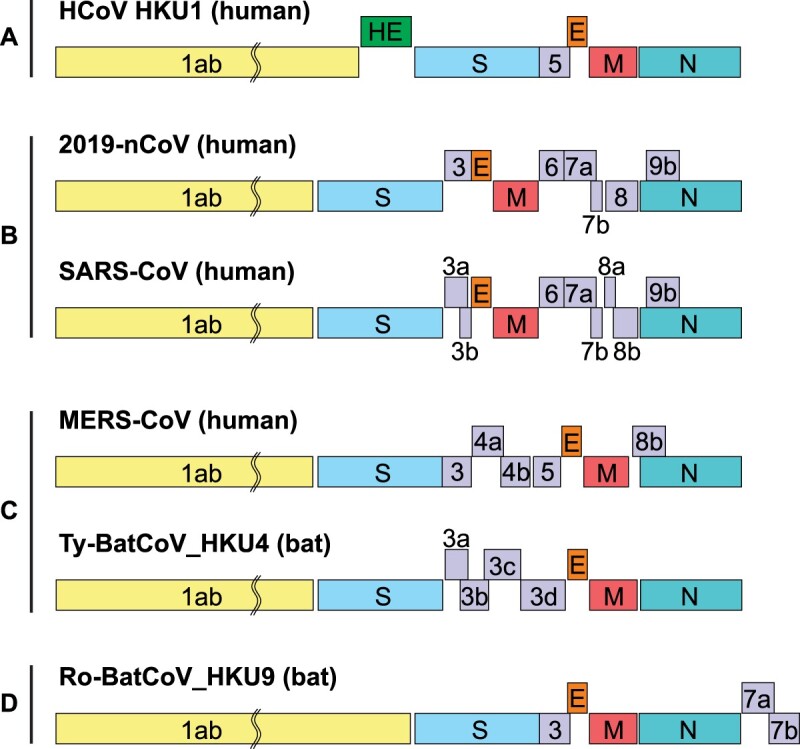
Betacoronavirus genome organization. The betacoronavirus genome comprises of the 5'-untranslated region (5'-UTR), open reading frame (orf) 1a/b (yellow box) encoding non-structural proteins (nsp) for replication, structural proteins including spike (blue box), envelop (orange box), membrane (red box), and nucleocapsid (cyan box) proteins, accessory proteins (purple boxes) such as orf 3, 6, 7a, 7b, 8 and 9b in the 2019-nCoV (HKU-SZ-005b) genome, and the 3'-untranslated region (3'-UTR). Examples of lineages A to D betacoronaviruses include human coronavirus (HCoV) HKU1 (lineage A), 2019-nCoV (HKU-SZ-005b) and SARS-CoV (lineage B), MERS-CoV and Tylonycteris bat CoV HKU4 (lineage C), and Rousettus bat CoV HKU9 (lineage D). The length of nsps and orfs are not drawn in scale.
There are 12 putative, functional open reading frames (orfs) expressed from a nested set of 9 subgenomic mRNAs carrying a conserved leader sequence in the genome, 9 transcription-regulatory sequences, and 2 terminal untranslated regions. The 5′- and 3′-UTRs are 265 and 358 nucleotides long, respectively. The 5′- and 3 ′-UTR sequences of 2019-nCoV are similar to those of other βCoVs with nucleotide identities of ⩾83.6%. The large replicase polyproteins pp1a and pp1ab encoded by the partially overlapping 5′-terminal orf1a/b within the 5′ two-thirds of the genome is proteolytic cleaved into 16 putative non-structural proteins (nsps). These putative nsps included two viral cysteine proteases, namely, nsp3 (papain-like protease) and nsp5 (chymotrypsin-like, 3C-like, or main protease), nsp12 (RNA-dependent RNA polymerase [RdRp]), nsp13 (helicase), and other nsps which are likely involved in the transcription and replication of the virus (Table 2). There are no remarkable differences between the orfs and nsps of 2019-nCoV with those of SARS-CoV (Table 3). The major distinction between SARSr-CoV and SARS-CoV is in orf3b, Spike and orf8 but especially variable in Spike S1 and orf8 which were previously shown to be recombination hot spots.
Table 2. Putative functions and proteolytic cleavage sites of 16 nonstructural proteins in orf1a/b as predicted by bioinformatics.
| NSP | Putative function/domain | Amino acid position | Putative cleave site |
|---|---|---|---|
| nsp1 | suppress antiviral host response | M1 – G180 | (LNGG'AYTR) |
| nsp2 | unknown | A181 – G818 | (LKGG'APTK) |
| nsp3 | putative PL-pro domain | A819 – G2763 | (LKGG'KIVN) |
| nsp4 | complex with nsp3 and 6: DMV formation | K2764 – Q3263 | (AVLQ'SGFR) |
| nsp5 | 3CL-pro domain | S3264 – Q3569 | (VTFQ'SAVK) |
| nsp6 | complex with nsp3 and 4: DMV formation | S3570 – Q3859 | (ATVQ'SKMS) |
| nsp7 | complex with nsp8: primase | S3860 – Q3942 | (ATLQ'AIAS) |
| nsp8 | complex with nsp7: primase | A3943 – Q4140 | (VKLQ'NNEL) |
| nsp9 | RNA/DNA binding activity | N4141 – Q4253 | (VRLQ'AGNA) |
| nsp10 | complex with nsp14: replication fidelity | A4254 – Q4392 | (PMLQ'SADA) |
| nsp11 | short peptide at the end of orf1a | S4393 – V4405 | (end of orf1a) |
| nsp12 | RNA-dependent RNA polymerase | S4393 – Q5324 | (TVLQ'AVGA) |
| nsp13 | helicase | A5325 – Q5925 | (ATLQ'AENV) |
| nsp14 | ExoN: 3′–5′ exonuclease | A5926 – Q6452 | (TRLQ'SLEN) |
| nsp15 | XendoU: poly(U)-specific endoribonuclease | S6453 – Q6798 | (PKLQ'SSQA) |
| nsp16 | 2'-O-MT: 2'-O-ribose methyltransferase | S6799 – N7096 | (end of orf1b) |
Table 3. Amino acid identity between the 2019 novel coronavirus and bat SARS-like coronavirus or human SARS-CoV.
| Amino acid identity (%) | 2019-nCoV | 2019-nCoV |
|---|---|---|
| vs. bat-SL-CoVZXC21 | vs. SARS-CoV | |
| NSP1 | 96 | 84 |
| NSP2 | 96 | 68 |
| NSP3 | 93 | 76 |
| NSP4 | 96 | 80 |
| NSP5 | 99 | 96 |
| NSP6 | 98 | 88 |
| NSP7 | 99 | 99 |
| NSP8 | 96 | 97 |
| NSP9 | 96 | 97 |
| NSP10 | 98 | 97 |
| NSP11 | 85 | 85 |
| NSP12 | 96 | 96 |
| NSP13 | 99 | 100 |
| NSP14 | 95 | 95 |
| NSP15 | 88 | 89 |
| NSP16 | 98 | 93 |
| Spike | 80 | 76 |
| Orf3a | 92 | 72 |
| Orf3b | 32 | 32 |
| Envelope | 100 | 95 |
| Membrane | 99 | 91 |
| Orf6 | 94 | 69 |
| Orf7a | 89 | 85 |
| Orf7b | 93 | 81 |
| Orf8/Orf8b | 94 | 40 |
| Nucleoprotein | 94 | 94 |
| Orf9b | 73 | 73 |
Spike
Spike glycoprotein comprised of S1 and S2 subunits. The S1 subunit contains a signal peptide, followed by an N-terminal domain (NTD) and receptor-binding domain (RBD), while the S2 subunit contains conserved fusion peptide (FP), heptad repeat (HR) 1 and 2, transmembrane domain (TM), and cytoplasmic domain (CP). We found that the S2 subunit of 2019-nCoV is highly conserved and shares 99% identity with those of the two bat SARS-like CoVs (SL-CoV ZXC21 and ZC45) and human SARS-CoV (Figure 2). Thus the broad spectrum antiviral peptides against S2 would be an important preventive and treatment modality for testing in animal models before clinical trials [18]. Though the S1 subunit of 2019-nCoV shares around 70% identity to that of the two bat SARS-like CoVs and human SARS-CoV (Figure 3(A)), the core domain of RBD (excluding the external subdomain) are highly conserved (Figure 3(B)). Most of the amino acid differences of RBD are located in the external subdomain, which is responsible for the direct interaction with the host receptor. Further investigation of this soluble variable external subdomain region will reveal its receptor usage, interspecies transmission and pathogenesis. Unlike 2019-nCoV and human SARS-CoV, most known bat SARSr-CoVs have two stretches of deletions in the spike receptor binding domain (RBD) when compared with that of human SARS-CoV. But some Yunnan strains such as the WIV1 had no such deletions and can use human ACE2 as a cellular entry receptor. It is interesting to note that the two bat SARS-related coronavirus ZXC21 and ZC45, being closest to 2019-nCoV, can infect suckling rats and cause inflammation in the brain tissue, and pathological changes in lung & intestine. However, these two viruses could not be isolated in Vero E6 cells and were not investigated further. The two retained deletion sites in the Spike genes of ZXC21 and ZC45 may lessen their likelihood of jumping species barriers imposed by receptor specificity.
Figure 2.

Comparison of protein sequences of Spike stalk S2 subunit. Multiple alignment of Spike S2 amino acid sequences of 2019-nCoV HKU-SZ-005b (accession number MN975262), bat SARS-like coronavirus isolates bat-SL-CoVZXC21 and bat-SL-CoVZXC45 (accession number MG772934.1 and MG772933.1, respectively) and human SARS coronavirus (accession number NC004718) was performed and displayed using CLUSTAL 2.1 and BOXSHADE 3.21 respectively. The black boxes represent the identity while the grey boxes represent the similarity of the four amino acid sequences.
Figure 3.
Comparison of protein sequences of A. Spike globular head S1, and B. S1 receptor-binding domain (RBD) subunit. Multiple alignment of Spike S1 amino acid sequences of 2019-nCoV HKU-SZ-005b (accession number MN975262), bat SARS-like coronavirus isolates bat-SL-CoVZXC21, bat-SL-CoVZXC45, bat-SL-CoV-YNLF_31C, bat-SL-CoV-YNLF_34C and bat SL-CoV HKU3-1 (accession number MG772934.1 and MG772933.1, KP886808, KP886809 and DQ022305, respectively), human SARS coronavirus GZ02 and Tor2 (accession number AY390556 and AY274119, respectively) and Paguma SARS-CoV (accession number AY515512) was performed and displayed using CLUSTAL 2.1 and BOXSHADE 3.21, respectively. The black background represents the identity while the grey background represents the similarity of the amino acid sequences. Orange box indicates the region of signal peptide, while green and blue boxes indicate the core domain and receptor binding domain respectively. Sequences of RBD, highlighted in (A) were used for comparison. External subdomain variable region of 2019-nCoV HKU-SZ-005b was predicted by comparison of amino acid similarity and published structural analysis [17]. Purple box indicates the external subdomain region.


Orf3b
A novel short putative protein with 4 helices and no homology to existing SARS-CoV or SARS-r-CoV protein was found within Orf3b (Figure 4). It is notable that SARS-CoV deletion mutants lacking orf3b replicate to levels similar to those of wild-type virus in several cell types [19], suggesting that orf3b is dispensable for viral replication in vitro. But orf3b may have a role in viral pathogenicity as Vero E6 but not 293T cells transfected with a construct expressing Orf3b underwent necrosis as early as 6 h after transfection and underwent simultaneous necrosis and apoptosis at later time points [20]. Orf3b was also shown to inhibit expression of IFN-β at synthesis and signalling [21]. Subsequently, orf3b homologues identified from three bat SARS-related-CoV strains were C-terminally truncated and lacked the C-terminal nucleus localization signal of SARS-CoV [22]. IFN antagonist activity analysis demonstrated that one SARS-related-CoV orf3b still possessed IFN antagonist and IRF3-modulating activities. These results indicated that different orf3b proteins display different IFN antagonist activities and this function is independent of the protein's nuclear localization, suggesting a potential link between bat SARS-related-CoV orf3b function and pathogenesis. The importance of this new protein in 2019-nCoV will require further validation and study.
Figure 4.

Analysis of orf3b. A. Multiple alignment of orf3b protein sequence between 2019-nCoV (HKU-SZ-005b), SARS-CoV and SARS-related CoV. B. A novel putative short protein found in orf3b.
Orf8
orf8 is an accessory protein found in the Betacoronavirus lineage B coronaviruses. Human SARS-CoVs isolated from early-phase patients, all civet SARS-CoVs, and other bat SARS-related CoVs contain full-length orf8 [23]. However, a 29-nucleotide deletion, which causes the split of full length of orf8 into putative orf8a and orf8b, has been found in all SARS-CoV isolated from mid- and late- phase human patients [24]. In addition, we have previously identified two bat SARS-related-CoV (Bat-CoV YNLF_31C and YNLF_34C) and proposed that the original SARS-CoV full-length orf8 is acquired from these two bat SARS-related-CoV [25]. Since the SARS-CoV is the closest human pathogenic virus to the 2019-nCoV, we performed phylogenetic analysis and multiple alignments to investigate the orf8 amino acid sequences. The orf8 protein sequences used in the analysis derived from early phase SARS-CoV that includes full-length orf8 (human SARS-CoV GZ02), the mid- and late-phase SARS-CoV that includes the split orf8b (human SARS-CoV Tor2), civet SARS-CoV (paguma SARS-CoV), two bat SARS-related-CoV containing full-length orf8 (bat-CoV YNLF_31C and YNLF_34C), 2019-nCoV, the other two closest bat SARS-related-CoV to 2019-nCoV SL-CoV ZXC21 and ZC45), and bat SARS-related-CoV HKU3-1 (Figure 5(A)). As expected, orf8 derived from 2019-nCoV belongs to the group that includes the closest genome sequences of bat SARS-related-CoV ZXC21 and ZC45. Interestingly, the new 2019-nCoV orf8 is distant from the conserved orf8 or orf8b derived from human SARS-CoV or its related viruses derived from civet (paguma SARS-CoV) and bat (bat-CoV YNLF_31C and YNLF_34C). This new orf8 of 2019-nCoV does not contain known functional domain or motif. An aggregation motif VLVVL (amino acid 75–79) has been found in SARS-CoV orf8b (Figure 5(B)) which was shown to trigger intracellular stress pathways and activates NLRP3 inflammasomes [26], but this is absent in this novel orf8 of 2019-nCoV. Based on a secondary structure prediction, this novel orf8 has a high possibility to form a protein with an alpha-helix, following with a beta-sheet(s) containing six strands (Figure 5(C)).
Figure 5.

Analysis of orf8 to show novel putative protein. (A) Phylogenetic analysis of orf8 amino acid sequences of 2019-nCoV HKU-SZ-005b (accession number MN975262), bat SARS-like coronavirus isolates bat-SL-CoVZXC21 and bat-SL-CoVZXC45 (accession number MG772934.1 and MG772933.1, respectively) and human SARS coronavirus (accession number AY274119) was performed using the neighbour-joining method with bootstrap 1000. The evolutionary distances were calculated using the JTT matrix-based method. (B) Multiple alignment was performed and displayed using CLUSTAL 2.1 and BOXSHADE 3.21, respectively. The black background represents the identity while the grey background represents the similarity of the amino acid sequences. (C) Structural analysis of Orf8 was performed using PSI-blast-based secondary structure PREDiction (PSIPRED). Predicted helix structure (h) and strand (s) were boxed with red and yellow respectively.
Phylogenetic relationship among 2019-nCoV and other βCoVs
The genome of 2019-nCoV has overall 89% nucleotide identity with bat SARS-related-CoV SL-CoVZXC21 (MG772934.1), and 82% with human SARS-CoV BJ01 2003 (AY278488) and human SARS-CoV Tor2 (AY274119). The phylogenetic trees constructed using the amino acid sequences of orf1a/b and the 4 structural genes (S, E, M, and N) were shown (Figure 6(A–E)). For all these 5 genes, the 2019-nCoV was clustered with lineage B βCoVs. It was most closely related to the bat SARS-related CoVs ZXC21 and ZC45 found in Chinese horseshoe bats (Rhinolopus sinicus) collected from Zhoushan city, Zhejiang province, China between 2015 and 2017. Thus this novel coronavirus should belong to the genus Betacoronavirus, subgenus Sabecovirus (previously lineage 2b of Group 2 coronavirus). SARS-related coronaviruses have been found continuously especially in horseshoe bat species in the last 13 years. Between 2003 and 2018, 339 complete SARS-related coronavirus genomes have been sequenced, including 274 human SARS-CoV, 18 civet SARS coronavirus, and 47 bat SARS-related coronaviruses mainly from Rhinolophus bat species. Together, they formed a distinct subclade among other lineage B βCoVs. These results suggested that the 2019-nCoV might have also originated from bats. But we cannot ascertain whether another intermediate or amplification animal host infected by 2019-nCoV could be found in the epidemiological market, just as in the case of Paguma civets for SARS-CoV.
Figure 6.
Phylogenetic tree construction by the neighbour joining method was performed using MEGA X software, with bootstrap values being calculated from 1000 trees using amino acid sequences of (A) orf1ab polypeptide; (B) Spike glycoprotein; (C) Envelope protein; (D) Membrane protein; (E) Nucleoprotein.





RNA secondary structures
As shown in Figure 7(A–C), the SARS-CoV 5′-UTR contains SL1, SL2, SL3, SL4, S5, SL5A, SL5B, SL5C, SL6, SL7, and SL8. The SL3 contains trans–cis motif [27]. The SL1, SL2, SL3, SL4, S5, SL5A, SL5B, and SL5C structures were similar among the 2019-nCoV, human SARS-CoV and the bat SARS-related ZC45. In the 2019-nCoV, part of the S5 found was inside the orf1a/b (marked in red), which was similar to SARS-CoV. In bat SARS-related CoV ZC45, the S5 was not found inside orf1a/b. The 2019-nCoV had the same SL6, SL7, and SL8 as SARS-CoV, and an additional stem loop. Bat SARS-related CoV ZC45 did not have the SARS-COV SL6-like stem loop. Instead, it possessed two other stem loops in this region. All three strains had similar SL7 and SL8. The bat SARS-like CoV ZC45 also had an additional stem loop between SL7 and SL8. Overall, the 5′-UTR of 2019-nCoV was more similar to that of SARS-CoV than the bat SARS-related CoV ZC 45. The biological relevance and effects of virulence of the 5′-UTR structures should be investigated further. The 2019-nCoV had various 3′-UTR structures, including BSL, S1, S2, S3, S4, L1, L2, L3, and HVR (Figure 7(D–F)). The 3′-UTR was conserved among 2019-nCoV, human SARS-CoV and SARS-related CoVs [27].
Figure 7.
Secondary structure prediction and comparison in the 5′-untranslated region (UTR) and 3′-UTR using the RNAfold WebServer (with minimum free energy and partition function in Fold algorithms and basic options. The SARS 5′- and 3′- UTR was used as a reference to adjust the prediction results.(A) SARS-CoV 5'-UTR; (B) 2019-nCoV (HKU-SZ-005b) 5'-UTR; (C) ZC45 5'-UTR; (D) SARS-CoV 3'-UTR; (E) 2019-nCoV (HKU-SZ-005b) 3'-UTR; (F) ZC45 3'-UTR.


In summary, 2019-nCoV is a novel lineage B Betacoronavirus closely related to bat SARS-related coronaviruses. It also has unique genomic features which deserves further investigation to ascertain their roles in viral replication cycle and pathogenesis. More animal sampling to determine its natural animal reservoir and intermediate animal host in the market is important. This will shed light on the evolutionary history of this emerging coronavirus which has jumped into human after the other two zoonotic Betacoroanviruses, SARS-CoV and MERS-CoV.
Acknowledgements
The funding sources had no role in the study design, data collection, analysis, interpretation, or writing of the report.
Correction Statement
This article was originally published with errors, which have now been corrected in the online version. Please see Correction (http://dx.doi.org/10.1080/22221751.2020.1737364)
Funding Statement
This study was partly supported by the donations of Michael Seak-Kan Tong, Respiratory Viral Research Foundation Limited, Hui Ming, Hui Hoy and Chow Sin Lan Charity Fund Limited, Chan Yin Chuen Memorial Charitable Foundation, Marina Man-Wai Lee, and the Hong Kong Hainan Commercial Association South China Microbiology Research Fund; and funding from the Consultancy Service for Enhancing Laboratory Surveillance of Emerging Infectious Diseases and Research Capability on Antimicrobial Resistance for Department of Health of the Hong Kong Special Administrative Region Government; the Theme-Based Research Scheme (T11/707/15) of the Research Grants Council, Hong Kong Special Administrative Region; Sanming Project of Medicine in Shenzhen, China (No. SZSM201911014); and the High Level-Hospital Program, Health Commission of Guangdong Province, China.
Disclosure statement
No potential conflict of interest was reported by the author(s).
References
- 1.Chan JF, To KK, Tse H, et al. . Interspecies transmission and emergence of novel viruses: lessons from bats and birds. Trends Microbiol. 2013 Oct;21(10):544–555. doi: 10.1016/j.tim.2013.05.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cheng VC, Lau SK, Woo PC, et al. . Severe acute respiratory syndrome coronavirus as an agent of emerging and reemerging infection. Clin Microbiol Rev. 2007 Oct;20(4):660–694. doi: 10.1128/CMR.00023-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chan JF, Lau SK, To KK, et al. . Middle East respiratory syndrome coronavirus: another zoonotic betacoronavirus causing SARS-like disease. Clin Microbiol Rev. 2015 Apr;28(2):465–522. doi: 10.1128/CMR.00102-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Woo PC, Lau SK, Chu CM, et al. . Characterization and complete genome sequence of a novel coronavirus, coronavirus HKU1, from patients with pneumonia. J Virol. 2005 Jan;79(2):884–895. doi: 10.1128/JVI.79.2.884-895.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Peiris JS, Lai ST, Poon LL, et al. . Yuen KY; SARS study group. coronavirus as a possible cause of severe acute respiratory syndrome. Lancet. 2003 Apr 19;361(9366):1319–1325. doi: 10.1016/S0140-6736(03)13077-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yeung ML, Yao Y, Jia L, et al. . MERS coronavirus induces apoptosis in kidney and lung by upregulating Smad7 and FGF2. Nat Microbiol. 2016 Feb 22;1:16004. doi: 10.1038/nmicrobiol.2016.4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.World Health Organization. Novel coronavirus . [cited 2020 Jan 16]. Available from: https://www.who.int/westernpacific/emergencies/novel-coronavirus.
- 8.World Health Organization. Novel Coronavirus – Thailand (ex-China) . [cited 2020 Jan 16]. Available from: https://www.who.int/csr/don/14-january-2020-novel-coronavirus-thailand-ex-china/en/.
- 9.South China Morning Post. Wuhan pneumonia: Japan confirms Chinese man had new coronavirus . [cited 2020 Jan 16]. Available from https://www.scmp.com/news/asia/east-asia/article/3046301/wuhan-pneumonia-japan-confirms-first-case-new-china-coronavirus.
- 10.Huang C, Wang Y, Li X, et al. . Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. 2020. DOI: 10.1016/S0140-6736(20)30183-5. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chan JF, Yuan S, Kok KH, et al. . A familial cluster of pneumonia associated with the 2019 novel coronavirus indicating person-to-person transmission: a study of a family cluster. Lancet. 2020. DOI: 10.1016/S0140-6736(20)30154-9 [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Saitou N, Nei M.. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol. 1987 Jul;4(4):406–425. [DOI] [PubMed] [Google Scholar]
- 13.Felsenstein J. Confidence limits on phylogenies: an approach using the bootstrap. Evolution. 1985 Jul;39(4):783–791. doi: 10.1111/j.1558-5646.1985.tb00420.x [DOI] [PubMed] [Google Scholar]
- 14.Zuckerkandl E, Pauling L.. Evolutionary divergence and convergence in proteins. In: Bryson V, Vogel HJ, editors. Evolving genes and proteins. New York: Academic Press; 1965. p. 97–166. [Google Scholar]
- 15.Kumar S, Stecher G, Li M, et al. . MEGA x: Molecular evolutionary Genetics analysis across computing platforms. Mol Biol Evol. 2018 Jun 1;35(6):1547–1549. doi: 10.1093/molbev/msy096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Buchan DWA, Jones DT.. The PSIPRED protein analysis Workbench: 20 years on. Nucleic Acids Res. 2019;47(W1):W402–W407. doi: 10.1093/nar/gkz297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang Q, Qi J, Yuan Y, et al. . Bat origins of MERS-CoV supported by bat coronavirus HKU4 usage of human receptor CD26. Cell Host Microbe. 2014 Sep 10;16(3):328–337. doi: 10.1016/j.chom.2014.08.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xia S, Yan L, Xu W, et al. . A pan-coronavirus fusion inhibitor targeting the HR1 domain of human coronavirus spike. Sci Adv. 2019 Apr 10;5(4):eaav4580. doi: 10.1126/sciadv.aav4580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yount B, Roberts RS, Sims AC, et al. . Severe acute respiratory syndrome coronavirus group-specific open reading frames encode nonessential functions for replication in cell cultures and mice. J Virol. 2005 Dec;79(23):14909–14922. doi: 10.1128/JVI.79.23.14909-14922.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Khan S, Fielding BC, Tan TH, et al. . Over-expression of severe acute respiratory syndrome coronavirus 3b protein induces both apoptosis and necrosis in Vero E6 cells. Virus Res. 2006 Dec;122(1-2):20–27. doi: 10.1016/j.virusres.2006.06.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kopecky-Bromberg SA, Martinez-Sobrido L, Frieman M, et al. . Severe acute respiratory syndrome coronavirus open reading frame (orf) 3b, orf 6, and nucleocapsid proteins function as interferon antagonists. J Virol. 2007 Jan;81(2):548–557. doi: 10.1128/JVI.01782-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhou P, Li H, Wang H, et al. . Bat severe acute respiratory syndrome-like coronavirus ORF3b homologues display different interferon antagonist activities. J Gen Virol. 2012 Feb;93(Pt 2):275–281. doi: 10.1099/vir.0.033589-0 [DOI] [PubMed] [Google Scholar]
- 23.Song HD, Tu CC, Zhang GW, et al. . Cross-host evolution of severe acute respiratory syndrome coronavirus in palm civet and human. Proc Natl Acad Sci U S A. 2005 Feb 15;102(7):2430–2435. doi: 10.1073/pnas.0409608102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Oostra M, de Haan CA, Rottier PJ.. The 29-nucleotide deletion present in human but not in animal severe acute respiratory syndrome coronaviruses disrupts the functional expression of open reading frame 8. J Virol. 2007;81:13876–13888. doi: 10.1128/JVI.01631-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lau SK, Feng Y, Chen H, et al. . Severe acute respiratory syndrome (SARS) coronavirus ORF8 protein Is acquired from SARS-related coronavirus from Greater horseshoe bats through recombination. J Virol. 2015 Oct;89(20):10532–10547. doi: 10.1128/JVI.01048-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shi CS, Nabar NR, Huang NN, et al. . SARS-Coronavirus Open reading frame-8b triggers intracellular stress pathways and activates NLRP3 inflammasomes. Cell Death Discov. 2019;5:101. doi: 10.1038/s41420-019-0181-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yang D, Leibowitz JL.. The structure and functions of coronavirus genomic 3’ and 5’ ends. Virus Res. 2015 Aug 3;206:120–133. doi: 10.1016/j.virusres.2015.02.025 [DOI] [PMC free article] [PubMed] [Google Scholar]