SARS — beginning to understand a new virus (original) (raw)
Key Points
- A new infectious disease, called severe acute respiratory syndrome (SARS), appeared in southern China in 2002. During the period from November 2002 to the summer of 2003, the World Health Organization recorded 8098 probable SARS cases and 774 deaths in 29 countries.
- A previously unknown coronavirus was isolated from FRhK-4 and Vero E6 cells inoculated with clinical specimens from patients. A virus with close homology to SARS-CoV was isolated from palm civets and racoon dogs, which are used as food in southern China
- In less than a month from the first indication that a coronavirus might be implicated in the disease, the nucleotide sequence of the virus was available, and diagnostic tests were set up.
- The phylogenetic analysis of the SARS-CoV genome revealed that the virus is distinct from the three known groups of coronaviruses and represents an early split-off from group 2.
- The development of antiviral drugs or vaccines is being investigated. Viral enzymes essential for virus replication, such as the RNA-dependent RNA polymerase (RdRp), the 3C-like cystein protease (3Clpro) and the helicases are the most attractive targets for antiviral molecules. Of the possible vaccine targets, the spike (S) protein represents the most promising one.
- So far, β-interferon is the only licensed drug available, which has been reported to interfere with virus replication in vitro. Should SARS return during the next winter, we will still need to rely mostly on quarantine measures to contain it.
Abstract
The 114-day epidemic of the severe acute respiratory syndrome (SARS) swept 29 countries, affected a reported 8,098 people, left 774 patients dead and almost paralysed the Asian economy. Aggressive quarantine measures, possibly aided by rising summer temperatures, successfully terminated the first eruption of SARS and provided at least a temporal break, which allows us to consolidate what we have learned so far and plan for the future. Here, we review the genomics of the SARS coronavirus (SARS-CoV), its phylogeny, antigenic structure, immune response and potential therapeutic interventions should the SARS epidemic flare up again.
Main
A new infectious disease, known as severe acute respiratory syndrome (SARS), appeared in the Guangdong province of southern China in 2002. It is mainly characterized by flu-like symptoms, including high fevers exceeding 38°C or 100.4°F, myalgia, dry non-productive dyspnea, lymphopaenia and infiltrate on chest radiography. In 38% of all cases, the resulting pneumonia led to acute breathing problems requiring artificial respirators1. The overall mortality rate was about 10%, but varied profoundly with age — although SARS affected relatively few children and generally appeared to be milder in the paediatric age group, the mortality rate in the elderly was as high as 50%2,3,4.
Although accurate information about the precise origin of the disease is not available, the Chinese Ministry of Health reported an outbreak of unexplained pneumonia to the World Health Organization (WHO) on 11 February 2003. During the period from 16 November 2002 to 9 February 2003, 305 cases and five deaths due to atypical pneumonia, which were originally thought to be caused by Chlamydia pneumoniae, occurred in the southern Chinese province of Guangdong5. A Chinese doctor who had been treating patients in Guangdong spread the infection outside of mainland China when he travelled to Hong Kong on 21 February 2003. There, while resident on the ninth floor of the Hotel Metropole, the patient developed symptoms and was transferred to the hospital on 22 February where he died the following day. Subsequently, ten secondary-infected guests of the hotel (eight from the ninth floor and two from the eleventh and fourteenth floors) boarded planes and carried the infection to Singapore, Vietnam, Canada and the United States, making SARS the first epidemic to be transmitted by air travel. When the epidemic finally waned after more than 100 days, the WHO counted a cumulative number of 8,098 probable SARS cases and 774 deaths worldwide, in a geographical area spanning 29 countries6.
Carlo Urbani, a WHO infectious disease expert who had been called to Hanoi, was the first to realize that the Chinese doctor in Hong Kong had suffered from a previously unknown disease and alerted the authorities. Following unprecedented collaboration between laboratories and scientists worldwide, a previously unidentified coronavirus was isolated from FRhK-4 and Vero E6 cells that were inoculated with clinical (nasopharyngeal, oropharyngeal and sputum) specimens from patients1,7,8. The association of the virus with the disease was confirmed when macaques that were inoculated with the virus developed symptoms similar to those observed in human cases of SARS9,10. It is likely that scientists from the Chinese Academy of Military Medical Science had observed what seemed to be coronavirus particles in an electron micrograph by 26 February 2003. However, with only a few samples available for analysis, they did not feel confident enough to challenge the authorities and did not publish their findings.
Close contact with very sick patients facilitates person-to-person transmission of the virus — apparently, SARS-CoV spreads in droplets but its efficiency of infection seems to be low, with an infectivity index of about 3 (Refs 11,12). In some cases, however, a single person can infect a high number of people but, as yet, there is no satisfactory explanation for this so-called 'superspreader' phenomenon2.
A virus with close homology to SARS-CoV was isolated from palm civets and racoon dogs, which are considered delicacies in southern China, indicating that the virus could have jumped recently from these mammals to man13. However, a second search by another group failed to reveal any trace of SARS-CoV in more than 60 animal species, including 76 palm civets14. Although unlikely, the possibility that humans infected these SARS-positive animals cannot be formally excluded. In the meantime, over 30 different SARS-CoV isolates have been sequenced, which will hopefully allow us to trace the chain of transmission back to its origin. The SARS epidemic, which caused global panic and huge economical damage to the Asian economy, was contained primarily by aggressive quarantine measures. Additional beneficial factors could include the attenuation of the virus following prolonged passage through the human population, or the onset of summer, as higher temperatures generally decrease the incidence of at least some respiratory infections.
A natural reservoir for the virus could serve as the launch pad for another SARS outbreak during the next winter–spring season. If we are lucky, however, the ability of this virus to cross the species barrier could be the result of a rare series of events that is unlikely to be repeated. A retrospective serological study did not detect anti-SARS-CoV antibodies in a normal population, indicating that this outbreak is the first introduction of SARS-CoV into the human population. At present, accidental infections of researchers handling the pathogen seem to pose the greatest risk for a renewed spread of the virus as a recent case in Singapore has shown15. For now, the transmission of this emerging infectious disease has been stopped, but there is no information on when, or if, SARS-CoV will re-emerge in the human population.
Coronaviruses and their genomes
Coronaviruses were named after their corona solis-like appearance in the electron microscope, which is caused by the club-shaped peplomers that radiate outwards from the viral envelope (Fig. 1a). The spherical capsid contains a positive-strand RNA genome of about 30 kb — the largest of its kind. Coronaviruses have been subdivided into three groups on the basis of serological and genetic properties16. Their broad host range extends from man to turkey; where they are typically associated with respiratory, enteric, hepatic and central nervous system diseases. In man, they cause mainly upper-respiratory-tract infections, and are responsible for a large proportion of all common colds.
Figure 1. Morphology of the SARS coronavirus.
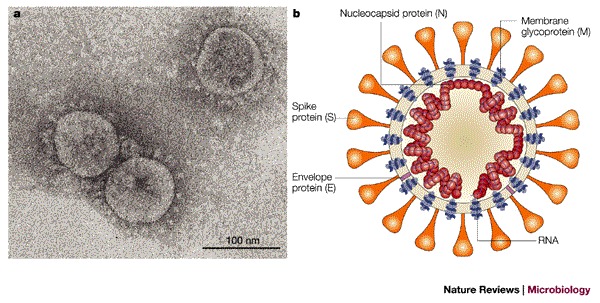
a | Electron micrograph of the virus that was cultivated in Vero cells (Image courtesy of Dr L. Kolesnikova, Institute of Virology, Marburg, Germany). Large, club-shaped protrusions consisting of spike protein form a crown-like corona that gives the virus its name. b | Schematic representation of the virus. A lipid bilayer comprising the spike protein, the membrane glycoprotein and the envelope protein cloaks the helical nucleocapsid, which consists of the nucleocapsid protein that is associated with the viral RNA. In the case of coronaviruses, the lipid envelope is derived from intracellular membranes.
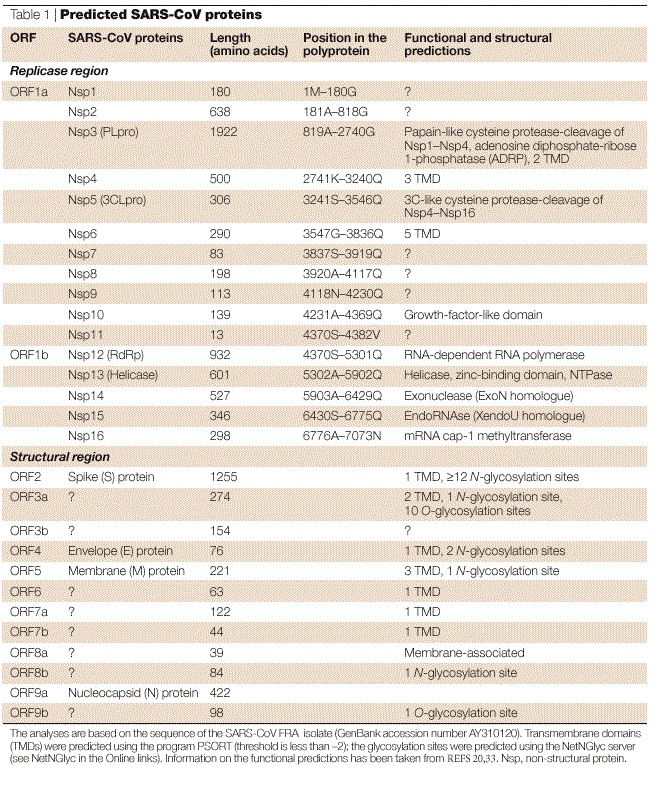
Sequence analysis revealed the organization of the 29,740 base (FRA isolate; GenBank acc.no. AY310120; see SARS coronavirus FRA in the Online links) genome of the SARS-CoV (Fig. 2) to show the characteristic features of coronaviruses17,18,19,20 (Fig. 3). Nucleotides 1–72 contain a predicted RNA leader sequence preceding an untranslated region (UTR) spanning 192 nucleotides. Two overlapping open reading frames (ORF1a and ORF1b), which encompass approximately two-thirds of the genome (nucleotides 265–21485), are downstream of the UTR. A translational read-through by a −1 ribosomal frameshift mechanism allows the translation of the overlapping reading frames into a single polyprotein20. Virus-encoded proteinases, namely the papain-like cysteine protease (PLpro) and the 3C-like cysteine protease (3CLpro), cleave the polyprotein into individual polypeptides, which provide all the proteins that are required for replication and transcription20,21. Wheareas group 2 coronaviruses contain two paralogous PLpros (PL1pro and PL2pro), only one is found in the ORF1a of the SARS-CoV genome. ORF1b encodes a helicase with ATPase and DNA (and possibly also RNA) duplex-unwinding activities, at least when it is expressed in Escherichia coli20. Computational analysis predicted that the carboxy-terminus of ORF1b encodes a mRNA cap-1 methyltransferase22. Some assumptions about the function of the remaining proteins can be made by analogy with other coronaviruses, although our knowledge about the biological functions of these proteins is also incomplete (see Table 1 for information on the encoded proteins). Together with host factors, some of these proteins might form the viral replication–transcription machinery that is associated with the membranous structures in the infected cells23,24. (The peculiar features of the replication cycle of coronaviruses are described in Box 1.) The remaining 3′ part of the genome encodes four structural proteins that are arranged in the same order in all coronaviruses: S, spike protein; E, envelope protein; M, membrane glycoprotein; and N, nucleocapsid protein. Furthermore, the structural protein region of the SARS-CoV genome contains several genes that encode additional non-structural proteins that are known as 'accessory genes' (Fig. 3). Some of these molecules seem to be dispensable for virus viability both in vitro and in vivo; their deletion creates viruses that are attenuated25,26,27. These accessory genes differ significantly among the three coronavirus groups — they are also referred to as group-specific genes. The SARS-CoV has eight predicted ORFs of unknown function in this region of the genome. It lacks the haemagglutinin esterase (HE) gene, which is encoded by almost all of the group 2 viruses.
Figure 2. Genome structure of SARS coronavirus.
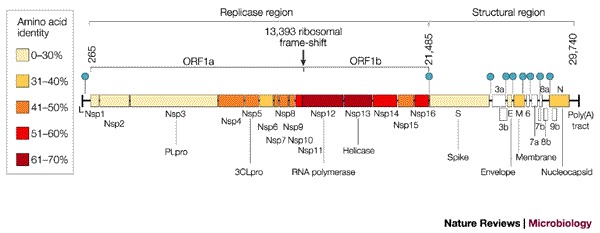
Replicase and structural regions are shown together with the predicted cleavage products in ORF1a and ORF1b. The position of the leader sequence (L), the 3′ poly(A) tract and the ribosomal frameshift site between ORF1a and ORF1b are also indicated. Each box represents a protein product (Nsp, non-structural protein). Colours indicate the level of amino-acid identity with the best-matching protein of other coronaviruses (Table 2). The SARS-CoV accessory genes are white. Filled circles indicate the positions of the nine transcription-regulatory sequences (TRSs) that are specific for SARS-CoV (5′ACGAAC3′).
Figure 3. Comparison of coronavirus genome structures.
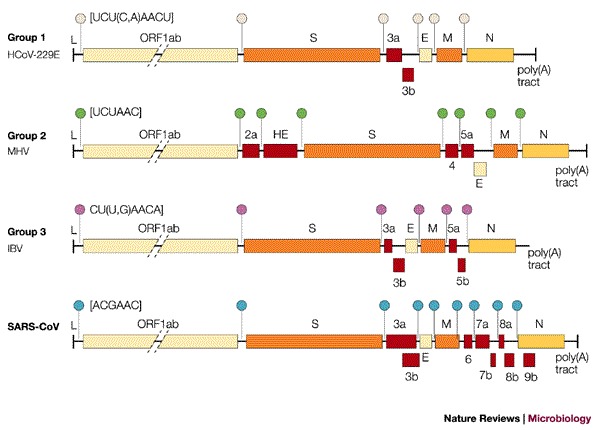
Genome organization of coronavirus representatives of group 1 (human coronavirus 229E, HCoV-229E), group 2 (mouse hepatitis virus, MHV) and group 3 (avian infectious bronchitis virus, IBV; SARS-CoV). Red boxes represent the accessory genes. The positions of the leader sequence (L) and poly(A) tract are indicated; circles of different colour represent group-specific transcription-regulatory sequences (TRS).
Table 1.
Predicted SARS-CoV proteins
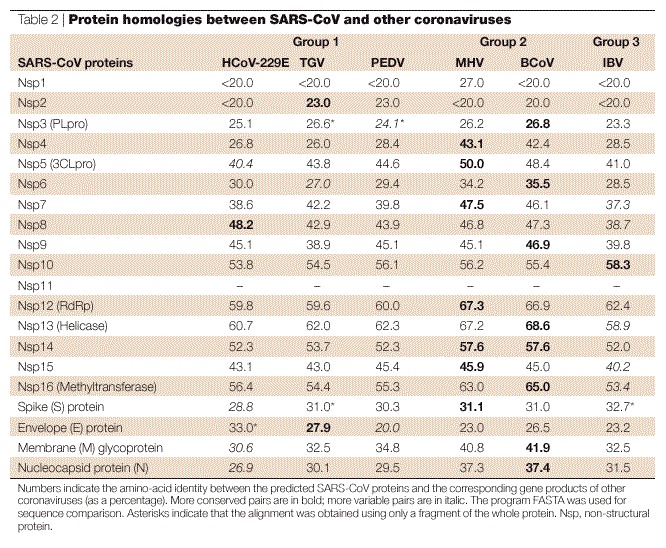
Finally, at the 3′ end of the genome, a second 340-nucleotide UTR, which is followed by a poly(A) tract, was found. This 3′ UTR contains a 32-nucleotide stem–loop II-like motif (s2m) motif, which has also been reported in astroviruses, in one equine rhinovirus and avian infectious bronchitis virus (IBV)28. A typical feature of coronaviruses is the presence of a transcription-regulatory sequence (TRS) that is important in RNA transcription and regulation (Figs 2 and 3). This short motif is usually found at the 3′ end of the leader RNA and, with a few exceptions, precedes each translated ORF29. When Thiel and colleagues20 isolated one genomic and eight subgenomic RNAs from the FRA strain and sequenced their 5′ ends, they identified a conserved sequence (5′ACGAAC3′) that was located in front of nine predicted ORFs, and which fitted the description of a TRS (Figs 2 and 3). By contrast, Marra et al.17 and Rota et al.18 proposed different TRSs (5′CUAAAC3′ and 5′AAACGAAC3′, respectively), but these sequences do not precede all predicted genes and no experimental evidence for their function has been provided. Although the overall organization of the SARS-CoV genome is similar to other coronaviruses (Fig. 3), the amino-acid conservation of the encoded proteins is usually low (Fig. 2; Table 2).
Table 2.
Protein homologies between SARS-CoV and other coronaviruses
Clinical isolates
Helped by the groundwork laid by previous generations of coronavirus researchers, the SARS epidemic has been the first infectious disease outbreak to fully benefit from the revolutionary technologies of the post-genomic era. Less than 1 month after the initial identification of the virus as the infectious agent of SARS, two independent genome sequences of the virus had been obtained17,18. Within 3 months, the genome sequences of 20 independent clinical isolates were made available in the GenBank database (see Box 2 for details). Comparative analysis of these isolates revealed more than than 99% sequence conservation. The few differences, however, have allowed a straightforward organization of all viral isolates into two families: those originating from mainland China and Hong Kong and those originating from the index case in the Hong Kong hotel30 (Fig. 4). Perhaps the most interesting observation was made in the human isolate GZ01, a strain that originated from Guangdong. Although all other human SARS-CoV genomes lack a stretch of 29-nucleotides in the 3′ end domain of ORF8a, this sequence is present in the GZ01 isolate. The additional 29-nucleotide segment in this strain fuses ORF8a and ORF8b into a single ORF, known as ORF8*, which encodes a 122-amino-acid protein. SARS-CoV-like strains isolated from mammals in China have been found to contain the same 29-nucleotide segment13. This observation raises the intriguing hypothesis that the 29-nucleotide deletion that has been observed in most human isolates could have increased the fitness of the virus in human hosts and allowed the spread to the human population.
Figure 4. Molecular relationship of 20 SARS genomes.
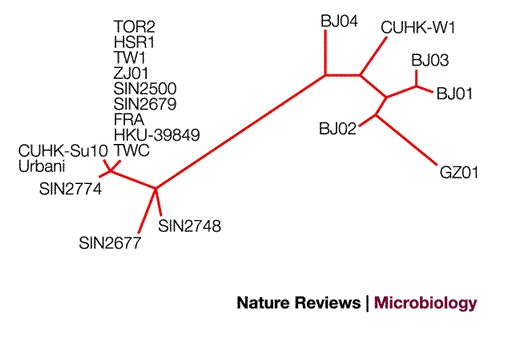
The unrooted tree was obtained through the alignment of whole-genome sequences considering only sequence variants that occurred at least twice. The analysis was performed using the maximum likelihood criterion as implemented in the Phylip package.
Lacking a proof-reading mechanism, RNA viruses are generally characterized by a high mutability. The high mutation rate results in continuously evolving viral species, which allow the virus to escape host defences. Additionally, coronaviruses have a high frequency of RNA recombination that has the theoretical potential of accelerating the emergence of new viral species29. Owing to the limited number of sequenced isolates, however, it is too early to draw any reliable conclusions concerning the mutation rate of SARS-CoV. The most interesting amino-acid changes that have been reported so far are two recurrent non-conservative amino-acid substitutions (Gly to Asp and Ile to Thr) in the antigenic domain S1 of the spike protein30. Gly and Ile can be found in the Hong Kong index case group, whereas Asp and Thr are found in the mainland isolates. Another, non-conservative substitution (Gly to Arg) in the S1 domain is only found in strains GZ01 and BJ02. It is tempting to speculate that these mutations represent adaptations to the new host or its immune response. Additional information on ongoing variability studies can be obtained directly from the SARS Coronavirus Resource (see Online links).
Phylogenetic analysis
The most important question following identification of the SARS-CoV was whether it represents a completely new group, a variant of one of the three known groups or a combination of these groups. Phylogenetic analysis based on the first available 300 and 405 nucleotides of the highly conserved polymerase gene7,31 indicated that SARS-CoV was distinct from the three known groups. Marra and Rota also reached the same conclusion17,18, and proposed that SARS-CoV be placed in its own group (Fig. 5a). Snijder et al. used rooted phylogenetic trees to recreate coronavirus evolution and included equine torovirus (EToV) as an outgroup. Their analysis of ORF1b, the most conserved region in the SARS-CoV genome, indicates that the SARS-CoV represents an early split-off from group 2 (Ref. 32). Our laboratory took a different approach to understanding the phylogenesis of the SARS-CoV. Reasoning that increased sequence variability should contain more information, we analysed less conserved proteins, such as the PLpro, spike protein, membrane glycoprotein and nucleocapsid protein. For each protein, a consensus sequence was generated for the three known groups of coronaviruses. In all cases, the SARS-CoV showed a statistically significant relationship with group 2 coronaviruses (Fig. 5b), which indicated that it is more closely related to members of group 2 and might share a common ancestor with them. Moreover, this conclusion is corroborated by the striking observation that 19 out of 20 cysteine residues in the S1 domain of the SARS-CoV spike protein are spatially conserved when compared with the group 2 consensus sequence, whereas only 5 cysteines are conserved when compared with group 1 or 3 consensus sequences (Fig. 6). This analysis supports the conclusion that the SARS-CoV virus split early from other group 2 viruses and has evolved independently for a long period of time.
Figure 5. Relationship between SARS-CoV and other coronaviruses using different phylogenetic strategies.
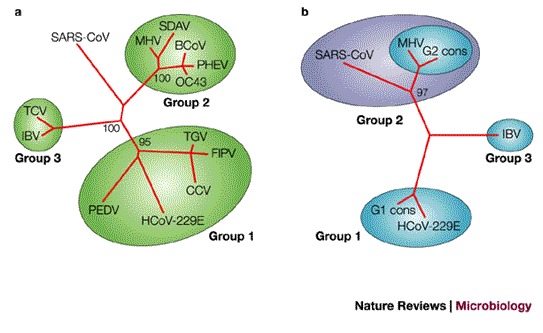
a | Unrooted tree obtained by comparing the well-conserved polymerase protein sequence. According to this approach, SARS-CoV belongs to a new group. The tree has been constructed using the protein sequences of the RNA-dependent RNA polymerase of the following coronaviruses: porcine epidemic diarrhea virus (PEDV), human coronavirus 229E (HCoV-229E), canine coronavirus (CCV), feline infectious peritonitis virus (FIPV), transmissible gastroenteritis virus (TGV), mouse hepatitis virus (MHV), bovine coronavirus (BCoV), syaloacryoadenitis virus of rats (SDAV), human coronavirus OC43 (OC43), haemagglutinating encephalomyelitis virus of swine (PHEV), turkey coronavirus (TCV), avian infectious bronchitis virus (IBV) and SARS-CoV. b | Tree obtained using the sequence of the S1 domain of the spike protein. The multiple sequence alignment was constructed using consensus sequences generated from group 1 and group 2 coronaviruses (G1 cons and G2 cons), the sequence of IBV (group 3) and of SARS-CoV. The neighbour-joining algorithm was used to build the tree51. Numbers represent the result of a bootstrap analysis performed with 100 replicates.
Figure 6. The S1 domain of SARS-CoV spike is structurally related to group 2 coronaviruses.
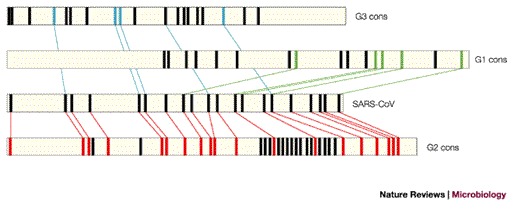
Schematic representation of cysteine positions in the S1 domains of group 1, 2 and 3 coronaviruses, compared with the SARS-CoV spike protein. Horizontal bars represent the S1 amino-acid sequences (in the case of SARS-CoV and IBV) or the consensus profiles (generated from group 1, (G1 cons) and from group 2 (G2 cons)). The bars are drawn to scale. Relative cysteine positions are indicated by rectangular bars. Only cysteines that are conserved within each consensus are reported. Coloured lines connect cysteines that are conserved between the SARS-CoV S1 domain and the consensus sequence generated from the group 1 (green), group 2 (red) and IBV S1 sequences (blue).
Protein targets
Although very little is known about the SARS-CoV proteins themselves, homologies with known coronavirus proteins can be used to predict the features of several SARS-CoV proteins that could be interesting targets for antiviral drugs or vaccines. Viral enzymes that are essential for virus replication are the most attractive candidates for the development of small, antiviral molecules, whereas some of the structural proteins represent obvious targets for vaccine development.
On the basis of the X-ray crystal structures of 3CLpro from transmissible gastroenteritis coronavirus (TGV)33 and human coronavirus 229E, a three-dimensional model of the corresponding SARS virus protein has been proposed34. The model can be used to reduce the number of compounds to be tested to find an effective protease inhibitor. Indeed, an active form of this protease has already been expressed in E.coli, which can be immediately used for drug screening. The helicase, which is already available in recombinant form, represents another attractive target for high-throughput screens20. PLpro is another potential candidate for antiviral drugs, although no homologous structures are available. Given its fundamental role in virus biology, the RNA-dependent RNA polymerase (RdRp) is high on the list of promising targets.
Of the different possible vaccine targets, the S glycoprotein is the most attractive candidate for exploitation. This protein forms the large surface projections that are characteristic of coronaviruses (Fig. 1), and which are most likely to be composed of homotrimers. The heavily glycosylated 1,255 amino-acid protein35 contains an amino-terminal, bulbous head (S1), which comprises the receptor-binding domain and is also believed to be responsible for the host and tissue tropism of the virus36,37,38. On the basis of bioinformatics and molecular modelling methods, Yu et al. proposed a putative human aminopeptidase N (hAPN) binding site in the S1 domain of the SARS-CoV spike protein39. All group 1 coronaviruses can use APN as a receptor40, whereas the group 2 mouse hepatitis virus (MHV) uses glycoproteins belonging to the carcinoembryonic antigen family as receptors on their target cells41. Whether the hAPN binding domain is the receptor in vivo remains to be shown. The protruding S1 domain is adjacent to the S2 domain, which consists of a stem, a transmembrane region and a short cytoplasmic tail. Analogous to other coronavirus S proteins, the SARS-CoV spike protein contains two heptad repeats that are located in the S2 domain. It is proposed that these repeats might trigger the fusion of the viral envelope with the cell membrane, as has been shown recently for MHV42. In some group 2 and group 3 viruses, the spike is cleaved during maturation into two subunits, S1 and S2, which stay non-covalently attached. The exact role of this cleavage process is unclear as it does not seem to influence infectivity; however, it may enhance fusion activity43,44,45,46,47. The mature SARS-CoV seems to contain uncleaved spike protein, but cleavage after binding to the target cell cannot be excluded. To conclude, the spike protein is a target for the development of agents that block the virus from binding to its cellular receptor and is the docking site for peptides that might inhibit fusion. Several additional targets for further study have been identified. First, the 76-amino-acid E protein — computer analysis has predicted a long transmembrane domain close to the N-terminus and two N-terminal glycosylation sites with a very low level of amino-acid similarity to other coronaviruses. Second, the M glycoprotein, which is a 221-residue polypeptide that consists of a short N-terminal ectodomain with a N-glycosylation site, three transmembrane segments and a C-terminus located on the interior side of the viral envelope, and which closely resembles M glycoproteins from other group 2 viruses. Third, the N protein, which is a 397-residue-long phosphoprotein that interacts with viral genomic RNA to form the helical nucleocapsid, and which has a low level of conservation with other coronaviruses.
The future for prevention and treatment
With the notable exception of β-interferon, which has been reported to interfere with the replication of the SARS virus in vitro48, no licensed drug or vaccine is available. Large-scale screening of existing antivirals or big chemical libraries for potential replication inhibitors has not been very successful. At present, the only promising substance is glycyrrhizin, a component of liquorice roots, which also has activity against HIV. This compound was identified during a small-scale experiment involving only a small number of known antiviral compounds49. The development of assays that are based on viral enzyme activity and which are amenable to high-throughput screening of existing and new chemical libraries, will likely identify effective compounds in the near future. Nevertheless, unless these putative compounds are already licensed or are existing products in the advanced stages of development, they will not be available to treat patients in the near future. Cellular proteins that are essential for virus replication should also not be overlooked as possible targets — new technologies such as double-stranded small interfering RNAs (siRNAs) have considerable future potential, but as yet, are still riddled with practical difficulties. At present, researchers are working on the development of efficacious delivery systems for siRNAs. And following the successful conclusion of this research, they will still need to prove their potential in animal models of infection.
Antibodies that are able to neutralize viral infection are also likely to be an effective way to prevent and cure this disease. Passive immunotherapy using sera from convalescent patients was initially proposed as treatment for disease. Given the excellent track record of human monoclonal antibodies in the treatment of cancer and infectious diseases, this should prove a fruitful area for therapeutic development. Indeed, monoclonal antibodies obtained from immortalized B lymphocytes of SARS convalescent patients have been shown to neutralize virus infection in vitro and to prevent virus replication in a mouse model of SARS-CoV infection (A. Lanzavecchia and R.R., unpublished observations).
Of course, a safe and effective vaccine would be the ideal solution, because not only would it prevent the disease in vaccinated people but a vaccine would also curtail the spread of the virus. Although only a short period of time passed since SARS-CoV was identified as the infectious agent that was responsible for the epidemic, candidate vaccines based on killed virus are already available. Their efficacy still needs to be shown, but our laboratory (and possibly others) are in the process of testing vaccines on the basis of inactivated SARS virus in pre-clinical models. In addition to the traditional approach, a number of newer technologies are being used. These include subunit vaccines containing recombinant spike protein expressed in mammalian cells or yeast, either alone or in combination with other SARS-CoV antigens. Alternatively, these antigens could be delivered by DNA immunization by non-replicating viruses, or viral vectors that are based on adenovirus, canarypox, modified vaccinia virus Ankara (MVA) or alphavirus. In particular, the devlopment of non-replicating coronavirus-like particles that mimic the structure of native virions could prove promising in the search for a successful vaccine as they display a large repertoire of antigenic sites and discontinuous epitopes. However, each of these approaches, including passive immunotherapy, need to be carefully evaluated as some vaccines that have been developed against feline coronavirus actually exacerbated the disease when vaccinated animals were challenged with the wild-type virus50.
Killed-virus vaccines that are ready to be tested in Phase I clinical trials are likely to be available soon. Under normal circumstances, a vaccine takes 6–8 years of clinical development after entering Phase I clinical trials. These timelines could be shortened considerably should the disease burden and a state of medical emergency induce the regulatory agencies to 'fast track' the approval process.
In conclusion, the development of therapeutic strategies against this new coronavirus seems to be possible with the available technologies and is not an unreachable goal to the same level as HIV or hepatitis C virus. Given enough time and economic pressure, antiviral drugs, human monoclonal antibodies, vaccines and siRNAs that are active against SARS-CoV are all likely to become available. The remaining question is whether we will have the time to develop effective therapies before another epidemic emerges in the human population. Should SARS return this winter, we will still need to rely primarily on quarantine measures to contain the disease. Equally important is the development of technologies that allow the rapid and simple diagnosis of SARS.
Just as worrying, however, is a scenario in which the virus does not re-emerge for a couple of years, causing the economic incentive for companies to invest in SARS to disappear, so none of the above measures will have been developed and implemented.
Box 1 | Life cycle of coronaviruses.
Coronaviruses contain the largest RNA genomes described so far — the positive-sense, single-stranded RNA molecule has a 5′ cap and a 3′ poly(A) tail. The life cycle of a coronavirus starts when the spike (S) protein, which forms the distinctive, eponymous crown that is observed with coronaviruses, interacts with a receptor through its S1 domain. The entry, which is probably mediated by the S2 domain, occurs by membrane fusion. The RNA genome is then released into the cytoplasm where replication takes place. The host translation machinery translates the overlapping open reading frames ORF1a and ORF1b by a ribosomal frame-shifting mechanism to produce a single polyprotein. Cleavage by virally encoded proteinases yields the components that are necessary to assemble the viral replication complex, which synthesizes full-length negative-strand RNA. In addition, a discontinuous transcription strategy during negative-strand synthesis produces a nested set of sub-genomic negative-sense RNAs. In this process, the transcription regulatory sequence (TRS), which is present at the 3′ end of the leader sequence and, with a few exceptions, upstream of each translated gene, is important. It is postulated to fuse the 3′ ends of the nascent subgenomic minus strands to the antisense leader sequence. These discontinuously synthesized minus strands then act as templates for the synthesis of positive-sense mRNAs. An alternative hypothesis proposes that these mRNA molecules are generated by discontinuous transcription during positive-strand synthesis (the figure shows the positive-sense mRNA products). In almost all cases, only the most 5′ ORF is translated. Nucleocapsid (N) protein and genomic RNA assemble in the cytoplasm to form the helical nucleocapsid. This core structure acquires its envelope by budding through intracellular membranes between the endoplasmic reticulum (ER) and the golgi apparatus. The membrane (M), envelope (E) and S proteins, all of which will be accommodated by the lipid bilayer, are transported through the ER to the budding compartment, where the nucleocapsid probably interacts with the M protein to trigger assembly. During the transport of the virus through the golgi apparatus, sugar moieties are modified and in some, but not all, coronaviruses the S protein is cleaved into S1 and S2 domains. Any S protein that is not incorporated into the virions is transported to the cell surface. Finally, the virus is released from the host cell by fusion of virion-containing vesicles with the plasma membrane. The common leader sequence on the 5′ end of each mRNA is shown in red.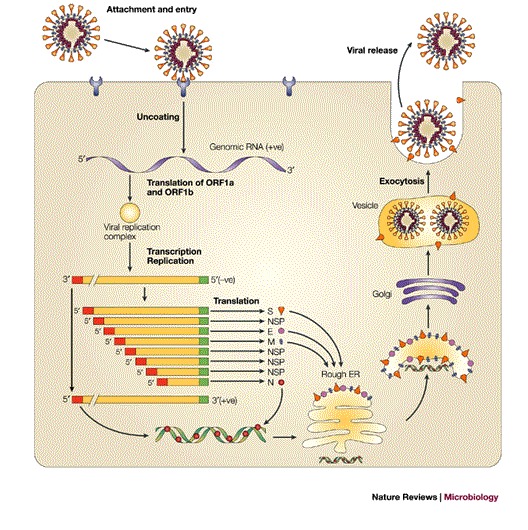
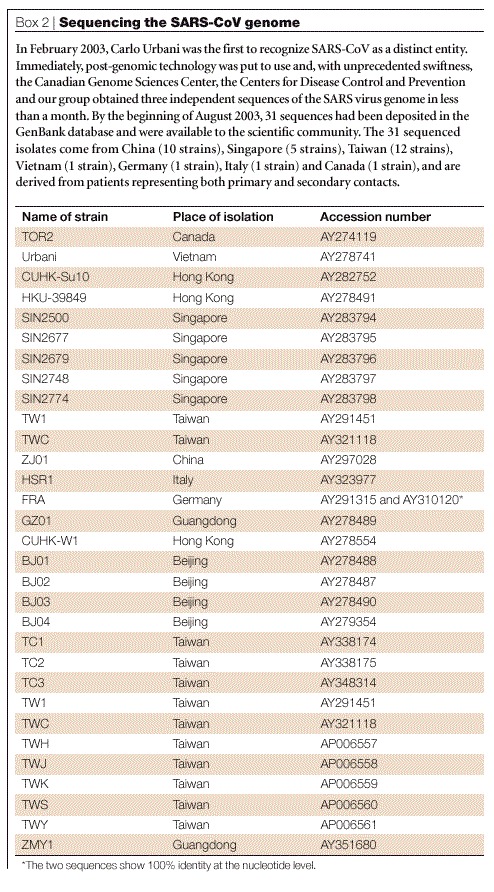
Box 2 | Sequencing the SARS-CoV genome.
In February 2003, Carlo Urbani was the first to recognize SARS-CoV as a distinct entity. Immediately, post-genomic technology was put to use and, with unprecedented swiftness, the Canadian Genome Sciences Center, the Centers for Disease Control and Prevention and our group obtained three independent sequences of the SARS virus genome in less than a month. By the beginning of August 2003, 31 sequences had been deposited in the GenBank database and were available to the scientific community. The 31 sequenced isolates come from China (10 strains), Singapore (5 strains), Taiwan (12 strains), Vietnam (1 strain), Germany (1 strain), Italy (1 strain) and Canada (1 strain), and are derived from patients representing both primary and secondary contacts. (Table 3)
Table 3.
SARS – BEGINNING TO UNDERSTAND A NEW VIRUS
Acknowledgements
The authors are grateful to the Fonds der Chemischen Industrie and the Chiron Vaccines Cellular Microbiology and Bioinformatics Unit for their continuous support.
Biographies
Konrad Stadler studied biochemistry at the University of Vienna, Austria, before he did his Ph.D. work on TBE virus at the Institute of Virology, University of Vienna. Subsequently, he was a postdoctoral fellow at the Institute of Virology, University of Utrecht, The Netherlands, working on the coronavirus MHV. At present, he is working at the Department of Immunology/Virology of Chiron S.r.l., Siena, Italy.
Vega Masignani graduated with a degree in pharmaceutical chemistry and a Ph.D. in biotechnology from the University of Siena, Italy, with a thesis on the computational approach to the development of a novel Neisseria meningitidis vaccine. At present, she is a staff scientist at the Cellular Microbiology and Bioinformatics Unit of Chiron Vaccines, carrying out research in the field of computer analysis applied to microbial pathogenesis.
Dr. Markus Eickmann studied biological science at the University of Marburg, Germany (1987–1993), followed by a doctoral thesis at the Institut für Virologie, Marburg. Since 1996, his research has focused on the structure and function of viral glycoproteins of several viruses, for example, human cytomegalovirus, Borna disease virus, influenza and SARS coronavirus.
Sergio Abrignani is a M.D., Ph.D., interested in immune response to viruses. In the last years, he has been working on the hepatitis C virus, focusing in particular on host–pathogen interactions. He is Vice President of Immunology and Virology Research at Chiron Vaccines.
Hans-Dieter Klenk graduated in 1963 from the Medical School of the University of Cologne, Germany. In 1967, he received a degree in biochemistry at the University of Tuebingen, Germany. He was a postdoctoral fellow at Rockefeller University in New York from 1967 to 1970 and subsequently, an associate professor at the University of Giessen, Germany. At present, he is professor and chairman of the Department of Virology at the University of Marburg, Germany. His main research interests include influenza virus and filoviruses.
Rino Rappuoli is responsible for Chiron Vaccines Research. After earning his Ph.D. in biological sciences from the University of Siena, he completed his education with stages at Washington University, Rockefeller University and Harvard Medical School. His field of expertise focuses on vaccines, bacterial pathogenesis and infectious diseases. He has developed the first recombinant bacterial vaccine (against pertussis) and a conjugate vaccine against meningococcus C, and at present is developing a vaccine against group B meningococcus using the genome-based approach known as reverse vaccinology.
Related links
DATABASES
Entrez
The Protein Data Bank
FURTHER INFORMATION
Accession codes
Accessions
GenBank/EMBL/DDBJ
Competing interests
S. Abrignani, R. Rappuoli, K. Stadler and V. Masignani are employed by Chiron Corporation.
References
- 1.Peiris JSM, et al. Coronavirus as a possible cause of severe acute respiratory syndrome. Lancet. 2003;361:1319–1325. doi: 10.1016/S0140-6736(03)13077-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Donnelly CA, et al. Epidemiological determinants of spread of causal agent of severe acute respiratory syndrome in Hong Kong. Lancet. 2003;361:1761–1766. doi: 10.1016/S0140-6736(03)13410-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hon KL, et al. Clinical presentations and outcome of severe acute respiratory syndrome in children. Lancet. 2003;361:1701–1703. doi: 10.1016/S0140-6736(03)13364-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Peiris JSM, et al. Clinical progression and viral load in a community outbreak of coronavirus-associated SARS pneumonia: a prospective study. Lancet. 2003;361:1767–1772. doi: 10.1016/S0140-6736(03)13412-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.WHO. Acute respiratory syndrome in China. [online], (cited 15 Oct 2003), <http://www.who.int/csr/don/2003_2_20/en/> (2003).
- 6.WHO. Summary of probable SARS cases with onset of illness from 1 November 2002 to 31 July 2003. [online], (cited 15 Oct 2003), <http://www.who.int/csr/sars/country/table2003_09_23/en/> (2003).
- 7.Drosten C, et al. Identification of a novel coronavirus in patients with severe acute respiratory syndrome. N. Engl. J. Med. 2003;348:1967–1976. doi: 10.1056/NEJMoa030747. [DOI] [PubMed] [Google Scholar]
- 8.Ksiazek TG, et al. A Nnvel coronavirus associated with severe acute respiratory syndrome. N. Engl. J. Med. 2003;348:1953–1966. doi: 10.1056/NEJMoa030781. [DOI] [PubMed] [Google Scholar]
- 9.Fouchier RA, et al. Aetiology: Koch's postulates fulfilled for SARS virus. Nature. 2003;423:240. doi: 10.1038/423240a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kuiken T, et al. Newly discovered coronavirus as the primary cause of severe acute respiratory syndrome. Lancet. 2003;362:263–270. doi: 10.1016/S0140-6736(03)13967-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Riley S, et al. Transmission dynamics of the etiological agent of SARS in Hong Kong: impact of public health interventions. Science. 2003;300:1961–1966. doi: 10.1126/science.1086478. [DOI] [PubMed] [Google Scholar]
- 12.Lipsitch M, et al. Transmission dynamics and control of severe acute respiratory syndrome. Science. 2003;300:1966–1970. doi: 10.1126/science.1086616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Guan Y, et al. Isolation and characterization of viruses related to the SARS coronavirus from animals in southern China. Science. 2003;302:276–278. doi: 10.1126/science.1087139. [DOI] [PubMed] [Google Scholar]
- 14.Normile D, Enserink M. SARS in China: tracking the roots of a killer. Science. 2003;301:297–299. doi: 10.1126/science.301.5631.297. [DOI] [PubMed] [Google Scholar]
- 15.WHO. Severe acute respiratory syndrome (SARS) in Singapore. [online], (cited 15 Oct 2003), <http://www.who.int/csr/don/2003_09_24/en/> (2003).
- 16.Enjuanes L, et al. Virus Taxonomy. 2002. pp. 835–849. [Google Scholar]
- 17.Marra MA, et al. The genome sequence of the SARS-associated coronavirus. Science. 2003;300:1399–1404. doi: 10.1126/science.1085953. [DOI] [PubMed] [Google Scholar]
- 18.Rota PA, et al. Characterization of a novel coronavirus associated with severe acute respiratory syndrome. Science. 2003;300:1394–1399. doi: 10.1126/science.1085952. [DOI] [PubMed] [Google Scholar]
- 19.Zeng FY, et al. The complete genome sequence of severe acute respiratory syndrome coronavirus strain HKU-39849 (HK–39) Exp. Biol. Med. 2003;228:866–873. doi: 10.1177/15353702-0322807-13. [DOI] [PubMed] [Google Scholar]
- 20.Thiel V, et al. Mechanisms and enzymes involved in SARS coronavirus genome expression. J. Gen. Virol. 2003;84:2305–2315. doi: 10.1099/vir.0.19424-0. [DOI] [PubMed] [Google Scholar]
- 21.Thiel V, Herold J, Schelle B, Siddell SG. Viral replicase gene products suffice for coronavirus discontinuous transcription. J. Virol. 2001;75:6676–6681. doi: 10.1128/JVI.75.14.6676-6681.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.von Grotthuss M, Wyrwicz LS, Rychlewski L. mRNA cap-1 methyltransferase in the SARS genome. Cell. 2003;113:701–702. doi: 10.1016/S0092-8674(03)00424-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lai MM, Cavanagh D. The molecular biology of coronaviruses. Adv. Virus Res. 1997;48:1–100. doi: 10.1016/S0065-3527(08)60286-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.van der Most RG, Spaan WJM. The Coronaviridae. 1995. pp. 11–31. [Google Scholar]
- 25.de Haan CAM, Masters PS, Shen X, Weiss S, Rottier PJM. The group-specific murine coronavirus genes are not essential, but their deletion, by reverse genetics, is attenuating in the natural host. Virology. 2002;296:177–189. doi: 10.1006/viro.2002.1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sola I, et al. Engineering the transmissible gastroenteritis virus genome as an expression vector inducing lactogenic immunity. J. Virol. 2003;77:4357–4369. doi: 10.1128/JVI.77.7.4357-4369.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sarma JD, Scheen E, Seo SH, Koval M, Weiss SR. Enhanced green fluorescent protein expression may be used to monitor murine coronavirus spread in vitro and in the mouse central nervous system. J. Neurovirol. 2002;8:381–391. doi: 10.1080/13550280260422686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jonassen CM, Jonassen TO, Grinde B. A common RNA motif in the 3′ end of the genomes of astroviruses, avian infectious bronchitis virus and an equine rhinovirus. J. Gen. Virol. 1998;79:715–718. doi: 10.1099/0022-1317-79-4-715. [DOI] [PubMed] [Google Scholar]
- 29.Lai MM, Holmes KV. Fields' Virology. 2001. pp. 1163–1185. [Google Scholar]
- 30.Ruan YJ, et al. Comparative full-length genome sequence analysis of 14 SARS coronavirus isolates and common mutations associated with putative origins of infection. Lancet. 2003;361:1779–1785. doi: 10.1016/S0140-6736(03)13414-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ksiazek TG, et al. A novel coronavirus associated with severe acute respiratory syndrome. N. Engl. J. Med. 2003;348:1953–1966. doi: 10.1056/NEJMoa030781. [DOI] [PubMed] [Google Scholar]
- 32.Snijder EJ, et al. Unique and conserved features of genome and proteome of SARS-coronavirus, an early split-off from the coronavirus group 2 lineage. J. Mol. Biol. 2003;331:991–1004. doi: 10.1016/S0022-2836(03)00865-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Anand K, et al. Structure of coronavirus main proteinase reveals combination of a chymotrypsin fold with an extra α-helical domain. EMBO J. 2002;21:3213–3224. doi: 10.1093/emboj/cdf327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Anand K, Ziebuhr J, Wadhwani P, Mesters JR, Hilgenfeld R. Coronavirus main proteinase (3CLpro) structure: basis for design of anti-SARS drugs. Science. 2003;300:1763–1767. doi: 10.1126/science.1085658. [DOI] [PubMed] [Google Scholar]
- 35.Krokhin O, et al. Mass Spectrometric Characterization of proteins from the SARS virus: a preliminary report. Mol. Cell Proteomics. 2003;2:346–356. doi: 10.1074/mcp.M300048-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cavanagh D. The Coronaviridae. 1995. pp. 73–113. [Google Scholar]
- 37.Kuo L, Godeke GJ, Raamsman MJB, Masters PS, Rottier PJM. Retargeting of coronavirus by substitution of the spike glycoprotein ectodomain: crossing the host cell species barrier. J. Virol. 2000;74:1393–1406. doi: 10.1128/JVI.74.3.1393-1406.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Haijema BJ, Volders H, Rottier PJM. Switching species tropism: an effective way to manipulate the feline coronavirus genome. J. Virol. 2003;77:4528–4538. doi: 10.1128/JVI.77.8.4528-4538.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yu XJ, et al. Putative hAPN receptor binding sites in SARS-CoV spike protein. Acta Pharmacol. Sin. 2003;24:481–488. [PubMed] [Google Scholar]
- 40.Tresnan DB, Holmes KV. Feline aminopeptidase N is a receptor for all group I coronaviruses. Adv. Exp. Med. Biol. 1998;440:69–75. doi: 10.1007/978-1-4615-5331-1_9. [DOI] [PubMed] [Google Scholar]
- 41.Holmes KV, Zelus BD, Schickli JH, Weiss SR. Receptor specificity and receptor-induced conformational changes in mouse hepatitis virus spike glycoprotein. Adv. Exp. Med. Biol. 2001;494:173–181. doi: 10.1007/978-1-4615-1325-4_29. [DOI] [PubMed] [Google Scholar]
- 42.Bosch BJ, van der Zee R, de Haan CAM, Rottier PJM. The coronavirus spike protein is a class I virus fusion protein: structural and functional characterization of the fusion core complex. J. Virol. 2003;77:8801–8811. doi: 10.1128/JVI.77.16.8801-8811.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bos EC, Luytjes W, Spaan WJ. The function of the spike protein of mouse hepatitis virus strain A59 can be studied on virus-like particles: cleavage is not required for infectivity. J. Virol. 1997;71:9427–9433. doi: 10.1128/jvi.71.12.9427-9433.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gombold JL, Hingley ST, Weiss SR. Fusion-defective mutants of mouse hepatitis virus A59 contain a mutation in the spike protein cleavage signal. J. Virol. 1993;67:4504–4512. doi: 10.1128/jvi.67.8.4504-4512.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Taguchi F. Fusion formation by the uncleaved spike protein of murine coronavirus JHMV variant cl-2. J. Virol. 1993;67:1195–1202. doi: 10.1128/jvi.67.3.1195-1202.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hingley ST, Leparc-Goffart I, Seo SH, Tsai JC, Weiss SR. The virulence of mouse hepatitis virus strain A59 is not dependent on efficient spike protein cleavage and cell-to-cell fusion. J. Neurovirol. 2002;8:400–410. doi: 10.1080/13550280260422703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Stauber R, Pfleiderera M, Siddell S. Proteolytic cleavage of the murine coronavirus surface glycoprotein is not required for fusion activity. J. Gen. Virol. 1993;74:183–191. doi: 10.1099/0022-1317-74-2-183. [DOI] [PubMed] [Google Scholar]
- 48.Cinatl J, et al. Treatment of SARS with human interferons. Lancet. 2003;362:293–294. doi: 10.1016/S0140-6736(03)13973-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cinatl J, et al. Glycyrrhizin, an active component of liquorice roots, and replication of SARS-associated coronavirus. Lancet. 2003;361:2045–2046. doi: 10.1016/S0140-6736(03)13615-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Olsen CW. A review of feline infectious peritonitis virus: molecular biology, immunopathogenesis, clinical aspects, and vaccination. Vet. Microbiol. 1993;36:1–37. doi: 10.1016/0378-1135(93)90126-R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Saitou N, Nei M. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 1987;4:406–425. doi: 10.1093/oxfordjournals.molbev.a040454. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.