Central Nervous System Primitive Neuroectodermal Tumors: A Clinicopathologic and Genetic Study of 33 Cases (original) (raw)
Abstract
Central nervous system (CNS) primitive neuroectodermal tumors (PNETs) include supratentorial, brain stem, and spinal cord tumors with medulloblastoma‐like histopathology. The prognostic impact of various pathologic and genetic features has not been thoroughly investigated. After re‐diagnosis of three infantile cases as atypical teratoid/rhabdoid tumor (AT/RT), 33 remaining CNS PNETs were retrieved for clinicopathologic and fluorescence in situ hybridization studies. Anaplastic and/or large cell features were seen in 18 of 33 (55%) examples and survival was decreased in these patients (P = 0.036). MYCN or MYCC gene amplifications were noted in about half, with a trend towards decreased survival (P = 0.112). Polysomies of chromosomes 2 and 8 were each individually associated with decreased survival in children, with an even stronger association when combined (P = 0.013). Neither EWS gene rearrangements, nor AT/RT‐like 22q deletions were encountered. We conclude that in CNS PNET: (i) routine application of INI1 immunohistochemistry helps rule out AT/RT, particularly in infants; (ii) MYC gene amplifications (especially MYCN) are common; (iii) involvement of CNS parenchyma by Ewing sarcoma/peripheral PNET is rare enough that EWS gene testing is not necessary unless significant dural involvement is present; and (iv) both anaplastic/large cell features and polysomies of 2 and 8 are associated with more aggressive clinical behavior.
Keywords: anaplasia, medulloblastoma, fluorescence in situ hybridization, primitive neuroectodermal tumor, prognosis
INTRODUCTION
Although a highly contentious topic, the recent fourth edition of the World Health Organization (WHO) classification of tumors of the central nervous system (CNS) made substantial revisions to the category of embryonal tumors based on the most currently available data and consensus opinions by experts in the field. The CNS primitive neuroectodermal tumors (CNS PNET) are now defined as “a heterogeneous group of tumors occurring predominantly in children and adolescents that may arise in the cerebral hemispheres, brain stem, or spinal cord, and are composed of undifferentiated or poorly differentiated neuroepithelial cells which may display divergent differentiation along neuronal, astrocytic and ependymal lines”(35). This new terminology includes previously termed “supratentorial primitive neuroectodermal tumors” (sPNETs), as well as histologically and clinically similar tumors of the brain stem and spinal cord (ie, CNS PNET, NOS). Accepted CNS PNET variants also include CNS neuroblastoma, ganglioneuroblastoma, medulloepithelioma and ependymoblastoma, based on their distinctive patterns of differentiation (35). The even rarer subtype, embryonal tumor with abundant neuropil and true rosettes (ETANTR) is also discussed within the CNS PNET chapter. ETANTR is a distinctive embryonal tumor with extensive neuronal and ependymal differentiation, showing some overlapping features with both CNS neuroblastoma and ependymoblastoma; they have been primarily encountered in infants with poor outcome 11, 16, 27. In contrast, other primitive round cell neoplasms, such as pineoblastoma, were considered sufficiently unique to retain separate diagnostic headings.
CNS PNETS occur predominantly in infants and children with a mean age of 5.5 years, but sporadic adult cases have also been reported 30, 42. The presence of large cell and anaplastic features, similar to those encountered in medulloblastomas, have occasionally been noted in CNS PNETs as well, although to our knowledge, no formal studies have been performed on their potential clinical significance 12, 39.
Despite shared histopathologic features with medulloblastomas, CNS PNETs are considerably more aggressive and have different genetic characteristics 17, 18, 43. For instance, isochromosome 17q is the most frequent genetic abnormality in medulloblastoma, but has only been reported once in a supratentorial PNET and once in a case of ETANTR 7, 15. In addition to differences in 17q gain, comparative genomic hybridization studies indicate that losses at chromosome 10 are present in medulloblastoma, whereas losses at 16p and 19p are more frequently encountered in CNS PNETs (23). Other reported molecular alterations in CNS PNET include: RASSIF1A promoter methylation (22), CDKN2A deletions (41), p14 ARF methylation (24), transcriptional silencing of DLC‐1 (38), expression of the Neuro D family of basic helix‐loop helix transcription factors and expression of the related neurogenic transcription factor, achaete scute (35).
In a recent study of malignant gliomas with primitive neuroectodermal tumor‐like components, MYCN or MYCC gene amplifications were seen in 43% of cases within the primitive elements (40). MYCN gene expression is also a well known biomarker, associated with advanced tumor stage in peripheral neuroblastomas 4, 46. Of even greater relevance, MYCN and MYCC gene amplifications have been reported in both medulloblastomas (especially those with anaplastic or large cell features) and CNS PNETs, though the latter tumor type has rarely been studied using these markers; in most cases, these alterations have been associated with aggressive clinical behavior and shortened patient survival 14, 19, 31, 44.
Lastly, the differential for primitive tumors of the CNS has recently expanded, given the recognition of peripheral type Ewing sarcoma/PNETs (EWS/pPNET) presenting in intracranial and intraspinal sites 9, 29, 34, 37. Virtually all of these cases have been meningeal‐based, often mimicking meningioma radiologically. Nevertheless, the possibility of occasional intraparenchymal EWS/PNETs has not been explored genetically (ie, evidence of EWS gene rearrangements) and they could be difficult to distinguish from CNS PNETs based on morphology alone. Given that the relationship between survival and histologic or genetic features has not been fully explored in CNS PNETs, we assessed clinicopathologic and fluorescence in situ hybridization (FISH) parameters on archived cases from our surgical pathology and consult files at a single medical center.
MATERIALS AND METHODS
Patient/tumor cohort
After approval by the Washington University in St. Louis Human Research Program, a retrospective study was conducted using a combination of archival in‐house pathology material and consultation cases submitted to the senior author between 1988 and July 2008. All non‐cerebellar CNS parenchymal tumors with the term PNET in the diagnosis were pulled for potential inclusion. Cases diagnosed as medulloblastoma or pineoblastoma were excluded. All available slides were reviewed and graded for anaplastic/large cell features using the previously established criteria for medulloblastomas (12). Additional immunohistochemistry was obtained as needed, particularly for older cases with more limited workup. Of particular interest was the subsequent inclusion of INI1 (BAF47; 1:25, BD Biosciences, Chicago, IL, USA) immunohistochemistry in all cases, as this useful antibody only became commercially available within the last few years. Initially, 36 tumors were identified, but nine were felt to have overlapping morphologic features with atypical teratoid/rhabdoid tumor (AT/RT), three of which showed loss of INI1 expression (ages 1.5–3.5 years), and were therefore re‐diagnosed as AT/RT, being excluded from further study. One of these three excluded cases had also undergone FISH analysis, which showed evidence of 22q loss (see later discussion and Figure 4D). Clinical data were obtained by review of medical records and were supplemented by dates of death derived from the Social Security Death Index (http://ssdi.genealogy.rootsweb.com/cgi‐bin/ssdi.cgi).
Figure 4.
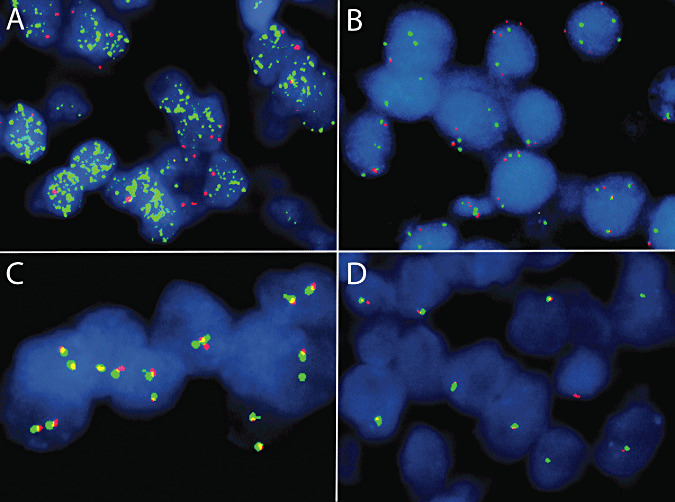
Representative fluorescence in situ hybridization results, including high‐level MYCN gene amplification (A; centromere 2 in red, MYCN in green; there are innumerable green signals), polysomy 8 (B; centromere 8 in green, MYCC in red; there are three to four green and red signals in most nuclei), and a lack of split signals using the Ewing sarcoma (EWS) break‐apart probe set (C,D; centromeric portion of EWS in red and telomeric portion in green; two fused yellow or red‐green signals are evident in most tumor cells). In most examples of central nervous system primitive neuroectodermal tumors, the majority of tumor cells showed the normal two copies of EWS (C), whereas an excluded case of atypical teratoid/rhabdoid tumor showed only one copy, consistent with a 22q deletion or monosomy 22 (D).
Immunohistochemistry and FISH
Immunohistochemical and FISH studies were performed and interpreted using standard techniques as previously described 5, 32, 40. Portions of the immunohistochemical and FISH studies had already been performed as part of the routine diagnostic workup, but this was supplemented for missing data in all cases with available paraffin blocks or archived unstained sections. As part of the diagnostic definition, all cases contained at least a subpopulation of small primitive cells with expression of one or more neuronal markers, most commonly synaptophysin. The most commonly applied markers were synaptophysin, neurofilament protein, Neu‐N and glial fibrillary acidic protein (GFAP). In consult cases, immunohistochemistry had also often been performed in part by the referring pathologists using standard techniques.
The DNA probes utilized for FISH were commercially available and consisted of the EWSR1 dual color break‐apart set (combines a 500 kb Spectrum Orange‐labeled probe on the centromeric side of the 7 kb EWSR1 breakpoint region between exons 7 and 10 of the EWS gene with an 1100 kb Spectrum Green‐labeled probe localizing just telomeric to this breakpoint region), centromere enumerating probe 2 (CEP2; labeled in Spectrum Orange) paired with the MYCN region on 2p24.1 (Spectrum Green), and CEP8 (Spectrum Green) paired with MYCC region on 8q24.12–q24.13 (Spectrum Orange) (Vysis/Abbott Laboratories, Downers Grove, IL, USA).
Statistics
Survival curves were generated using the Kaplan–Meier method. Statistical analysis for association between age and survival was computed using the Cox regression model. P values were derived via the log–rank test. These statistical analyses were performed using SPSS for Windows (version 17, SPSS Inc., Chicago, IL, USA). Fisher exact test was used to assess the potential relationships between presence of anaplastic/large cell features and either MYC gene amplifications or polysomies (chromosomal gains). All statistical tests were two‐sided and P < 0.05 was considered significant.
RESULTS
Clinical findings
Demographic and radiologic findings are summarized in Table 1, with representative neuroimaging shown in Figure 1. The age of patients at diagnosis ranged from 6 months to 62 years, and the overall male : female ratio was 1.2:1. The majority of patients were children (n = 27; ages 0.5–16.5 years; median of 5.5 years), whereas all six adult patients (ages 20–61 years; median of 27 years) had supratentorial tumors that were submitted in consultation to the senior author. Presenting symptoms included headaches, seizures, dizziness, nausea/vomiting, cranial nerve palsies, blurred vision, somnolence, weakness and sensory abnormalities. The neuroimaging studies were available for review in only 11 patients and the majority of tumors (n = 8) were heterogeneously enhancing (Figure 1) or ring‐enhancing masses. However, three cerebral tumors (one frontal and two temporal tumors) were non‐enhancing. Many of these tumors appeared more discrete than is typically seen with high‐grade diffuse gliomas, although none of the imaging features were considered sufficiently specific to provide a confident pre‐operative diagnosis of CNS PNET. The tumors involved the cerebral hemisphere in 28 (Figure 1A,B), brainstem in three (Figure 1C,D), thalamus in one and spinal cord in one patient.
Table 1.
Demographic and radiographic features of patients.
| Pediatric | Adult | |
|---|---|---|
| Age | 0.5–18 years (median = 5.5 years) | 20–61 years (median = 27 years) |
| Sex | 15 male : 12 female (ratio = 1.25) | 2 male : 4 female (ratio = 0.5) |
| Survival time | 2 months–15 years (median = 32 months) | Not available |
| Location | ||
| Cerebral hemisphere | 22/27 (81%) | 6/6 (100%) |
| Brain stem | 3/27 (11%) | 0 |
| Thalamus | 1/27 (4%) | 0 |
| Spinal cord | 1/27 (4%) | 0 |
| Imaging findings (n = 11) | ||
| Size (largest dimension) | 3–7 cm (5 ± 1.6 cm) | Not available |
| Contrast enhancement | 8/11 + (72%) | Not available |
Figure 1.
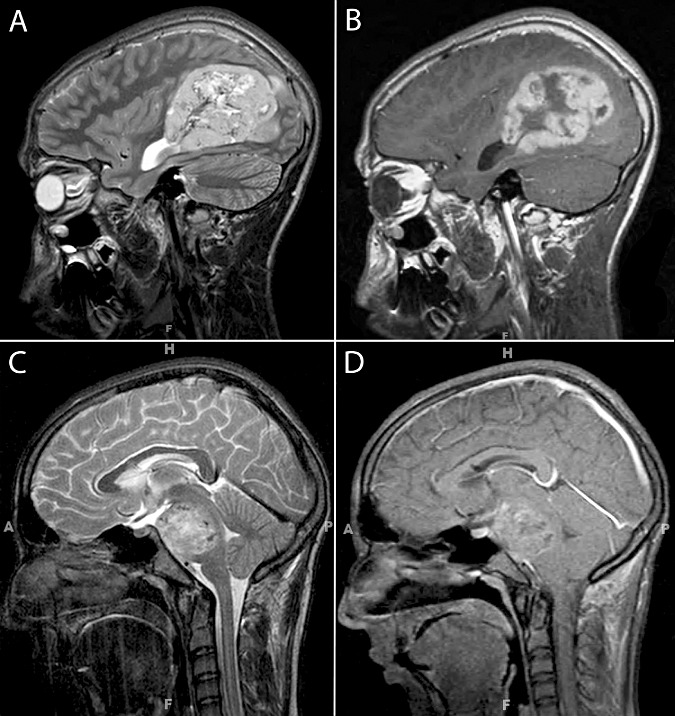
Representative T2‐weighted (A,C) and post‐contrast T1‐weighted (B,D) images from supratentorial (A,B) and brainstem (C,D) examples of central nervous system primitive neuroectodermal tumors. The tumors appear relatively discrete and show heterogeneous contrast enhancement with patchy hypodensities suggestive of necrosis.
Follow up data were available for 22 patients (21 pediatric and one adult patients), of which 16 patients (73%) had died. Survival times ranged from 2 months to 15 years (median = 32 months) from the first diagnosis of CNS PNET. The 3‐year survival was 33%.
Histologic and immunohistochemical features
Histopathologic features are summarized in Table 2 and illustrated in 2, 3. Using current WHO criteria, all cases qualified for the diagnosis of CNS PNET, the majority of which would have been not otherwise specified. However, several had features of CNS PNET subtypes, including one ganglioneuroblastoma, one ependymoblastoma and one ETANTR, the latter of which was previously published as a case report (11). There were no examples of medulloepithelioma. All cases were highly cellular and composed mostly of small (slightly larger in anaplastic/large cells), primitive appearing cells with high N/C ratios, often with a predominantly solid central component and variably infiltrative peripheral margins (Figure 2). Nuclear molding was common and mitotic/pyknotic indices were typically high. In classic (non‐anaplastic) examples, nuclei were oval to carrot shaped and hyperchromatic, associated with minimal discernable cytoplasm. Homer Wright (neuroblastic) rosettes (Figure 2D) were noted in five (18%) pediatric and two (33%) adult patients. Other histologic features such as desmoplastic/nodular pattern (Figure 2E) (3.5%), true rosettes (Figure 2F) (7%), sarcomatous features (3.5%) and ganglion cell differentiation (3.5%) were only encountered rarely in pediatric patients and in none of the adult patients. Large cell (increased cell and nuclear size with oval‐shaped nuclei and macronucleoli; Figure 3A) and anaplastic (increased cell and nuclear size with irregular, pleomorphic hyperchromatic nuclei; Figure 3B) features such as those described in medulloblastomas were seen in 15 (55%) pediatric and three (50%) adult patients, often with both patterns encountered in the same tumors. In all such cases, the anaplastic features were considered moderate to severe using Eberhart criteria (12) and they were diffuse or at least widespread, involving large portions of the tumor. Tumors with only small foci of anaplasia or only slight anaplastic features were not scored as large cell/anaplastic.
Table 2.
Pathologic and genetic features in pediatric and adult patients. Abbreviations: EWS = Ewing sarcoma; GFAP = glial fibrillary acidic protein.
| Histologic features/protein expression | Pediatric (%) | Fluorescence in situ hybridization | Pediatric (%) |
|---|---|---|---|
| Anaplastic/Large cell features (n = 27) | 59 | MYCN amplification (n = 22) | 41 |
| Homer Wright Rosettes (n = 27) | 18 | MYCC amplification (n = 21) | 9.5 |
| GFAP (n = 26) | 77 | EWS rearrangement (n = 21) | 0 |
| Synaptophysin (n = 23) | 87 | Polysomy 2 (n = 23) | 43 |
| Neurofilament (n = 10) | 70 | Polysomy 8 (n = 23) | 52 |
| Neu‐N (n = 14) | 64 | Polysomy 22q (n = 21) | 5 |
| CD99 (n = 11) | 36 | ||
| CAM 5.2 (n = 9) | 33 |
| Histologic features/protein expression | Adults (%) | Fluorescence in situ hybridization | Adults (%) |
|---|---|---|---|
| Anaplasia/large cell features (n = 6) | 50 | MYCN amplification (n = 5) | 0 |
| Homer Wright rosettes (n = 6) | 33 | MYCC amplification (n = 5) | 16 |
| GFAP (n = 5) | 60 | EWS rearrangement (n = 6) | 0 |
| Synaptophysin (n = 6) | 100 | Polysomy 2 (n = 5) | 100 |
| Neurofilament (n = 5) | 40 | Polysomy 8 (n = 5) | 100 |
| Polysomy 22q (n = 6) | 33 |
Figure 2.
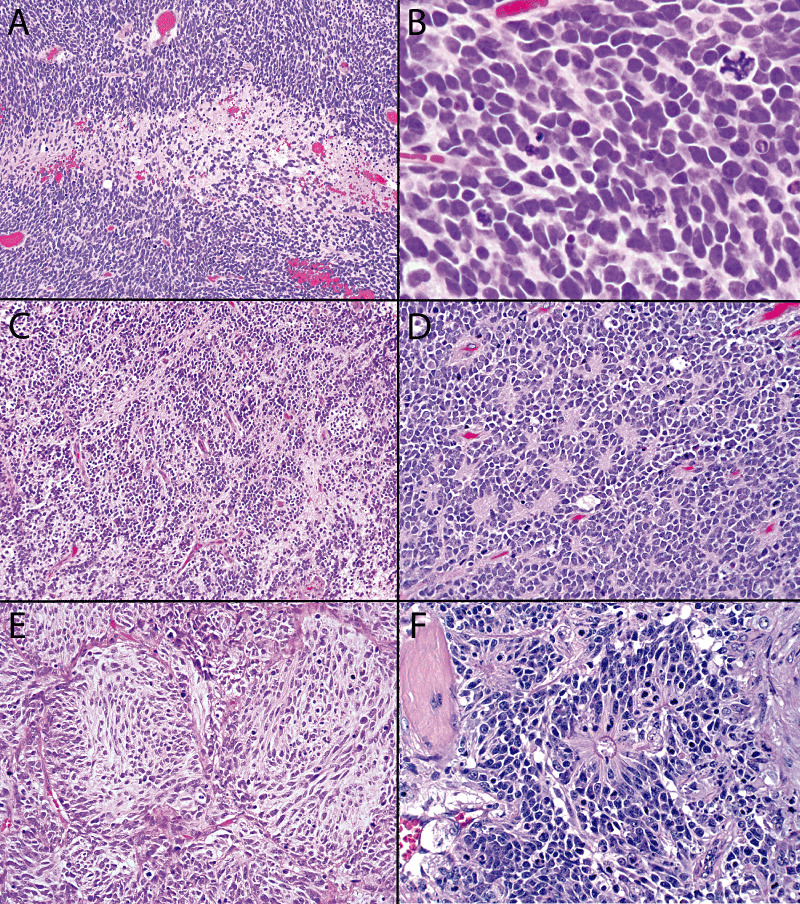
Examples of classical central nervous system primitive neuroectodermal tumor variants (ie, without anaplastic or large cell features). Hypercellularity with patchy necrosis is seen at low magnification (A), with higher magnification revealing oval to carrot‐shaped hyperchromatic nuclei with minimal discernable cytoplasm, numerous mitotic figures and pyknotic nuclei, and occasional atypical mitoses (B). Tumor‐associated neuropil was appreciated in a subset of cases, either as the deposition of a patchy delicate fibrillary matrix (C) or associated with Homer Wright (neuroblastic) rosettes (D). Less common features included a nodular growth pattern (E) and ependymoblastic (true) rosettes (F).
Figure 3.
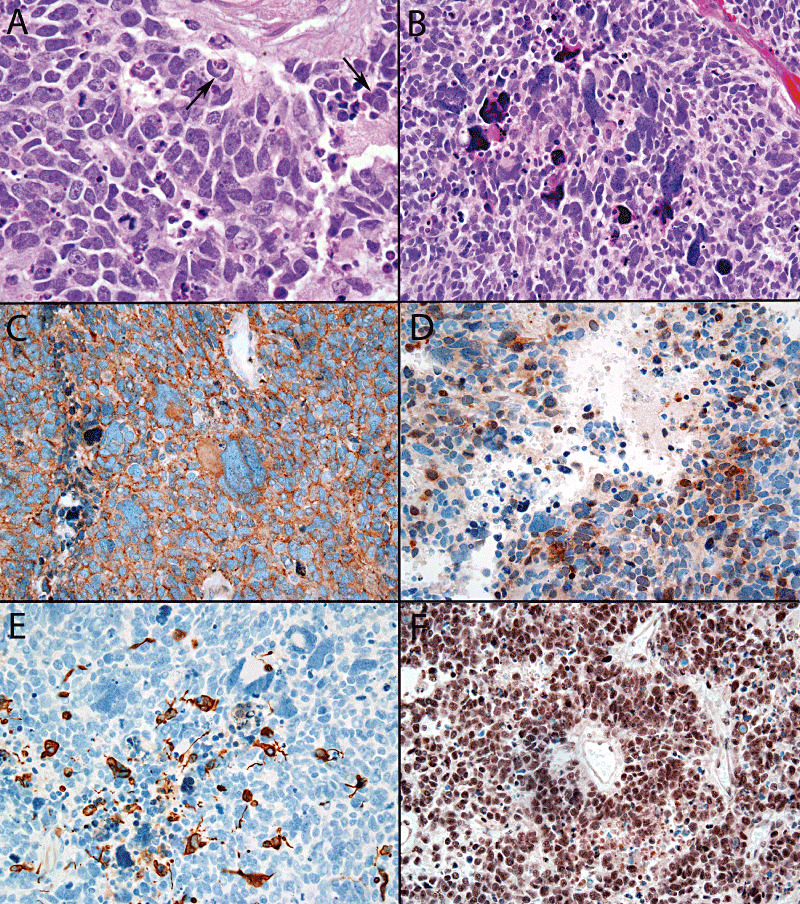
Large cell features included enlarged vesicular nuclei, prominent nucleoli, and cell wrapping (arrows) (A), whereas anaplasia was defined by the presence of enlarged pleomorphic, sometimes bizarre or multinucleated tumor cells (B). The tumor displayed in panel B expressed neuronal markers, displaying nearly diffuse synaptophysin (C) and patchy Neu‐N positivity (D). Scattered glial fibrillary acidic protein‐positive tumor cells were also present (E). In contrast to atypical teratoid/rhabdoid tumor, the large cell neoplasm displayed in A retained BAF47 (INI‐1) expression in tumor nuclei (F).
Among the neuronal markers, synaptophysin was most commonly performed (28 cases) and was positive in 87% of pediatric and all adult cases (Figure 3C). The pattern of expression of synaptophysin was focal in half of these cases and extensive in the rest. Other positive neuronal markers included neurofilament protein (70%) and Neu‐N (64%) (Figure 3D). GFAP was performed in the majority of cases and was at least focally positive in 77% of pediatric and 60% of adult patients. Most cases showed scattered positive tumor cells (Figure 3E). The EWS/PNET‐associated marker, CD99 showed membrane pattern expression in 36% of pediatric tumors, although it was typically not as diffuse as one would expect in EWS/pPNET. As per study definition, BAF47 (INI‐1) expression was retained in tumor nuclei of all CNS PNET cases (Figure 3F).
FISH
FISH was performed in 30 patients (24 pediatric and all six adult patients). The results are summarized in 2, 3, with representative cases illustrated in Figure 4. Among pediatric tumors, 11 (50%) showed MYC gene amplifications, including nine (41%) MYCN (Figure 4A) and two (9.5%) MYCC subtypes. In adults, only one tumor showed an MYCC amplification. No cases showed amplification of both genes simultaneously. Polysomies of chromosomes 2, 8 (Figure 4B) and 22 were seen in 43%, 52% and 5% of pediatric tumors respectively. All adult tumors showed polysomies of 2 and 8, with two additionally demonstrating polysomy 22. The EWS break‐apart FISH did not show any split red and green signals that suggest EWS gene rearrangements in any of the 27 pediatric and adult tumors studied (Figure 4C). There was also no evidence of 22q deletion in any of these cases, although one of the excluded cases of AT/RT had evidence of 22q loss (Figure 4D).
Table 3.
Prognostic factors for survival in pediatric patients. Abbreviations: EWS = Ewing sarcoma; LC = large cell features.
| Factor | Distribution (%) | Survival (P value) |
|---|---|---|
| Age (1 year increase) | 0.5–18 years | >0.90* |
| Male (vs. female) | 15/27 (55) | >0.90 |
| Anaplasia/large cell features | 16/27 (59) | 0.053 |
| Fluorescence in situ hybridization | ||
| MYC amplification | 11/22 (50) | 0.112 |
| _MYC_N | 9/22 (41) | |
| _MYC_C | 2/22 (9) | |
| EWS rearrangement | 0/21 (0) | Not available |
| Polysomy 2 | 10/23 (43) | 0.025 |
| Polysomy 8 | 11/23 (52) | 0.040 |
| Polysomy 2 and 8 | 9/20 (45) | 0.013 |
| Polysomy 22q | 2/21 (5%) | Not available |
Survival analyses
Patients with anaplastic or large cell features had reduced overall survival (P = 0.036) (Figure 5A). When survival was estimated separately in the pediatric population, this association approached, but did not quite reach statistical significance (P = 0.053). Associations between patient survival and genetic alterations in tumors were also analyzed in pediatric patients. Although there was a trend towards decreased survival times in patients of MYC amplified tumors, this did not reach statistical significance (P = 0.11) (Figure 5B). However, polysomies of chromosomes 2 and 8 were each associated with worse survival (P = 0.025 and 0.04, respectively). In tumors with both combined polysomies of 2 and 8, the association was even stronger (P = 0.013) (Figure 5C). There was no obvious association between anaplastic/large cell features and MYC gene amplifications (P = 0.7), although polysomies were considerably more common in the CNS PNETs with anaplastic/large cell features (P = 0.01).
Figure 5.
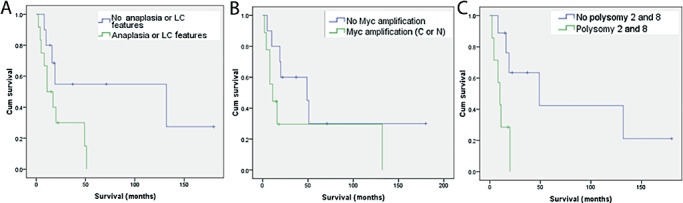
Kaplan–Meier survival curves in of central nervous system primitive neuroectodermal tumor patients based on the presence versus absence of anaplastic/large cell features (A), MYC gene amplifications (B) and combined polysomies of chromosomes 2 and 8 (C).
DISCUSSION
CNS PNETs constitute a heterogeneous and rare group of tumors. The current 2007 WHO classification scheme lumps these non‐cerebellar tumors together because of similar clinical behavior (35). As such, poorly differentiated brain stem and spinal cord tumors are now grouped with their supratentorial counterparts, whereas pineoblastoma retains a separate diagnostic heading. Some have studied the biologic characteristics of CNS PNET in conjunction with medulloblastomas, the latter shown to have significantly different clinical and genetic characteristics 7, 14, 19, 23, 24, 41, 43. To our knowledge, ours is one of the larger series investigating the histopathologic and genetic characteristics of CNS PNETs, particularly using the new WHO definitions.
The overall survival in pediatric population in our study was 33%, which is comparable with the 29%–57% rates reported in the literature 1, 8, 10, 12, 26. In a further analogy with medulloblastomas occurring in the cerebellum, we found that by routine histology, anaplastic and/or large cell features may be encountered. In fact, they were even more common in CNS PNETs, with roughly 54% of cases displaying such features. Whether or not this might explain in part, the considerably worse prognosis of the latter is unclear from this study alone. Nonetheless, as with medulloblastomas, we found shortened survival for CNS PNET patients whose tumors harbored anaplastic/large cell features (P = 0.036). This difference did not quite reach statistical significance when considering pediatric patients alone (P = 0.053), although it was close and potentially could have reached significance in a slightly larger cohort.
One of the important genetic findings of this study was the frequent presence of MYC gene amplifications (especially MYCN) in CNS PNETs. Several studies have investigated MYC gene alterations as a prognostic marker in human medulloblastomas 3, 14, 19, 44, 47. All indicate a low frequency of less than 10% for MYCC amplification, with even rarer MYCN amplifications. Association of such MYC amplifications with poor prognosis has been noted 2, 3, 25. In one study, MYCC messenger RNA expression did not always correlate with MYCC amplification, but was found to be an independent prognostic of progression free survival (19). Others have found MYCC and MYCN amplifications to be most common in the anaplastic/large cell medulloblastomas, which are already associated with a particularly poor prognosis 6, 13, 33, 36, 45. In the supratentorial or CNS PNET, these genes have only rarely been assessed, with amplifications occasionally reported (28). Our findings indicate that such amplifications are in fact common in CNS PNETs, detected in roughly half of all pediatric cases. In contrast to medulloblastomas, MYCN was more frequent than MYCC amplifications and the gene amplifications were not tightly associated with the presence of anaplastic/large cell features on histology. Although there was a trend towards shortened survival in the presence of MYC gene amplifications, our data did not reach statistical significance. In contrast, polysomies of chromosomes 2 and 8 were also associated with decreased survival times and these differences were found to be statistically significant. The latter were also more closely linked to the presence of anaplastic/large cell features, a finding that is not too surprising as large pleomorphic tumor nuclei are often polyploid or highly aneuploid. However, the biological explanation for the association between survival time and polysomies of 2 and 8 is less clear. Nonetheless, our data suggest that FISH analysis may provide diagnostically and prognostically useful information in the workup of pediatric CNS PNETs.
An important differential diagnosis for CNS PNET, particularly in children of younger than 3 years old is AT/RT (21). Diagnosis of AT/RT is often confirmed by the lack of nuclear expression of INI1. During our initial review of cases with diagnosis of primitive neuroectodermal tumor, we encountered a subgroup of nine cases containing epithelioid or rhabdoid features. The INI1 immunostain revealed lack of nuclear expression in three of these tumors (33%). All three patients expired within 6 months of diagnosis despite an aggressive treatment. These three tumors were therefore re‐diagnosed as AT/RT and excluded from further study. Of interest, Haberler et al reported a subset of primitive neuroectodermal tumors without classic rhabdoid features, but loss of INI1 nuclear expression (20). In their series, this subgroup showed aggressive clinical behavior and poor response to treatment similar to AT/RT in general. The findings from this and our study suggest that INI1 protein analysis should be performed in all primitive appearing pediatric embryonal CNS tumors without distinctive features, especially in infants, given that most AT/RTs occur in patients less than 3 years of age.
At the other end of the age spectrum, one must always be wary of a CNS PNET diagnosis in an older adult. Nonetheless, they clearly can occur in this age group, analogous to the situation with adult medulloblastomas. In our series, we included six adult examples, all of which were challenging cases submitted in consultation to one of the authors (A. Perry). It is worth noting that malignant gliomas that secondarily develop foci resembling PNET have recently been reported (40). Therefore, it is critical to examine the specimen carefully for a component resembling diffuse glioma. The latter most often resembles glioblastoma or gliosarcoma, although lower grade astrocytic and even oligodendroglial neoplasms may be encountered as well. The PNET‐like foci in such cases often form distinct nodules within the background of a diffuse glioma, but otherwise resemble CNS PNET morphologically, immunohistochemically, clinically (increased risk of CSF dissemination), and even genetically, in terms of MYC gene amplifications. On the other hand, glioma‐like alterations, such as 10q loss or 1p/19q codeletions are often found throughout both components, suggesting that the glioma came first, with PNET‐like foci developing later. In our adult cases of CNS PNET, no diffuse glioma component was seen and although each of these included large resection specimens, the possibility that a small focus of glioma was not sampled or that it was “overrun” by the PNET‐like element cannot be entirely excluded. Unfortunately, there was insufficient clinical followup in our small cohort of adult cases to accurately assess prognostic variables and determine if they differ significantly from their pediatric counterparts.
One of the characteristic genetic features of peripheral PNETs is a translocation that results in the fusion of the EWS gene with an ETS transcription factor, leading to oncogenic activation of the EWS gene (48). Given recent reports of EWS/pPNET within the cranial and spinal cavities, we searched for the presence EWS gene rearrangements in our series of CNS PNET, but did not encounter any in either the pediatric or adult tumors. Of importance, the immunohistochemical expression of CD99 in a third of our cases poses a potential diagnostic pitfall and highlights the importance of genetic confirmation, rather than reliance on this immunostain alone. Whereas most of our cases did not display the characteristic diffuse membrane pattern immunoreactivity of EWS/pPNET, some showed sufficient expression to create diagnostic confusion. A more important diagnostic clue is the typical meningeal involvement of EWS/pPNET cases reported within the craniospinal vault 9, 29, 34, 37. Given this rare involvement of CNS parenchyma by EWS/pPNET and the lack of gene rearrangements in our series, we propose that EWS gene testing is only needed if significant dural involvement is present. On the other hand, another interesting finding was that none of our CNS PNETs showed any evidence of 22q loss using this probe set, a feature that helps separate them from AT/RTs, since such deletions are common in the latter.
In summary, the current study supports the following conclusions regarding CNS PNET: (i) routine application of INI1 immunohistochemistry helps rule out AT/RT, particularly for infants or young children; although most such cases display rhabdoid or epithelioid cells, some may be composed predominantly or entirely of small primitive cells; (ii) medulloblastoma‐like anaplastic/large cell features are common in CNS PNETs; this is probably associated with a more aggressive phenotype, although larger series are needed to further substantiate this conclusion; (iii) MYC gene amplifications (especially MYCN) are common in this tumor type using FISH analysis, a finding that may be diagnostically useful; (iv) despite overlapping histology, the diagnostic consideration of EWS/pPNET in CNS parenchymal tumors is sufficiently rare that EWS gene testing is not necessary unless significant dural involvement is present; nonetheless, AT/RT‐like chromosome 22q deletions are not a common feature of CNS PNETs and this may be useful in the differential diagnosis; and (v) the presence of polysomies of chromosomes 2 and 8 on FISH studies are associated with shortened overall patient survival, a finding of potential prognostic value.
ACKNOWLEDGMENTS
The authors are grateful to Drs. Vivienne Tobias, Elizabeth J. Cochran, Ryan Miller, Sandro Casavilca Zambrano and Terri Haddix for sharing their cases in consultation and providing additional clinical information.
REFERENCES
- 1.Albright AL, Wisoff JH, Zeltzer P, Boyett J, Rorke LB, Stanley P_et al_ (1995) Prognostic factors in children with supratentorial (nonpineal) primitive neuroectodermal tumors. A neurosurgical perspective from the Children's Cancer Group. Pediatr Neurosurg 22:1–7. [DOI] [PubMed] [Google Scholar]
- 2.Aldosari N, Bigner SH, Burger PC, Becker L, Kepner JL, Friedman HS, McLendon RE (2002) MYCC and MYCN oncogene amplification in medulloblastoma. A fluorescence in situ hybridization study on paraffin sections from the Children's Oncology Group. Arch Pathol Lab Med 126:540–544. [DOI] [PubMed] [Google Scholar]
- 3.Badiali M, Pession A, Basso G, Andreini L, Rigobello L, Galassi E, Giangaspero F (1991) N‐myc and c‐myc oncogenes amplification in medulloblastomas. Evidence of particularly aggressive behavior of a tumor with c‐myc amplification. Tumori 77:118–121. [DOI] [PubMed] [Google Scholar]
- 4.Bordow SB, Norris MD, Haber PS, Marshall GM, Haber M (1998) Prognostic significance of MYCN oncogene expression in childhood neuroblastoma. J Clin Oncol 16:3286–3294. [DOI] [PubMed] [Google Scholar]
- 5.Bridge RS, Rajaram V, Dehner LP, Pfeifer JD, Perry A (2006) Molecular diagnosis of Ewing sarcoma/primitive neuroectodermal tumor in routinely processed tissue: a comparison of two FISH strategies and RT‐PCR in malignant round cell tumors. Mod Pathol 19:1–8. [DOI] [PubMed] [Google Scholar]
- 6.Brown HG, Kepner JL, Perlman EJ, Friedman HS, Strother DR, Duffner PK_et al_ (2000) “Large cell/anaplastic” medulloblastomas: a Pediatric Oncology Group Study. J Neuropathol Exp Neurol 59:857–865. [DOI] [PubMed] [Google Scholar]
- 7.Burnett ME, White EC, Sih S, Von Haken MS, Cogen PH (1997) Chromosome arm 17p deletion analysis reveals molecular genetic heterogeneity in supratentorial and infratentorial primitive neuroectodermal tumors of the central nervous system. Cancer Genet Cytogenet 97:25–31. [DOI] [PubMed] [Google Scholar]
- 8.Cohen BH, Zeltzer PM, Boyett JM, Geyer JR, Allen JC, Finlay JL_et al_ (1995) Prognostic factors and treatment results for supratentorial primitive neuroectodermal tumors in children using radiation and chemotherapy: a Children's Cancer Group randomized trial. J Clin Oncol 13:1687–1696. [DOI] [PubMed] [Google Scholar]
- 9.Dedeurwaerdere F, Giannini C, Sciot R, Rubin BP, Perilongo G, Borghi L_et al_ (2002) Primary peripheral PNET/Ewing's sarcoma of the dura: a clinicopathologic entity distinct from central PNET. Mod Pathol 15:673–678. [DOI] [PubMed] [Google Scholar]
- 10.Dirks PB, Harris L, Hoffman HJ, Humphreys RP, Drake JM, Rutka JT (1996) Supratentorial primitive neuroectodermal tumors in children. J Neurooncol 29:75–84. [DOI] [PubMed] [Google Scholar]
- 11.Dunham C, Sugo E, Tobias V, Wills E, Perry A (2007) Embryonal tumor with abundant neuropil and true rosettes (ETANTR): report of a case with prominent neurocytic differentiation. J Neurooncol 84:91–98. [DOI] [PubMed] [Google Scholar]
- 12.Eberhart CG, Burger PC (2003) Anaplasia and grading in medulloblastomas. Brain Pathol 13:376–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Eberhart CG, Kratz J, Wang Y, Summers K, Stearns D, Cohen K_et al_ (2004) Histopathological and molecular prognostic markers in medulloblastoma: c‐myc, N‐myc, TrkC, and anaplasia. J Neuropathol Exp Neurol 63:441–449. [DOI] [PubMed] [Google Scholar]
- 14.Fruhwald MC, O'Dorisio MS, Rush LJ, Reiter JL, Smiraglia DJ, Wenger G_et al_ (2000) Gene amplification in PNETs/medulloblastomas: mapping of a novel amplified gene within the MYCN amplicon. J Med Genet 37:501–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fuller C, Fouladi M, Gajjar A, Dalton J, Sanford RA, Helton KJ (2006) Chromosome 17 abnormalities in pediatric neuroblastic tumor with abundant neuropil and true rosettes. Am J Clin Pathol 126:1–7. [DOI] [PubMed] [Google Scholar]
- 16.Gessi M, Giangaspero F, Lauriola L, Gardiman M, Scheithauer BW, Halliday W_et al_ (2009) Embryonal tumors with abundant neuropil and true rosettes: a distinctive CNS primitive neuroectodermal tumor. Am J Surg Pathol 33:211–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Geyer JR, Zeltzer PM, Boyett JM, Rorke LB, Stanley P, Albright AL_et al_ (1994) Survival of infants with primitive neuroectodermal tumors or malignant ependymomas of the CNS treated with eight drugs in 1 day: a report from the Childrens Cancer Group. J Clin Oncol 12:1607–1615. [DOI] [PubMed] [Google Scholar]
- 18.Geyer JR, Sposto R, Jennings M, Boyett JM, Axtell RA, Breiger D_et al_ (2005) Multiagent chemotherapy and deferred radiotherapy in infants with malignant brain tumors: a report from the Children's Cancer Group. J Clin Oncol 23:7621–7631. [DOI] [PubMed] [Google Scholar]
- 19.Grotzer MA, Hogarty MD, Janss AJ, Liu X, Zhao H, Eggert A_et al_ (2001) MYC messenger RNA expression predicts survival outcome in childhood primitive neuroectodermal tumor/medulloblastoma. Clin Cancer Res 7:2425–2433. [PubMed] [Google Scholar]
- 20.Haberler C, Laggner U, Slavc I, Czech T, Ambros IM, Ambros PF_et al_ (2006) Immunohistochemical analysis of INI1 protein in malignant pediatric CNS tumors: lack of INI1 in atypical teratoid/rhabdoid tumors and in a fraction of primitive neuroectodermal tumors without rhabdoid phenotype. Am J Surg Pathol 30:1462–1468. [DOI] [PubMed] [Google Scholar]
- 21.Ho DM, Hsu CY, Wong TT, Ting LT, Chiang H (2000) Atypical teratoid/rhabdoid tumor of the central nervous system: a comparative study with primitive neuroectodermal tumor/medulloblastoma. Acta Neuropathol 99:482–488. [DOI] [PubMed] [Google Scholar]
- 22.Inda MM, Castresana JS (2007) RASSF1A promoter is highly methylated in primitive neuroectodermal tumors of the central nervous system. Neuropathology 27:341–346. [DOI] [PubMed] [Google Scholar]
- 23.Inda MM, Perot C, Guillaud‐Bataille M, Danglot G, Rey JA, Bello MJ_et al_ (2005) Genetic heterogeneity in supratentorial and infratentorial primitive neuroectodermal tumours of the central nervous system. Histopathology 47:631–637. [DOI] [PubMed] [Google Scholar]
- 24.Inda MM, Munoz J, Coullin P, Fauvet D, Danglot G, Tunon T_et al_ (2006) High promoter hypermethylation frequency of p14/ARF in supratentorial PNET but not in medulloblastoma. Histopathology 48:579–587. [DOI] [PubMed] [Google Scholar]
- 25.Jay V, Edwards V, Hoving E, Rutka J, Becker L, Zielenska M, Teshima I (1999) Central neurocytoma: morphological, flow cytometric, polymerase chain reaction, fluorescence in situ hybridization, and karyotypic analyses. Case report. J Neurosurg 90:348–354. [DOI] [PubMed] [Google Scholar]
- 26.Johnston DL, Keene DL, Lafay‐Cousin L, Steinbok P, Sung L, Carret AS_et al_ (2008) Supratentorial primitive neuroectodermal tumors: a Canadian pediatric brain tumor consortium report. J Neurooncol 86:101–108. [DOI] [PubMed] [Google Scholar]
- 27.Judkins AR, Ellison DW (2008) Ependymoblastoma: dear, damned, distracting diagnosis, farewell!. Brain Pathol doi: 10.1111/j.1750‐3639.2008.00253.x (e‐pub ahead of print). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kagawa N, Maruno M, Suzuki T, Hashiba T, Hashimoto N, Izumoto S, Yoshimine T (2006) Detection of genetic and chromosomal aberrations in medulloblastomas and primitive neuroectodermal tumors with DNA microarrays. Brain Tumor Pathol 23:41–47. [DOI] [PubMed] [Google Scholar]
- 29.Kazmi SA, Perry A, Pressey JG, Wellons JC, Hammers Y, Palmer CA (2007) Primary Ewing sarcoma of the brain: a case report and literature review. Diagn Mol Pathol 16:108–111. [DOI] [PubMed] [Google Scholar]
- 30.Kim DG, Lee DY, Paek SH, Chi JG, Choe G, Jung HW (2002) Supratentorial primitive neuroectodermal tumors in adults. J Neurooncol 60:43–52. [DOI] [PubMed] [Google Scholar]
- 31.Lamont JM, McManamy CS, Pearson AD, Clifford SC, Ellison DW (2004) Combined histopathological and molecular cytogenetic stratification of medulloblastoma patients. Clin Cancer Res 10:5482–5493. [DOI] [PubMed] [Google Scholar]
- 32.Leonard JR, Cai DX, Rivet DJ, Kaufman BA, Park TS, Levy BK, Perry A (2001) Large cell/anaplastic medulloblastomas and medullomyoblastomas: clinicopathological and genetic features. J Neurosurg 95:82–88. [DOI] [PubMed] [Google Scholar]
- 33.Lo KC, Rossi MR, Eberhart CG, Cowell JK (2007) Genome wide copy number abnormalities in pediatric medulloblastomas as assessed by array comparative genome hybridization. Brain Pathol 17:282–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mazur MA, Gururangan S, Bridge JA, Cummings TJ, Mukundan S, Fuchs H_et al_ (2005) Intracranial Ewing sarcoma. Pediatr Blood Cancer 45:850–856. [DOI] [PubMed] [Google Scholar]
- 35.McLendon RE, Judkins AR, Eberhart CG, Fuller GN, Sarkar C, Ng HK (2007) Central nervous system primitive neuroectodermal tumours. In: WHO Classification of Tumours of the Central Nervous System. Louis DN, Ohgaki H, Wiestler OD, Cavenee WK (eds), pp. 141–146. IARC: Lyon. [Google Scholar]
- 36.Min HS, Lee YJ, Park K, Cho BK, Park SH (2006) Medulloblastoma: histopathologic and molecular markers of anaplasia and biologic behavior. Acta Neuropathol 112:13–20. [DOI] [PubMed] [Google Scholar]
- 37.Mobley BC, Roulston D, Shah GV, Bijwaard KE, McKeever PE (2006) Peripheral primitive neuroectodermal tumor/Ewing's sarcoma of the craniospinal vault: case reports and review. Hum Pathol 37:845–853. [DOI] [PubMed] [Google Scholar]
- 38.Pang JC, Chang Q, Chung YF, Teo JG, Poon WS, Zhou LF_et al_ (2005) Epigenetic inactivation of DLC‐1 in supratentorial primitive neuroectodermal tumor. Hum Pathol 36:36–43. [DOI] [PubMed] [Google Scholar]
- 39.Perry A (2002) Medulloblastomas with favorable versus unfavorable histology: how many small blue cell tumor types are there in the brain? Adv Anat Pathol 9:345–350. [DOI] [PubMed] [Google Scholar]
- 40.Perry A, Miller CR, Gujrati M, Scheithauer BW, Zambrano SC, Jost SC_et al_ (2009) Malignant gliomas with primitive neuroectodermal tumor‐like components: a clinicopathologic and genetic study of 53 cases. Brain Pathol 19:81–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pfister S, Remke M, Toedt G, Werft W, Benner A, Mendrzyk F_et al_ (2007) Supratentorial primitive neuroectodermal tumors of the central nervous system frequently harbor deletions of the CDKN2A locus and other genomic aberrations distinct from medulloblastomas. Genes Chromosomes Cancer 46:839–851. [DOI] [PubMed] [Google Scholar]
- 42.Pickuth D, Leutloff U (1996) Computed tomography and magnetic resonance imaging findings in primitive neuroectodermal tumours in adults. Br J Radiol 69:1–5. [DOI] [PubMed] [Google Scholar]
- 43.Russo C, Pellarin M, Tingby O, Bollen AW, Lamborn KR, Mohapatra G_et al_ (1999) Comparative genomic hybridization in patients with supratentorial and infratentorial primitive neuroectodermal tumors. Cancer 86:331–339. [DOI] [PubMed] [Google Scholar]
- 44.Scheurlen WG, Schwabe GC, Joos S, Mollenhauer J, Sorensen N, Kuhl J (1998) Molecular analysis of childhood primitive neuroectodermal tumors defines markers associated with poor outcome. J Clin Oncol 16:2478–2485. [DOI] [PubMed] [Google Scholar]
- 45.Stearns D, Chaudhry A, Abel TW, Burger PC, Dang CV, Eberhart CG (2006) c‐myc overexpression causes anaplasia in medulloblastoma. Cancer Res 66:673–681. [DOI] [PubMed] [Google Scholar]
- 46.Warnat P, Oberthuer A, Fischer M, Westermann F, Eils R, Brors B (2007) Cross‐study analysis of gene expression data for intermediate neuroblastoma identifies two biological subtypes. BMC Cancer 7:89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wasson JC, Saylors RL 3rd, Zeltzer P, Friedman HS, Bigner SH, Burger PC_et al_ (1990) Oncogene amplification in pediatric brain tumors. Cancer Res 50:2987–2990. [PubMed] [Google Scholar]
- 48.Whang‐Peng J, Freter CE, Knutsen T, Nanfro JJ, Gazdar A (1987) Translocation t(11; 22) in esthesioneuroblastoma. Cancer Genet Cytogenet 29:155–157. [DOI] [PubMed] [Google Scholar]