Effects of Once-Weekly Exenatide on Cardiovascular Outcomes in Type 2 Diabetes (original) (raw)
. Author manuscript; available in PMC: 2022 Dec 27.
Published in final edited form as: N Engl J Med. 2017 Sep 14;377(13):1228–1239. doi: 10.1056/NEJMoa1612917
Abstract
BACKGROUND
The cardiovascular effects of adding once-weekly treatment with exenatide to usual care in patients with type 2 diabetes are unknown.
METHODS
We randomly assigned patients with type 2 diabetes, with or without previous cardiovascular disease, to receive subcutaneous injections of extended-release exenatide at a dose of 2 mg or matching placebo once weekly. The primary composite outcome was the first occurrence of death from cardiovascular causes, nonfatal myocardial infarction, or nonfatal stroke. The coprimary hypotheses were that exenatide, administered once weekly, would be noninferior to placebo with respect to safety and superior to placebo with respect to efficacy.
RESULTS
In all, 14,752 patients (of whom 10,782 [73.1%] had previous cardiovascular disease) were followed for a median of 3.2 years (interquartile range, 2.2 to 4.4). A primary composite outcome event occurred in 839 of 7356 patients (11.4%; 3.7 events per 100 person-years) in the exenatide group and in 905 of 7396 patients (12.2%; 4.0 events per 100 person-years) in the placebo group (hazard ratio, 0.91; 95% confidence interval [CI], 0.83 to 1.00), with the intention-to-treat analysis indicating that exenatide, administered once weekly, was noninferior to placebo with respect to safety (P<0.001 for noninferiority) but was not superior to placebo with respect to efficacy (P = 0.06 for superiority). The rates of death from cardiovascular causes, fatal or nonfatal myocardial infarction, fatal or nonfatal stroke, hospitalization for heart failure, and hospitalization for acute coronary syndrome, and the incidence of acute pancreatitis, pancreatic cancer, medullary thyroid carcinoma, and serious adverse events did not differ significantly between the two groups.
CONCLUSIONS
Among patients with type 2 diabetes with or without previous cardiovascular disease, the incidence of major adverse cardiovascular events did not differ significantly between patients who received exenatide and those who received placebo. (Funded by Amylin Pharmaceuticals; EXSCEL ClinicalTrials.gov number, NCT0144338.)
The risk of death from any cause among persons with type 2 diabetes is up to twice that of the general population,1 and the risk of death from cardiovascular causes is up to four times that of the general population.2,3 Improved glycemic control has been shown to improve microvascular outcomes,4,5 but a beneficial effect on macrovascular outcomes is less certain.6 In trials that assessed cardiovascular outcomes associated with the use of newer glucose-lowering agents, no effect on major adverse cardiovascular events (MACE) was shown with three dipeptidyl peptidase 4 (DPP-4) inhibitors (saxagliptin, alogliptin, and sitagliptin)7–9 and one exendin-4–based glucagon-like peptide-1 (GLP-1) receptor agonist (lixisenatide),10 but a lower risk of MACE was shown with two GLP-1 receptor agonists (liraglutide and semaglutide)11,12 and two sodium-glucose cotransporter 2 (SGLT-2) inhibitors (empagliflozin and canagliflozin).13,14
A once-weekly, injectable, extended-release formulation of exenatide, an exendin-4–based GLP-1 receptor agonist approved for the treatment of type 2 diabetes, has been shown to lower blood glucose and induce modest decreases in body weight, blood pressure, and lipid levels15,16 but has also been shown to increase heart rate.17,18 In accordance with regulatory guidance,19,20 the Exenatide Study of Cardiovascular Event Lowering (EXSCEL) assessed the long-term cardiovascular safety and efficacy of exenatide, administered once weekly, in patients with type 2 diabetes who had a wide range of cardiovascular risk.
METHODS
TRIAL OVERSIGHT
We conducted this pragmatic, randomized, double-blind, placebo-controlled, event-driven trial at 687 sites in 35 countries. The design of this academically led trial has been reported previously.21 A diagram showing the overall design is provided in Figure S1 in the Supplementary Appendix, available with the full text of this article at NEJM.org. The trial was conducted jointly by the Duke Clinical Research Institute and the University of Oxford Diabetes Trials Unit in collaboration with the sponsor, Amylin Pharmaceuticals, a wholly owned subsidiary of AstraZeneca. The protocol, which was approved by the ethics committee at each participating site, and the statistical analysis plan are available at NEJM.org. The statistical analyses were performed by the Duke Clinical Research Institute, independent of the sponsor. Details of the trial organization and a complete list of the investigators are provided in the Supplementary Appendix.
All the patients provided written informed consent. Our trial was designed and overseen by a steering committee composed of nine investigators and two employees of the sponsor. An independent data and safety monitoring committee, who had access to unblinded data, performed regular safety surveillance. All the authors had access to the final trial results and vouch for the accuracy and completeness of the data and analyses and for the fidelity of the trial to the protocol. The manuscript, drafted by the second and third authors, was revised and approved by all the authors, who assume responsibility for its accuracy and completeness and made the decision to submit the manuscript for publication.
TRIAL POPULATION
Adults with type 2 diabetes (defined as a glycated hemoglobin level of 6.5 to 10.0% [48 to 96 mmol per mole]) were eligible for participation in the trial. The trial was designed such that approximately 70% of enrolled patients would have had previous cardiovascular events and 30% would not have had previous cardiovascular events. Previous cardiovascular events were defined as a history of major clinical manifestation of coronary artery disease, ischemic cerebrovascular disease, or atherosclerotic peripheral arterial disease. Patients were permitted to receive up to three oral glucose-lowering agents or to receive insulin, either alone or in combination with up to two oral glucose-lowering agents. Key exclusion criteria were a history of two or more episodes of severe hypoglycemia (defined as hypoglycemia for which a patient received third-party assistance) during the preceding 12 months, end-stage kidney disease or an estimated glomerular filtration rate (eGFR) at entry of less than 30 ml per minute per 1.73 m2 of body-surface area, a personal or family history of medullary thyroid carcinoma or multiple endocrine neoplasia type 2, a baseline calcitonin level of greater than 40 ng per liter, or previous treatment with a GLP-1 receptor agonist.
RANDOMIZATION AND TRIAL REGIMEN
Patients were randomly assigned in a 1:1 ratio to receive subcutaneous injections of extended-release exenatide at a dose of 2 mg or matching placebo once weekly. An interactive voice-response system assigned patients on the basis of computer-generated block randomization within each site, with stratification according to history of cardiovascular disease. Patients were required to discontinue the trial regimen if they had two or more occurrences of severe hypoglycemia between trial visits (despite adjustment of other glucose-lowering agents), if they had irreversible kidney dysfunction (confirmed by two consecutive eGFR values of <30 ml per minute per 1.73 m2) or received renal-replacement therapy, or if they were found to have an elevated calcitonin level (>40 ng per liter at baseline or ≥50 ng per liter thereafter). Calcitonin levels were measured at a central laboratory at baseline and annually thereafter. Other laboratory values were obtained from usual-care blood sampling, which was consistent with the pragmatic design of the trial. In order to minimize potential confounding effects of differential glycemic levels on trial outcomes, the use of open-label glucose-lowering agents (including DPP-4 inhibitors but not including GLP-1 receptor agonists) was encouraged to promote glycemic equipoise between the two trial groups and to help patients reach clinically appropriate glycated hemoglobin targets.
OUTCOMES
The primary outcome was defined as the first occurrence of any component of the composite outcome of death from cardiovascular causes, nonfatal myocardial infarction, or nonfatal stroke (three-component MACE outcome), in a time-to-event analysis. Secondary outcomes included death from any cause, death from cardiovascular causes, and the first occurrence of nonfatal or fatal myocardial infarction, nonfatal or fatal stroke, hospitalization for acute coronary syndrome, and hospitalization for heart failure, in time-to-event analyses. An independent clinical events classification committee whose members were unaware of the trial-group assignments adjudicated all the components of the primary composite outcome, secondary outcomes, ventricular arrhythmias that led to intervention, neoplasms, and pancreatitis; these events are defined in the Clinical Event Definitions section in the Supplementary Appendix.
Additional outcomes that were prespecified in the protocol included a composite of death from cardiovascular causes or hospitalization for heart failure, the first confirmed revascularization procedure, initiation of the first interventional glucose-lowering medication received during the trial other than the trial regimen, and absolute values and changes from baseline in glycated hemoglobin level, body weight, blood pressure, and lipid levels. Prespecified events of clinical interest for which information was collected systematically at every follow-up visit, regardless of seriousness, were pancreatitis, neoplasm, severe hypoglycemia, and expected cardiovascular or diabetes-related complications. Information on other nonserious adverse events was not collected (details are provided in Section 10.3 in the protocol).
Patients were followed for adverse events until the end of the trial or 70 days after discontinuation of the trial regimen. For patients who were either lost to follow-up or had withdrawn consent, vital status was ascertained during the 70-day washout period by searches conducted with the use of local or national electronic health records, death registries, or other publicly available sources (as permitted by local ethics approvals).
STATISTICAL ANALYSIS
Information on sample size and power calculations has been published previously21; we estimated that with 1360 patients with a confirmed primary composite outcome event, the trial would have 85% power to detect a risk of a primary composite outcome event that was 15% lower with once-weekly administration of exenatide than with placebo, at a two-sided alpha level of 0.05. The time-to-event analyses were performed with the use of a Cox proportional-hazards model for primary, secondary, and exploratory outcomes in the intention-to-treat population, stratified according to history of cardiovascular disease, with trial regimen as an explanatory variable. The intention-to-treat population included all patients who underwent randomization. The Kaplan–Meier method was used to calculate event rates. The primary safety hypothesis was that exenatide, administered once weekly, would be noninferior to placebo for the primary outcome, with a noninferiority margin of 1.3 for the upper limit of the two-sided 95% confidence interval of the hazard ratio. The primary efficacy hypothesis was that exenatide, administered once weekly, would be superior to placebo, with a superiority margin of less than 1.0 for the upper limit of the two-sided 95% confidence interval. The statistical analysis plan prespecified the use of hierarchical testing in the following order: noninferiority for the primary composite outcome, superiority for the primary composite outcome, superiority for all-cause mortality, superiority for each component of the cardiovascular composite (tested with the use of the Hochberg procedure), and finally, if all three components were superior, superiority for hospitalization for acute coronary syndrome and for hospitalization for heart failure (tested with the use of the Hochberg procedure). In accordance with the hierarchical testing plan, if a significant difference was not found for an outcome, formal hypothesis testing was not to be conducted for the remaining ordered outcomes.
Sensitivity analyses of the primary efficacy outcome were performed in the per-protocol population, which included all randomized patients who received at least one dose of the trial regimen and had no major protocol violations (see the protocol for further details). The safety analyses were performed in patients who underwent randomization and received at least one dose of exenatide or placebo.
Baseline characteristics were summarized as means and standard deviations, medians and interquartile ranges, or percentages. The primary composite outcome was analyzed in prespecified subgroups that were defined according to baseline characteristics, including age at randomization, sex, race, geographic region, type of glucose-lowering therapy, duration of diabetes, history or no history of a cardiovascular event or heart failure, body-mass index, glycated hemoglobin level, and kidney function. Repeated measures, such as glycated hemoglobin level and body weight, were analyzed with the use of longitudinal models with mixed effects, with differences between the trial groups estimated by least-squares mean differences with 95% confidence intervals. Analyses were conducted with the use of SAS software, version 9.4 (SAS Institute).
RESULTS
TRIAL PATIENTS
In all, 14,752 patients (of whom 10,782 [73.1%] had previous cardiovascular disease) underwent randomization from June 18, 2010, through September 16, 2015, and were included in the intention-to-treat population; 7356 patients were assigned to receive exenatide and 7396 to receive placebo once weekly (Fig. S2 in the Supplementary Appendix).22 The planned closeout of follow-up of the patients was from December 5, 2016, to May 11, 2017, after the prespecified required minimum of 1360 patients were confirmed to have had a primary composite outcome event.
The demographic, disease, and clinical characteristics of the patients did not differ significantly between the groups, nor did the use of glucose-lowering agents or medications to reduce the risk of cardiovascular disease, with the exception of lipid-lowering medications and SGLT-2 inhibitors (Table S1 in the Supplementary Appendix).22 At baseline, the median length of time that patients had had diabetes was 12.0 years (interquartile range, 7.0 to 18.0), the median glycated hemoglobin level was 8.0% (interquartile range, 7.3 to 8.9) (63.9 mmol per mole [interquartile range, 56.3 to 73.8]), and 2389 patients (16.2%) had a history of heart failure.
A total of 14,187 patients (96.2%) completed the trial, and vital status was obtained for 98.8% of the patients. The median duration of follow-up was 3.2 years (interquartile range, 2.2 to 4.4; maximum, 6.8) and was similar in the two groups. The median duration of exposure to the trial regimen was 2.4 years (interquartile range, 1.4 to 3.8) in the exenatide group and 2.3 years (interquartile range, 1.2 to 3.6) in the placebo group. The mean percentage of time that participants received the trial regimen (i.e., the duration of time that participants received the trial regimen relative to the duration of time that they were expected to receive the regimen during the trial) was 76.0% and 75.0%, respectively. Premature discontinuation of the trial regimen was primarily the result of decision by the patient (Table S2 in the Supplementary Appendix).
CHANGES IN RISK FACTORS
At 6 months, the mean glycated hemoglobin level was 0.7 percentage points lower in the exenatide group than in the placebo group (95% confidence interval [CI], −0.7 to −0.6). This difference narrowed during the course of the trial (overall least-squares mean difference, −0.53%; 95% CI, −0.57 to −0.50; P<0.001) (Fig. 1). Overall least-squares mean values were also lower with exenatide than with placebo with respect to body weight (difference of −1.27 kg), systolic blood pressure (−1.57 mm Hg), low-density lipoprotein cholesterol (−1.5 mg per deciliter [−0.04 mmol per liter]), and triglycerides (−1.8 mg per deciliter [−0.02 mmol per liter]); values were higher in the exenatide group than in the placebo group with respect to diastolic blood pressure (difference of 0.25 mm Hg) and heart rate (difference of 2.51 beats per minute) (Fig. 1, and Figs. S3 through S5 in the Supplementary Appendix).
Figure 1. Effects of Once-Weekly Exenatide.
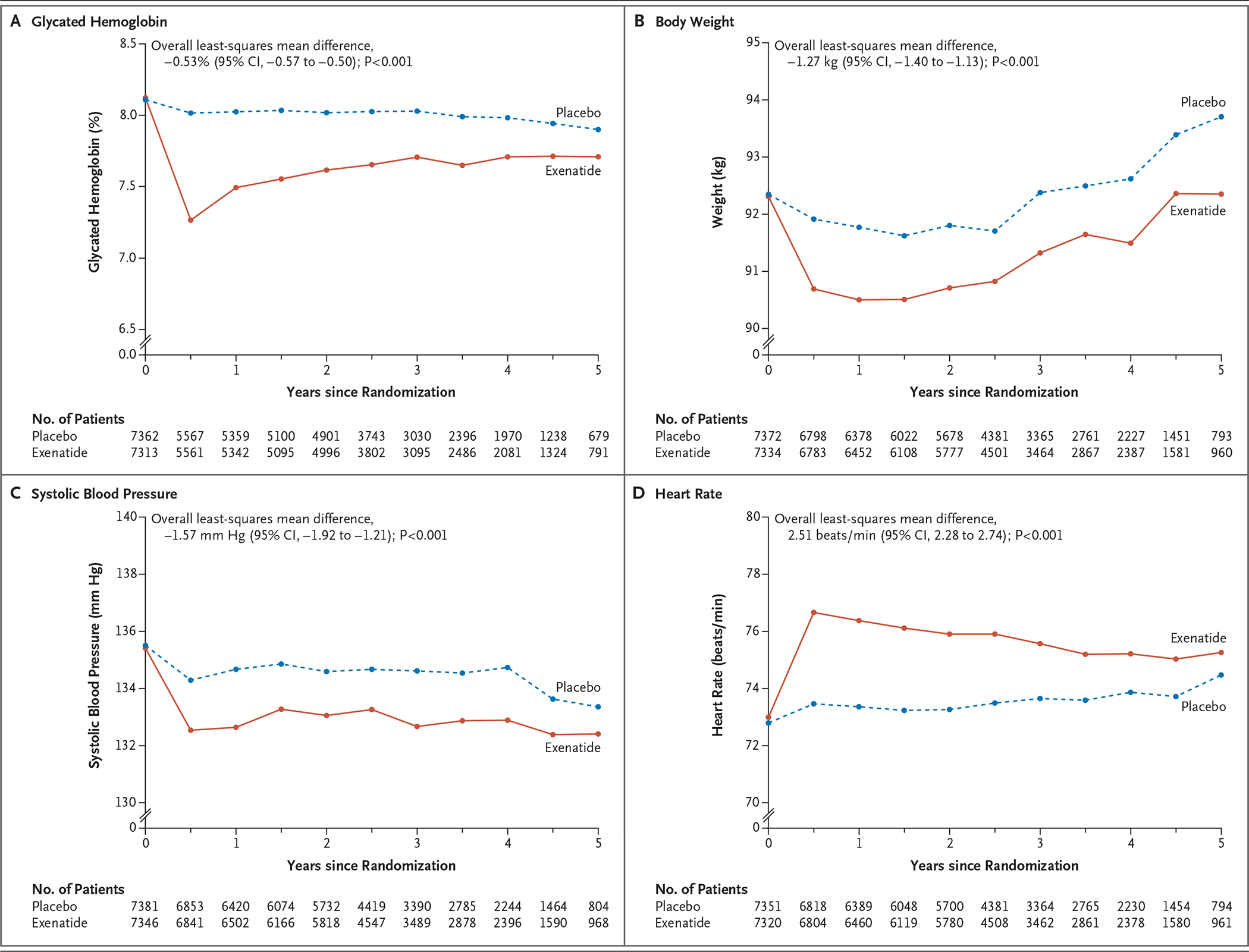
Shown are the effects of once-weekly administration of exenatide on glycated hemoglobin levels, body weight, systolic blood pressure, and heart rate. Overall least-squares mean differences were estimated from the model that included only the patients in whom a baseline value and at least one postbaseline value were obtained.
PRIMARY AND SECONDARY CARDIOVASCULAR OUTCOMES
A primary composite outcome event occurred in 839 of 7356 patients (11.4%; 3.7 events per 100 person-years) in the exenatide group and in 905 of 7396 patients (12.2%; 4.0 events per 100 person-years) in the placebo group (hazard ratio, 0.91; 95% CI, 0.83 to 1.00), with the intention-to-treat analysis indicating that exenatide, administered once weekly, was noninferior to placebo with respect to safety (P<0.001 for noninferiority) and was not superior to placebo with respect to efficacy (P = 0.06 for superiority) (Table 1 and Fig. 2, and Fig. S6 in the Supplementary Appendix). There was no evidence of heterogeneity among the three components of the composite outcome (Table S3 in the Supplementary Appendix). The per-protocol primary analysis resulted in a hazard ratio that was similar to that of the intention-to-treat analysis (hazard ratio, 0.95; 95% CI, 0.85 to 1.07; P<0.001 for noninferiority, P = 0.39 for superiority), also without evidence of heterogeneity among the composite components.
Table 1.
Rates of the Primary Composite Outcome and Key Secondary Outcomes.*
| Outcome | Exenatide (N = 7356) | Placebo (N = 7396) | Hazard Ratio (95% CI)† | ||
|---|---|---|---|---|---|
| Patients with Event | Incidence Rate of First Event | Patients with Event | Incidence Rate of First Event | ||
| no. (%) | no. of events/100 patient-yr | no. (%) | no. of events/100 patient-yr | ||
| Primary composite outcome | 839 (11.4) | 3.7 | 905 (12.2) | 4.0 | 0.91 (0.83–1.00) |
| Secondary outcomes | |||||
| Death from any cause | 507 (6.9) | 2.0 | 584 (7.9) | 2.3 | 0.86 (0.77–0.97) |
| Death from cardiovascular causes‡ | 340 (4.6) | 1.4 | 383 (5.2) | 1.5 | 0.88 (0.76–1.02) |
| Fatal or nonfatal myocardial infarction | 483 (6.6) | 2.1 | 493 (6.7) | 2.1 | 0.97 (0.85–1.10) |
| Fatal myocardial infarction§ | 17 (0.2) | — | 13 (0.2) | — | 1.29 (0.63–2.66) |
| Fatal or nonfatal stroke | 187 (2.5) | 0.8 | 218 (2.9) | 0.9 | 0.85 (0.70–1.03) |
| Fatal stroke§ | 18 (0.2) | — | 25 (0.3) | — | 0.71 (0.39–1.30) |
| Hospitalization for heart failure | 219 (3.0) | 0.9 | 231 (3.1) | 1.0 | 0.94 (0.78–1.13) |
| Hospitalization for acute coronary syndrome | 602 (8.2) | 2.6 | 570 (7.7) | 2.5 | 1.05 (0.94–1.18) |
Figure 2. Trial Outcomes.
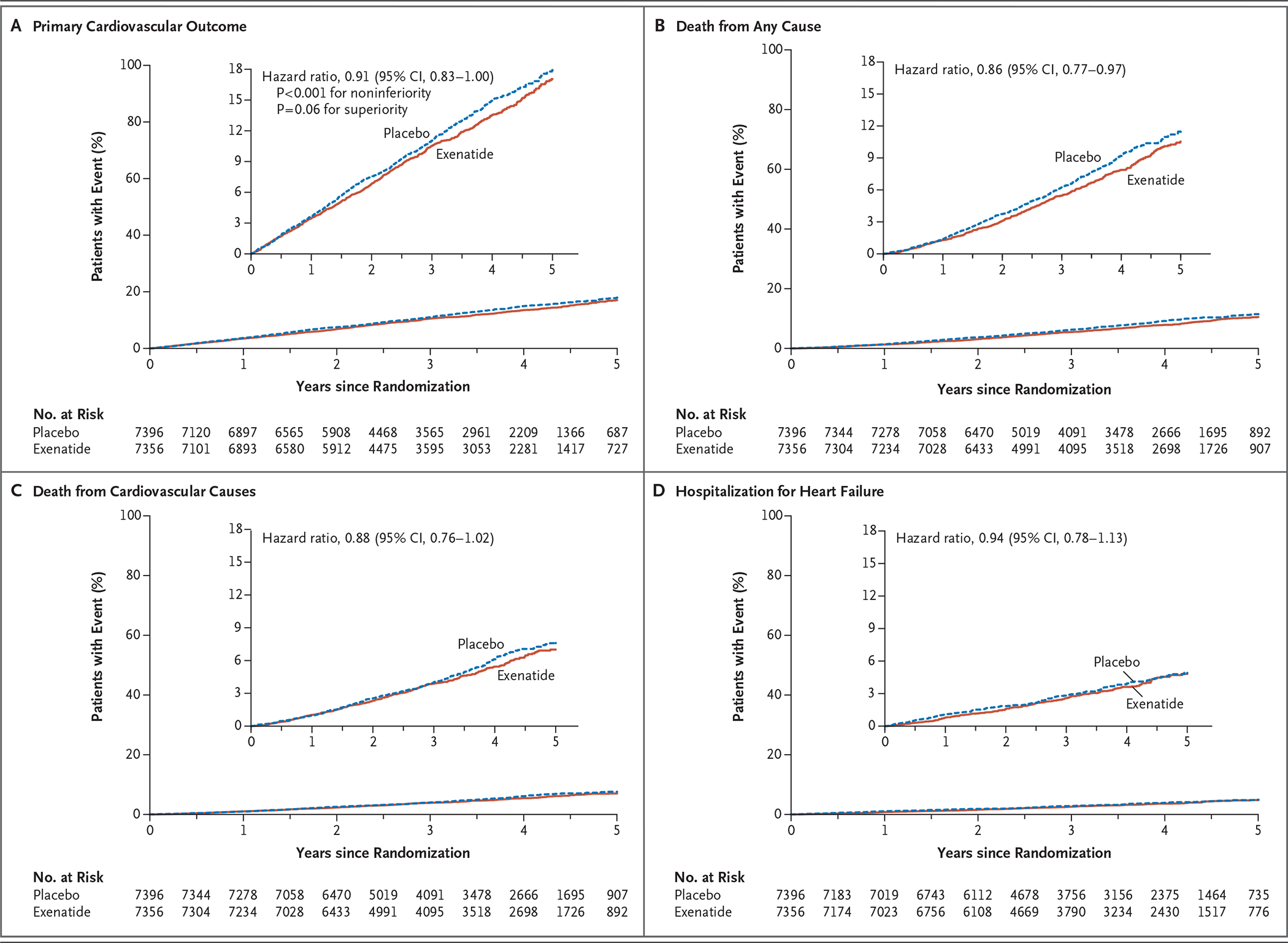
Shown are the rates of the primary cardiovascular outcome (a composite of death from cardiovascular causes, nonfatal myocardial infarction, or nonfatal stroke), death from any cause, death from cardiovascular causes, and hospitalization for heart failure in the exenatide and placebo groups. The inset in each panel shows the same data on an enlarged y axis.
The risk of death from any cause was 6.9% in the exenatide group and 7.9% in the placebo group (hazard ratio, 0.86; 95% CI, 0.77 to 0.97); this difference was not considered to be statistically significant on the basis of the hierarchical testing plan (Table 1 and Fig. 2). Causes of death were adjudicated as cardiovascular in 45.4% of the patients in the exenatide group and in 41.3% of the patients in the placebo group, as noncardiovascular in 32.9% and 34.4% of the patients, respectively, and as unknown in 21.7% and 24.3% of the patients (Table S4 in the Supplementary Appendix). The rates of the first fatal or nonfatal myocardial infarction, fatal or nonfatal stroke (Figs. S7 and S8 in the Supplementary Appendix), and other secondary outcomes did not differ significantly between the two groups. In univariate analyses of the primary composite outcome in 16 prespecified subgroups that were defined according to baseline characteristics, only the subgroup of patients characterized by a baseline age of younger than 65 years versus 65 years or older showed heterogeneity (P = 0.005, unadjusted for multiplicity) (Fig. 3). Results of the prespecified sensitivity analyses for the primary composite outcome are shown in Figure S9 in the Supplementary Appendix.
Figure 3. Primary Composite Outcome According to Prespecified Subgroups.
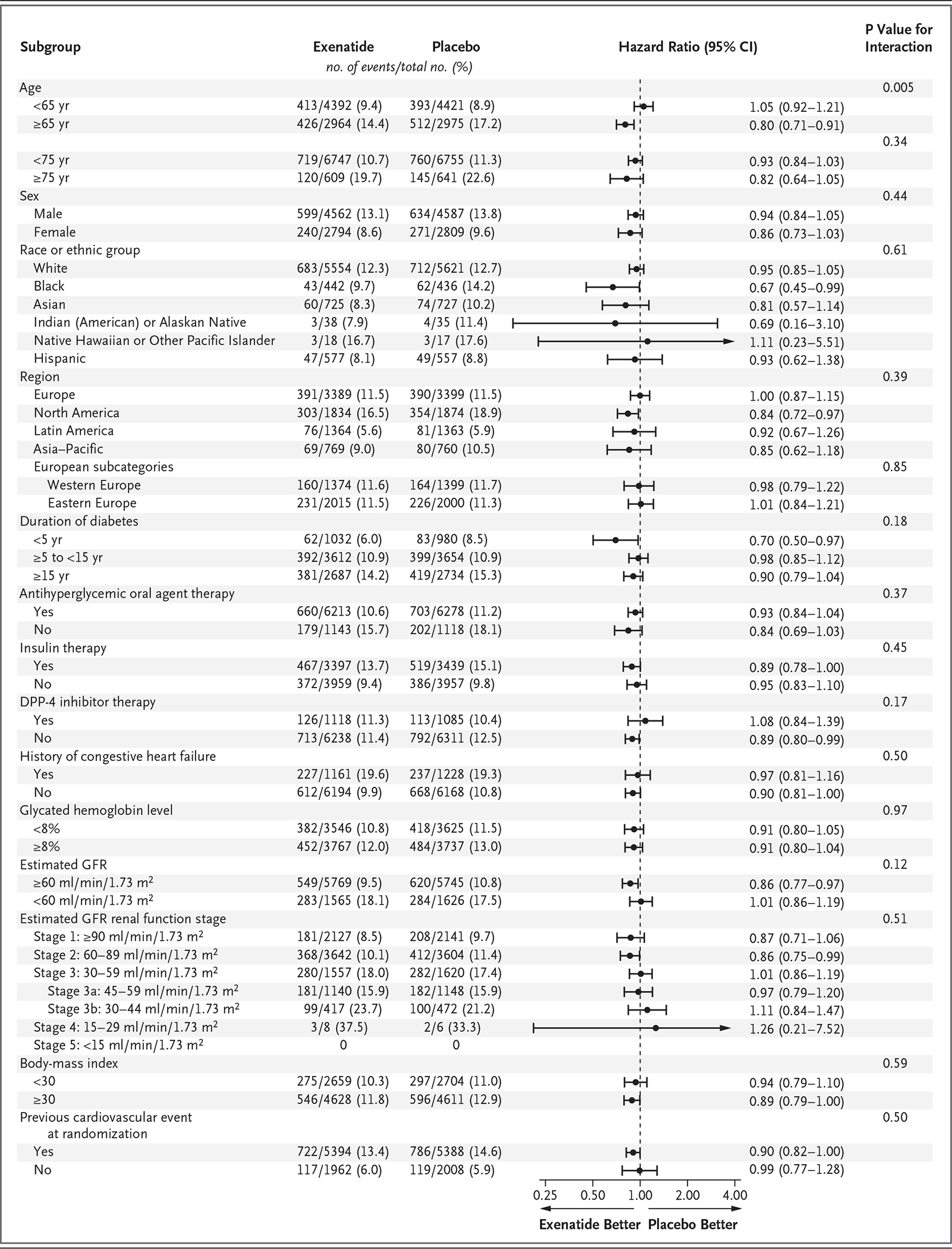
Race or ethnic group was reported by the patient. The body-mass index is the weight in kilograms divided by the square of the height in meters. DPP-4 denotes dipeptidyl peptidase 4, and GFR glomerular filtration rate.
ADDITIONAL EFFICACY OUTCOMES
The rate of cardiovascular or peripheral revascularization procedures was similar in the two groups (hazard ratio, 0.94; 95% CI, 0.85 to 1.05). Patients randomly assigned to exenatide, administered once weekly, had a lower risk of receiving an additional cointerventional glucose-lowering agent than patients in the placebo group (hazard ratio, 0.67; 95% CI, 0.63 to 0.71; P<0.001) and a lower risk of initiating long-term insulin therapy (hazard ratio, 0.61; 95% CI, 0.54 to 0.68; P<0.001), with less open-label use of SGLT-2 inhibitors (in 6.5% vs. 9.4% of the patients) and GLP-1 receptor agonists (2.5% vs. 3.6%) during follow-up (Table S5 in the Supplementary Appendix). New concomitant cardiovascular and other medications administered during follow-up are listed in Table S6 in the Supplementary Appendix.
SAFETY OUTCOMES
No clinically relevant between-group differences were seen in the incidence of serious adverse events or events of clinical interest (Table 2, and Table S7 in the Supplementary Appendix). Confirmed events of acute pancreatitis were uncommon, and the number of patients who had confirmed acute pancreatitis was similar in the exenatide group and the placebo group (26 and 22, respectively), as were the number of patients who had cancers overall (355 and 361) and the numbers of patients who had pancreatic cancer (15 and 16) and medullary thyroid carcinoma (2 and 1). More patients in the exenatide group than in the placebo group had thyroid papillary carcinomas (10 vs. 4).
Table 2.
Serious Adverse Events and Events of Clinical Interest.*
| Event | Exenatide (N = 7344) | Placebo (N = 7372) |
|---|---|---|
| Serious adverse events — no. of patients (%) † | ||
| Any serious adverse event | 1234 (16.8) | 1222 (16.6) |
| Serious adverse event related to trial regimen | 56 (0.8) | 38 (0.5) |
| Serious adverse event that resulted in permanent discontinuation of trial regimen | 108 (1.5) | 104 (1.4) |
| Events of clinical interest | ||
| Adjudicated pancreatitis‡ | ||
| Patients with event — no. (%) | 26 (0.4) | 22 (0.3) |
| Severity of event — no. of patients (%) | ||
| Mild | 23 (0.3) | 20 (0.3) |
| Severe | 2 (<0.1) | 2 (<0.1) |
| Unknown | 1 (<0.1) | 0 |
| First-event rate per 100 patient-years | 0.11 | 0.09 |
| No. of events | 28 | 23 |
| No. of events per patient | ||
| 1 | 24 | 21 |
| 2 | 2 | 1 |
| ≥3 | 0 | 0 |
| Event rate per 100 patient-years | 0.12 | 0.10 |
| Adjudicated charter-defined cancerठ| ||
| Patients with event — no. (%) | 355 (4.8) | 361 (4.9) |
| First-event rate per 100 patient-years | 1.5 | 1.6 |
| No. of events | 429 | 442 |
| Event rate per 100 patient-years | 1.8 | 1.9 |
| Adjudicated cancers of interest — no. (%)‡ | ||
| Patients with medullary thyroid carcinoma | 2 (<0.1) | 1 (<0.1) |
| Patients with pancreatic cancer | 15 (0.2) | 16 (0.2) |
| Severe hypoglycemia¶ | ||
| Patients with event — no. (%) | 247 (3.4) | 219 (3.0) |
| First-event rate per 100 patient-years | 1.0 | 0.9 |
| No. of events | 404 | 450 |
| Event rate per 100 patient-years | 1.6 | 1.8 |
The rate of severe hypoglycemia did not differ significantly between the two groups, either when measured as the first event only (1.0 events per 100 patient-years in the exenatide group and 0.9 events per 100 patient-years in the placebo group) or when recurrent events were included (1.6 events per 100 patient-years and 1.8 events per 100 patient-years, respectively; risk ratio, 0.85; 95% CI, 0.67 to 1.08) (Table 2).
DISCUSSION
In this pragmatic, multinational, cardiovascular outcomes trial, which was performed in a usual-care setting among patients with type 2 diabetes, with or without previous cardiovascular disease, the addition of once-weekly extended-release exenatide to usual care was compared with usual care alone for the management of diabetes and cardiovascular risk factors over a median of 3.2 years of follow-up; the results showed that exenatide was noninferior to placebo with respect to cardiovascular safety but was not superior to placebo with respect to efficacy. The risk of death from any cause was 6.9% in the exenatide group and 7.9% in the placebo group (hazard ratio, 0.86; 95% CI, 0.77 to 0.97); this difference was not considered to be statistically significant on the basis of the hierarchical testing plan. Events of acute pancreatitis, pancreatic cancer, and medullary thyroid carcinoma were uncommon, with similar rates in the two groups.
The pragmatic design of the trial included integration with usual care and wide-ranging eligibility criteria. For example, patients with any degree of cardiovascular risk who were at least 18 years of age (with no upper age limit) were eligible. To further augment the potential generalizability of any findings, we evaluated the cardiovascular effect of once-weekly extended-release exenatide in the usual-care setting by maintaining the focus of management of diabetes and cardiovascular risk with the usual-care provider. There was no run-in period to preferentially enhance adherence to the trial regimen. All open-label medications for diabetes were permitted (except for GLP-1 receptor agonists), including SGLT-2 and DPP-4 inhibitors.
In the Liraglutide Effect and Action in Diabetes: Evaluation of Cardiovascular Outcome Results (LEADER) trial,11 a randomized trial in which patients with type 2 diabetes (mean glycated hemoglobin level, 8.7%) who had established cardiovascular disease (81% of enrolled patients) or cardiovascular risk factors were assigned to receive liraglutide or placebo, the risk of a three-component MACE outcome event was lower with liraglutide than with placebo (hazard ratio, 0.87; 95% CI, 0.78 to 0.97). Similar results were observed in the Trial to Evaluate Cardiovascular and Other Long-term Outcomes with Semaglutide in Subjects with Type 2 Diabetes (SUSTAIN-6),12 a randomized trial in which patients with type 2 diabetes (mean glycated hemoglobin level, 8.7%) who had established cardiovascular disease (83% of enrolled patients) or cardiovascular risk factors were assigned to receive semaglutide or placebo; the risk of a three-component MACE outcome was lower with semaglutide than with placebo (hazard ratio, 0.74; 95% CI, 0.58 to 0.95). In the LEADER trial, the risk of death from any cause was 15% lower with liraglutide than with placebo (similar to the 14% difference seen in our trial), but no such difference was seen with semaglutide in SUSTAIN-6. In our trial, statistical significance for the primary composite outcome was not achieved, but the direction and magnitude of the cardiovascular outcomes observed were not inconsistent with those seen in the LEADER and SUSTAIN-6 trials. In contrast, the Evaluation of Lixisenatide in Acute Coronary Syndrome (ELIXA) trial,10 which compared lixisenatide with placebo in patients who had had a recent acute coronary syndrome and who were followed for a median of 2.1 years, showed no significant difference between the groups in a four-component MACE outcome, nor in the rate of death from any cause.
The lack of cardiovascular efficacy in the current trial may be related to multiple factors. The median follow-up time in our trial was shorter than that in the LEADER trial (3.2 years vs. 3.8 years), as was the duration of exposure to the trial regimen (2.4 years vs. 3.5 years); in addition, the baseline glycated hemoglobin level in our trial was lower than that in the LEADER trial (8.0% vs. 8.7%), and the rate of discontinuation of the trial regimen was higher. The effect of exenatide, administered once weekly, on modifiable cardiovascular risk factors was modest. The disproportionate use in the placebo group of diabetes therapies known to reduce cardiovascular risk, such as SGLT-2 inhibitors13,14 and GLP-1 receptor agonists,11 may have preferentially resulted in lower event rates in the placebo group. Also, the four GLP-1 receptor agonists evaluated to date may not all be bioequivalent.
We did not observe any specific safety issues during our trial; there was no adverse signal with respect to heart failure, despite the higher mean heart rate in the exenatide group than in the placebo group, and events of acute pancreatitis and pancreatic cancer were rare, with similar rates in the two groups. Studies in animals have suggested a higher incidence of thyroid C-cell adenomas and carcinomas with once-weekly administration of extended-release exenatide than with placebo,23 but that finding has not been replicated in humans. In our trial, medullary thyroid carcinoma was reported in two patients in the exenatide group, as compared with one patient in the placebo group; all three patients in whom medullary thyroid carcinomas occurred had elevated calcitonin levels at baseline.
A major limitation of our trial was the rate of premature discontinuation of the trial regimen, which was driven primarily by patient decision. We speculate that probable factors for discontinuation were the complexity of the first-generation injection device that was used24 and the fact that our trial had no run-in period. Another possible limitation was that usual-care regimens were not standardized and therefore may have introduced variability. The difference between the two groups in the rate of death from any cause may have been influenced by the modest between-group difference in glycated hemoglobin levels.
In summary, our results show that once-weekly administration of extended-release exenatide in patients with type 2 diabetes at a wide range of cardiovascular risk appeared not to cause an increase in their overall cardiovascular risk.
Supplementary Material
Appendix
Acknowledgments
Supported by Amylin Pharmaceuticals, a wholly owned subsidiary of AstraZeneca.
We thank the patients, without whom this trial would not have been possible; Karen Hannan, Irene Kennedy, Rishi Patel, and Lynne Durborow for operational leadership; Phil Smith of the Duke Clinical Research Institute for statistical assistance; Peter Hoffmann of the Duke Clinical Research Institute for editorial assistance; and the academic partners and contract research organizations who provided assistance (PAREXEL International and the Canadian VIGOUR Centre).
Footnotes
Disclosure forms provided by the authors are available with the full text of this article at NEJM.org.
Contributor Information
Rury R. Holman, Diabetes Trials Unit, Oxford Centre for Diabetes, Endocrinology, and Metabolism, University of Oxford, Oxford, United Kingdom
M. Angelyn Bethel, Diabetes Trials Unit, Oxford Centre for Diabetes, Endocrinology, and Metabolism, University of Oxford, Oxford, United Kingdom
Robert J. Mentz, Duke Clinical Research Institute, Duke University School of Medicine, Durham, North Carolina
Vivian P. Thompson, Duke Clinical Research Institute, Duke University School of Medicine, Durham, North Carolina
Yuliya Lokhnygina, Duke Clinical Research Institute, Duke University School of Medicine, Durham, North Carolina
John B. Buse, Division of Endocrinology, University of North Carolina School of Medicine, Chapel Hill, North Carolina
Juliana C. Chan, Department of Medicine and Therapeutics, Chinese University of Hong Kong, Hong Kong
Jasmine Choi, AstraZeneca Research and Development, Gaithersburg, MD
Stephanie M. Gustavson, AstraZeneca Research and Development, Gaithersburg, MD
Nayyar Iqbal, AstraZeneca Research and Development, Gaithersburg, MD
Aldo P. Maggioni, Associazione Nazionale Medici Cardiologi Ospedalieri (ANMCO) Research Center, Florence, Italy
Steven P. Marso, Department of Cardiology, University of Texas Southwestern Medical Center, Dallas
Peter Öhman, AstraZeneca Research and Development, Gaithersburg, MD
Neha J. Pagidipati, Duke Clinical Research Institute, Duke University School of Medicine, Durham, North Carolina
Neil Poulter, International Centre for Circulatory Health, Imperial College London, London, United Kingdom
Ambady Ramachandran, India Diabetes Research Foundation and Dr. A. Ramachandran’s Diabetes Hospitals, Chennai, India
Bernard Zinman, Lunenfeld–Tanenbaum Research Institute, Mount Sinai Hospital and University of Toronto, Toronto
Adrian F. Hernandez, Duke Clinical Research Institute, Duke University School of Medicine, Durham, North Carolina
REFERENCES
- 1.Tancredi M, Rosengren A, Svensson A-M, et al. Excess mortality among persons with type 2 diabetes. N Engl J Med 2015;373:1720–32. [DOI] [PubMed] [Google Scholar]
- 2.Centers for Disease Control and Prevention. National diabetes statistics report, 2014. (https://stacks.cdc.gov/view/cdc/23442).
- 3.The Emerging Risk Factors Collaboration. Diabetes mellitus, fasting blood glucose concentration, and risk of vascular disease: a collaborative meta-analysis of 102 prospective studies. Lancet 2010;375:2215–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.UK Prospective Diabetes Study (UKPDS) Group. Intensive blood-glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33). Lancet 1998;352:837–53. [PubMed] [Google Scholar]
- 5.Zoungas S, Arima H, Gerstein HC, et al. Effects of intensive glucose control on microvascular outcomes in patients with type 2 diabetes: a meta-analysis of individual participant data from randomised controlled trials. Lancet Diabetes Endocrinol 2017;5:431–7. [DOI] [PubMed] [Google Scholar]
- 6.Turnbull FM, Abraira C, Anderson RJ, et al. Intensive glucose control and macrovascular outcomes in type 2 diabetes. Diabetologia 2009;52:2288–98. [DOI] [PubMed] [Google Scholar]
- 7.Scirica BM, Bhatt DL, Braunwald E, et al. Saxagliptin and cardiovascular outcomes in patients with type 2 diabetes mellitus. N Engl J Med 2013;369:1317–26. [DOI] [PubMed] [Google Scholar]
- 8.White WB, Cannon CP, Heller SR, et al. Alogliptin after acute coronary syndrome in patients with type 2 diabetes. N Engl J Med 2013;369:1327–35. [DOI] [PubMed] [Google Scholar]
- 9.Green JB, Bethel MA, Armstrong PW, et al. Effect of sitagliptin on cardiovascular outcomes in type 2 diabetes. N Engl J Med 2015;373:232–42. [DOI] [PubMed] [Google Scholar]
- 10.Pfeffer MA, Claggett B, Diaz R, et al. Lixisenatide in patients with type 2 diabetes and acute coronary syndrome. N Engl J Med 2015;373:2247–57. [DOI] [PubMed] [Google Scholar]
- 11.Marso SP, Daniels GH, Brown-Frandsen K, et al. Liraglutide and cardiovascular outcomes in type 2 diabetes. N Engl J Med 2016;375:311–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Marso SP, Bain SC, Consoli A, et al. Semaglutide and cardiovascular outcomes in patients with type 2 diabetes. N Engl J Med 2016;375:1834–44. [DOI] [PubMed] [Google Scholar]
- 13.Zinman B, Wanner C, Lachin JM, et al. Empagliflozin, cardiovascular outcomes, and mortality in type 2 diabetes. N Engl J Med 2015;373:2117–28. [DOI] [PubMed] [Google Scholar]
- 14.Neal B, Perkovic V, Mahaffey KW, et al. Canagliflozin and cardiovascular and renal events in type 2 diabetes. N Engl J Med 2017377:644–57. [DOI] [PubMed] [Google Scholar]
- 15.Blonde L, Pencek R, MacConell L. Association among weight change, glycemic control, and markers of cardiovascular risk with exenatide once weekly: a pooled analysis of patients with type 2 diabetes. Cardiovasc Diabetol 2015;14:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Knop FK, Brønden A, Vilsbøll T. Exenatide: pharmacokinetics, clinical use, and future directions. Expert Opin Pharmacother 2017;18:555–71. [DOI] [PubMed] [Google Scholar]
- 17.Henry RR, Klein EJ, Han J, Iqbal N. Efficacy and tolerability of exenatide once weekly over 6 years in patients with type 2 diabetes: an uncontrolled open-label extension of the DURATION-1 study. Diabetes Technol Ther 2016;18:677–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Grimm M, Han J, Weaver C, et al. Efficacy, safety, and tolerability of exenatide once weekly in patients with type 2 diabetes mellitus: an integrated analysis of the DURATION trials. Postgrad Med 2013;125:47–57. [DOI] [PubMed] [Google Scholar]
- 19.European Medicines Agency. Guideline on clinical investigation of medicinal products in the treatment or prevention of diabetes mellitus. May 14, 2012 (http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2012/06/WC500129256.pdf).
- 20.Department of Health and Human Services. Guidance for industry: diabetes mellitus — evaluating cardiovascular risk in new antidiabetic therapies to treat type 2 diabetes. December 2008 (https://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM071627.pdf).
- 21.Holman RR, Bethel MA, George J, et al. Rationale and design of the EXenatide Study of Cardiovascular Event Lowering (EXSCEL) trial. Am Heart J 2016;174:103–10. [DOI] [PubMed] [Google Scholar]
- 22.Mentz RJ, Bethel MA, Gustavson S, et al. Baseline characteristics of patients enrolled in the Exenatide Study of Cardiovascular Event Lowering (EXSCEL). Am Heart J 2017;187:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.European Medicines Agency. Assessment report for Bydureon. 2011. (http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/002020/WC500108239.pdf).
- 24.Lorenzi G, Schreiner B, Osther J, Boardman M. Application of adult-learning principles to patient instructions: a usability study for an exenatide once-weekly injection device. Clin Diabetes 2010;28:157–62. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix