The Zinc Finger Protein A20 Inhibits TNF-induced NF-κB–dependent Gene Expression by Interfering with an RIP- or TRAF2-mediated Transactivation Signal and Directly Binds to a Novel NF-κB–inhibiting Protein ABIN (original) (raw)
Abstract
The zinc finger protein A20 is a tumor necrosis factor (TNF)– and interleukin 1 (IL-1)-inducible protein that negatively regulates nuclear factor-kappa B (NF-κB)–dependent gene expression. However, the molecular mechanism by which A20 exerts this effect is still unclear. We show that A20 does not inhibit TNF- induced nuclear translocation and DNA binding of NF-κB, although it completely prevents the TNF- induced activation of an NF-κB–dependent reporter gene, as well as TNF-induced IL-6 and granulocyte macrophage–colony stimulating factor gene expression. Moreover, NF-κB activation induced by overexpression of the TNF receptor–associated proteins TNF receptor–associated death domain protein (TRADD), receptor interacting protein (RIP), and TNF recep- tor–associated factor 2 (TRAF2) was also inhibited by expression of A20, whereas NF-κB activation induced by overexpression of NF-κB–inducing kinase (NIK) or the human T cell leukemia virus type 1 (HTLV-1) Tax was unaffected. These results demonstrate that A20 inhibits NF-κB–dependent gene expression by interfering with a novel TNF-induced and RIP- or TRAF2-mediated pathway that is different from the NIK–IκB kinase pathway and that is specifically involved in the transactivation of NF-κB. Via yeast two-hybrid screening, we found that A20 binds to a novel protein, ABIN, which mimics the NF-κB inhibiting effects of A20 upon overexpression, suggesting that the effect of A20 is mediated by its interaction with this NF-κB inhibiting protein, ABIN.
Keywords: A20, ABIN, nuclear factor-κB, tumor necrosis factor, TRAF
Tumor necrosis factor (TNF)1 is a pleiotropic cytokine that acts as an important mediator of inflammation and immune responses by modulating the expression of many inflammatory proteins (Barnes and Karin, 1997). Induction of specific transcriptional programs by TNF is mediated by the activation of the transcription factor nuclear factor-kappa B (NF-κB). Moreover, an antiapoptotic function for NF-κB has been described recently (for review see Van Antwerp et al., 1998). TNF-induced activation of NF-κB is initiated by the recruitment of a number of proteins to both TNF receptors, with the TNF-R55 being far more effective than the TNF-R75 (Vandenabeele et al., 1995). The TNF-R55 recruits by its death domain the death domain–containing adapter protein TRADD (TNF receptor–associated death domain protein) (Hsu et al., 1995), which then interacts with the death domain–containing proteins FADD/MORT1 and receptor interacting protein (RIP) (Hsu et al., 1996a,b). The former initiates signaling leading to apoptosis (Boldin et al., 1996; Muzio et al., 1996), whereas the latter recently has been shown to be crucial for NF-κB activation by TNF (Kelliher et al., 1998). Moreover, TRADD can also recruit the TNF receptor–associated factor 2 (TRAF2) via a direct interaction or via the interaction of TRAF2 with RIP (Hsu et al., 1996a). TRAF2 belongs to a larger family whose members have been shown to be involved in NF-κB activation by several members of the TNF receptor family, including TNF-R55, TNF-R75, CD30, and CD40, but also by the interleukin 1 (IL-1) receptor (Cao et al., 1996; Hsu et al., 1996b; Ishida et al., 1996; Lee et al., 1996; Ng et al., 1998).
Studies with TRAF2-deficient cells recently have suggested that there is a lot of redundancy within the TRAF family (Yeh et al., 1997). TRAFs are believed to provide the signal to NF-κB activation by directly associating with the NF-κB–inducing kinase (NIK) (Malinin et al., 1997). NIK associates with and activates at least two kinases, IκB kinase (IKK)α and IKKβ, which phosphorylate the inhibitory subunit IκB leading to its degradation and the migration of NF-κB to the nucleus (Mercurio et al., 1997; Zandi et al., 1997). In addition to the NIK–IκB pathway, TRAF2 also initiates the activation of JNK and p38 mitogen-activated protein (MAP) kinases, which also have been shown to be important regulators of gene expression (Beyaert et al., 1996; Reinhard et al., 1997; Carpentier et al., 1998).
Besides IκB, NF-κB–dependent gene expression is also negatively regulated by the zinc finger protein A20 (Cooper et al., 1996; Jäättelä et al., 1996). The latter can be induced by different stimuli including: TNF, bacterial lipopolysaccharide, IL-1, tetradecanoic phorbol acetate (TPA), activation of the B cell surface receptor CD40, adhesion to extracellular matrix proteins, as well as overexpression of the human T cell leukemia virus type I (HTLV-1) Tax and the Epstein-Barr virus LMP1 gene product (Dixit et al., 1990; Laherty et al., 1992, 1993; Yurochko et al., 1992; Sarma et al., 1995). The expression of A20 is itself under the control of NF-κB (Krikos et al., 1992), suggesting that A20 is involved in the negative feedback regulation of NF-κB activation. However, the molecular mechanism of A20-mediated inhibition of NF-κB activation is at present unclear. Here we show that A20 inhibits TNF-induced NF-κB–dependent gene expression by interfering with a TRAF2-mediated signaling pathway that is different from the NIK–IκB or the p38 MAP kinase pathway, and which is specifically involved in the transactivation of NF-κB. Furthermore, we demonstrate that the A20 binding inhibitor of NF-κB activation (ABIN), a protein that interacts with the functional COOH-terminal part of A20, has similar NF-κB inhibiting effects as A20, suggesting that A20 can inhibit NF-κB activation via its interaction with ABIN.
Materials and Methods
Cloning of the mA20 Gene
L929r2 murine fibrosarcoma cells (Vanhaesebroeck et al., 1991) were treated for 4 h with 1,000 IU/ml mTNF and 10 μg/ml cycloheximide. Poly(A)+-mRNA was extracted and the first strand cDNA was synthesized using random primers (De Valck et al., 1996). Transformed MC1061 bacteria, containing the L929r2 cDNA library, were grown or lifted on C/P lift membranes (Bio-Rad Laboratories). The membranes were soaked for 7 min in denaturing solution (1.5 M NaCl, 0.5 M NaOH), twice for 3 min in neutralizing solution (1.5 M NaCl, 0.5 M Tris-HCl, pH 7.2, 1 mM EDTA) and washed in 2× SSC. DNA was fixed on the membranes by UV illumination and baking for 2 h at 80°C. The filters were incubated in prehybridization solution (1 mM EDTA, 0.5 M NaHPO4, pH 7.2, 7% SDS) at 60°C. Hybridization was done with the [32P]dCTP-labeled StuI-XhoI fragment of hA20 cDNA. Filters were washed at room temperature with a buffer containing 1 mM EDTA, 40 mM NaHPO4, pH 7.2, and 5% SDS, and subjected to autoradiography. Nine individual clones giving a positive signal were obtained. The plasmid of one of these clones contained a cDNA insert of ∼3.7 kb, including besides the complete coding sequence of mA20 also part of the 5′ and 3′ untranslated region.
Reagents
Recombinant mTNF and hTNF were produced by Escherichia coli and purified to at least 99% homogeneity. The preparations used had a specific biological activity of 1.4 × 108 IU/mg and 8.8 × 106 IU/mg purified protein, respectively, as determined with the international standard (code 88/532 and code 87/650; National Institute for Biological Standards and Control). Recombinant murine IL-1β was produced by E. coli and purified to at least 99% homogeneity. The preparations used had a specific biological activity of 3.65 × 108 IU/mg purified protein, as determined with the international standard (code 93/668). TPA was purchased from Sigma Chemical Co. Recombinant green fluorescent protein (rGFP) and polyclonal rabbit antiserum directed against rGFP originated from CLONTECH Laboratories and polyclonal phosphospecific p38 MAP kinase antibody and polyclonal p38 MAP kinase antibody were purchased from New England Biolabs.
Expression Plasmids
The cDNAs encoding mutant GFP (S65T) and a fusion protein of GFP (S65T) followed by murine A20 were cloned in the eukaryotic expression plasmid pCAGGS (Niwa et al., 1991). A20 was cloned as a blunted NcoI-EcoRI fragment in the SmaI site of the multiple cloning site inserted before the stop codon of GFP (pCAGGS-GFP/A20). The eukaryotic expression plasmids for ABIN were made by inserting the corresponding PCR fragment in frame with an NH2-terminal E-tag into pCAGGS. PCR fragments encoding TRADD and RIP were cloned in the pCDNA1 vector in frame with a COOH-terminal E-tag. The PCR fragment encoding CD40 was cloned in the plasmid pCDNA3 (pCDNA3-CD40) and was a gift of Dr. S. Pype (Catholic University of Leuven, Leuven, Belgium). Expression plasmids encoding TRAF2 and NIK were gifts of Dr. D. Goeddel (Tularik, San Francisco, CA) and Dr. D. Wallach (Weizmann Institute of Science, Rehovot, Israel), respectively. The plasmids phIL6LUC and p(κB)3LUC have been described previously (Plaisance et al., 1997). The plasmid pNFconluc, encoding the luciferase reporter gene driven by a minimal NF-κB responsive promoter was a gift of Dr. A. Israel (Institut Pasteur, Paris, France). The plasmids pAP1-luc and pSRE-luc were obtained from Stratagene. The plasmid pUT651, encoding β-galactosidase, was obtained from Eurogentec and the plasmid pIEX encoding the HTLV-1 Tax under control of the CMV promoter was obtained from Dr. K.-T. Jeang (National Institutes of Health, Bethesda, MD) (Semmes and Jeang, 1992). An expression plasmid for p300 (pCMV-p300) was provided by Dr. R. Eckner (Institute for Molecular Biology, Zurich, Switzerland) (Eckner et al., 1994). The yeast plasmid pAS2-A20 was described earlier (De Valck et al., 1996).
Cell Culture Conditions
L929sA and L929r2 murine fibrosarcoma cells and human embryonic kidney cells 293T were grown as described (Vanhaesebroeck et al., 1991; De Valck et al., 1997). Stable transfection of L929sA cells with the expression vectors was carried out by the calcium phosphate–DNA precipitation method. The pSV2neo-plasmid containing the neomycin resistance gene was coprecipitated as a selection marker (Southern and Berg, 1982). Selection of transfected clones was obtained by adding G418 (600 μg/ml) to the growth medium of the cells. Cells were mycoplasma-free, as judged by a DNA fluorochrome assay.
IL-6 Assay and Granulocyte Macrophage Colony Stimulating Factor (GM-CSF) Assay
L929sA transfectants were seeded in a 6-well plate at 6 × 105 cells/ 500 μl medium/well. After 24 h, cells were either stimulated for 6 h with 1,000 IU/ml mTNF or left untreated. Supernatants were harvested and assayed for IL-6 or GM-CSF in a 7TD1 or a FDCp1 cell proliferation assay, respectively (DeLamarter et al., 1985; Van Snick et al., 1986).
NF-κB Gel Retardation Assay
Cells in a 6-well plate were stimulated with 1,000 IU/ml mTNF for 5, 15, or 30 min, and nuclear fractions were prepared as described by Dignam et al. (1983). NF-κB DNA binding was analyzed by incubating 10 μg nuclear proteins for 30 min with the NF-κB specific 32P-labeled oligonucleotide probe (5′-agctATGTGGGATTTTCCCATGAGCagct-3′) containing the NF-κB site from the promoter of the mIL-6 gene. Protein–DNA complexes were separated on a 4% native polyacrylamide gel as described previously (Beyaert et al., 1996).
Transient Transfections and Reporter Gene Assays
L929sA cells were seeded at a density of 2 × 106 cells/75-cm2 flask. 24 h later, cells were transfected by the DNA–calcium phosphate cotransfection method with the plasmids pUT651 and phIL6LUC or p(κB)3LUC. The plasmids phIL6LUC and p(κB)3LUC contain the luciferase gene after the complete hIL-6 promoter or the minimal hIL-6 promoter preceded by three copies of the NF-κB recognition site, respectively. 48 h after transfection, cells were trypsinized and seeded at 1.2 × 105 cells/24-well. At 66 h after transfection, cells were either noninduced or induced for 6 h with 1,000 IU/ml hTNF. Inducible promoter activity was determined by measuring the luciferase and β-galactosidase activity present in cell extracts as described previously (De Valck et al., 1996). Transfection of 293T cells proceeded similarly, except that 293T cells were plated in 6-well plates at 4 × 105 cells per well and transiently transfected with 800 ng expression plasmids as well as 100 ng of pNFconluc and 100 ng pUT651. For measurements of AP1- and serum responsive element (SRE)–mediated transcription, cells were transfected with 100 ng of the plasmids pAP1-luc or pSRE-luc instead of 100 ng pNFconluc.
p38 MAP Kinase Phosphorylation Analysis
106 L929sA cells were seeded in 6-well plates. The next day, cells were either untreated or treated with 1,000 IU/ml mTNF for 5, 15, or 30 min. Cell lysates were prepared by directly adding SDS sample buffer (62.5 mM Tris-HCl, pH 6.8, 2% SDS, 10% glycerol, 50 mM DTT, 0.1% bromophenol blue) to the cells. Proteins were separated by 15% SDS-PAGE and blotted on a nitrocellulose filter. Phosphorylated p38 MAP kinase was detected with a polyclonal phosphospecific p38 MAP kinase antibody and enhanced chemiluminescence (Amersham International). The total amount of p38 MAP kinase present in the same cell extracts was revealed with a polyclonal p38 MAP kinase specific antibody.
Yeast-based Two-Hybrid Screening
The yeast two-hybrid system was purchased from CLONTECH Laboratories. The screening of a L929r2 cDNA library with pAS2-A20 was described previously (De Valck et al., 1997). Yeast colonies expressing interacting proteins were selected by growth on minimal media lacking Trp, Leu, and His, in the presence of 5 mM 3-amino-1,2,4-triazole and by screening for β-galactosidase activity. Plasmid DNA was extracted from the positive colonies and the pGAD424 vectors encoding candidate A20-interacting proteins were recovered by electroporation in the E. coli strain HB101 and growth on media lacking Leu.
Cell Transfection, Coimmunoprecipitation, and Western Blotting
2 × 106 human embryonic kidney 293T cells were plated on 10-cm petri dishes and transiently transfected by calcium phosphate–DNA coprecipitation. 24 h after transfection, cells were lysed in 500 μl of lysis buffer (50 mM Hepes, pH 7.6, 250 mM NaCl, 0.1% NP-40, 5 mM EDTA). Lysates were incubated with 5 μl of rabbit anti-GFP antibody (CLONTECH Laboratories) and immunocomplexes were immobilized on protein A–Trisacryl (Pierce Chemical Co.). The latter were washed twice with lysis buffer and twice with lysis buffer containing 1 M NaCl. Coprecipitating proteins were separated by SDS-PAGE and analyzed by Western blotting with mouse anti–E-tag antibody (Pharmacia Biotech).
Localization of GFP and GFP/A20 and Immunolocalization of ABIN
Localization of GFP/A20 was analyzed by means of GFP fluorescence. Therefore, L929sA cells stably expressing GFP or GFP/A20 were seeded on coverslips in 6-well plates and microscopically analyzed for GFP fluorescence (emission at 510 nm) at an excitation wavelength of 485 nm.
To identify the subcellular localization of ABIN, 4 × 105 293T cells were seeded on coverslips in 6-well plates and transfected with 1 μg plasmid DNA. 24 h after transfection, cells were fixed on the coverslips with 3% paraformaldehyde. Upon permeabilization with 1% Triton X-100, the cells were incubated for 2 h with mouse anti–E-tag antibody (1/1,000) followed by a second incubation with anti-mouse Ig antibody coupled to biotin (1/1,000; Amersham). After a subsequent incubation with streptavidin coupled to Texas red (Amersham), fluorescence can be analyzed via fluorescence microscopy (Axiophot; Zeiss), using a filter set with excitation at 543 nm and emission at 600 nm. In the same cells, fluorescence of GFP can be detected at a different wavelength, namely excitation at 485 nm and emission at 510 nm.
Results
A20 Inhibits TNF-induced Expression of IL-6 GM-CSF, and an NF-κB–dependent Reporter Gene
A20 has been described as a TNF inducible inhibitor of NF-κB activation, mainly on the basis of its effect on NF-κB–dependent expression of a reporter gene (Cooper et al., 1996; Jäättelä et al., 1996). Therefore, we first analyzed its effect on the TNF-induced expression of IL-6 and GM-CSF in control (L929SA-neo) and in L929sA cells overexpressing either a fusion protein of A20 with GFP (L929SA-GFP/A20) or GFP as such (L929SA-GFP). Expression of these cytokines has been shown to be NF-κB– dependent (Schreck et al., 1990; Zhang et al., 1990). Compared with the amount of IL-6 and GM-CSF found in the supernatant of TNF-treated L929SA-neo and L929SA-GFP cells, their levels were considerably decreased in L929SA-GFP/A20 cells (Table I). A similar inhibitory effect of A20 on TNF-induced gene activation could be seen when GFP-A20 expressing L929sA cells were transiently transfected with an expression plasmid containing the luciferase reporter gene under control of either the hIL-6 promoter (phIL6LUC) or a minimal promoter with three NF-κB–binding sites [p(κB)3LUC], and analyzed for luciferase expression upon stimulation with 1,000 IU/ml mTNF for 6 h (partially shown in Fig. 1 A). These results demonstrate that A20 interferes with the NF-kB–dependent expression of a reporter gene as well as with the expression of endogenous genes that are NF-κB regulated.
Table I.
Effect of A20 Expression on TNF-induced Expression of IL-6 and GM-CSF in L929sA Transfectants
| IL-6 | GM-CSF | |||
|---|---|---|---|---|
| −mTNF | +mTNF | −mTNF | +mTNF | |
| pg/ml | pg/ml | |||
| L929SA-NEO | <5 | 6561 | <14 | 691 |
| <5 | 6150 | <14 | 760 | |
| L929SA-GFP | <5 | 2187 | <14 | 810 |
| <5 | 2560 | <14 | 840 | |
| L929SA-GFP/A20 | <5 | 15 | <14 | 92 |
| <5 | 45 | <14 | 115 |
Figure 1.


A20 inhibits NF-κB–dependent promoter activity without affecting NF-κB binding to DNA. (A) L929sA clones stably expressing the neomycin resistance gene alone or in combination with the plasmids encoding GFP or GFP/A20 as indicated were transiently transfected with a luciferase reporter gene under the control of three NF-κB recognition sequences [p(κB)3LUC]. Cells were either untreated (open bars) or treated with 1,000 IU/ ml mTNF (filled bars) for 6 h. Promoter activity is expressed as the luciferase (luc) activity relative to the β-galactosidase (gal) activity in order to correct for differences in transfection efficiency. Data from a representative experiment (total number of experiments, 2) are expressed as the mean value (n = 3) with SD < 10%. (B) The same transfectants were either untreated (−) or treated with 1,000 IU/ml mTNF (+) for 30 min. Nuclear extracts were analyzed for active NF-κB in an electrophoretic mobility shift assay as described in Materials and Methods.
A20 Does Not Prevent the Nuclear Translocation and DNA Binding of NF-κB
NF-κB activation involves the release of the inhibitory protein IκB from NF-κB in the cytosol, leading to the nuclear translocation and binding of NF-κB to its specific recognition sequence in DNA (Henkel et al., 1993). The latter can be analyzed in a gel shift assay in which binding of active NF-κB to an NF-κB specific DNA probe leads to a slower migration of this probe in a nondenaturing polyacrylamide gel. Nuclear fractions prepared from L929SA-neo, L929SA-GFP, and L929SA-GFP/A20 cells that were either unstimulated or stimulated for 30 min with mTNF were analyzed in such an assay. Remarkably, although A20 completely prevented NF-κB–dependent gene expression as described above, no clear differences in TNF-induced DNA binding of NF-κB were observed (Fig. 1 B). Also, stimulation of these cells for shorter periods, 5 or 15 min, did not reveal an effect of A20 (data not shown). Nevertheless, TNF-induced nuclear translocation and DNA binding of NF-κB could be completely abolished by pretreatment with the proteasome inhibitor MG132 that inhibits NF-κB activation by preventing IκB degradation (Chen et al., 1995; data not shown). These results indicate that A20 inhibits NF-κB–dependent gene induction by specifically interfering with an NF-κB transactivation signal, without affecting the nuclear translocation and DNA binding of NF-κB.
By the presence of seven zinc finger structures in A20, it has been originally suggested that A20 may bind to DNA and directly interfere with the transcriptional machinery (Opipari et al., 1990). However, more recently, it has been demonstrated that A20 transiently expressed in 293T cells exclusively localized in the cytoplasm (Vincenz and Dixit, 1996). In our L929sA cells stably expressing GFP/A20 fusion proteins, a similar speckled cytoplasmic staining was observed via GFP fluorescence and confocal microscopy (Fig. 2). In contrast, GFP as such was detected both in the cytoplasm and the nucleus. Furthermore, treatment of the cells with TNF did not induce a detectable relocalization of A20 (data not shown). These results suggest that A20 cannot directly interfere with NF-κB transcriptional activity, but indicate that A20-mediated inhibition of NF-κB transactivation is a cytosolic event.
Figure 2.
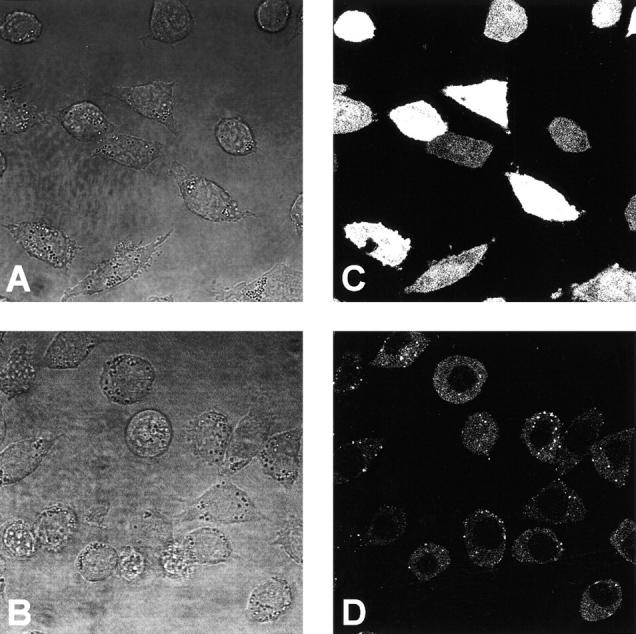
Subcellular localization of GFP and the fusion protein GFP/A20. Cells were seeded in coverglass chambers (Nunc) and GFP fluorescence was analyzed by excitation at 490 nm and emission between 510 and 525 nm using confocal microscopy (Zeiss). Transmission (A and B) and corresponding fluorescence images (C and D) are shown for the cell lines L929SA-GFP (A and C) and L929SA-GFP/A20 (B and D). No fluorescence was observed in L929SA-neo cells (data not shown).
A20 Inhibits a TNF-induced NF-κB Transactivation Pathway That Is Initiated at the Level of RIP/TRAF2 and That Is Independent of NIK
TNF-induced activation of NF-κB involves the recruitment of several cytoplasmic signaling proteins to the TNF-R55 (for review see Darnay and Aggarwal, 1997), which is the main signaling receptor for TNF. Moreover, overexpression of some of these proteins is sufficient to induce NF-κB activation. To investigate at which step A20 interferes with NF-κB activation, we tested the effect of A20 on NF-κB–dependent gene expression induced by TNF treatment or by overexpression of the TNF receptor associating proteins TRADD, RIP, TRAF2, and NIK in 293T cells. As expected, A20 completely prevented NF-κB– dependent luciferase expression induced by TNF (Fig. 3). A similar protective effect of A20 could be observed when NF-κB was activated by overexpression of TRADD, RIP, or TRAF2. However, NF-κB activation by overexpression of downstream acting NIK was not sensitive to A20. These results clearly demonstrate that A20 interferes with an RIP- or TRAF2-initiated NF-κB transactivation signal, which is different from the NIK-mediated pathway leading to nuclear translocation of NF-κB.
Figure 3.

Effect of A20 on NF-κB activation induced by TNF or overexpression of TRADD, RIP, TRAF2, and NIK in 293T cells. Cells were transfected with 1 μg plasmid DNA consisting of 300 ng empty plasmid or plasmids encoding TRADD, RIP, TRAF2, or NIK, 100 ng pUT651, 100 ng pNFconluc, 200 ng pCAGGS-GFP, or pCAGGS-GFP/A20, as well as 300 ng carrier plasmid DNA. Cells not expressing the NF-κB inducing proteins were left untreated or were treated for 6 h with 1,000 IU/ml hTNF. All cells were lysed 24 h after transfection and luciferase and β-galactosidase activities were measured. Promoter activity is expressed as the luciferase (luc) activity relative to the β-galactosidase (gal) activity. Data from a representative experiment (total number of experiments, 3) are expressed as the mean value (n = 3) with SD < 10%.
Inhibition of NF-κB Activation by A20 Is Stimulus-dependent
Recently, it was shown that activation of NF-κB induced by the HTLV transactivator Tax protein is mediated by NIK and both IKKα and IKKβ (Chu et al., 1998; Uhlik et al., 1998; Yin et al., 1998). In contrast, overexpression of a TRAF2 dominant negative mutant was not able to block Tax-induced NF-κB activation, excluding a role for TRAF2 in this pathway (Geleziunas et al., 1998; Yin et al., 1998). Therefore, we tested the effect of GFP/A20 overexpression on Tax-induced expression of an NF-κB–dependent reporter gene in 293T cells. Whereas TNF-induced NF-κB activation was completely prevented by A20, the latter could not block the gene-inducing effects mediated by Tax (Fig. 4 A). These results further indicate that A20 interferes with TNF-induced NF-κB–dependent gene expression at the level of an RIP- or TRAF2-initiated NF-κB transactivation pathway that is independent of NIK.
Figure 4.
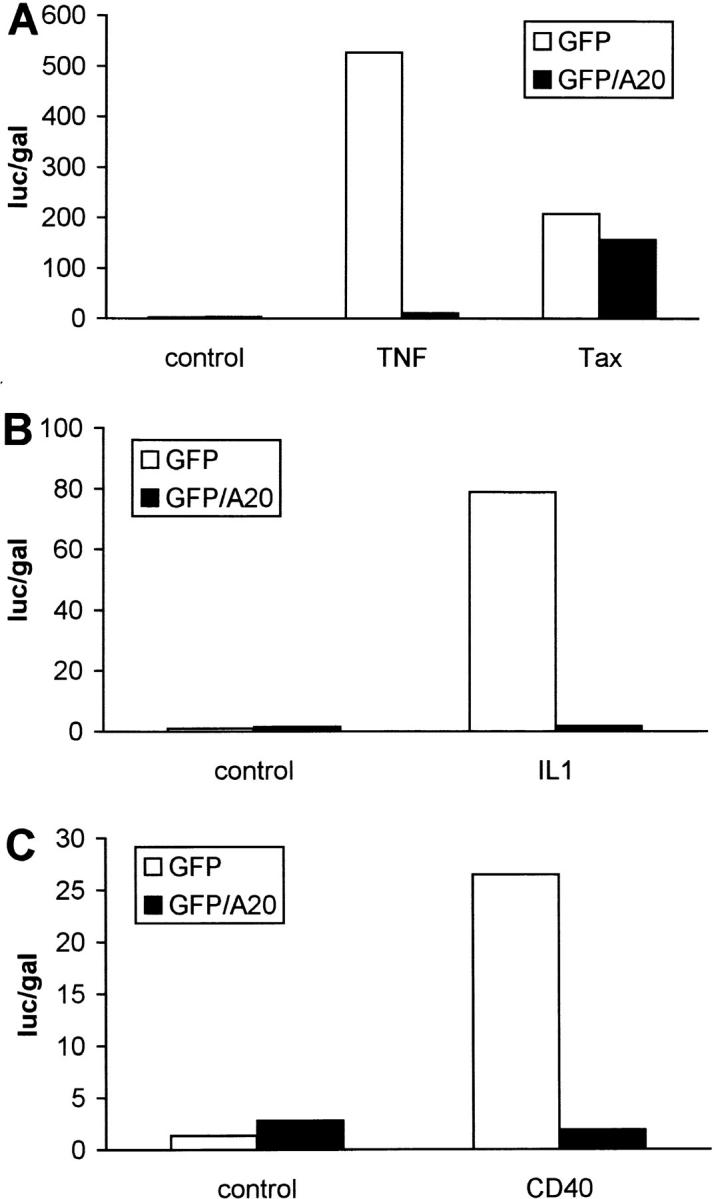
Effect of A20 on NF-κB activation by HTLV-1 Tax (A), IL-1 (B), or CD40 (C) in 293T cells. Cells were transfected with reporter gene constructs and GFP/A20 as described in the legend of Fig. 3. In A, NF-κB activation was induced by stimulation with 1,000 IU/ml hTNF for 6 h or by coexpression of an expression plasmid for HTLV-1 Tax (pIEX). In B, NF-κB activation was induced by stimulation with 7,000 IU/ml IL-1β for 6 h, whereas in C NF-κB activation was induced by coexpression of an expression plasmid for CD40 (pCDNA3-CD40). All cells were lysed 24 h after transfection and analyzed for luciferase expression as described in Materials and Methods. Data from a representative experiment (total number of experiments, 3) are expressed as the mean (n = 3) with SD < 10%.
To further substantiate this hypothesis, we also analyzed the effect of A20 on the activation of NF-κB by a number of other stimuli for which NF-κB activation has been shown to be mediated by TRAF2 or TRAF family members. IL-1 engages TRAF6 in its signaling pathway (Cao et al., 1996), whereas CD40 initiates NF-κB activation via TRAF2, TRAF5, and TRAF6 (Hu et al, 1994; Ishida et al., 1996a,b). Overexpression of A20 completely prevented IL-1–induced NF-κB activation in 293T cells (Fig. 4 B), which confirms previously published data (Jäättelä et al., 1996). Similarly, NF-κB activation induced by overexpression of CD40 was also abolished when A20 was coexpressed (Fig. 4 C). These results are in agreement with the above suggested role for a TRAF-initiated signaling pathway as target for A20.
Surprisingly, A20 also inhibited NF-κB activation by the protein kinase C activator TPA (Fig. 5). The latter also activates AP1- and SRE-dependent gene expression in 293T cells. Therefore, we analyzed whether A20 could also prevent the TPA-induced expression of a luciferase reporter gene whose expression is controlled by AP1- or SRE-binding proteins. However, in contrast to NF-κB–dependent gene expression, the TPA-induced activation of AP1- or SRE-dependent transcription was not sensitive to A20. These results demonstrate that A20 can also act as a specific inhibitor of NF-κB–dependent gene expression induced by TPA. It remains to be seen whether TRAFs or downstream signaling proteins are also involved in NF-κB activation by this stimulus.
Figure 5.

Effect of A20 on TPA-induced activation of gene expression mediated by NF-κB, AP1, or SRE. 293T cells were transfected with 100 ng pUT651 and 100 ng NFconluc, pAP1-luc, or pSRE-luc, in combination with 100 ng pCAGGS-GFP or pCAGGS-GFP/A20. Cells were either left untreated or treated with 200 ng/ml TPA for 6 h. Cells were analyzed for luciferase expression as described in Materials and Methods. Data from a representative experiment (total number of experiments, 2) are expressed as the mean (n = 3) with SD < 10%.
A20 Does Not Prevent the TNF-induced Activation of p38 MAP Kinase
Recently, we and others demonstrated an important role for TNF-induced activation of p38 MAP kinase in the transactivation of NF-κB (Beyaert et al., 1996; Bergmann et al., 1998; Vanden Berghe et al., 1998). Moreover, TNF-induced p38 MAP kinase activation was shown to be mediated by TRAF2 (Carpentier et al., 1998). To investigate whether A20 inhibited the TNF-induced transactivation of NF-κB by preventing p38 MAP kinase activation, we analyzed the TNF-induced activation of p38 MAP kinase in GFP/A20–transfected versus GFP-transfected L929SA cells. p38 MAP kinase activation was revealed by immunodetection of phosphorylated p38 MAP kinase with p38 MAP kinase phosphospecific antibodies. TNF induced a transient phosphorylation of p38 MAP kinase in L929SA-GFP as well as in L929SA-GFP/A20 cells (Fig. 6). Time kinetics of TNF-induced p38 MAP kinase phosphorylation/ activation were similar in both cell lines, although in some experiments dephosphorylation occurred slightly faster in A20-expressing cells. These results make it unlikely that A20 prevents NF-κB transactivation by inhibiting the TNF-induced activation of p38 MAP kinase.
Figure 6.

A20 has no effect on TNF-induced p38 MAP kinase activation. L929sA-GFP and L929sA-GFP/A20 cells were left untreated or treated with 1,000 IU/ml mTNF during 5, 15, or 30 min and immediately lysed in SDS lysis buffer. Total lysates were separated on SDS-PAGE and activated p38 MAP kinase was detected by Western blotting using polyclonal antibodies recognizing specifically phosphorylated p38 MAP kinase. As a control for the amount of protein loaded, total expression of p38 MAP kinase was revealed with polyclonal anti-p38 MAP kinase antibodies and found to be identical in all samples (data not shown).
NF-κB Activation Cannot Be Rescued from A20 Inhibition by CBP/p300 Overexpression
CREB-binding protein (CBP) and p300 are coactivators that link transcriptional activators to the basal transcriptional apparatus. Both CBP and p300 were shown to act as coactivators of NF-κB–dependent gene expression by a direct interaction with the p65 subunit (Gerritsen et al., 1997; Perkins et al., 1997). The latter interaction was recently shown to be enhanced by phosphorylation of p65 by protein kinase A (Zhong et al., 1998). To analyze whether A20 would prevent NF-κB transactivation by affecting the interaction between p65 and CBP/p300, we analyzed if TNF-induced NF-κB–dependent reporter gene expression could be rescued from A20 inhibition by overexpression of p300 in 293T cells. Basal and TNF-induced NF-κB activity was increased approximately threefold when p300 was coexpressed (Fig. 7), which is in agreement with a coactivator function for p300. However, p300 could not rescue TNF-induced NF-κB activation from inhibition by A20. These results make it unlikely that A20 prevents TNF-induced NF-κB–dependent gene expression by abolishing the transactivation of NF-κB by CBP/p300.
Figure 7.

Effect of overexpression of p300 on TNF-induced NF-κB activation and its inhibition by A20. 293T cells were transfected with 100 ng pUT651 and 100 ng pNFconluc in combination with or without 100 ng pCMV-p300. 100 ng pCAGGS-GFP, pCAGGS-GFP/A20, or empty vector pCAGGS were cotransfected as indicated. 24 h after transfection, cells were either left untreated or treated with 1,000 IU/ml hTNF for 6 h. All cells were lysed and analyzed for luciferase expression as described in Materials and Methods. Data from a representative experiment (total number of experiments, 3) are expressed as the mean (n = 3) with SD < 10%.
A20 Binds to a Novel NF-κB Inhibitory Protein ABIN
Although it is known that the NF-κB inhibiting potential of A20 resides in its COOH-terminal zinc finger–containing domain (Song et al., 1996; Natoli et al., 1998; Heyninck et al., 1999), the underlying mechanism is still unclear. Therefore, we used the yeast two-hybrid system to screen an L929r2 cDNA library for A20 interacting proteins that might be involved in the negative regulation of NF-κB. From 1.3 × 106 transformants, 10 clones expressed A20-interacting proteins, including A20 itself (De Valck et al., 1996) and 14-3-3 proteins (De Valck et al., 1997). Three clones contained COOH-terminal fragments of the same cDNA encoding a protein that we named ABIN. Full-length ABIN cDNA was subsequently isolated from the L929r2 cDNA library by colony hybridization with a fragment cloned by two-hybrid analysis as a probe. Several cDNAs were isolated and in the longest cDNA stop codons were identified in all three frames 5′ of a potential initiator methionine. Two different splice variants were found of ∼2,800 and 2,600-nt long. Northern blot analysis revealed that ABIN is expressed in all murine tissues tested as an mRNA of ∼2,800 bp that is in accordance with the length of the cloned full-length cDNA (data not shown). In contrast to A20, ABIN mRNA is constitutively expressed in both TNF-sensitive and TNF-resistant subclones derived from the parental cell line L929s (Vanhaesebroeck et al., 1991), irrespective of TNF stimulation.
The two splice variants of the ABIN cDNA contained an open reading frame of 1,941 and 1,782 nucleotides respectively, initiating at two different methionines [ABIN (1-647) and ABIN (54-647)] (GenBank/EMBL/DDBJ accession numbers AJ242777 and AJ242778). These cDNAs encode proteins of 72 and 68 kD, containing an amphiphatic helix with four consecutive repeats of a leucine followed by six random amino acid residues, which is characteristic of a leucine zipper structure. Also, full-length ABIN(1-647) and ABIN(54-647) bound A20 in a yeast two-hybrid assay, confirming the original interaction found with the 3′ COOH-terminal fragments ABIN(390-599), ABIN(249-647), and ABIN(312-647). The latter all contain the putative leucine zipper protein interaction motif (397-420). In addition, ABIN also interacted with A20 in mammalian cells since ABIN was able to coimmunoprecipitate with A20 in 293T cells that were transiently transfected with an expression plasmid for chimeric GFP-A20 protein and ABIN with an NH2-terminal E-tag (Fig. 8). Interestingly, interaction of ABIN with A20 required the COOH-terminal, zinc finger–containing part of A20 [A20(369-775)]. This domain was shown previously to be required for dimerization of A20 and for the interaction of A20 with 14-3-3 proteins (De Valck et al., 1996, 1997). Furthermore, overexpression of this domain is sufficient for the NF-κB inhibiting effects (Song et al., 1996; Natoli et al., 1998; Heyninck et al., 1999). In contrast, the NH2-terminal part of A20 has been shown to interact with TRAF2 (Song et al., 1996), suggesting that A20 acts as an adapter protein between TRAF2 and ABIN. The interaction between A20 and ABIN was not influenced by stimulation with TNF (data not shown). To characterize the subcellular distribution of ABIN, we transiently transfected GFP-A20 and E-tagged ABIN cDNA in 293T cells and analyzed their expression by means of GFP fluorescence and immunofluorescence via the anti–E-tag antibody, respectively. ABIN colocalized with A20 throughout the cytoplasm, both in unstimulated and in TNF-stimulated cells (data not shown).
Figure 8.

Coimmunoprecipitation of A20 and ABIN after transient transfection of the encoding plasmids of E-tagged ABIN and GFP, GFP-A20, GFP-A20(369-775), GFP-A20(1-368), or an empty expression vector as a negative control in 293T cells. Immunoprecipitation (top) was performed with anti-GFP antibody and Western blot detection with anti–E-tag antibody. To control expression levels of ABIN, 10 μl of lysates was subjected to SDS-PAGE and Western blot detection with anti–E-tag antibody (bottom).
Database similarity searches (BLAST) showed that ABIN is the murine homologue of the human cDNA encoding a human immunodeficiency virus (HIV) Nef–associating factor, NAF1 (Fukushi et al., 1999). HIV–Nef contributes substantially to disease pathogenesis by augmenting virus replication and markedly perturbing T cell function. Interestingly, the effect of Nef on host cell activation has been explained in part by its interaction with specific cellular proteins involved in signal transduction (for review see Harris, 1996), of which ABIN might be an example.
ABIN Mimics the NF-κB Inhibiting Effects of A20
Since both A20 and Nef were shown previously to block the signal transduction pathway leading to NF-κB activation upon stimulation with TNF or IL-1 and T cell receptor stimulation, respectively (Niederman et al., 1992; Bandres et al., 1994; Jäättelä et al., 1996; Song et al., 1996), we investigated the effect of ABIN on NF-κB–dependent reporter gene expression in transiently transfected 293T cells. GFP and GFP-A20 served as negative and positive controls, respectively. Similar to A20, both splice variants of ABIN were able to block TNF-induced NF-κB activation in these cells, with the shorter NH2-terminal truncated isoform being slightly more effective (Fig. 9 A). Overexpression of a combination of suboptimal doses of A20 and ABIN, on their own not sufficient to inhibit NF-κB activation, diminished NF-κB activation upon stimulation with TNF considerably (Fig. 9 B). This suggests that ABIN might mediate the NF-κB inhibiting effect of A20. Furthermore, cotransfection of expression plasmids encoding the TNF receptor–associated signaling proteins TRADD, RIP, TRAF2, and NIK together with the expression plasmid encoding ABIN, showed that the latter completely inhibited NF-κB activation induced by TRADD or RIP, and partially TRAF2-induced NF-κB activation. In contrast, no effect of ABIN was observed when NF-κB–dependent reporter gene expression was induced by NIK or more directly by overexpression of the p65 subunit of NF-κB (Fig. 9 C). Similarly, Tax-induced NF-κB activation that is TRAF-independent and is insensitive to A20, was also not affected by coexpression of ABIN (Fig. 9 D). These results suggest that ABIN, like A20, inhibits TNF-induced NF-κB activation at the level of RIP or TRAF2 proteins, preceding the activation of the NIK–IκB kinase steps.
Figure 9.




(A) Effect of two splice variants of ABIN [ABIN(1-647) and ABIN(54-647)] on the activation of NF-κB, measured by reporter gene activity. 293T cells were transiently transfected with 100 ng pUT651, 100 ng pNFconluc, and 200 ng expression plasmid and stimulated with TNF (1,000 IU/ml) during 6 h. As a control, plasmids encoding GFP and GFP-A20 were transfected. (B) Effect of transient transfection of suboptimal quantities of expression plasmids encoding A20 (5 ng) and ABIN (20 ng) on TNF-mediated NF-κB induction in 293T cells. (C) Effect of ABIN on NF-κB activation in 293T cells induced by overexpression of TRADD, RIP, TRAF2, NIK, or p65 after transfection of 300 ng of their encoding plasmids, together with 100 ng pUT651, 100 ng pNFconluc, and 500 ng pCAGGS-ABIN. (D) Effect of ABIN on NF-κB activation induced by overexpression of Tax in 293T cells. Cells were transfected with 1 μg plasmid DNA consisting of 300 ng empty plasmid or plasmid encoding Tax (pIEX), 100 ng pUT651, 100 ng pNFconluc, 200 ng pCAGGS or pCAGGS-ABIN, and 300 ng carrier plasmid DNA. All cells were lysed 30 h after transfection. In all experiments, cell extracts were analyzed for luciferase and β-galactosidase activity and the data are plotted as luc/gal, which is representative of NF-κB activity. Each value is the mean (n = 3) with SD < 10%.
Discussion
The zinc finger protein A20 is encoded by an immediate early response gene and acts as an inhibitor of NF-κB– dependent gene expression induced by different stimuli including TNF and IL-1 (Cooper et al., 1996; Jäättelä et al., 1996; Song et al., 1996). Here we show that the TNF-induced expression of GM-CSF and IL-6, as well as the TNF-induced expression of a luciferase reporter gene that is expressed under control of the complete hIL-6 promoter or the minimal hIL-6 promoter preceded by 3 NF-κB recognition sequences, are clearly inhibited in L929sA cells stably transfected with A20. These results are consistent with the fact that NF-κB is required for IL-6 and GM-CSF gene transcription (Schreck et al., 1990; Zhang et al., 1990). Surprisingly, gel retardation assays revealed that overexpression of A20 had no effect on the TNF-induced nuclear translocation and DNA binding of NF-κB. Also the constitution of the NF-κB complex was not altered in cells overexpressing A20, and consisted in both cases of a p65 and a p50 subunit as revealed by gel supershift assays (data not shown). Therefore, the inhibition of NF-κB– dependent gene expression by A20 cannot be explained by an A20-induced alteration in the subunits of NF-κB. Ferran et al. (1998) showed recently that A20 acts upstream of IκB degradation and prevented the nuclear translocation of NF-κB. The reason for the discrepancy with our results is still unclear. Because activation of NF-κB is an early response after stimulation with TNF, we analyzed NF-κB translocation at 5, 15, and 30 min after TNF stimulation, whereas results of Ferran et al. were obtained 2 h after TNF stimulation. The latter is quite late and might already be regulated by secondary factors that are A20-sensitive. Moreover, NF-κB activation at later times is also regulated by TNF-induced negative regulatory proteins such as IκBα and A20 whose expression is itself under the control of NF-κB, further raising the complexity of NF-κB activation at later time points (Krikos et al., 1992; LeBail et al., 1993). Alternatively, we cannot exclude cell type–dependent differences.
Until now, the mechanism by which A20 blocks the activation of NF-κB–dependent gene regulation was not known. Up to now, the NIK–IκB kinase pathway has been assumed to be responsible for NF-κB activation upon TNF treatment. However, our finding that A20 has no effect on the translocation of NF-κB to the nucleus argues against this pathway as the target for A20. Moreover we demonstrated that A20 could inhibit RIP- and TRAF2- but not NIK-induced NF-κB–dependent reporter gene activation, suggesting that A20 interferes with NF-κB activation upstream of NIK. These conclusions were confirmed by the inability of A20 to block Tax-induced NF-κB activation since the latter was shown to activate NF-κB by directly interfering with the downstream kinases NIK, IKKα, and IKKβ, independent of TRAF2 (Chu et al., 1998; Uhlik et al., 1998; Yin et al., 1998).
The above results suggest that TNF-induced NF-κB– dependent gene activation requires at least two different pathways: an NIK-mediated pathway leading to translocation of NF-κB to the nucleus, and an NIK-independent pathway leading to transactivation of NF-κB. Our results demonstrate that A20 specifically interferes with the NF-κB transactivation pathway. A20 has also been shown to interact with the TNF receptor–associated protein TRAF2 (Song et al., 1996). Interestingly, TRAF2 as well as some other members of the TRAF protein family, including TRAF5 and TRAF6, were shown to play a positive role in NF-κB activation induced by different cytokines, such as TNF, IL-1, and CD40 ligand, via their interaction with the NF-κB inducing kinase NIK (Cao et al., 1996; Malinin et al., 1997). The fact that A20 directly associates with TRAF2, as well as our observation that A20 not only prevents NF-κB activation by TNF but also by IL-1 and CD40 overexpression, points to a role of a TRAF-mediated signaling pathway as a target for A20. However, gene knockout studies have recently shown that TRAF2 is not absolutely required for NF-κB activation by TNF, although this probably is a consequence of redundancy within the TRAF protein family (Yeh et al., 1997). Alternatively, RIP may be more important than TRAF2 in mediating activation of NF-κB upon TNF stimulation (Kelliher et al., 1998).
The nature of the RIP/TRAF–initiated NF-κB transactivation signal is still unclear. Recently, an important role in the transactivating potential of NF-κB upon TNF stimulation was demonstrated for p38 MAP kinase (Beyaert et al., 1996; Bergmann et al., 1998; Vanden Berghe et al., 1998). This kinase becomes activated by stimulation of cells with TNF as well as by overexpression of TRAF2. In contrast, overexpression of NIK did not induce the phosphorylation of p38 MAP kinase, indicating that a separate pathway initiating at TRAF2 leads to the activation of p38 MAP kinase (Carpentier et al., 1998). Similar to the effect of A20 overexpression, inhibition of p38 MAP kinase with the specific inhibitor SB203580 also prevented NF-κB– dependent gene expression without altering the translocation of NF-κB to the nucleus (Beyaert et al., 1996; Bergmann et al., 1998). As these results suggested that A20 might interfere with the TRAF2-p38 MAP kinase pathway, we investigated if A20 was able to prevent the TNF-induced activation of p38 MAP kinase. However, no significant effect of A20 on p38 MAP kinase phosphorylation, which is a marker for its activation, could be observed. Although these results indicate that A20 does not act upstream of p38 MAP kinase activation, it is still possible that A20 interferes with the TRAF2-p38 MAP kinase/ NF-κB transactivation pathway downstream of p38 MAP kinase. The validation of this possibility awaits the identification of the p38 MAP kinase substrate that is involved in NF-κB transactivation. Alternatively, our results might also fit with the existence of another RIP- or TRAF2-initiated pathway that contributes to NF-κB–dependent transcription, and which is blocked by A20.
Our observation that A20 also prevents NF-κB activation by TPA indicates that the A20-sensitive pathway might also be activated by protein kinase C, at least in some cell lines. Similar results were obtained by Cooper et al. (1996). In contrast, stable expression of A20 has been reported to be unable to prevent TPA-induced NF-κB activation in breast carcinoma MCF cells (Jäättelä et al., 1996). These controversial results might reflect a cell type specific effect or differences in A20 expression levels upon stable and transient transfection. It should be mentioned that a role for protein kinase C in TNF-induced NF-κB transactivation has been suggested recently based on the inhibition with a protein kinase C inhibitor (Bergmann et al., 1998). Whether protein kinase C functions downstream of TRAF2 or in a totally separate pathway remains to be established. In any case, the protein kinase C–mediated pathway sensitive to A20 seems to regulate specifically NF-κB–dependent gene expression, as TPA-induced transcription that is controlled by an AP1-responsive element or a SRE was not sensitive to A20. The latter result, as well as our finding that Tax-mediated NF-κB activation is not affected by A20, also excludes an aspecific effect of A20 on the general transcription machinery.
The transcription activating potential of NF-κB has been primarily attributed to the p65 subunit, whose transactivating potential resides in its COOH-terminal portion (Ballard et al., 1992; Schmitz et al., 1994). Furthermore, the p65 subunit becomes phosphorylated during the activation of NF-κB upon TNF stimulation (Naumann and Scheidereit, 1994; Schmitz et al., 1995). Indeed, a p65 phosphorylating activity was found in the IκB kinase complex (Mercurio et al., 1997). Moreover, it was also shown that IκB is associated with the protein kinase A catalytic subunit that can phosphorylate the p65 in its rel homology domain resulting in enhanced activity of NF-κB (Zhong et al., 1997). Recently, p65 phosphorylation was shown to promote an interaction between p65 and the coactivators CBP/p300 (Zhong et al., 1998). The latter were previously shown to synergistically enhance the transcription activating potential of NF-κB (Gerritsen et al., 1997; Perkins et al., 1997). However, it is unlikely that A20 interferes with protein kinase A or another signaling pathway leading to the engagement of the coactivators CBP/p300 in the transactivation of NF-κB because we were unable to rescue NF-κB activation from A20 inhibition by overexpression of CBP/p300. Moreover, activation of the protein kinase A catalytic subunit requires degradation of IκB and the activation of the NIK–IκB kinase pathway which is, however, not modulated by A20. Also, A20 did not interfere with NF-κB–dependent gene expression obtained by overexpression of the p65 subunit as such (Cooper et al., 1996).
In endothelial cells, TRAF2 has been recently shown to translocate to the nucleus, where it might directly regulate transcription (Min et al., 1998). Because A20 can bind to TRAF2 (Song et al., 1996), and exclusively resides in the cytosol, A20 might prevent nuclear localization of TRAF2.
Screening of a cDNA library for A20 interacting proteins by the yeast two-hybrid system has revealed some isoforms of the 14-3-3 proteins that interact with the COOH-terminal zinc finger domain of A20 (Vincenz and Dixit, 1996; De Valck et al., 1997). 14-3-3 proteins were shown to function as adapter proteins between A20 and c-Raf. Moreover, 14-3-3 also functioned as a chaperone in these studies (Vincenz and Dixit, 1996). However, by mutation analysis we previously demonstrated that the interaction of 14-3-3 proteins with A20 is not involved in the effect of A20 on NF-κB activation (De Valck et al., 1997). By the yeast two-hybrid screening system, we also identified ABIN as a novel A20-interacting leucine zipper protein. The interaction of ABIN with A20 was confirmed in human cells and shown to map to the functional COOH-terminal zinc finger–containing domain of A20. Upon overexpression, ABIN potently inhibits NF-κB activation induced by TNF. Furthermore, also ABIN interferes with TNF-induced NF-κB activation at the level of RIP/ TRAF2. Therefore, the ability of A20 to block TNF-mediated NF-κB activation is likely to involve the binding of the NF-κB inhibitory protein ABIN to the COOH-terminal zinc finger domain of A20. Moreover, the fact that A20 can also interact via its NH2-terminal domain with TRAF1 and TRAF2 (Song et al., 1996) suggests that A20 can recruit ABIN to the TRAF2 complex in the TNF signaling pathway.
In conclusion, A20 appears to prevent NF-κB–dependent gene expression by specifically interfering in the cytosol with a novel RIP/TRAF2–initiated transactivation pathway, thus inhibiting the TNF-induced expression of several cytokines and proinflammatory proteins. Since A20 also inhibits NF-κB activation by IL-1 and CD40, which all signal to NF-κB activation via members of the TRAF family, further identification of TRAF-mediated NF-κB transactivation signals may provide means of achieving more specific antiinflammatory treatments.
Acknowledgments
The authors thank Dr. D. Wallach, Dr. D. Goeddel, Dr. K.-T. Jeang, Dr. R. Eckner, and Dr. S. Pype for providing expression plasmids. Dr. S. Zini is thanked for assistance in cloning of the A20 gene, S. Dewaele for construction of an L929r2 cDNA library, A. Meeus and W. Burm for technical assistance, and R. Cocquyt and F. Molemans (all from University of Ghent, Ghent, Belgium) for DNA sequencing.
Abbreviations used in the paper
ABIN
A20 binding inhibitor of NF-κB activation
CBP
CREB (cAMP responsive element binding protein) binding protein
GFP
green fluorescent protein
GM-CSF
granulocyte macrophage–colony stimulating factor
HTLV-1
human T cell leukemia virus type 1
IKK
IκB kinase
IL
interleukin
MAP kinase
mitogen-activated protein kinase
NF-κB
nuclear factor-kappa B
NIK
NF-κB–inducing kinase
RIP
receptor interacting protein
SRE
serum response element
TNF
tumor necrosis factor
TPA
tetradecanoic phorbol acetate
TRADD
TNF receptor–associated death domain protein
TRAF
TNF receptor–associated factor
Footnotes
R. Beyaert, G. Haegeman, and R. Contreras are a postdoctoral research assistant and research directors with the Fonds voor Wetenschappelijk Onderzoek-Vlaanderen, respectively. Research was supported by the Interuniversitaire Attractiepolen, the Fonds voor Wetenschappelijk Onderzoek-Vlaanderen, the Sportvereniging tegen Kanker, European Community–Biomed2 grant and a European Community–Training and Mobility Research grant.
D. De Valck's present address is Laboratory of Skeletal Developments and Joint Disorders, Catholic University of Leuven, B-3000 Leuven, Belgium. W. Van Criekinge's present address is Devgen, Technologiepark, B-9052 Zwijnaarde, Belgium.
References
- Ballard DW, Dixon EP, Peffer NJ, Bogerd H, Doerre S, Stein B, Greene WC. The 65-kDa subunit of human NF-kappa B functions as a potent transcriptional activator and a target for v-Rel–mediated repression. Proc Natl Acad Sci USA. 1992;89:1875–1879. doi: 10.1073/pnas.89.5.1875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bandres JC, Ratner L. Human immunodeficiency virus type 1 Nef protein down-regulates transcription factors NF-κB and AP-1 in human T cells in vitro after T-cell receptor stimulation. J Virol. 1994;68:3243–3249. doi: 10.1128/jvi.68.5.3243-3249.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes PJ, Karin M. Nuclear factor-κB: a pivotal transcription factor in inflammatory diseases. N Engl J Med. 1997;336:1066–1071. doi: 10.1056/NEJM199704103361506. [DOI] [PubMed] [Google Scholar]
- Bergmann M, Hart L, Lindsay M, Barnes PJ, Newton R. IκB degradation and nuclear factor-κB DNA binding are insufficient for interleukin-1β and tumor necrosis factor-α induced κB-dependent transcription. J Biol Chem. 1998;273:6607–6610. doi: 10.1074/jbc.273.12.6607. [DOI] [PubMed] [Google Scholar]
- Beyaert R, Cuenda A, Vanden W, Berghe, Plaisance S, Lee JC, Haegeman G, Cohen P, Fiers W. The p38/RK mitogen-activated protein kinase pathway regulates interleukin-6 synthesis in response to tumor necrosis factor. EMBO (Eur Mol Biol Organ) J. 1996;15:1914–1923. [PMC free article] [PubMed] [Google Scholar]
- Boldin MP, Goncharov TM, Goltsev YV, Wallach D. Involvement of MACH, a novel MORT1/FADD–interacting protease, in Fas/APO-1- and TNF receptor-induced cell death. Cell. 1996;85:803–815. doi: 10.1016/s0092-8674(00)81265-9. [DOI] [PubMed] [Google Scholar]
- Cao Z, Xiong J, Takeuchi M, Kurama T, Goeddel DV. TRAF6 is a signal transducer for interleukin-1. Nature. 1996;383:443–446. doi: 10.1038/383443a0. [DOI] [PubMed] [Google Scholar]
- Carpentier I, Declercq W, Malinin NL, Wallach D, Fiers W, Beyaert R. TRAF2 plays a dual role in NF-κB-dependent gene activation by mediating the TNF-induced activation of p38 MAPK and IκB kinase pathways. FEBS (Fed Eur Biochem Soc) Lett. 1998;425:195–198. doi: 10.1016/s0014-5793(98)00226-9. [DOI] [PubMed] [Google Scholar]
- Chen Z, Hagler J, Palombella VJ, Melandrini F, Scherer D, Ballard D, Maniatis T. Signal-induced site-specific phosphorylation targets IκBα to the ubiquitin-proteasome pathway. Genes Dev. 1995;9:1586–1597. doi: 10.1101/gad.9.13.1586. [DOI] [PubMed] [Google Scholar]
- Chu ZL, DiDonato JA, Hawiger J, Ballard DW. The tax oncoprotein of human T-cell leukemia virus type I activates with and persistently activates IκB kinases containing IKKα and IKKβ. J Biol Chem. 1998;273:15891–15894. doi: 10.1074/jbc.273.26.15891. [DOI] [PubMed] [Google Scholar]
- Cooper JT, Stroka DM, Brostjan C, Palmetshofer A, Bach FH, Ferran C. A20 blocks endothelial cell activation through a NF-κB-dependent mechanism. J Biol Chem. 1996;271:18068–18073. doi: 10.1074/jbc.271.30.18068. [DOI] [PubMed] [Google Scholar]
- Darnay BG, Aggarwal BB. Early events in TNF signaling: a story of associations and dissociations. J Leuk Biol. 1997;61:559–566. doi: 10.1002/jlb.61.5.559. [DOI] [PubMed] [Google Scholar]
- DeLamarter JF, Mermod JJ, Liang CM, Eliason JF, Thatcher DR. Recombinant murine GM-CSF from E. colihas biological activity and is neutralized by a specific antiserum. EMBO (Eur Mol Biol Organ) J. 1985;4:2575–2581. doi: 10.1002/j.1460-2075.1985.tb03973.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Valck D, Heyninck K, Van Criekinge W, Contreras R, Beyaert R, Fiers W. A20, an inhibitor of cell death, self-associates by its zinc finger domain. FEBS (Fed Eur Biochem Soc) Lett. 1996;384:61–64. doi: 10.1016/0014-5793(96)00283-9. [DOI] [PubMed] [Google Scholar]
- De Valck D, Heyninck K, Van Criekinge W, Vandenabeele P, Fiers W, Beyaert R. A20 inhibits NF-kappaB activation independently of binding to 14-3-3 proteins. Biochem Biophys Res Commun. 1997;238:590–594. doi: 10.1006/bbrc.1997.7343. [DOI] [PubMed] [Google Scholar]
- Dignam JD, Lebovitz RM, Roeder RG. Accurate transcription initiation by RNA polymerase II in a soluble extract from isolated mammalian nuclei. Nucleic Acids Res. 1983;11:1475–1489. doi: 10.1093/nar/11.5.1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixit VM, Green S, Sarma V, Holzman LB, Wolf FW, O'Rourke K, Ward PA, Prochownik EV, Marks RM. Tumor necrosis factor-alpha induction of novel gene products in human endothelial cells including a macrophage-specific chemotaxin. J Biol Chem. 1990;265:2973–2978. [PubMed] [Google Scholar]
- Eckner R, Ewen ME, Newsome D, Gerdes M, DeCaprio JA, Livingston DM. Molecular cloning and functional analysis of the adenovirus E1A-associated 300 kDa protein (p300) reveals a protein with properties of a transcriptional activator. Genes Dev. 1994;8:869–884. doi: 10.1101/gad.8.8.869. [DOI] [PubMed] [Google Scholar]
- Ferran C, Stroka DM, Badrichani AZ, Cooper JT, Wrighton CJ, Soares M, Grey ST, Bach FH. A20 inhibits NF-κB activation in endothelial cells without sensitizing to tumor necrosis factor-mediated apoptosis. Blood. 1998;91:2249–2258. [PubMed] [Google Scholar]
- Fukushi M, Dixon J, Kimura T, Tsurutani N, Dixon MJ, Yamamoto N. Identification and cloning of a novel cellular protein Naf1, Nef-associated factor 1, that increases cell surface CD4 expression. FEBS (Fed Eur Biochem Soc) Lett. 1999;442:83–88. doi: 10.1016/s0014-5793(98)01631-7. [DOI] [PubMed] [Google Scholar]
- Geleziunas R, Ferrell S, Lin X, Mu Y, Cunningham ET, Grant M, Connelly MA, Hambor JE, Marcu KB, Green WC. Human T-cell leukemia virus type 1 Tax induction of NF-κB involves activation of the IκB kinase α (IKKα) and IKKβ cellular kinases. Mol Cell Biol. 1998;18:5157–5165. doi: 10.1128/mcb.18.9.5157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerritsen ME, Williams AJ, Neish AS, Moore S, Shi Y, Collins T. CREB-binding protein/p300 are transcriptional coactivators of p65. Proc Natl Acad Sci USA. 1997;94:2927–2932. doi: 10.1073/pnas.94.7.2927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris M. From negative factor to a critical role in virus pathogenesis: the changing fortunes of Nef. J Gen Virol. 1996;77:2379–2392. doi: 10.1099/0022-1317-77-10-2379. [DOI] [PubMed] [Google Scholar]
- Henkel T, Machleidt T, Alkalay I, Kronke M, Ben-Neriah Y, Baeuerle PA. Rapid proteolysis of I kappa B-alpha is necessary for activation of transcription factor NF-kappa B. Nature. 1993;365:182–185. doi: 10.1038/365182a0. [DOI] [PubMed] [Google Scholar]
- Heyninck K, Beyaert R. The cytokine-inducible zinc finger protein A20 inhibits IL-1-induced NF-κB activation at the level of TRAF6. FEBS (Fed Eur Biochem Soc) Lett. 1999;442:147–150. doi: 10.1016/s0014-5793(98)01645-7. [DOI] [PubMed] [Google Scholar]
- Hsu H, Xiong J, Goeddel DV. The TNF receptor 1-associated protein TRADD signals cell death and NF-kappa B activation. Cell. 1995;81:495–504. doi: 10.1016/0092-8674(95)90070-5. [DOI] [PubMed] [Google Scholar]
- Hsu H, Huang J, Shu HB, Baichwal V, Goeddel DV. TNF-dependent recruitment of the protein kinase RIP to the TNF receptor-1 signaling complex. Immunity. 1996a;4:387–396. doi: 10.1016/s1074-7613(00)80252-6. [DOI] [PubMed] [Google Scholar]
- Hsu H, Shu HB, Pan MG, Goeddel DV. TRADD-TRAF2 and TRADD-FADD interactions define two distinct TNF receptor 1 signal transduction pathways. Cell. 1996b;84:299–308. doi: 10.1016/s0092-8674(00)80984-8. [DOI] [PubMed] [Google Scholar]
- Hu HM, O'Rourke K, Boguski MS, Dixit VM. A novel RING finger protein interacts with the cytoplasmic domain of CD40. J Biol Chem. 1994;269:30069–30072. [PubMed] [Google Scholar]
- Ishida T, Mizushima SI, Azuma S, Kobayashi N, Tojo T, Suzuki K, Aizawa S, Watanabe T, Mosialos G, Kieff E, Yamamoto T, Inoue J. Identification of TRAF6, a novel tumor necrosis factor receptor-associated factor protein that mediates signaling from an amino-terminal domain of the CD40 cytoplasmic region. J Biol Chem. 1996a;271:28745–28748. doi: 10.1074/jbc.271.46.28745. [DOI] [PubMed] [Google Scholar]
- Ishida T, Tojo T, Aoki T, Kobayashi N, Ohishi T, Watanabe T, Yamamoto T, Inoue J-I. TRAF5, a novel tumor necrosis factor receptor-associated factor family protein, mediates CD40 signaling. Proc Natl Acad Sci USA. 1996b;93:9437–9442. doi: 10.1073/pnas.93.18.9437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jäättelä M, Mouritzen H, Elling F, Bastholm L. A20 zinc finger protein inhibits TNF and IL-1 signaling. J Immunol. 1996;156:1166–1173. [PubMed] [Google Scholar]
- Kelliher MA, Grimm S, Ishida Y, Kuo F, Stanger BZ, Leder P. The death domain kinase RIP mediates the TNF-induced NF-kappaB signal. Immunity. 1998;8:297–303. doi: 10.1016/s1074-7613(00)80535-x. [DOI] [PubMed] [Google Scholar]
- Krikos A, Laherty CD, Dixit VM. Transcriptional activation of the tumor necrosis factor alpha-inducible zinc finger protein, A20, is mediated by kappa B elements. J Biol Chem. 1992;267:17971–17976. [PubMed] [Google Scholar]
- Laherty CD, Hu HM, Opipari AW, Wang F, Dixit VM. The Epstein-Barr virus LMP1 gene product induces A20 zinc finger protein expression by activating nuclear factor kappa B. J Biol Chem. 1992;267:24157–24160. [PubMed] [Google Scholar]
- Laherty CD, Perkins ND, Dixit VM. Human T cell leukemia virus type I Tax and phorbol 12-myristate 13-acetate induce expression of the A20 zinc finger protein by distinct mechanisms involving nuclear factor kappa B. J Biol Chem. 1993;268:5032–5039. [PubMed] [Google Scholar]
- LeBail O, Schmidt-Ullrich R, Israël A. Promoter analysis of the gene encoding for IκBα/MAD-3 inhibitor of NF-κB: positive regulation by members of the rel/NF-κB family. EMBO (Eur Mol Biol Organ) J. 1993;12:5043–5049. doi: 10.1002/j.1460-2075.1993.tb06197.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SY, Lee SY, Kandala G, Liou ML, Liou HC, Choi Y. CD30/TNF receptor-associated factor interaction: NF-kappa B activation and binding specificity. Proc Natl Acad Sci USA. 1996;93:9699–9703. doi: 10.1073/pnas.93.18.9699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malinin NL, Boldin MP, Kovalenko AV, Wallach D. MAP3K-related kinase involved in NF-kappaB induction by TNF, CD95, and IL-1. Nature. 1997;385:540–544. doi: 10.1038/385540a0. [DOI] [PubMed] [Google Scholar]
- Mercurio F, Zhu H, Murray BW, Shevchenko A, Bennett BL, Li J, Young DB, Barbosa M, Mann M, Manning A, Rao A. IKK-1 and IKK-2: cytokine-activated IkappaB kinases essential for NF-kappaB activation. Science. 1997;278:860–866. doi: 10.1126/science.278.5339.860. [DOI] [PubMed] [Google Scholar]
- Min W, Bradley JR, Galbraith JJ, Jones SJ, Ledgerwood EC, Pober JS. The N-terminal domains target TNF-receptor-associated factor-2 to the nucleus and display transcriptional regulatory activity. J Immunol. 1998;161:319–324. [PubMed] [Google Scholar]
- Muzio M, Chinnaiyan AM, Kischkel FC, O'Rourke K, Shevchenko A, Ni J, Scaffidi C, Bretz JD, Zhang M, Gentz R, et al. FLICE, a novel FADD-homologous ICE/CED-3-like protease, is recruited to the CD95 (Fas/APO-1) death–inducing signaling complex. Cell. 1996;14:817–827. doi: 10.1016/s0092-8674(00)81266-0. [DOI] [PubMed] [Google Scholar]
- Natoli G, Costanzo A, Guido F, Moretti F, Bernardo A, Burgio VL, Agresti C, Levreno M. Nuclear factor κB-independent cytoprotective pathways originating at tumor necrosis factor receptor-associated factor 2. J Biol Chem. 1998;273:31262–31272. doi: 10.1074/jbc.273.47.31262. [DOI] [PubMed] [Google Scholar]
- Naumann M, Scheidereit C. Activation of NF-kappa B in vivo is regulated by multiple phosphorylations. EMBO (Eur Mol Biol Organ) J. 1994;13:4597–4607. doi: 10.1002/j.1460-2075.1994.tb06781.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng PW, Janicke RU, Porter AG. Mutations which abolish phosphorylation of the TRAF-binding domain of TNF receptor 2 enhance receptor-mediated NF-kappa B activation. Biochem Biophys Res Commun. 1998;244:756–762. doi: 10.1006/bbrc.1998.8323. [DOI] [PubMed] [Google Scholar]
- Niederman TM, Garcia JV, Hastings WR, Luria S, Ratner L. Human immunodeficiency virus type 1 Nef protein inhibits NF-κB induction in human T cells. J Virol. 1992;66:6213–6219. doi: 10.1128/jvi.66.10.6213-6219.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niwa H, Yamamura K, Miyazaki J. Efficient selection for high-expression transfectants with a novel eukaryotic vector. Gene. 1991;108:193–199. doi: 10.1016/0378-1119(91)90434-d. [DOI] [PubMed] [Google Scholar]
- Opipari AW, Boguski MS, Dixit VM. The A20 cDNA induced by tumor necrosis factor α encodes a novel type of zinc finger protein. J Biol Chem. 1990;265:14705–14708. [PubMed] [Google Scholar]
- Perkins ND, Felzien LK, Betts JC, Leung K, Beach DH, Nabel GJ. Regulation of NF-kappaB by cyclin-dependent kinases associated with the p300 coactivator. Science. 1997;275:523–527. doi: 10.1126/science.275.5299.523. [DOI] [PubMed] [Google Scholar]
- Plaisance S, Vanden W, Berghe, Boone E, Fiers W, Haegeman G. Recombination signal sequence binding protein Jkappa is constitutively bound to the NF-kappaB site of the interleukin-6 promoter and acts as a negative regulatory factor. Mol Cell Biol. 1997;17:3733–3743. doi: 10.1128/mcb.17.7.3733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinhard C, Shamoon B, Shyamala V, Williams LT. Tumor necrosis factor alpha-induced activation of c-jun N-terminal kinase is mediated by TRAF2. EMBO (Eur Mol Biol Organ) J. 1997;16:1080–1092. doi: 10.1093/emboj/16.5.1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarma V, Lin Z, Clark L, Rust BM, Tewari M, Noelle RJ, Dixit VM. Activation of the B-cell surface receptor CD40 induces A20, a novel zinc finger protein that inhibits apoptosis. J Biol Chem. 1995;270:12343–12346. doi: 10.1074/jbc.270.21.12343. [DOI] [PubMed] [Google Scholar]
- Schmitz, M.L., M.A. dos Santos Silva, H. Altmann, M. Czisch, T.A. Holak, and P.A. Baeuerle. 1994. Structural and functional analysis of the NF-kappa B p65 C terminus. An acidic and modular transactivation domain with the potential to adopt an alpha-helical conformation. J. Biol. Chem. 269:25613– 25620. [PubMed]
- Schmitz, M.L., M.A. dos Santos Silva, and P.A. Baeuerle. 1995. Transactivation domain 2 (TA2) of p65 NF-kappa B. Similarity to TA1 and phorbol ester-stimulated activity and phosphorylation in intact cells. J. Biol. Chem. 270: 15576–15584. [DOI] [PubMed]
- Schreck R, Baeuerle PA. NF-kappa B as inducible transcriptional activator of the granulocyte-macrophage colony-stimulating factor gene. Mol Cell Biol. 1990;10:1281–1286. doi: 10.1128/mcb.10.3.1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semmes OJ, Jeang K-T. Mutational analysis of human T-cell leukemia virus type I Tax: regions necessary for function determined with 47 mutant proteins. J Virol. 1992;66:7183–7192. doi: 10.1128/jvi.66.12.7183-7192.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song HY, Rothe M, Goeddel DV. The tumor necrosis factor-inducible zinc finger protein A20 interacts with TRAF1/TRAF2 and inhibits NF-kappaB activation. Proc Natl Acad Sci USA. 1996;93:6721–6725. doi: 10.1073/pnas.93.13.6721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southern PJ, Berg J. Transformation of mammalian cells to antibiotic resistance with a bacterial gene under control of the SV40 early region promoter. J Mol Appl Genet. 1982;1:327–341. [PubMed] [Google Scholar]
- Uhlik M, Good L, Xiao G, Harhaj EW, Zandi E, Karin M, Sun S. NF-κB-inducing kinase and IκB kinase participate in human T-cell leukemia virus I Tax-mediated NF-κB activation. J Biol Chem. 1998;273:21132–21136. doi: 10.1074/jbc.273.33.21132. [DOI] [PubMed] [Google Scholar]
- Van Antwerp DJ, Martin SJ, Verma IM, Green DR. Inhibition of TNF-induced apoptosis by NF-κB. Trends Cell Biol. 1998;8:107–110. doi: 10.1016/s0962-8924(97)01215-4. [DOI] [PubMed] [Google Scholar]
- Vandenabeele P, Declercq W, Beyaert R, Fiers W. Two tumour necrosis factor receptors: structure and function. Trends Cell Biol. 1995;5:392–399. doi: 10.1016/s0962-8924(00)89088-1. [DOI] [PubMed] [Google Scholar]
- Vanden Berghe, W., S. Plaisance, E. Boone, K. De Bosscher, M.L. Schmitz, W. Fiers, and G. Haegeman. p38 and extracellular signal-regulated kinase mitogen-activated protein kinase pathways are required for nuclear factor-κB p65 transactivation mediated by tumor necrosis factor. J Biol Chem. 1998;273:3285–3290. doi: 10.1074/jbc.273.6.3285. [DOI] [PubMed] [Google Scholar]
- Vanhaesebroeck B, Van Bladel S, Lenaerts A, Suffys P, Beyaert R, Lucas R, Van Roy F, Fiers W. Two discrete types of tumor necrosis factor-resistant cells derived from the same cell line. Cancer Res. 1991;51:2469–2477. [PubMed] [Google Scholar]
- Van Snick J, Cayphas S, Vink A, Uyttenhove C, Coulie PG, Rubira MR, Simpson RJ. Purification and NH2-terminal amino acid sequence of a T-cell-derived lymphokine with growth factor activity for B-cell hybridomas. Proc Natl Acad Sci USA. 1986;83:9679–9683. doi: 10.1073/pnas.83.24.9679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincenz C, Dixit VM. 14-3-3 proteins associate with A20 in an isoform-specific manner and function both as chaperone and adapter molecules. J Biol Chem. 1996;271:20029–20034. doi: 10.1074/jbc.271.33.20029. [DOI] [PubMed] [Google Scholar]
- Yeh WC, Shahinian A, Speiser D, Kraunus J, Billia F, Wakeham A, de la Pompa JL, Ferrick D, Hum B, Iscove N, et al. Early lethality, functional NF-kappaB activation, and increased sensitivity to TNF-induced cell death in TRAF2-deficient mice. Immunity. 1997;7:715–725. doi: 10.1016/s1074-7613(00)80391-x. [DOI] [PubMed] [Google Scholar]
- Yin MJ, Christerson LB, Yamamotot Y, Kwak YT, Xu S, Mercurio F, Barbosa M, Cobb MH, Gaynor RB. HTLV-I Tax protein binds to MEKK1 to stimulate IκB kinase activity and NF-κB activation. Cell. 1998;93:875–884. doi: 10.1016/s0092-8674(00)81447-6. [DOI] [PubMed] [Google Scholar]
- Yurochko AD, Liu DY, Eierman D, Haskill S. Integrins as a primary signal transduction molecule regulating monocyte immediate-early gene induction. Proc Natl Acad Sci USA. 1992;89:9034–9038. doi: 10.1073/pnas.89.19.9034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zandi E, Rothwarf DM, Delhase M, Hayakawa M, Karin M. The IkappaB kinase complex (IKK) contains two kinase subunits, IKKalpha and IKKbeta, necessary for IkappaB phosphorylation and NF-kappaB activation. Cell. 1997;91:243–252. doi: 10.1016/s0092-8674(00)80406-7. [DOI] [PubMed] [Google Scholar]
- Zhang YH, Lin JX, Vilcek J. Interleukin-6 induction by tumor necrosis factor and interleukin-1 in human fibroblasts involves activation of a nuclear factor binding to a kappa B-like sequence. Mol Cell Biol. 1990;10:3818–3823. doi: 10.1128/mcb.10.7.3818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong H, SuYang H, Erdjument-Bromage H, Tempst P, Ghosh S. The transcriptional activity of NF-kappaB is regulated by the IkappaB-associated PKAc subunit through a cyclic AMP-independent mechanism. Cell. 1997;89:413–424. doi: 10.1016/s0092-8674(00)80222-6. [DOI] [PubMed] [Google Scholar]
- Zhong H, Voll RE, Ghosh S. Phosphorylation of NF-κB p65 by PKA stimulates transcriptional activity by promoting a novel bivalent interaction with the coactivator CBP/p300. Mol Cell. 1998;1:661–671. doi: 10.1016/s1097-2765(00)80066-0. [DOI] [PubMed] [Google Scholar]