Differential Susceptibility to Hepatic Inflammation and Proliferation in AXB Recombinant Inbred Mice Chronically Infected with Helicobacter hepaticus (original) (raw)
Abstract
Helicobacter hepaticus is a naturally occurring pathogen of mice and has been used to develop models of chronic hepatitis, liver cancer, and, more recently, inflammatory bowel disease, in selected mouse strains. A/JCr mice are particularly susceptible to **H. hepaticus**-induced hepatitis and subsequent development of liver neoplasms, whereas C57BL/6 mice are resistant. In this study, we inoculated nine AXB recombinant inbred (RI) mouse strains, derived from A/J and C57BL/6 mice, with H. hepaticus to determine the genetic basis of resistance to Helicobacter-induced liver disease. Mice were surveyed 14 months after inoculation by culture and PCR for H. hepaticus colonization of the liver and cecum, and microscopic morphometric evaluations of the liver were performed to quantify and correlate the severity of inflammation, apoptosis, and proliferation. Analysis of variance of hepatic inflammation demonstrated significant variation among the RI strains (P < 0.0001), and the strain distribution pattern suggested a multigenic basis of disease resistance. Quantitative trait analysis using linear regression suggested possible linkage to loci on mouse chromosome 19. Hepatocellular and biliary epithelial apoptosis and proliferation indices, including proliferation of oval cells, were markedly increased and correlated with severity of inflammation. Prevalence of hepatic neoplasia was also increased in susceptible RI strains. These findings demonstrate a genetic basis for susceptibility to **Helicobacter**-induced disease and provide insight into its pathogenesis.
Helicobacter hepaticus is a well-described, naturally occurring pathogen of mice that causes chronic hepatitis and cancer and has been used as a model for the study of both hepatic carcinogenesis and the biology of _Helicobacter_-induced gastrointestinal disease. Like Helicobacter pylori, the organism is a gram-negative, urease-positive, helical bacterium that is transmitted through fecal-oral contamination and results in chronic immune cell stimulation, inflammation, and neoplasia in susceptible animals. 1-3 In A/J mice, infection causes severe hepatitis and the eventual development of preneoplastic altered foci, hyperplasia, adenoma, and carcinoma in the liver. In contrast, C57BL/6 mice infected with H. hepaticus are highly resistant to the development of disease and have minimal or no liver lesions. 2 As with _H. pylori_-induced gastritis in people, the particular host factors in A/J and C57BL/6 mice that alter the pathogenesis and severity of disease expression after infection with H. hepaticus are poorly understood. Several studies have implicated an imbalance or dysregulation in immunity in response to the bacteria similar to that seen in H. pylori gastritis. 3-6 None, however, have defined the genetic basis for predisposition or protection against _Helicobacter_-induced disease.
H. hepaticus preferentially colonizes the mucosa of the cecum and colon of mice and, in the A/J strain, directly infects the liver and gallbladder, causing chronic active hepatitis. Liver lesions typically have a peribiliary and perivascular distribution, although foci of parenchymal inflammation and necrosis also occur, particularly in severely affected animals. Significant changes associated with inflammation and hepatocellular damage are hyperplasia of oval cells, Kupffer cells, and, less commonly, Ito cells. 2,3,7 Within 14–20 months after infection, the majority of A/J mice develop hepatic neoplasia. 2,3 The progression from chronic active hepatitis to hyperplasia and neoplasia is similar to the spectrum of gastric disease seen in humans infected with H. pylori and may involve similar pathogenic mechanisms. In humans, for example, direct cell damage from bacteria-elaborated toxins, 8,9 autoimmunity, 5 and production of reactive oxygen species from infiltrative leukocytes 10 have been implicated in the pathogenesis of _H. pylori_-induced stomach lesions. Disruption of the balance between physiological apoptosis and cell replacement proliferation also appears to play a role in disease and development of gastric neoplasia. 6 H. hepaticus, likewise, excretes a soluble toxin that can damage cells in vitro, 9 and autoimmunity with production of auto-antibodies against heat shock proteins, as well as the generation of reactive oxygen species from within the liver, has been shown to be part of the pathogenesis of H. hepaticus disease. 4,11,12 Furthermore, the development of a nonprotective, Th1-like immune response, similar to that seen in humans with H. pylori, has recently been demonstrated in A/J mice experimentally infected with H. hepaticus. 13
The differential susceptibility of A/J and C57BL/6 inbred mice to _H. hepaticus_-induced disease provides a powerful tool for analyzing both the genetic means of resistance to Helicobacter and the pathophysiology of disease expression through the use of recombinant inbred (RI) animals derived from these two strains. AXB RI strains are derived from female A/J and male C57BL/6 matings that give rise to genetically variable F2 offspring. Randomly selected F2 breeding pairs then undergo 20 successive generations of brother/sister matings to generate a set of distinct but related RI strains. 14 These RI strains have been used to study host mechanisms of disease resistance and susceptibility to several other infectious agents, including mouse hepatitis virus, Mycobacterium spp., Plasmodium chabaudi, Toxoplasma gondii, and Legionella pneumophila. 15-20 These studies contributed significantly to the identification of the Ity/Bcg locus, which confers protection against Salmonella typhimurium and Mycobacterium spp. in mice, 16 and led to the recognition of NRAMP (natural resistance-associated macrophage protein) as the gene product of resistance of the Bcg locus. 21,22 In this study, we utilized nine strains of RI AXB mice to analyze the genetics of susceptibility and resistance to _Helicobacter_-induced hepatitis over a 14-month period and further characterize strain-related differences in the kinetics of hepatic proliferation and apoptosis as part of the pathogenesis of bacterial-induced liver neoplasia in mice.
Materials and Methods
Experimental Design
Ten mice (five males and five females) from each of nine AXB RI strains (Jackson Laboratories, Bar Harbor, ME) were inoculated at 10 weeks of age with H. hepaticus (ATCC 51449). The mice were inoculated by oral gavage with 10 8 organisms in 0.2 ml broth media three times at 2-day intervals. Feces were cultured for H. hepaticus 3 weeks after inoculation to verify infection. The recombinant inbred mice were euthanized 14 months after inoculation. Twenty male mice from the progenitor strains (A/J and C57BL6) served as controls, half of which were inoculated in the same manner as the test animals and half of which were sham inoculated with media alone. All were euthanized 6 months after infection. Animals were housed in Association for Assessment and Accreditation of Laboratory Animal Care-approved facilities at the Massachusetts Institute of Technology.
Helicobacter hepaticus Isolation
Bacterial isolation was accomplished using blood agar plates at 37°C under microaerobic conditions in vented jars containing N2, H2, and CO2 (80:10:10). Freshly collected fecal samples were placed in 0.5 ml phosphate-buffered saline (PBS), homogenized, and left at room temperature (RT) for 1 hour to allow particulates to settle. The supernatant was filtered through a 0.45-μm syringe filter (Arodisc; Gelman Sciences, Ann Arbor, MI), then cultured on blood agar plates. In addition, cecum samples were taken at the time of necropsy and frozen at −70°C until they were processed for culture and/or polymerase chain reaction (PCR). The tissue was placed in Brucella broth with 5% fetal calf serum and homogenized. The homogenate was filtered through a 0.45-μm filter before culture on blood agar plates. Characteristic colonies were gram stained, and bacteria were examined for morphology. In addition, the bacteria were assessed for catalase, urease, and oxidase activity, as well as for resistance to cephalothin and nalidixic acid. 1
DNA Extraction
DNA was extracted from approximately 50 mg of liver tissue from all animals and from 50 mg of cecal tissue from culture-negative animals, when frozen tissue was available, with a High Pure PCR Template Preparation kit (Boehringer Mannheim, Indianapolis, IN). In addition, when frozen cecal tissue was not available, DNA was extracted from Carnoy’s fixed, paraffin-embedded tissue with an Oncor EX-WAX DNA Extraction Kit. Two 25-μm-thick sections were cut from each tissue block and placed in a 1.5-ml microcentrofuge tube; the DNA was extracted according to the manufacturer’s instructions (Oncor, Gaithersburg, MD).
Polymerase Chain Reaction (PCR) Analysis
PCR was performed on DNA extracted from cecal tissues that were culture-negative and on liver samples from all animals for which tissue was available. Primer sequences specific for H. hepaticus (GCA TTT GAA ACT GTT ACT CTG and CTG TTT TCA AGC TCC CC) were used to amplify a 417-bp DNA fragment of the 16s rRNA. A 50-μl reaction containing 5 μl of the sample DNA was used for PCR at 35 cycles of 94°C for 1 minute, 60°C for 2 minutes, and 72°C for 2 minutes, followed by 6 minutes of extension at 72°C.
Pathology
A complete necropsy was performed on each mouse. Standard samples of the liver were collected as described elsewhere. 23 The samples consisted of a longitudinal section cut from each lobe (left lateral and middle, right middle and quadrate) from the apical or free margin to the hilus. In addition to the liver, samples of the small intestine, cecum, and colon were taken for microscopic evaluation. Samples were immersion fixed in 10% neutral buffered formalin for 24 hours, embedded in paraffin, cut into 5-μm sections, and stained with hematoxylin-eosin or by Warthin-Starry methods.
Morphometric Analysis of Inflammation
Eighty of the 90 AXB RI mice and 20 of the 20 parental controls (A/J and C57BL, infected and uninfected) that entered the study were examined. The 10 mice that were not evaluated died before reaching the 14-month postinoculation time point and were excluded from the study.
The severity and extent of hepatic inflammation in AXB recombinant inbreds and parental controls were evaluated morphometrically as follows: the surface area of liver affected with inflammation was quantified using a 10 × 10 ocular grid. At ×200 magnification, each box in the 10 × 10 grid covered an area of 50 μm2. The inflammatory foci within the three largest sections of standard liver lobe samples were identified, and the number of 50 μm 2 boxes in the 10 × 10 ocular grid filled by inflammatory cells in each focus of inflammation was counted to determine the surface area of inflammation. For inflammatory foci that contained scattered, disparate infiltrates of leukocytes, a total of six leukocytes was counted as one box. If a 50 μm 2 box was not completely filled by inflammatory cells, it was not counted unless it contained at least six leukocytes. Both portal and nonportal areas were assessed for the number of 50 μm 2 boxes in the 10 × 10 grid filled with inflammatory cells (ie, surface area of inflammation) in the three standard liver lobes for each mouse. Scores were standardized for size of liver lobe sections for each mouse by counting the total number of portal areas within the liver lobes as an estimate of liver section surface area, then dividing the inflammatory scores (total number of 50 μm 2 boxes occupied by leukocytes) by the area estimate (total number of portal areas). All slides were read without knowledge of the strain. Thus the data depicted in Figure 3 ▶ are the surface area of liver affected by inflammation per portal area in square microns.
Figure 3.

Geometric means and standard errors for the morphometric scores of hepatic inflammation in the nine AXB recombinant inbred mouse strains. The surface area affected by inflammation was determined for each mouse by examination of three liver sections prepared from samples collected in a standardized manner. The number of 50 μm 2 boxes in an ocular grid filled with inflammatory cells was counted, and the area of those counted boxes was calculated. The number of portal areas contained within the three liver sections was used as an estimate of total surface area of the three liver sections. The area calculated from the boxes filled with inflammatory cells (surface area of inflammation) was divided by the number of portal areas counted (total surface area estimate) to give the area affected by inflammation per portal area in square microns.
Because the data were not normally distributed (more specifically, they were positively skewed), a log transformation was performed before statistical analyses. The mean inflammation score was the phenotype used for linkage analysis.
Transferase-Mediated dUTP-Biotin Nick End-Labeling (TUNEL) Staining
The terminal deoxynucleotidyl transferase-mediated dUTP-biotin nick end-labeling method (ApopTag; Oncor) was used to stain apoptotic cells according to the manufacturer’s directions; staining was visualized with diaminobenzidine (DAB) chromogen on liver sections of selected subsets of mice (five animals each from strains 1, 5, 8, and 13). Positive staining was correlated with morphological changes of nuclear condensation and cytoplasmic fragmentation. Positively stained hepatocytes, bile duct epithelia, oval cells, or leukocytes were counted, and the surface area of liver sections was determined using Optimas 6 image software (Media Cybernetics, Silver Spring, MD) on scanned images. The number of positively stained cells was expressed per unit area of liver section to give the apoptotic index. The data were then log transformed before analysis.
Immunohistochemistry
Proliferating cell nuclear antigen (PCNA) staining was performed on liver sections fixed in Carnoy’s fixative. Tissues from the same subset of mice used to determine the apoptotic index were used for PCNA staining. Briefly, sections were rehydrated, rinsed in PBS, heated to 94°C in 0.1 mol/L citrate buffer (pH 6.0) for 20 minutes, cooled to room temperature, blocked with Peroxobloc (Zymed Laboratories, San Francisco, CA) for 1 minute, rinsed in PBS, and then treated with CAS block (Zymed Laboratories) for 30 minutes. Mouse anti-PCNA diluted 1:200 in PBS with 10% goat serum was added and incubated at room temperature for 1 hour. Sections were rinsed in PBS and then treated with biotinylated anti-mouse IgG (1:400 dilution) (Zymed Laboratories) for 30 minutes, rinsed again in PBS, and then incubated with streptavidin peroxidase (1:500 dilution) (Zymed Laboratories). Sections were rinsed in PBS, followed by staining with DAB chromogen (Kirkegaard and Perry Laboratories, Gaithersburg, MD) or aminoethyl carbazole (AEC) (Zymed Laboratories). The tissues were dehydrated in alcohol and xylene and coverslipped. The area of the liver lobes was determined as described for the apoptosis index, and the proliferation index was similarly expressed as the number of positively stained cells per unit area. The data were log transformed before statistical analysis.
Oval cell staining was done on formalin-fixed tissue sections. The sections were prepared as described for PCNA staining, then exposed to rat anti-mouse A6 monoclonal antibody diluted 1:60 in PBS with 10% goat serum for 1 hour at RT and rinsed with PBS (antibody provided by Dr. Natlya Engelhardt). 24 Secondary antibody (biotinylated goat anti-rat IgG) at a 1:50 dilution was applied for 30 minutes. Sections were rinsed in PBS and then incubated with streptavidin peroxidase (1:500 dilution). Sections were again rinsed in PBS, followed by staining with DAB chromogen. The oval cell index was calculated as described for the apoptotic and proliferation indices, and again, the data were log transformed before analysis.
Lymphocytes were typed by staining with anti-CD3, which identifies T cells, and anti-CD79α, which is present on B cells. Tissue sections were rehydrated, then for the CD3 antibody they were blocked with Peroxoblock for 20 minutes and rinsed with PBS, incubated in 200 U pronase/50 ml PBS for 10 minutes, and rinsed extensively with water, then PBS. For CD79α detection, tissue sections were boiled in 0.1 mol/L citrate buffer for 5 minutes, cooled to RT, boiled for another 5 minutes, incubated with Peroxoblock (Zymed Laboratories) for 20 minutes, then rinsed with PBS. Primary antibody (anti-CD3 or anti-CD79α) was applied at a 1:100 dilution. Sections were washed with PBS, and secondary antibody (biotinylated goat anti-rabbit IgG or rabbit anti-mouse IgG) was applied at a 1:100 dilution for 30 minutes. Sections were again rinsed thoroughly with PBS, and streptavidin peroxidase was applied for 30 minutes. Sections were rinsed in PBS, followed by staining with DAB chromogen.
Statistical Analysis
Statistical analyses were done with Data Desk and Stata statistical software. An analysis of variance was performed for each trait to determine the effect of strain. When the strain effect proved to be significant, post hoc pairwise comparisons were performed to identify which strains differed significantly in that particular trait. In each instance of multiple post hoc pairwise comparisons, a Bonferroni correction was used to maintain an experiment-wise error rate of 0.05. Two-sample _t_-tests were performed when animals were compared that fell into only two groups, positive and negative for H. hepaticus, for example. All tests were considered significant at an α level of 0.05.
Linkage Analysis
Interval mapping using linear regression was performed with Map Manager QT2698 and the AXB/BXA RI database. 25,26
Results
Colonization with H. hepaticus
Three weeks after H. hepaticus inoculation, H. hepaticus was isolated from 100% of the fecal samples collected. The culture and PCR results for the cecal tissue collected 14 months after inoculation, along with the PCR results for H. hepaticus colonization of livers, are presented in Table 1 ▶ . The results indicate a marked reduction in cecal and liver colonization in two of the AXB RI strains. The proportion of ceca and livers that tested positive for H. hepaticus within a single strain ranged from 10% to 100%. The presence of H. hepaticus in the livers was also verified by visualizing the bacteria in Warthin-Starry silver-stained liver sections. Bacteria were seen in the extracellular spaces between hepatocytes (Figure 1H) ▶ .
Table 1.
Colonization Status of the Cecum and Liver 14 Months after Inoculation Relative to Severity of Hepatic Inflammation in AXB Recombinant Inbred Strains of Mice
| Strain no. | Cecal H.h. status (+/total) | Liver H.h. status (+/total) | Mean inflammation per portal area |
|---|---|---|---|
| 8 | 7 /9 | 7 /10 | 5092 |
| 1 | 9 /10 | 5 /8 | 4973 |
| 12 | 7 /8 | 3 /7 | 3116 |
| 10 | 8 /9 | 8 /9 | 2831 |
| 2 | 3 /10 | 7 /10 | 2599 |
| 6 | 4 /5 | 4 /4 | 2344 |
| 5 | 2 /10 | 1 /10 | 1396 |
| 13 | 9 /9 | 6 /8 | 1111 |
| 4 | 2 /7 | 1 /7 | 676 |
Figure 1.
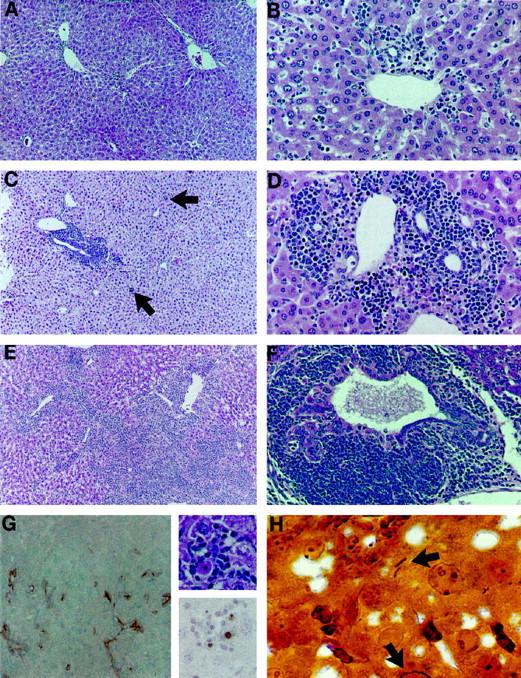
Representative photomicrographs of AXB RI strains demonstrating differential severity, distribution, and morphology of chronic inflammation 14 months after inoculation with H. hepaticus. A and B: Characteristic lesions of mild hepatitis with involvement of portal areas in relatively resistant RI strains 4 and 5, respectively. H&E, ×57 and ×114. C and D: Progressive chronic inflammation centered around veins and bile ductules in RI strains 2 and 10, respectively. Inflammation is chiefly lymphocytic, including numerous plasma cells but also contains scattered neutrophils and eosinophils and involves areas of parenchyma (arrows). H&E, ×57 and ×114. E and F: Severe lesions from RI strains 8 and 1, respectively. Inflammation has spread through the liver and is often associated with biliary epithelial hyperplasia and dysplasia. H&E, ×57 and ×95, respectively. G, Left: A RI strain 1 animal with severe hepatitis and high PCNA proliferation index also had pronounced oval cell proliferation, shown by immunostaining for A6 antibody. AEC, hematoxylin counterstain, ×95. G, Right: Hepatic parenchymal inflammation centered around apoptotic cells from AXB RI strain 8. H&E, ×370 (top), and TUNEL immunohistochemistry with hematoxylin counterstain, ×370 (bottom). H: Warthin-Starry stained liver section from RI strain 8, showing intercellular H. hepaticus organisms (arrows).
Two-sample _t_-tests were performed to compare the severity of inflammation in animals that tested positive for H. hepaticus colonization 14 months after inoculation with that in animals that tested negative. A single _t_-test was performed for each of the following contrasts: positive versus negative in the cecum, positive versus negative in the liver, and positive in both the liver and cecum versus not positive in both. The _t_-tests demonstrated that the inflammation scores were significantly greater in the group that was positive for H. hepaticus colonization than in the group that was negative, whether the bacteria were present in the cecum, the liver, or both (P = 0.036, P = 0.004, and P = 0.002, respectively), establishing a direct correlation between persistent H. hepaticus colonization and hepatitis.
AXB strains four and five had among the lowest mean inflammation scores and the lowest rates of infection in both the cecum and the liver. However, the ceca in strain 13, also a strain with minimal inflammation, all cultured positive for H. hepaticus.
Pathology
AXB mouse strains developed variable degrees of hepatitis characteristic of chronic H. hepaticus infection (Figure 1, A–F) ▶ , and, in some strains, preneoplastic altered foci, hyperplasia, and neoplasia were also present. Hepatitis consisted of mild to severe inflammatory cell infiltrates, chiefly in portal areas around bile ducts and ductules and within the walls of intralobular and portal veins. In severely affected animals, portal lesions had extended through the limiting plate. Parenchymal lesions comprised random, variably-sized aggregates of leukocytes, often centered around necrotic or apoptotic hepatocytes. Inflammatory cells were mostly lymphocytes and included some plasma cells, with lesser numbers of macrophages, neutrophils, and occasional eosinophils. Fibrosis with deposition of collagen was evident in some inflamed portal areas; however, oval cell hyperplasia was more commonly associated with inflammation and hepatic damage. In some cases, duplication of bile ducts and dysplasia of ductular epithelium were also present. Eosinophilic or clear cell altered hepatic foci, nodular hepatocellular hyperplasia, and neoplasia were present in strains particularly susceptible to _H. hepaticus_-induced hepatitis (Table 2) ▶ . Neoplasms included hepatocellular adenomas (one in strain 2 and one in strain 1), a hepatocellular carcinoma (strain 6), and a hemangiosarcoma (strain 1) (Figure 2, A–D) ▶ . Furthermore, in two mice in strain 12, lymphosarcoma was present in the liver as well as in mesenteric lymph nodes and large intestine. The neoplasms were distributed equally among males and females.
Table 2.
Number of Mice with Preneoplastic and Neoplastic Hepatic Lesions Relative to Severity of Hepatic Inflammation
| Strain no. | Nodular hyperplasia | Altered foci | Neoplasia | Total (%) | Mean inflammation per portal area |
|---|---|---|---|---|---|
| 8 | 3 | 1 | 0 | 4 (40) | 5092 |
| 1 | 0 | 2 | 2 | 4 (40) | 4973 |
| 12 | 2 | 0 | 2 | 4 (50) | 3116 |
| 10 | 0 | 0 | 0 | 0 (0) | 2831 |
| 2 | 2 | 0 | 1 | 3 (30) | 2599 |
| 6 | 0 | 0 | 1 | 1 (20) | 2344 |
| 5 | 0 | 0 | 0 | 0 (0) | 1396 |
| 13 | 0 | 0 | 0 | 0 (0) | 1111 |
| 4 | 0 | 0 | 0 | 0 (0) | 676 |
Figure 2.
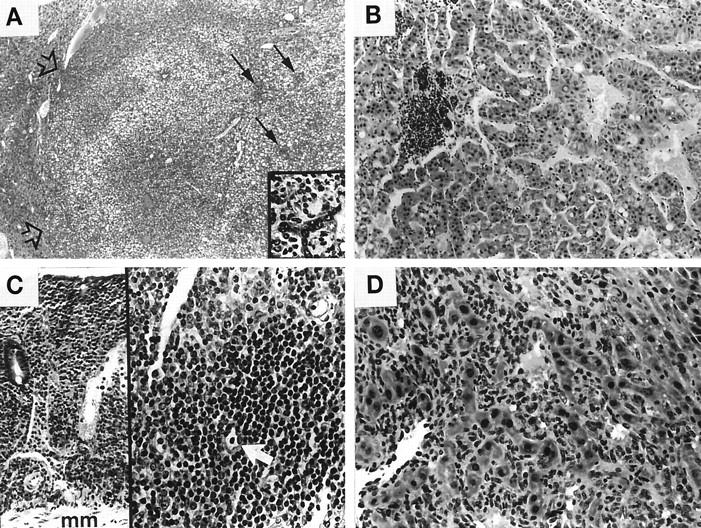
A: Photomicrograph of AXB RI strain 1 male with hepatocellular adenoma that has compressed adjacent liver parenchyma (open arrows); variable degrees of cytoplasmic vacuolization and residual multifocal inflammation (solid arrows) are present within the neoplasm. H&E, ×31. The inset shows an area of lymphocytic inflammation surrounding poorly organized biliary epithelium within neoplasm (lower arrow). H&E, ×165. B: Hepatocellular carcinoma with irregular cords and clusters of large, pleomorphic hepatocytes and foci of lymphocytic inflammation from AXB RI strain 6 male. H&E, ×100. C: AXB RI strain 12 female with B-cell lymphosarcoma that has diffusely infiltrated an area of large intestine from the muscularis mucosa (mm) to the superficial epithelium (left). H&E, ×78. The same animal had lymphosarcoma within the liver (right). A remaining bile ductule is present with the neoplasm (white arrow). H&E, ×160. D: AXB RI strain 1 female with severe hepatitis and development of poorly differentiated hemangiosarcoma lining sinusoids and vascular channels. H&E, ×160.
Typhlocolitis has been reported in immunodeficient and A/J mice, both naturally and experimentally infected with H. hepaticus. 27-29 Rectal prolapse is often the first observed clinical sign of H. hepaticus infection in a variety of mouse strains. 30 In the current study, three animals in one strain, strain 2, had a prolapsed rectum and were examined for entero- and typhlocolitis. Although severe inflammation was apparent in the prolapsed tissue, the remainder of the gastrointestinal tract contained only mild or no inflammation.
Morphometric Analysis
The severity of inflammation in the positive and negative parental control animals confirmed previous findings suggesting that C57BL/6 mice are resistant and A/J mice are susceptible to the effects of H. hepaticus. C57BL/6 mice had very low levels of inflammation in both _H. hepaticus_-infected and uninfected groups (P = 0.88), whereas the A/J mice had marked inflammation in the _H. hepaticus_-infected group but minimal liver lesions in the uninfected negative control group (P = 0.04) (data not shown).
The graph in Figure 3 ▶ depicts the mean surface area of liver affected by inflammation for the nine RI strains. An analysis of variance of the inflammation scores revealed a main effect for strain, with a P value of <0.0001. Post hoc pairwise comparisons of the strains, using a Bonferroni correction that held the experiment-wise error rate to 0.05, revealed that there were significant differences between individual strains. The inflammation in strains 1 and 8 was significantly greater than that seen in strains 4, 5, and 13. In addition, the inflammation observed in strains 2, 10, and 12 was more severe, although not significantly so, than that seen in strains 4, 5, and 13, and less severe than the inflammation in strains 1 and 8. This continuum of inflammation scores suggests that multiple genes are involved in resistance to H. hepaticus infection.
Two-sample _t_-tests indicated that there was no effect of gender, when the data were collapsed across strain (P = 0.69) or when each strain was considered individually (P ranged from 0.11 to 0.88).
Immunohistochemistry
Morphometric immunohistochemical analyses for apoptosis, hepatic proliferation, and oval cell proliferation demonstrated statistically significant differences among AXB strains, and all parameters correlated with severity of inflammation. Immunohistochemical TUNEL staining coupled with morphological evaluation of liver sections demonstrated apoptosis of hepatocytes, bile duct epithelium, and leukocytes, including both infiltrative inflammatory cells and Kupffer cells. Apoptotic hepatocytes were often encircled by inflammatory cells (Figure 1G) ▶ , as were bile ducts and ductules with apoptotic epithelium. The apoptotic indices for hepatocytes alone and for all cell types counted (hepatocytes, biliary epithelium, and leukocytes) were significantly different between strains (P = 0.0012 and P = 0.0006, respectively). High levels of hepatic inflammation, therefore, were associated with increased apoptosis.
PCNA staining was primarily localized to hepatocytes, leukocytes, and, less commonly, biliary epithelium. The proliferation index had a pattern among the strains similar to that of the apoptotic index. Again, the strains with the greatest amount of inflammation had the greatest number of proliferating cells within the liver. The differences in the proliferation index for hepatocytes alone among strains was not significant, although it approached significance with a P value of 0.0919, and the difference in the proliferation index for all cell types counted was significant (P = 0.0493).
Immunohistochemical staining of AXB livers with A6 antibody demonstrated oval cells near portal areas and, occasionally, deeper within midzonal regions of hepatic lobules (Figure 1G) ▶ . Increases in oval cell numbers were significantly different between AXB strains (P = 0.006) and correlated positively with inflammatory scores in AXB strains and within individual animals. The graphs in Figures 4, 5, and 6 ▶ ▶ ▶ depict the proliferation, apoptotic, and oval cell indices for each of the cell types counted in the four strains evaluated.
Figure 4.

The apoptotic index for the four AXB recombinant inbred strains assessed for apoptosis. The TUNEL procedure was performed on liver sections from four RI strains with the most extreme inflammation phenotypes. The positively stained cells in liver sections were counted and categorized, the total surface area of the liver sections was determined with image analysis software, and the number of positive cells was divided by the total surface area. The data were then log transformed to obtain the apoptotic index. The graphs represent the mean apoptotic indices for hepatocytes (A), bile duct epithelial cells (B), Kupffer cells (C), lymphocytes (D), and all cell types combined (E).
Figure 5.

The proliferation index for the four AXB recombinant inbred strains assessed for proliferation. Liver sections from four RI strains with the most extreme inflammation phenotypes were immunostained with PCNA antibody. The positively stained cells in liver sections were counted and categorized, the total surface area of the liver sections was determined with image analysis software, and the number of positive cells was divided by the total surface area. The data were then log transformed to obtain the proliferation index. The graphs represent the mean proliferation indices for hepatocytes (A), bile duct epithelial cells (B), Kupffer cells (C), lymphocytes (D), and all cell types combined (E).
Figure 6.

The oval cell index for the four AXB recombinant inbred strains assessed for oval cell hyperplasia. Liver sections from four RI strains with the most extreme inflammation phenotypes were immunostained with A6 antibody, which is specific for oval cells. The positively stained cells in liver sections were counted, the surface area of the liver sections was determined with image analysis software, and the number of positive cells was divided by the total surface area. The data were then log transformed to obtain the oval cell index. The graphs represent the mean oval cell indices for the four RI strains.
Immunostaining of the two lymphosarcomas involving the liver was positive for CD79α and negative for CD3.
Linkage Analysis
Quantitative trait loci linkage analysis did not identify any significant correlations with known marker loci, suggesting that there is not a single major gene responsible for conferring resistance to H. hepaticus. However, based on the guidelines proposed by Lander and Kruglyak 31 for reporting suggestive loci, the analysis did reveal possible linkage to chromosome 19 at marker loci D19Mit34 and D19Mit36.
Discussion
Host, pathogen, and environmental factors collectively cause _Helicobacter_-associated gastrointestinal disease. In H. pylori gastritis, pathogen strain differences, superinfection with complementary Helicobacter strains, and bacterial inoculation doses can determine the outcome of infection. The role of environmental factors, such as dietary salt levels, stress, and hygiene, have likewise been shown to be important in the development of disease. 32-35 The significance of host factors, although directly implicated by several studies of _H. pylori_-infected people, are poorly understood. 36-38 In particular, the genetic basis for resistance to prolonged colonization and development of symptomatic gastritis and neoplasia is unknown. In this study, we utilized recombinant inbred mice derived from susceptible (A/J) and resistant (C57BL/6) mice to assess the contribution of host factors in the pathogenesis of liver disease caused by H. hepaticus, a bacterium with many similarities to H. pylori.
Morphometric hepatic inflammatory scores from experimentally infected AXB RI strains demonstrated not only a statistically significant difference in susceptibility to _H. hepaticus_-induced liver disease, but also indicated that susceptibility was likely to be a complex trait with a polygenic basis. Other infectious disease studies using RI mice have similarly pointed to the additive influence of multiple genes in determining disease susceptibility. In DBA/2 congenic animals that were developed following preliminary BXD RI studies, for instance, genes encoding alloantigens of the NKR-P1 receptor and the fifth component of complement were linked to resistance to mousepox virus. 39,40 A notable exception to polygenic influence was the discovery of the Bcg locus on mouse chromosome 1, where resistance was found to be encoded by a single gene, Nramp. 21,22 In our study, the variability in liver lesions seen after infection with H. hepaticus infection was not likely to be due to the effects of a single major gene. Linkage analysis suggested that loci on chromosome 19 (D19Mit34 and D19Mit36) may contribute, in part, to _Helicobacter_-induced disease in AXB RI mice. Interestingly, a number of immunologically important genes are located on chromosome 19, including CD5, which has been shown to be a phenotypic marker of autoreactive B lymphocytes in mice. 41
Unlike H. pylori, which selectively colonizes gastric mucosa in humans, H. hepaticus naturally colonizes the cecum and colon in susceptible strains of mice and causes chronic hepatitis and neoplasia. The mechanisms by which H. hepaticus induces liver disease are unclear. In A/J mice with severe hepatic disease, the liver is often directly colonized by H. hepaticus. However, it is possible that bacteria within the cecum or colon under favorable circumstances may excrete a soluble toxin that reaches the liver via portal circulation to contribute to hepatic damage. Autoimmunity, probably triggered after the initial onset of liver inflammation, has also been suggested as being involved in the pathogenesis of hepatitis. 11 In addition, H. hepaticus has recently been shown to possess genes that code for a cytolethal distending toxin (CDT) (manuscript in preparation). As a virulence factor, the H. hepaticus CDT is similar to that found in other bacteria, including Escherichia coli and Campylobacter jejuni, in that it induces cell cycle arrest in the G2/M phase. 42,43
In our study, colonization results of cecum and liver coupled with hepatic inflammatory scores in the nine AXB RI strains of mice suggested genes that confer resistance to H. hepaticus in mice likely manifest their effects on several different levels. For example, in two out of three of the most resistant AXB RI strains, H. hepaticus was present in the large intestine of only 4/17 animals (23%) 14 months after inoculation, whereas in the two most susceptible AXB RI strains, it was found in the intestines of 16/19 animals (84%). From these data, it appears that intestinal mucosal immunity may have been an important mechanism in limiting colonization by H. hepaticus and, possibly, limiting spread to the hepatobiliary system. However, an additional mechanism of resistance, such as systemic immunity or altered sensitivity to bacterial toxins, may also have been operative, as evidenced by AXB RI strain 13, which had high levels of H. hepaticus colonization in both the large intestine (100%) and the liver (75%) but very low hepatic inflammatory scores and no neoplasia. Liver colonization without hepatitis has previously been reported in inbred mice with H. hepaticus. 44 Perhaps some variation in lymphocyte antigen receptor repertoire, specific immune cell subsets within the liver, or quantitative expression in the Th1/Th2 cytokine axis influenced recognition and responsiveness to H. hepaticus, rendering the bacteria more commensal than pathogenic in these animals. Alternatively, these mice may have been relatively insensitive to H. hepaticus toxins or other unrecognized virulence factors.
In _H. pylori_-induced gastritis, both immune-mediated damage and direct cytotoxicity result in mucosal epithelial necrosis and/or apoptosis and contribute to disease. 45,46 In _H. hepaticus_-induced hepatitis, production of cytotoxins and generation of reactive oxygen species during inflammation have been documented. 9,12 Apoptosis has also been implicated in the pathogenesis of Helicobacter liver disease in B6C3F1 inbred mice. 44 In our study, we found a statistically significant correlation between inflammation and apoptosis and markedly higher apoptosis levels in susceptible AXB RI strains, suggesting that programmed cell death induced directly or indirectly by H. hepaticus contributed to hepatocellular loss. In a recent publication, 12 cytochrome P450 induction and production of reactive oxygen species were shown to occur in vivo after infection with H. hepaticus; both could be initiators of hepatocyte apoptosis. The role of toxins is less clear, although the newly discovered CDT in H. hepaticus may act to initiate apoptosis by preventing cell division. Based on the morphology of apoptosis staining in our study, it appears that leukocytes may also be involved in programmed cell death. Previous immunophenotypic studies have shown that inflammatory cell infiltrates in _H. hepaticus_-induced hepatitis in A/J mice contain T and B lymphocytes. 13 It is likely that natural killer (NK) cells or T cells expressing the NK1.1 receptor are also present. 47 If lymphocytes, especially NK cells, do contribute to hepatocyte apoptosis, differences in antigen receptor expression, such as NK1.1 or NKR-P1, may account for the low level of disease in AXB RI strain 13 as well as the ability of SCID mice to develop hepatitis. 27,28
Increased cell proliferation as determined by immunostaining for PCNA or BrdU has been demonstrated in both H. pylori gastritis and H. hepaticus hepatitis. 6,7,44 Similarly, we found statistically significant elevations in hepatocyte staining in AXB RI mice that corresponded to severity of inflammation. High levels of PCNA staining were also seen in biliary epithelium, Kupffer cells, and, particularly in animals with severe hepatitis, in lymphocytes that had infiltrated the liver. Notably, two mice from AXB RI strain 12 developed lymphosarcoma that involved the liver, mesenteric lymph nodes, and large intestine, and both neoplasms were of B-cell origin, as are _H. pylori_-associated MALT lymphomas in people. 48,49 An additional significant finding was the presence of oval cell hyperplasia in susceptible AXB RI strains. Typically, oval cell proliferation has been linked to chronic, ongoing hepatic damage, usually from carcinogens. 50 More recently, however, proinflammatory cytokines produced by leukocytes have been recognized as important mitogens for oval cells. 51 In _H. hepaticus_-induced hepatitis, both direct cell damage and inflammatory cell-derived cytokines probably contributed to oval cell proliferation. Interestingly, Helicobacter spp. has recently been linked to biliary disease in people. 52
H. hepaticus was originally discovered when a substantial increase in hepatocellular neoplasia was observed in male A/J mice. 1 In our study, the incidence of neoplasia was 8.5%, and, when combined with preneoplastic lesions, it increased to 20.7%. These figures are markedly higher than the background incidence of neoplasia in either of the parental (A/J and C57BL/6) strains. 2,53 As in previous reports, most of the neoplastic and preneoplastic lesions in AXB RI mice were of hepatocellular origin, although one mouse from AXB RI strain 1 with severe hepatitis developed a poorly differentiated hemangiosarcoma of the liver. Hemangiosarcoma has previously been recognized in _H. hepaticus_-infected mice 54 and could be related to the high occurrence of vascular inflammatory lesions. Most importantly, however, the incidence of neoplasia corresponded closely with AXB RI strain susceptibility to _H. hepaticus_-induced hepatitis. Unlike earlier studies, we did not observe a gender difference in either the severity of hepatitis or the incidence of liver neoplasia in AXB RI mice. Strain sample size may be responsible for the absence of a gender effect in our study. Alternatively, the gender effect may be linked to the Y chromosome in A/J mice and was not observed because the Y chromosome was contributed by male C57BL/6 mice in the original parental strain cross.
The link between infectious hepatitis and neoplasia has been well established in people with hepatitis C infection and in a number of animal models of viral hepatitis. However, the role of enteric bacteria, such as H. pylori and H. hepaticus, as risk factors for cancer has only recently been recognized. We assessed the effects of H. hepaticus infection in AXB RI strains and found significant differences among the strains, confirming the importance of host genetic factors in the development of hepatitis and neoplasia as well as the usefulness of RI strains in the study of Helicobacter pathogenesis. Thus, this study has provided the basis for more thorough investigation of the genetic factors involved in _Helicobacter_-induced disease.
Whereas H. pylori is the prototype of a bacterial etiology of cancer in people, H. hepaticus has proved to be an effective surrogate in mice and has led to a greater understanding of related pathogens and their role in human disease.
Footnotes
Address reprint requests to Dr. James G. Fox, Division of Comparative Medicine, Massachusetts Institute of Technology, 77 Massachusetts Avenue, Bldg. 16-820, Cambridge, MA 02139.
Supported by National Institutes of Health grants RR07036 and CA67529.
References
- 1.Fox JG, Dewhirst FE, Tully JG, Paster BJ, Yan L, Taylor NS, Collins MJ, Jr, Gorelick PL, Ward JM: Helicobacter hepaticus sp.nov., a microaerophilic bacterium isolated from livers and intestinal mucosal scrapings from mice. J Clin Microbiol 1994, 32:1238-1245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ward JM, Fox JG, Anver MR, Haines DC, George CV, Collins MJ, Jr, Gorelick PL, Nagashima K, Gonda MA, Gilden RV, Tully JG, Russell RJ, Benveniste RE, Paster BJ, Dewhirst FE, Donovan JC, Anderson LM, Rice JM: Chronic active hepatitis and associated liver tumors in mice caused by a persistent bacterial infection with a novel Helicobacter species. J Natl Cancer Inst 1994, 86:1222-1227 [DOI] [PubMed] [Google Scholar]
- 3.Ward JM, Anver MR, Haines DC, Benveniste RE: Chronic active hepatitis in mice caused by Helicobacter hepaticus. Am J Pathol 1994, 145:959-968 [PMC free article] [PubMed] [Google Scholar]
- 4.Canella KA, Diwan BA, Gorelick PL, Donovan PJ, Sipowicz MA, Kasprzak KS, Weghorst CM, Snyderwine EG, Davis CD, Keefer LK, Kyrtopoulos SA, Hecht SS, Wang M, Anderson LM, Rice JM: Liver tumorigenesis by Helicobacter hepaticus: considerations of mechanism. In Vivo 1996, 10:285-292 [PubMed] [Google Scholar]
- 5.Steininger H, Faller G, Dewald E, Brabletz T, Jung A, Kirchner T: Apoptosis in chronic gastritis and its correlation with antigastric autoantibodies. Virchows Arch 1998, 433:13-18 [DOI] [PubMed] [Google Scholar]
- 6.Jones NL, Shannon PT, Cutz E, Yeger H, Sherman PM: Increase in proliferation and apoptosis of gastric epithelial cells early in the natural history of Helicobacter pylori infection. Am J Pathol 1997, 151:1695-1703 [PMC free article] [PubMed] [Google Scholar]
- 7.Fox JG, Li X, Yan X, Cahill RJ, Hurley R, Lewis R, Murphy JC: Chronic proliferative hepatitis in A/JCr mice associated with persistent Helicobacter hepaticus infection: a model of _Helicobacter_-induced carcinogenesis. Infect Immun 1996, 64:1548-1558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Megraud F: Toxic factors of Helicobacter pylori. Eur J Gastroenterol Hepatol 1994, 6(Suppl):S5-S10 [PubMed] [Google Scholar]
- 9.Taylor NS, Fox JG, Yan L: In vitro hepatotoxic factor in Helicobacter hepaticus, H. pylori and other Helicobacter species. J Med Microbiol 1995, 42:48-52 [DOI] [PubMed] [Google Scholar]
- 10.Phull PS, Green CJ, Jacyna MR: A radical view of the stomach: the role of oxygen-derived free radicals and anti-oxidants in gastroduodenal disease. Eur J Gastroenterol Hepatol 1995, 7:265-274 [PubMed] [Google Scholar]
- 11.Ward JM, Benveniste RE, Fox CH, Battles JK, Gonda MA, Tully JG: Autoimmunity in chronic active Helicobacter hepatitis of mice: serum antibodies and expression of heat shock protein 70 in liver. Am J Pathol 1996, 148:509-517 [PMC free article] [PubMed] [Google Scholar]
- 12.Sipowicz MA, Chomarat P, Diwan BA, Anver MA, Awasthi YC, Ward JM, Rice JM, Kasprzak KS, Wild CP, Anderson LM: Increased oxidative DNA damage and hepatocyte overexpression of specific cytochrome P450 isoforms in hepatitis of mice infected with Helicobacter hepaticus. Am J Pathol 1997, 151:933-941 [PMC free article] [PubMed] [Google Scholar]
- 13.Whary MT, Morgan TJ, Dangler CA, Gaudes KJ, Taylor NS, Fox JG: Chronic active hepatitis induced by Helicobacter hepaticus in the A/JCr mouse is associated with a Th1 cell-mediated immune response. Infect Immun 1998, 66:3142-4148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nesbitt MN, Skamene E: Recombinant inbred mouse strains derived from A/J and C57BL/6J: a tool for the study of genetic mechanisms in host resistance to infection and malignancy. J Leuk Biol 1984, 36:357-364 [DOI] [PubMed] [Google Scholar]
- 15.Dindzans VJ, Skamane E, Levy GA: Susceptibility/resistance to mouse hepatitis virus strain 3 and macrophage procoagulant activity are genetically linked and controlled by two non-H-2-linked genes. J Immunol 1986, 137:2355-2360 [PubMed] [Google Scholar]
- 16.Skamene E, Gros P, Forget A, Kongshavn PAL, St. Charles C, Taylor BA: Genetic regulation of resistance to intracellular pathogens. Nature 1982, 297:506-510 [DOI] [PubMed] [Google Scholar]
- 17.Hoffmann EJ, Weidanz WP, Long CA: Susceptibility of CXB recombinant inbred mice to murine plasmodia. Infect Immun 1984, 43:981-985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stevenson MM, Skamene E: Murine malaria: resistance of AXB/BXA recombinant inbred mice to Plasmodium chabaudi. Infect Immun 1985, 47:452-456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McLeod R, Skamene E, Brown CR, Eisenhauer PB, Mack DG: Genetic regulation of early survival and cyst number after peroral Toxoplasma gondii infection of AXB/BXA recombinant inbred and B10 congenic mice. J Immunol 1989, 143:3031-3034 [PubMed] [Google Scholar]
- 20.Beckers MC, Yoshida S, Morgan K, Skamene E, Gros P: Natural resistance to infection with Legionella pneumophila: chromosomal localization of the Lgn1 susceptibility gene. Mamm Genome 1995, 8:540-545 [DOI] [PubMed] [Google Scholar]
- 21.Malo D, Vidal S, Hu J, Skamane E, Gros P: High resolution map in the vicinity of the host resistance locus Bcg. Genomics 1993, 16:655-663 [DOI] [PubMed] [Google Scholar]
- 22.Malo D, Vogan K, Vidal S, Hu J, Cellier M, Schurr E, Fuks A, Bumstead N, Morgan K, Gros P: Haplotype mapping and sequence analysis of the mouse Nramp gene predict susceptibility to infection with intracellular parasites. Genomics 1994, 23:51-61 [DOI] [PubMed] [Google Scholar]
- 23.Devor DE, Henneman JR, Kurata Y, Rehm S, Weghorst CM, Ward JM: Pathology procedures in laboratory animal carcinogenesis studies. Waalkes MP Ward JM eds. Carcinogenesis. 1994, :429-465 Raven Press New York [Google Scholar]
- 24.Bennoun M, Rissel M, Engelhardt N, Guillouzo A, Briand P, Weber-Benarous A: Oval cell proliferation in early stages of hepatocarcinogenesis in simian virus 40 large T transgenic mice. Am J Pathol 1993, 143:1326-1334 [PMC free article] [PubMed] [Google Scholar]
- 25.Manly KF, Olson JM: Overview of QTL mapping software and introduction to map manager QT. Mamm Genome 1999, 10:327-334 [DOI] [PubMed] [Google Scholar]
- 26.Manly KF: A Macintosh program for storage and analysis of experimental genetic mapping data. Mamm Genome 1993, 4:303-313 [DOI] [PubMed] [Google Scholar]
- 27.Li X, Fox JG, Whary MT, Yan L, Shames B, Zhao Z: SCID/NCr mice naturally infected with Helicobacter hepaticus develop progressive hepatitis, proliferative typhlitis, and colitis. Infect Immun 1998, 66:5477-5484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cahill RJ, Foltz CJ, Fox JG, Dangler CA, Powrie F, Schauer DB: Inflammatory bowel disease: an immunity-mediated condition triggered by bacterial infection with Helicobacter hepaticus. Infect Immun 1997, 65:3126-3131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Russell RJ, Haines DC, Anver MR, Battles JK, Gorelick PL, Blumenauer LL, Gonda MA, Ward JM: Use of antibiotics to prevent hepatitis and typhlitis in male SCID mice spontaneously infected with Helicobacter hepaticus. Lab Anim Sci 1995, 45:373-378 [PubMed] [Google Scholar]
- 30.Foltz CJ, Fox JG, Cahill R, Murphy JC, Yan L, Shames B, Schauer DB: Spontaneous inflammatory bowel disease in multiple mutant mouse lines: association with colonization by Helicobacter hepaticus. Helicobacter 1998, 3:69-78 [DOI] [PubMed] [Google Scholar]
- 31.Lander E, Kruglyak L: Genetic dissection of complex traits: guidelines for interpreting and reporting linkage results. Nature Genet 1995, 11:241-247 [DOI] [PubMed] [Google Scholar]
- 32.Ahn YO: Diet and stomach cancer in Korea. Int J Cancer 1997, 10(Suppl):7-9 [DOI] [PubMed] [Google Scholar]
- 33.Boeing H, Jedrychowski W, Wahrendorf J, Popiela T, Tobiasz-Adamczyk B, Kulig A: Dietary risk factors in intestinal and diffuse types of stomach cancer: a multicenter case-control study in Poland. Cancer Causes Control 1991, 2:227-233 [DOI] [PubMed] [Google Scholar]
- 34.Clemens J, Albert MJ, Rao M, Huda S, Qadri F, Van Loon FP, Pradhan B, Naficy A, Banik A: Sociodemographic, hygienic and nutritional correlates of Helicobacter pylori infection of young Bangladeshi children. Pediatr Infect Dis J 1996, 15:1113-1118 [DOI] [PubMed] [Google Scholar]
- 35.Malaty HM, Graham DY, Isaksson I, Engstrand L, Pedersen NL: Co-twin study of the effect of environment and dietary elements on acquisition of Helicobacter pylori infection. Am J Epidemiol 1998, 148:793-797 [DOI] [PubMed] [Google Scholar]
- 36.Shirin H, Moss SF: Helicobacter pylori induced apoptosis. Gut 1998, 43:592-594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ernst PB, Crowe SE, Reyes VE: How does Helicobacter pylori cause mucosal damage? The inflammatory response. Gastroenterology 1997, 113(Suppl 6):S35-S42 [DOI] [PubMed] [Google Scholar]
- 38.Go MF: What are the host factors that place an individual at risk for _Helicobacter pylori_-associated disease? Gastroenterology 1997, 113(Suppl 6):S15-S20 [DOI] [PubMed] [Google Scholar]
- 39.Brownstein DG, Gras L: Differential pathogenesis of lethal mousepox in congenic DBA/2 mice implicates natural killer cell receptor NKR-P1 in necrotizing hepatitis and the fifth component of complement in recruitment of circulating leukocytes to spleen. Am J Pathol 1997, 150:1407-1420 [PMC free article] [PubMed] [Google Scholar]
- 40.Brownstein DG, Pravin NB, Gras L, Jacoby RO: Chromosomal locations and gonadal dependence of genes that mediate resistance to ectromelia (mousepox) virus-induced mortality. J Virol 1991, 65:1946-1951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ye YL, Suen JL, Chen YY, Chiang BL: Phenotypic and functional analysis of activated B cells of autoimmune NZB × NZW F1 mice. Scand J Immunol 1998, 47:122-126 [DOI] [PubMed] [Google Scholar]
- 42.Pérès SY, Marchès O, Daigle F, Nougayrède JP, Hérault F, Tasca C, De Rycke J, Oswald E: A new cytolethal distending toxin (CDT) from Escherichia coli producing CNF2 blocks HeLa cell division in G2/M phase. Mol Microbiol 1997, 24:1095-1107 [DOI] [PubMed] [Google Scholar]
- 43.Whitehouse CA, Balbo PB, Pesci EC, Cottle DL, Mirabito PM, Pickett CL: Campylobacter jejuni cytolethal distending toxin causes a G2-phase cell cycle block. Infect Immun 1998, 66:1934-1940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nyska A, Maronpot RR, Eldridge SR, Haseman JK, Hailey JR: Alteration in cell kinetics in control B6C3F1 mice infected with Helicobacter hepaticus. Toxicol Pathol 1997, 25:591-596 [DOI] [PubMed] [Google Scholar]
- 45.Fan X, Crowe SE, Behar S, Gunasena H, Ye G, Haeberle H, Van Houten N, Gourley WK, Ernst PB, Reyes VE: The effect of class II major histocompatibility complex expression on adherence of Helicobacter pylori and induction of apoptosis in gastric epithelial cells: a mechanism for T helper cell type 1-mediated damage. J Exp Med 1998, 187:1659-1669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cover TL: The vacuolating cytotoxin of Helicobacter pylori. Mol Microbiol 1996, 20:241-246 [DOI] [PubMed] [Google Scholar]
- 47.Burton RC, Smart YC, Koo GC, Winn HJ: Studies on murine natural killer (NK) cells. V. Genetic analysis of NK cell markers. Cell Immunol 1991, 135:445-453 [DOI] [PubMed] [Google Scholar]
- 48.Spencer J, Wotherspoon AC: Gastric MALT lymphoma and Helicobacter pylori. Cancer Surv 1997, 30:213-231 [PubMed] [Google Scholar]
- 49.Yumoto N, Furukawa M, Kurosu K, Mikata A: A particular characteristic of IgH complementarity-determining region 3 suggests autoreactive B-cell origin of primary gastric B-cell lymphomas. Lab Invest 1998, 78:261-268 [PubMed] [Google Scholar]
- 50.Steinberg P, Steinbrecher R, Radaeva S, Schirmacher P, Dienes HP, Oesch F, Bannasch P: Oval cell lines OC/CDE 6 and OC/CDE 22 give rise to cholangio-cellular and undifferentiated carcinomas after transformation. Lab Invest 1994, 71:700-709 [PubMed] [Google Scholar]
- 51.Isfort RJ, Cody DB, Richards WG, Yoder BK, Wilkinson JE, Woychik RP: Characterization of growth factor responsiveness and alterations in growth factor homeostasis involved in the tumorigenic conversion of mouse oval cells. Growth Factors 1998, 15:81-94 [DOI] [PubMed] [Google Scholar]
- 52.Fox JG, Dewhirst FE, Shen Z, Feng Y, Taylor NS, Paster BK, Ericson RL, Lau CN, Correa P, Araya JC, Roa I: Hepatic Helicobacter species identified in bile and gallbladder tissue from Chileans with chronic cholecystitis. Gastroenterology 1998, 114:755-763 [DOI] [PubMed] [Google Scholar]
- 53.Becker FF: Morphological classification of mouse liver tumors based on biological characteristics. Cancer Res 1982, 42:3918-3923 [PubMed] [Google Scholar]
- 54.Hailey JR, Haeman JK, Bucher JR, Radovsky AE, Malarkey DE, Miller RT, Nyska A, Maronpot RR: Impact of Helicobacter hepaticus infection in B6C3F1 mice from twelve National Toxicology Program carcinogenesis studies. Toxicol Pathol 1998, 26:602-611 [DOI] [PubMed] [Google Scholar]