Denaturing Gradient Gel Electrophoresis Analysis of 16S Ribosomal DNA Amplicons To Monitor Changes in Fecal Bacterial Populations of Weaning Pigs after Introduction of Lactobacillus reuteri Strain MM53 (original) (raw)
Abstract
The diversity and stability of the fecal bacterial microbiota in weaning pigs was studied after introduction of an exogenous Lactobacillus reuteri strain, MM53, using a combination of cultivation and techniques based on genes encoding 16S rRNA (16S rDNA). Piglets (n = 9) were assigned to three treatment groups (control, daily dosed, and 4th-day dosed), and fresh fecal samples were collected daily. Dosed animals received 2.5 × 1010 CFU of antibiotic-resistant L. reuteri MM53 daily or every 4th day. Mean Lactobacillus counts for the three groups ranged from 1 × 109 to 4 × 109 CFU/g of feces. Enumeration of strain L. reuteri MM53 on MRS agar (Difco) plates containing streptomycin and rifampin showed that the introduced strain fluctuated between 8 × 103 and 5 × 106 CFU/g of feces in the two dosed groups. Denaturing gradient gel electrophoresis (DGGE) of PCR-amplified 16S rDNA fragments, with primers specific for variable regions 1 and 3 (V1 and V3), was used to profile complexity of fecal bacterial populations. Analysis of DGGE banding profiles indicated that each individual maintained a unique fecal bacterial population that was stable over time, suggesting a strong host influence. In addition, individual DGGE patterns could be separated into distinct time-dependent clusters. Primers designed specifically to restrict DGGE analysis to a select group of lactobacilli allowed examination of interspecies relationships and abundance. Based on relative band migration distance and sequence determination, L. reuteri was distinguishable within the V1 region 16S rDNA gene patterns. Daily fluctuations in specific bands within these profiles were observed, which revealed an antagonistic relationship between L. reuteri MM53 (band V1-3) and another indigenous Lactobacillus assemblage (band V1-6).
Gastrointestinal microbial ecology is experiencing a renaissance where classical culture-based microbial techniques are being supplemented, if not replaced by, molecular tools and techniques (20, 32). Theoretically, molecular methods based upon the 16S rDNA gene can now be used to identify all bacterial genus or species within gastrointestinal microbial communities whereas cultivation approaches are biased due to the inability of some bacteria to grow on selective media, excluding them from further analysis (1, 23). Currently, methods based on genes encoding 16S rRNA (16S rDNA) are being used to examine differences in the bacteria inhabiting the gastrointestinal tract of a variety of animal models, such as mice (15, 38), rabbits (39), chickens (19), pigs (17, 21, 22, 31), and humans (14, 33, 40). At present, molecular analysis of the porcine gastrointestinal microbiota has been limited to examination of samples that were cultivated prior to genotypic analysis (2, 6, 11, 22, 31, 34).
Classical methods used to study bacterial populations in the cecum, colon, and feces of pigs have found a wide range of characteristic genera, including Lactobacillus, Streptococcus, Peptococcus, Eubacterium, Clostridium, Bifidobacterium, and Bacteroides (reviewed by Stewart [29]). Of particular interest are the Lactobacillus populations within the gastrointestinal tract of the piglet, due to their purported benefits for gut function and health (29, 36). While, these Lactobacillus populations establish early in the piglet, succession occurs throughout the pig's lifetime, with lactobacilli remaining a predominant portion of the population (29, 31). Several reports indicate that lactobacilli exert antagonistic characteristics toward other bacteria and fungal species (6). Numerous species and strains of Lactobacillus, including L. reuteri, have been detected and isolated from pig intestine and feces using both conventional and molecular techniques (2, 17, 24). Several L. reuteri strains have been evaluated for characteristics necessary for use as probiotic or therapeutic supplements (7). L. reuteri strain MM53 was isolated originally from human breast milk and exhibits properties of an effective probiotic organism (24). L. reuteri MM53 possesses adhesive properties (25), secretes bacteriocin-like products (30), and exhibits preventative properties towards community-acquired or rotavirus-induced diarrhea (4).
The present study investigated the utility of PCR-denaturing gradient gel electrophoresis (DGGE) for monitoring changes in fecal bacterial populations after introduction of an exogenous Lactobacillus strain. L. reuteri MM53, selected for double antibiotic resistance, was introduced into the intestinal tract of piglets and detected using classical antibiotic plate counts. Molecular ecological analysis based on the DGGE technique, targeting the V3-16S rDNA region with bacterium-specific primers and the V1-16S rDNA region with _Lactobacillus_-specific primers, allowed us to establish profiles for predominant assemblages of fecal bacteria, as well as L. reuteri populations, without the need for cultivation based enumeration. This paper presents, for the first time, the application of DGGE to ecological analysis of the fecal bacteria of pigs. These techniques enable a more specific and detailed representation of the shifts in bacterial community structure in the intestinal tract and feces of pigs. The results obtained demonstrate the unique stability and repeatability of banding patterns in individual animals.
MATERIALS AND METHODS
Bacterial strain and culture conditions.
Antibiotic-resistant isolates of L. reuteri strain MM53 (BioGaia Biologics Inc., Raleigh, N.C.) were obtained by serial culturing on Lactobacillus MRS medium (Difco Inc., Detroit, Mich.) containing increasing concentrations of rifampin and streptomycin (Sigma Co., St. Louis, Mo.). Antibiotics were increased to final concentrations of 400 μg/ml for streptomycin and 40 μg/ml for rifampin. Background levels of resistant fecal bacteria were not detected at this antibiotic concentration. Prior to dosing, L. reuteri MM53 was incubated at 37°C in 100 ml of MRS broth containing streptomycin (400 μg/ml) and rifampin (40 μg/ml) for 24 h. Cells were enumerated by direct count and concentrated by centrifugation (3,440 × g, 10 min at 4°C). The cell pellet was resuspended in 2 ml of MRS broth, giving a total volume of 3 ml. Cell suspension aliquots containing 2.5 × 1010 CFU per 500 μl were mixed with individual fillings from Oreo cookies (n = 6). Pigs were dosed with one cookie within 30 min of inoculum preparation.
Test animals and sample collection.
Piglets were housed in individual pens and subjected to a 12 h light cycle at the University of Illinois Swine Research Facility. Pens were cleaned twice daily. Full-sibling piglets (n = 9) were weaned at 21 days and fed a nonmedicated weaning piglet diet (20% whey, 47% soy meal, 28% corn, and 4% trace minerals) for 7 days prior to the start and throughout the study. Piglets were allowed unrestricted access to feed and water. The experimental groups were (i) control group, fed untreated cookies daily; (ii) test group dosed daily with L. reuteri MM53; and (iii) test group dosed once every 4th day with L. reuteri MM53 and with untreated cookies on nondosing days. Piglets were weighed on days 0, 8, 14, and 21 of the trial. Experimental days 0 and 21 corresponded to 28 and 49 days of age, respectively.
Fresh fecal samples (approximately 100 g) were collected each morning. Samples were divided immediately into aliquots for DNA and plate count analyses, stored on dry ice, and transported to the laboratory within 30 min. Samples for DNA analysis were stored at −20°C until analyzed. Viable plate counts were performed immediately upon receipt of the samples. After completion of the study, piglets were fed a single 500-g aliquot of diet containing 1% (wt/wt) chromium oxide as a marker. Digesta transit times were estimated based on appearance and clearance of marker from feces in order to determine if reduction in fecal counts was less than (colonization), equal to (elimination), or greater than (antagonistic interaction) turnover or washout from the intestinal tract.
Viable plate counts of lactobacilli and antibiotic-resistant L. reuteri strain MM53.
Fresh fecal samples were serially diluted in anaerobic diluent (12) using standard anaerobic technique (3). A miniature-drop count method (five 20-μl drops per plate) was used to plate dilutions (10−1 to 10−8) on Lactobacillus MRS agar alone and Lactobacillus MRS agar containing streptomycin (400 μg/ml) and rifampin (40 μg/ml). Plates were incubated at 37°C for 48 h in an anaerobic cabinet. Random colonies selected daily from antibiotic plates were checked by light microscopy for cell morphology and Gram reaction to confirm recovery of L. reuteri.
Analysis of fecal samples by PCR-DGGE.
DNA extraction of fecal samples for DGGE analysis was performed as described by Tsai and Olson (35) and Simpson et al. (27). Two sets of primers were used for PCR amplification targeting different variable regions of the 16S rDNA. The 16S rDNA variable region 3 (V3) primer set, PCR amplification, and subsequent DGGE analysis have been described previously (16, 27). Briefly, the PCR mixture contained 125 ng of genomic DNA, 25 pmol of each primer, 4 μl of deoxynucleoside triphosphate mixture, 5 μl of 10× Ex Taq buffer, 5 μl of a 25% acetamide solution, and 0.5 μl of TaKaRa (Shuzo, Otsu, Japan) Ex Taq polymerase. The final volume was adjusted to 50 μl with sterile deionized water. The PCR amplification used touch-down cycling which consisted of lowering the annealing temperature every second cycle until it reached 55°C, at which temperature nine additional cycles were completed for a total of 29 cycles. Single-stranded DNA remaining from the PCR was degraded using mung bean nuclease (Stratagene, La Jolla, Calif.) as described previously (27). DGGE analysis for V3-16S rDNA variable region products (∼200 bp) was performed using gels containing a gradient of 35 to 60% denaturant. A 100% denaturing solution contains 40% (vol/vol) formamide and 7 M urea. Electrophoresis running time was 2 h at 150 V followed by 1 h at 200 V.
Several specific primers and probes have been designed for detection of different lactobacilli within V1 of the 16S rDNA (10, 36). The V1-16S rDNA primer set used in this study was designed to amplify Lactobacillus species that cluster in the L. reuteri phylogenetic group (L. fermentum, L. oris, L. pontis, and L. vaginalis) as defined by Schleifer and Ludwig (26). Primers were designed complementary to E. coli consensus positions 0092 (Lacto #1) and 0338 (Lacto #2). Primer Lacto #2 is the complement of the bacterium-specific probe S-D-Bact-0338-a-A-18, applied in a reverse orientation to obtain a 350-bp fragment. Primer sequences were, for Lacto #1 containing a GC clamp, 5′-CGCCCGCCGCGCGCGGCGGGCGGGGCGGGGGCACGGGGGGGTCGARCGMACTGGCCC-3′ and, for Lacto #2, 5′-GCTGCCTCCCGRAGGAGT-3′. PCR amplification of V1-16S rDNA sequence fragments was performed using the same amplification program described above for V3-16S rDNA region primers. However, the V1-16S rDNA region DGGE analysis was performed on optimized gels, using a gradient of 40 to 50% denaturant and electrophoresis running time adjusted to 2.5 h at 150 V followed by 1.5 h at 200 V.
Cloning and sequencing of V1-16S rDNA gene DGGE amplicons.
Representative bands were excised from DGGE gels and sequenced for identification. Recovery of DNA consisted of sterile excision of bands, washing excised polyacrylamide pieces three times with sterile deionized H2O for 1 h, three freeze-thaw cycles, and disruption of the acrylamide matrix with a mini-bead beater, reciprocating shaker (Biospec Products, Bartlesville, Okla.). After disruption, a 2-μl aliquot was removed and reamplified by PCR using the original Lacto primers. Resulting PCR products were purified using Chroma-Spin TE-100 columns (Clontech, Palo Alto, Calif.). Final eluants containing purified PCR fragments were diluted and sequenced at the W. M. Keck Center for Comparative and Functional Genomics, Biotechnology Center, University of Illinois. Sequencing was carried out using an Applied Biosystems Inc. (Foster City, Calif.) automated sequencing system. Analysis of nucleotide sequence data was performed using Sequencher 3.0 (Gene Codes Corp., Ann Arbor, Mich.), and sequences obtained were compared to Lactobacillus sequences stored in GenBank (National Center for Biotechnology Information, Bethesda MD) using GeneWorks 2.5.1 (IntelliGenetics Inc., Mountain View, Calif.).
Construction of intestinal bacterial standard ladder.
Standard ladders were created by individually PCR-amplifying DNA extracted from predominant intestinal bacterial strains using V3-16S rDNA DGGE primers. After confirmation of individual PCR product formation for each strain, products were combined in equal PCR product ratios and diluted 1:2 with DGGE loading buffer. This stock solution was used for all gels compared within a data set. The ladder consisted of the following organisms (n = 8) listed in order of migration distance: Bacteroides fragilis 25285T, Bifidobacterium bifidum (ATCC 29521), Escherichia coli (enteropathogenic E. coli; 2348T69), Fibrobacter succinogenes S85, Ruminococcus albus SY3, R. albus 7, R. albus 8, and Streptococcus bovis JB1. Each time a batch of standard ladder stock solution was amplified and mixed, aliquots were tested independently to ascertain the confidence intervals (i.e., fluctuations in band location or intensity due to variations in PCR amplification or mixing ratios). A single ladder stock was used for all gels analyzed and reported herein. Before being included in the data set, a gel was required to first fit within the calculated confidence interval for the standard ladder. The confidence interval was derived from comparisons of 12 triplicate standard runs under normal conditions using the formula 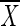 e ±
e ± 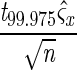 , where ς̂x is the pooled standard deviation for each lane, √
, where ς̂x is the pooled standard deviation for each lane, √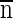 is the number of standards in the sample (i.e., the number of standard lanes per gel), and
is the number of standards in the sample (i.e., the number of standard lanes per gel), and 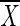 e is the average from previous standard runs. The confidence limit was set at ≥96%, equivalent to 1 standard deviation.
e is the average from previous standard runs. The confidence limit was set at ≥96%, equivalent to 1 standard deviation.
Staining and analysis of gel patterns.
Gels were developed by silver staining (16) and scanned using a GS-710 Calibrated Imaging Densitometer (Bio-Rad Inc., Hercules, Calif.). Gel patterns were analyzed using Diversity Database 2.1, part of the Discovery Series (Bio-Rad Inc.). Comparisons of DGGE pattern profiles were performed using Dice's similarity coefficient (_D_sc) analysis and Ward's algorithm. _D_sc values were compared based on presence or absence of bands; where bands were present, they were weighted based upon intensities. Dice's coefficient is defined as follows:
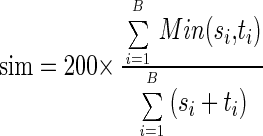
and dist = 100 − sim
where si equals the normalized density of the band assigned to the _i_th band type or si equals 0 if the lane does not have a band assigned to the _i_th type, B is the number of band types in the lane's band set, and ti represents two samples with lanes that are in the same band set. Min, sim, and dist represent mininum, similarity, and distance, respectively. Ward's algorithm is defined as follows:
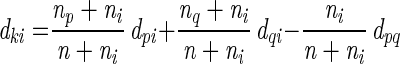
where p and q are indices indicating two clusters that are to be joined into a single cluster; k is the index of the cluster formed by joining clusters p and q; i is the index of any remaining clusters other than clusters p, q, or k; np is the number of samples in the _p_th cluster; nq is the number of clusters in the _q_th cluster, n is the number of clusters in the _k_th cluster formed by joining the _p_th and _q_th clusters (n = np + nq); and dpq is the distance between cluster p and cluster q as discussed by Sneath et al. and in the Diversity Database Manual (28). Tukey plots of _D_sc values were created using the Statview 5.0 graphics program (SAS Institute Inc., Cary, N.C.).
RESULTS
Animal observations.
Piglets remained healthy throughout the 20-day study period. Initial piglet weights prior to the trial averaged 10.9 ± 0.8 kg (unless otherwise noted, values are means ± standard deviations). Following completion of the trial, average weights of the control group, daily-dosed group, and 4th-day-dosed group were 28.3 ± 6.5, 24.8 ± 3.7, and 27.8 ± 2.1 kg, respectively. Body weights increased an average of 20 to 40% each week, and weight gain was similar among treatment groups. The mean digesta transit time was 32 h for all three treatment groups.
Quantitation of lactobacilli and L. reuteri MM53 based on viable plate counts.
Mean Lactobacillus counts for the control group ranged from 1 × 109 to 3 × 109 CFU/g of feces until day 12, when the levels decreased to 1.8 × 108 CFU/g of feces (Fig. 1A). The daily-dosed and 4th-day-dosed groups maintained mean Lactobacillus counts of 2 × 109 to 4 × 109 CFU/g of feces. Antibiotic plate counts from fecal samples confirmed that the introduced antibiotic-resistant L. reuteri strain survived passage through the intestinal tract (Fig. 1B). After termination of dosing on day 13, all piglets were monitored for an additional week, and L. reuteri MM53 could only be detected in feces for an additional 2 days in the daily-dosed and 4th-day-dosed groups. No antibiotic-resistant colonies were detected in controls or in the two dosing groups prior to introduction of the antibiotic-resistant strain. During the dosing period, the introduced strain fluctuated from 1 × 105 to 5 × 106 CFU/g of feces in the daily-dosed group. For the 4th-day-dosed group, antibiotic-resistant colonies persisted for only 2 days postdosing, with counts fluctuating between 8 × 103 and 6 × 105 CFU/g of feces, and reaching a maximum of 5 × 106 CFU/g of feces after the initial dose.
FIG. 1.

Total Lactobacillus (A) and L. reuteri MM53 (B) populations in daily fecal samples obtained from piglets for control (×), daily-dosed (●), and 4th-day-dosed (□) groups.
Analysis of bacterial diversity in fecal samples using PCR-DGGE.
Banding patterns for the V3-16S rDNA PCR amplicons are presented in Fig. 2. This gel image consists of one fecal sample from each of the nine piglets on day 15. The number of bands per lane varied from 11 to 25. The database set which was created for these samples contained a total of 35 unique bands, which were found in different combinations among the DGGE banding patterns. Occurrence of different bands from within the set was as follows: 9% of the band set occurred in >90% of the piglet samples, 28% occurred in >81% of the samples, and 41% occurred in >60% of the samples. Pattern comparisons were made between the groups using single-factor analysis of variance based on the number of bands present. When samples were blocked by period, i.e., before (days −1 and 0), during (days 1 through 13), and after (days 14 through 18) dosing, DGGE patterns did not differ among piglets before dosing. However, significant differences were seen for both dosing and postdosing time phases. Significant differences (P < 0.025) were found between the control and 4th-day-dosed groups and between the daily-dosed and 4th-day-dosed groups based on band numbers, which were 19 ± 3, 18 ± 4, and 14 ± 3, respectively. However, the control and the daily-dosed groups did not differ significantly (P < 0.05). Multiple blocked analyses of variance assessing dosing treatment, pig, or day effects did not detect significant effects.
FIG. 2.

PCR-DGGE profile generated from fecal samples obtained from individual piglets on day 15 using primers specific for the V3-16S rDNA region showing diversity of banding patterns present in each animal. Bacterial standard marker lanes are denoted as M. Lanes 1 to 3 are from the control group, lanes 4 to 6 are from the daily-dosed piglet group, and lanes 7 to 9 are from the 4th-day-dosed piglet group. Arrows indicate bands which are common to most piglet fecal samples.
Although some differences were noted in position, intensity, and number of bands present, the fecal bacterial DGGE analysis demonstrated relatively stable banding patterns throughout the collection period. Each animal had its own unique and repeatable profile, indicating that within-animal variation was less than between-animal variation. Several bands, (Fig. 2) were common in all samples. In addition, there were daily variations in band intensities within piglet samples and some appearance or disappearance of bands. However, for each individual animal, the overall 20-day pattern was sufficiently stable to observe shifts in individual bands representing temporal changes in bacterial populations.
Examination of successional changes in fecal bacterial populations.
An example of a typical DGGE gel banding pattern obtained for the V3-16S rDNA region from fecal samples of a daily-dosed piglet (E2) is presented in Fig. 3. Gels for other piglets showed similar DGGE pattern stability (data not shown), while individual animal patterns were significantly different from each other. This gel consists of three standard lanes (marked M) and 20 samples (consecutive sampling days −1 to 18). The stability of the pattern over time was evident not only by direct visual comparison, but also by calculation of _D_sc. The _D_sc values (n = 1,710) for all piglets ranged from 43 to 82% for the 10th to 90th percentiles and from 50 to 76% for the 25th to 75th percentiles, and means ranged from 60 to 70% as shown in Fig. 4. The _D_sc values indicated that all samples shared a large portion of the band set, while a few had unique bands as indicated by data points with low _D_sc values.
FIG. 3.

PCR-DGGE profile generated from fecal samples obtained from an individual piglet (E2) over the 20-day experimental period using primers specific for the V3-16S rDNA region showing stability of banding patterns within individual animal. Lanes −1 through 18 correspond to sampling days. Bacterial standard marker lanes are denoted as M.
FIG. 4.

Tukey plots of V3-16S rDNA region similarity coefficients (n = 190 each) calculated within individuals for fecal samples collected daily from piglets in control, daily-dosed, and 4th-day-dosed groups. Means are indicated as solid squares; the 25th, 50th, and 75th percentile data are indicated as the bottom, middle, and top edges of the boxes, respectively; the 10th and 90th percentiles are indicated as whiskers; and the extreme data points are indicated as circles. Letter and number designations on the x axis refer to individual animal identification numbers.
Since distinct pattern separations were observed within each group over time, cluster analysis was performed using Ward's algorithm. Figure 5 is an example of the cluster pattern formed when all piglets within the 4th-day-dosed group were analyzed. The samples separated into two time phase clusters which were designated as early (days −1 through 9) and late (days 10 through 18). The two time periods grouped together within a single major cluster for piglets F1 and F2; however, piglet F3 exhibited a separation of clusters, as was observed for the majority of piglets from the other two treatment groups (data not shown). This tight clustering of patterns according to individual, whether as a divided major cluster or as two separate but smaller clusters, demonstrated the stability of, and differences between, individual animal patterns. Examination of this dendrogram (Fig. 5), shows that only 3 of 60 profiles (5.0%) did not cluster with the appropriate piglet pattern group. Thus, the fecal sample from piglet F3 on day 17 clustered with the early phase of piglet F1 rather than with the late phase of F3; the fecal sample from piglet F1 on days −1 and 0 clustered with sample days 17 and 18; and the fecal sample from piglet F3 day −1 clustered with the late phase samples. These observations correlate with the pattern described in Fig. 6 for the 4th-day-dosed group.
FIG. 5.

Dendrogram illustrating the correlation between animal-to-animal variation in V3-16S rDNA region banding patterns. The three piglets (F1, F2, and F3) represented are from the 4th-day-dosing group. Numbers indicate the sampling day with individual animal designations shown within parentheses. Marker bars (●——●) indicate divisions between individual piglet pattern groups. Brackets indicate the two major time period pattern separations (early and late) that occur within each animal's pattern group. Dendrogram distances are in arbitrary units.
FIG. 6.

Successional changes in fecal bacterial populations for control, daily-dosed, and 4th-day-dosed groups based on cluster analysis of temporal PCR-DGGE banding patterns. Profiles were separated into start ( ), early (
), early ( ), middle (
), middle ( ), and late (
), and late ( ) time phases according to their cluster patterns generated using Ward's algorithm. The experimental dosing protocol was carried out as described in Materials and Methods between days 1 and 13 indicated by the vertical arrows. Dosing days for the 4th-day-dosed group are boxed.
) time phases according to their cluster patterns generated using Ward's algorithm. The experimental dosing protocol was carried out as described in Materials and Methods between days 1 and 13 indicated by the vertical arrows. Dosing days for the 4th-day-dosed group are boxed.
When banding patterns for all nine piglets were compared using a dendrogram generated using Ward's algorithm, a similar, but more complex, clustering pattern was evident. The two time periods evident from the within-group comparison were no longer as evident, and a more complex time phase separation could be discerned. The clusters separated into four time (± 1 day) phases: start (days −1 to 2), early (days 3 to 7), middle (days 8 to 13), and late (days 14 to 18). Clear distinctions in relatedness of patterns over time were observed within each treatment group and are summarized in Fig. 6. The patterns for control and daily-dosed piglets could be separated into the four logical and distinct time phases with the exception of pattern data derived from day 5, which clustered with the starting samples in both of these groups. The 4th-day-dosed piglets exhibited altered pattern groupings. The typical start cluster contained only days −1 and 0 but also included days 17 and 18 of the late-phase cluster. This shift in pattern clustering affected the timing of successional changes within the remaining three time phases for the 4th-day-dosed piglet group. Calculations based on analysis of all three groups demonstrated only 11 exceptions out of 180 samples (6.1%) which did not cluster with the corresponding piglet pattern group (data not shown).
Analysis of Lactobacillus diversity using PCR-DGGE.
As expected, the DGGE gel banding pattern observed for fecal lactobacilli was much simpler than that of the bacterial (V3 DGGE) banding patterns. The total number of bands was reduced to a maximum of eight, with an average of four (Fig. 7A). Several bands were excised from the DGGE gels for sequencing and are listed in Table 1 with their percent similarity to Lactobacillus sequences in GenBank. Bands 1 and 6 had 90 to 91% sequence similarity with unidentified Lactobacillus spp., which suggests that they are potentially uncultivated and, hence, uncharacterized species. However, the data must be interpreted with caution, as the V1 region of the 16s rDNA is highly variable, and the complete 16S rDNA gene sequence is required to make a precise phylogenetic assignment for each organism. The two bands detected for the L. reuteri MM53 control culture (Fig. 7A, lane 1) may be the result of multiple copies of the 16S rDNA. Both bands were sequenced and the second (V1-3), which corresponded to a higher percent homology (98 versus 95%) with the L. reuteri type strain (DSM 20016), was used to track L. reuteri populations on DGGE gels. When compared against the sequence of the type strain, there were 4 (V1-3)- and 15 (V1-2)-bp differences respectively, resulting in the two similarity values listed (Table 1).
FIG. 7.

(A) PCR-DGGE pattern obtained using _Lactobacillus_-specific V1-16S rDNA primers. A box indicates the L. reuteri band used to track changes in this population. Lanes: 1, pure culture L. reuteri MM53; 2, control piglet fecal sample; 3, daily-dosed piglet fecal sample. Band numbers correspond to different Lactobacillus species as follows: 1, Lactobacillus sp., 2, L. reuteri; 3, L. reuteri; 4, L. pontis; 5, L. panis; 6, Lactobacillus sp. (B) Contribution of L. reuteri band V1-3 intensities from V1-16S rDNA region DGGE gels expressed as a percentage of total density (V1 through V6) for the three treatment groups (control [▴], daily dosed [□], and 4th day dosed [●]). Dosing days for the 4th-day-dosed group are boxed, and the last dosing day is indicated by the dotted line.
TABLE 1.
Identification of Lactobacillus bands excised from V1-16S rDNA DGGE gels determined by sequence alignment
| Band no. | GenBank accession no. | Identity | % Similarity |
|---|---|---|---|
| 1 | AB016864 | Lactobacillus sp. | 91 |
| 2 | L23507 | L. reuteri (DSM 20016) | 95 |
| 3 | L23507 | L. reuteri (DSM 20016) | 98 |
| 4 | X76329 | L. pontis (LTH2587) | 96 |
| 5 | X94230 | L. panis | 96 |
| 6 | D79213 | Lactobacillus sp. | 90 |
The _D_sc values calculated for _Lactobacillus_-specific V1-16S rDNA DGGE patterns had a wider range than that observed for the V3-16S rDNA regions, although percent similarities were higher. The _D_sc values (n = 1,710) for all piglets ranged from 35 to 95% for the 10th to 90th percentiles and from 52 to 90% for the 25th to 75th percentiles, and means ranged from 70 to 85% as shown in Fig. 8. These high _D_sc values indicated that all samples shared a major portion of the band set, while a few had unique bands as indicated by low _D_sc values. All nine animals had _D_sc values between 95 and 100% similarity for the V1-16S rDNA region. The V3-16S rDNA region had _D_sc values between 85 and 90%, with only one animal having a data point at 95%. This closer relationship between patterns was most likely a statistical effect of a simplified (Fig. 7A) banding pattern (6 versus 35 bands).
FIG. 8.

Tukey plot of V1-16S rDNA region similarity coefficients (n = 190 each) calculated within individuals for fecal samples collected daily from piglets in control, daily-dosed, and 4th-day-dosed groups. Means are indicated as solid squares; the 25th, 50th, and 75th percentile data are indicated as the bottom, middle, and top edges of the boxes, respectively; the 10th and 90th percentiles are indicated as whiskers; and the extreme data points are indicated as circles. Letter and number designations on the x axis refer to individual piglet identification numbers.
Variations in mean band intensity of fecal L. reuteri populations (band 3, designated V1-3) (Fig. 7A) for each treatment group are presented in Fig. 7B. The control and daily-dosed piglets had L. reuteri bands which fluctuated between 10 and 25% of the total lane intensities, while the 4th-day-dosed piglets had bands with fluctuations in intensity between 20 and 35%. The control piglets exhibited a cyclic oscillation pattern, with peaks on days 2, 10 to 11, and 17, indicating a periodic fluctuation for this group. The daily-dosed animals exhibited two stable plateaus in the pattern; one region spanned 5 days (day −1 through day 4) at 10% of populations and a second region spanned 7 days (day 5 through day 11) at 20 to 25% of populations, based on band intensities. After discontinuing the daily treatments, a 2-day cyclic pattern emerged, with fluctuations ranging from 7 to 32%. For the 4th-day-dosed animals peaks were observed following dosing days. Peaks occurred on days 5, 7, 10, and 13, which corresponded with dosing days 5, 9, and 13. The last two peaks (days 16 and 18) occurred after dosing and followed the oscillation pattern of 2- to 3-day intervals observed for the previous periods. Comparisons of fluctuation patterns between the control and dosed piglets indicated a possible suppression of the normal shifts in the indigenous L. reuteri population, resulting from the introduction of the dosed strain.
An additional oscillation phenomenon related to the introduced strain was also demonstrated, with one of the other bands detected with the Lactobacillus primer set (band 6, designated V1-6) (Fig. 7A). In control piglets (Fig. 9A), the V1-6 population maintained 10 to 15% higher band intensities than that of V1-3, with the exception of day 9. For the 4th-day-dosed piglets (Fig. 9B), the initial levels of band V1-6 accounted for 28 to 38% of the population, but after introduction of L. reuteri MM53, these levels dropped to between 15 and 25%. A rebound occurred on day 4, with intensity indicating ∼50% of the population. However, following an upsurge in band V1-3 intensity to 33 to 70% (days 6 to 9), the V1-6 population decreased to approximately 15% and remained at those levels until the decline in abundance of V1-3 (day 10 to 12), at which point there was a resurgence to 45%. Following termination of dosing, this countercycle pattern was observed to continue through day 18. Counteroscillating patterns for bands V1-3 and V1-6 were observed in all nine animals throughout the dosing and postdosing periods.
FIG. 9.

Comparison of intensities of V1-3 band (L. reuteri) (▴) with V1-6 band (Lactobacillus sp. (●) in a control animal (D2) showing a counteroscillation pattern over the duration of the experiment (A) and for a 4th-day-dosed piglet (F2) showing a dosage-altered pattern (B). For clarity only two of the six possible bands are included in panels (A) and (B). Dosing days for the 4th-day-dosed piglets are boxed, and the last dosing day is indicated by the broken line.
DISCUSSION
For the purposes of this study, rifampin-streptomycin-resistant derivatives of L. reuteri MM53 were generated to facilitate enumeration from feces and to differentiate the administered L. reuteri strain from indigenous populations. The strain was easily detectable on antibiotic-selective plates, at a level of 103 CFU/g (wet weight) of feces. Murphy et al. (15) used rifampin resistance to evaluate establishment of L. salivarius strains in the murine gastrointestinal tract and feces. L. salivarius UCC118 was administered at a daily dose of 4 × 109 CFU and recovered in fecal samples at 1.7 × 106 to 2.1 × 106 CFU/g, which is similar to the range (1 × 106 to 5 × 106 CFU/g) of recovery in feces observed with L. reuteri MM53 in the present study. These studies demonstrate the power of antibiotic selectivity in tracking exogenous or probiotic organisms introduced into the intestinal tract. Pedersen and Tannock (21) suggested that in vivo transfer of antibiotic resistance genes to other intestinal or fecal bacteria could complicate detection of marker strains. The present experiments were not affected by this problem, since the marker strain was selected for double antibiotic resistance and no background colonies other than strain MM53 were detected on MRS plates supplemented with streptomycin (400 μg/ml) and rifampin (40 μg/ml) for the duration of the experiment.
Since detectable levels of colonization, as measured in fecal samples, did not persist longer than 2 days after termination of dosing, calculations were made based on washout as a result of digesta turnover. Digesta transit rates in piglets vary, with mean retention times from 30 to 70 h depending on diet and age (8, 29). Based on the estimated transit time for piglets in this study (mean, 32 h), the rapid reduction in the bacterial counts indicates an antagonistic interaction rather than simply dilution by turnover of gut contents. Based on turnover alone (digesta volume diluted by one half each 32 h), the 2-log reduction in CFU should have taken about 9 days and not the 2 days measured in this study.
Several recent studies have indicated that is not uncommon for introduced bacteria, particularly those under investigation for probiotic properties or preparations, to be undetectable 3 to 5 days after termination of treatment (7, 18, 19, 38). However, Murphy et al. (15) reported that although L. salivarius strains were no longer detectable in mouse feces 4 days posttreatment, the marked strains were found to have persisted in the ileo-cecal region of the small intestine 7 days after cessation of dosing. This study (15) suggested that enumeration of strains in excreted feces may not be an accurate reflection of colonization and persistence at specific sites within the intestinal tract. Since individual intestinal compartments were not examined for colonization in the current study, it is possible that colonization persisted for longer than 2 days but was below detection limits in feces (103 CFU/g of feces).
The cyclic pattern (between 1 and 2 logs in number) observed for antibiotic-resistant L. reuteri in the daily-dosed animals (Fig. 1B) is difficult to explain. Initially, this was thought to be related to the dosing and sampling schedule. However, the peaks on days 3, 7, and 12 are at 4- to 5-day rather than 1- to 2-day intervals and do not correspond to the peaks in L. reuteri bands determined by V1-16S rDNA DGGE analysis for the daily-dosed group (Fig. 7B). Thus, this cyclic pattern is more likely the result of antagonistic interactions based on the finding of rapid reduction in counts of the introduced strain at a rate much faster than could be accounted for simply by dilution or turnover of intestinal content, and the large cyclic fluctuations observed between specific bands in V1-16S rDNA DGGE profiles (Fig. 9). The large cyclic fluctuations decreased as the experiment progressed, which may be related to slower digesta transit as piglets aged. However, it is more likely the result of the rapid reduction in counts of the introduced L. reuteri strain MM53.
One of the current limitations to molecular microbial ecology techniques based on analysis of banding patterns (DGGE, temperature gradient gel electrophoresis [TGGE]) is the comparison of profiles between gels. Finding acceptable standards for gel comparisons is difficult, as molecular markers similar to those used with agarose or sodium dodecyl sulfate-polyacrylamide gels are not commercially available for DGGE. The construction of a standard ladder set allows putative identification of sample bands based on location and can be used by database programs to map gel curvatures. While the standard set constructed here was based on the V3-16S rDNA gene from common intestinal organisms, other regions are also suitable for this application. For example, Walter et al. (38) recently developed a Lactobacillus identification ladder utilizing the V2 to V3-16S rDNA region. Regardless of the specific application, the standard ladder constructed must first be calibrated, and subsequently the same ladder must be utilized for all gels included in a particular set of comparisons. In particular, the Diversity Database program requires the use of multiple standard lanes per gel, which are used to calculate gel contours based upon band migrations within the standard ladder. Mapping permits calculation of normalizing factors, which are used for comparisons between gels or to other data sets within the database. Additionally, the standard ladder bands for each gel are compared statistically with previously analyzed gel databases to determine if they are within acceptable confidence limits or if the gel should be discarded.
Comparisons of DGGE gel patterns in dendrogram (Fig. 5) or diagram (Fig. 6) format allowed visualization of successional changes. The separation of sampling days into two time phases (arbitrarily described as early and late) when comparing within each treatment group (three piglets; n = 60 fecal sample profiles) was clearly evident. However, when these gel patterns were compared as a dendrogram for the whole data set (nine piglets; n = 180 fecal sample profiles [data not presented]), four relatively stable time phases could be distinguished (start [days −1 to 2], early [days 3 to 7], middle [days 8 to 13], and late [days 14 to 18]). The aberrant grouping of first and last days of the 4th-day-dosed group indicated that the banding patterns were possibly reverting to the pattern observed prior to initiation of dosing. When these gel patterns were compared as a dendrogram for the whole data set (not presented), the grouping of individual patterns was still distinguishable as multiple smaller clusters, usually consisting of 5 to 7 days of closely associated samples. This suggests that when comparing data sets of increasing size, the clusters become more fragmented (randomly associated), rather than grouping strictly by phase or treatment.
Zoetendal et al. (40) reported individual pattern stability after analysis of four human fecal samples over a 6-month period using TGGE to determine bacterium population profiles. The relative stability and individuality of the patterns indicated that each individual harbored a specific and unique fecal bacterial community. These authors (40) concluded, after examination of several unrelated individuals of different ages and different dietary preferences, that the reasons for pattern uniqueness were likely to be found in host factors. The individuals (piglets) in the current experiment were full siblings and were fed the same diet, yet banding patterns were still unique for each piglet. These patterns were unique prior to, during, and after cessation of dosing, regardless of treatment group. Considering that the three variables noted above by Zoetendal et al. (40) were not present in the current analysis and that the same uniqueness was observed, we suggest that the data support an as-yet-unexplored host influence on the patterns generated. However, given the complexity of bacterium populations found in the intestinal tract, the majority of which are presumed metabolically active, and their impact upon the community and the host (5), the separation of predominant influences is likely to be difficult.
While individual piglets had stable and repeatable bacterial specific banding patterns over time, there were clear differences between individual patterns, which were unique for each animal. Differences were accounted for either by presence or absence of bands, or by differences in band intensities and were used to calculate the varied _D_sc values obtained from the data set. Similarities within patterns observed for individual piglets in our study, i.e., common predominant bands, suggested that while each individual piglet was unique, there were also common, possibly dominant, bacterial species which were present in more than one individual. Several studies have utilized different techniques to obtain fingerprints or profiles of individual intestinal or fecal bacterium populations from different animals or humans, while not necessarily investigating ecological succession. These techniques have included plasmid profiling (32), ribotyping (11), direct cloning (22), pulsed-field gel electrophoresis (20), and TGGE (40). Interestingly, regardless of the molecular technique applied, results demonstrated that individual subjects maintained a distinct and characteristic bacterial population or profile. Kimura et al. (9) not only recognized the unique and distinctive composition of the predominant bifidobacterial and lactobacillus populations present in human feces, but also that some Lactobacillus strains were characteristic of the specific human host.
_Lactobacillus_-specific PCR-DGGE analysis demonstrated that a background population of L. reuteri was present in the fecal samples of control piglets but that L. reuteri banding pattern intensities were significantly above background levels in the two dosed groups. At present, this technique is able to detect at the species but not strain level. This was due to primer specificity limitations based on limited sequence heterogeneity of the 16S rDNA V1 target region (38). The intergenic region between 16 and 23S rDNA genes of strain MM53 are being investigated for potential design of strain-specific primers. Tannock et al. (34) recently examined 28 Lactobacillus intestinal isolates using the 16S-23S intergenic region and found that there were sufficient differences to identify lactobacilli at the strain level. Three strains of L. reuteri obtained from the intestine of the rat, mouse, and pig were identifiable individually based on PCR-specific amplification. This indicates that specificity may be improved to the strain level after sequencing of the intergenic region from strain MM53.
Frequent inoculation of L. reuteri influenced the cyclic fluctuations in patterns for both the daily-dosed and 4th-day-dosed groups. However, following termination of dosing, patterns for both groups of dosed animals became similar to those of the controls, indicating that this was possibly a normal occurrence. The _D_sc values for the Lactobacillus data were higher overall than those for the bacterium-specific analysis, yet analysis of V1-16S rDNA data in dendrogram format did not yield time phase separations as found for the V3-16S rDNA region data. Comparison of patterns containing only 4 to 8 bands, where only a select portion of the Lactobacillus population is targeted, can be expected to be more closely related than comparisons of patterns with up to 32 bands, representing a larger portion of the fecal bacterial populations. This indicates that a minimum number (10 to 15) of bands is needed to generate adequate pattern comparisons.
The observation of an as-yet-undefined relationship between a Lactobacillus population (band V1-6) (Fig. 7A), detected by DGGE analysis of V1-16S rDNA fragments, which was inversely related to populations of L. reuteri MM53, demonstrates some of the possible dynamic interactions and relationships which can occur in the intestinal tract. While the true nature of this relationship is currently unknown, we can speculate that it is competitive, due to the opposing pattern changes and rapid L. reuteri disappearance after termination of dosing. The occurrence of L. reuteri as a less abundant population in the control piglets possibly indicates that this other Lactobacillus assemblage might influence the dynamic population cycles of L. reuteri. Cessation of dosing resulted in an altered banding pattern which reverted to that observed in control piglets. Fluctuations within the other detected Lactobacillus populations did not correlate with changes in L. reuteri populations.
In summary, this study describes the application of DGGE for monitoring changes in gastrointestinal and fecal microbiota of piglets after the introduction of an exogenous strain of L. reuteri. These results provide convincing evidence that each individual exhibits a unique bacterial community as demonstrated by stable and repeatable banding patterns. The use of bacterium-specific primers allowed examination of population changes, in relation to dosing or time. Examination of patterns using primers specific for a select group of Lactobacillus permitted a detailed look into interspecies relationships and abundance as influenced by dosing. L. reuteri was clearly distinguishable within this reduced pattern based upon relative distance calculations and sequence determinations unlike that of bacterial specific patterns. The overall significance of this study for the area of probiotic research and for intestinal health and function is that this technique will be useful for monitoring changes in the microbiota of individuals with intestinal disorders or after administration of probiotic supplements. Microbes beneficial to gut health and function can be administered and detected, and their influence on other bacteria can be monitored over time. Importantly, these techniques provide some of the tools essential for the study of host-microbe interactions.
ACKNOWLEDGMENTS
We gratefully acknowledge Kilby Willenburg and Lindsay A. Marek for assistance with collection and preparation of samples, Joanne Chee-Sanford for development of the DGGE bacterial standard ladder, and Steve Skerlos for assistance with the statistical analysis.
Support was provided by Illinois Council for Food and Agricultural Research (C-FAR), Campus Research Board of University of Illinois, and the Agricultural Experimental Station, University of Illinois at Urbana-Champaign.
REFERENCES
- 1.Amann R, Ludwig W, Schleifer K. Phylogenetic identification and in situ detection of individual microbial cells without cultivation. Microb Ecol. 1995;59:143–169. doi: 10.1128/mr.59.1.143-169.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Axelsson L, Lindgren S. Characterization and DNA homology of Lactobacillus strains isolated from pig intestine. J Appl Bacteriol. 1987;62:433–440. doi: 10.1111/j.1365-2672.1987.tb02673.x. [DOI] [PubMed] [Google Scholar]
- 3.Bryant M P. Commentary on the Hungate technique for culture of anaerobic bacteria. Am J Clin Nutr. 1972;25:1324–1328. doi: 10.1093/ajcn/25.12.1324. [DOI] [PubMed] [Google Scholar]
- 4.Casas I, Dobrogosz W. Lactobacillus reuteri: overview of a new probiotic for humans and animals. Microecol Ther. 1997;26:221–231. [Google Scholar]
- 5.Gaskins H R. Intestinal bacteria and their influence on swine growth. In: Lewis A J, Southern L L, editors. Swine Nutrition, in press. Boca Raton, Fla: CRC Press; 2000. [Google Scholar]
- 6.Henriksson A, André L, Conway P. Distribution of lactobacilli in the porcine gastrointestinal tract. FEMS Microbiol Ecol. 1995;16:55–60. [Google Scholar]
- 7.Jacobsen C N, Nielsen V R, Hayford A E, Møller P L, Michaelsen K F, Pærregaard A, Sandström B, Tvede M, Jakobsen M. Screening of probiotic activities of forty-seven strains of Lactobacillus spp. by in vitro techniques and evaluation of the colonization ability of five selected strains in humans. Appl Environ Microbiol. 1999;65:4949–4956. doi: 10.1128/aem.65.11.4949-4956.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jonsson E, Conway P. Probiotics for pigs. In: Fuller R, editor. Probiotics: the scientific basis. Vol. 1. New York, N.Y: Chapman and Hall; 1992. pp. 260–316. [Google Scholar]
- 9.Kimura K, McCartney A, McConnell M, Tannock G. Analysis of fecal populations of bifidobacteria and lactobacilli and investigation of the immunological responses of their human hosts to the predominant strains. Appl Environ Microbiol. 1997;63:3394–3398. doi: 10.1128/aem.63.9.3394-3398.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Klijn N, Weerkamp A, de Vos W. Identification of mesophilic lactic acid bacteria by using polymerase chain reaction-amplified variable regions of 16S rRNA and specific DNA probes. Appl Environ Microbiol. 1991;57:3390–3393. doi: 10.1128/aem.57.11.3390-3393.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Krause D, White B, Mackie R. Ribotyping of adherent Lactobacillus from weaning pigs: a basis for probiotic selection based on diet and gut compartment. Anaerobe. 1997;3:317–325. doi: 10.1006/anae.1997.0118. [DOI] [PubMed] [Google Scholar]
- 12.Mackie R I, Wilkins C A. Enumeration of anaerobic bacterial microflora of the equine gastrointestinal tract. Appl Environ Microbiol. 1988;54:2155–2160. doi: 10.1128/aem.54.9.2155-2160.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McCartney A, Wenzhi W, Tannock G. Molecular analysis of the composition of the bifidobacterial and Lactobacillus microflora of humans. Appl Environ Microbiol. 1996;62:4608–4613. doi: 10.1128/aem.62.12.4608-4613.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Morelli L, Cesena C, de Haën C, Gozzini L. Taxonomic Lactobacillus composition of feces from human newborns during the first few days. Microb Ecol. 1998;35:205–212. doi: 10.1007/s002489900076. [DOI] [PubMed] [Google Scholar]
- 15.Murphy L, Dunne C, Kiely B, Shanahan F, O'Sullivan G, Collins K. In vivo assessment of potential probiotic Lactobacillus salivarius strains: evaluation of their establishment, persistence, and localization in the murine gastrointestinal tract. Microb Ecol Health Dis. 1999;11:149–157. [Google Scholar]
- 16.Muyzer G, Brinkhoff T, Nübel U, Santegoeds C, Schäfer H, Wawer C. Denaturing gradient gel electrophoresis (DGGE) in microbial ecology. In: Akkermans A, van Elsas J D, de Bruijn F, editors. Molecular microbial ecology manual. 3.4.4. Boston, Mass: Kluwer Academic Publishers; 1998. pp. 1–27. [Google Scholar]
- 17.Naito S, Hayashidani H, Kaneko K, Ogawa M, Benno Y. Development of intestinal lactobacilli in normal piglets. J Appl Bacteriol. 1995;79:230–236. doi: 10.1111/j.1365-2672.1995.tb00940.x. [DOI] [PubMed] [Google Scholar]
- 18.Netherwood T, Bowden R, Harrison P, O'Donnell A, Parker D, Gilbert H. Gene transfer in the gastrointestinal tract. Appl Environ Microbiol. 1999;65:5139–5141. doi: 10.1128/aem.65.11.5139-5141.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Netherwood T, Gilbert H, Parker D, O'Donnell A. Probiotics shown to change bacterial community structure in the avian gastrointestinal tract. Appl Environ Microbiol. 1999;65:5134–5138. doi: 10.1128/aem.65.11.5134-5138.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.O'Sullivan D. Methods for analysis of the intestinal microflora. In: Tannock G, editor. Probiotics: a critical review. Wymondham, United Kingdom: Horizon Scientific Press; 1999. pp. 23–44. [Google Scholar]
- 21.Pedersen K, Tannock G. Colonization of the porcine gastrointestinal tract by lactobacilli. Appl Environ Microbiol. 1989;55:279–283. doi: 10.1128/aem.55.2.279-283.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pryde S, Richardson A, Stewart C, Flint H. Molecular analysis of the microbial diversity present in the colonic wall, colonic lumen, and cecal lumen of a pig. Appl Environ Microbiol. 1999;65:5372–5377. doi: 10.1128/aem.65.12.5372-5377.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Raskin L, Capman W, Sharp R, Poulsen L, Stahl D. Molecular ecology of gastrointestinal ecosystems. In: Mackie R I, White B A, Isaacson R E, editors. Gastrointestinal microbiology. Vol. 2. New York, N.Y: Chapman and Hall; 1997. pp. 243–298. [Google Scholar]
- 24.Reid G. The scientific basis for probiotic strains of Lactobacillus. Appl Environ Microbiol. 1999;65:3763–3766. doi: 10.1128/aem.65.9.3763-3766.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Roos S, Aleljung P, Robert N, Lee B, Wadstrom T, Lindberg M, Jonsson H. A collagen binding protein from Lactobacillus reuteri is part of an ABC transporter system. FEMS Microb Lett. 1996;144:33–38. doi: 10.1111/j.1574-6968.1996.tb08505.x. [DOI] [PubMed] [Google Scholar]
- 26.Schleifer K, Ludwig W. Phylogeny of the genus Lactobacillus and related genera. Syst Appl Microbiol. 1995;18:461–467. [Google Scholar]
- 27.Simpson J M, McCracken V J, White B A, Gaskins H R, Mackie R I. Application of denaturant gradient gel electrophoresis for the analysis of the porcine gastrointestinal microbiota. J Microbiol Methods. 1999;36:167–179. doi: 10.1016/s0167-7012(99)00029-9. [DOI] [PubMed] [Google Scholar]
- 28.Sneath P H, Sokal R R. Numerical taxonomy: the principles and practice of numerical classification. W. H. San Francisco, Calif: Freeman & Company; 1973. [Google Scholar]
- 29.Stewart C. Microorganisms in hindgut fermentors. In: Mackie R I, White B A, Isaacson R E, editors. Gastrointestinal microbiology. Vol. 2. New York, N.Y: Chapman and Hall; 1997. pp. 142–186. [Google Scholar]
- 30.Talarico T, Casas I, Chung T, Dobrogosz W. Production and isolation of reuterin, a growth inhibitor produced by Lactobacillus reuteri. Antimicrob Agents Chemother. 1988;32:1854–1858. doi: 10.1128/aac.32.12.1854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tannock G. Lactobacillus succession in the piglet digestive tract demonstrated by plasmid profiling. Appl Environ Microbiol. 1990;56:1310–1316. doi: 10.1128/aem.56.5.1310-1316.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tannock G. Analysis of the intestinal microflora: a renaissance. Antonie Leeuwenhoek. 1999;76:265–278. [PubMed] [Google Scholar]
- 33.Tannock G W, Munro K, Harmsen H J M, Welling G W, Smart J, Gopal P K. Analysis of the fecal microflora of human subjects consuming a probiotic containing Lactobacillus rhamnosus DR20. Appl Environ Microbiol. 2000;66:2578–2588. doi: 10.1128/aem.66.6.2578-2588.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tannock G W, Tilsala-Timisjarvi A, Rodtong S, Munro K, Ng J, Alatossava T. Identification of Lactobacillus isolates from the gastrointestinal tract, silage, and yoghurt by 16S-23S rRNA gene intergenic spacer region sequence comparisons. Appl Environ Microbiol. 1999;65:4264–4267. doi: 10.1128/aem.65.9.4264-4267.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tsai Y-L, Olson B. Detection of low numbers of bacterial cells in soils and sediments by polymerase chain reaction. Appl Environ Microbiol. 1992;58:754–757. doi: 10.1128/aem.58.2.754-757.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vaughan E, Mollet B, deVos W. Functionality of probiotics and intestinal lactobacilli: light in the intestinal tract tunnel. Curr Opin Biotechnol. 1999;10:505–510. doi: 10.1016/s0958-1669(99)00018-x. [DOI] [PubMed] [Google Scholar]
- 37.Vogel R, Böcker G, Stolz P, Ehrmann M, Fanta D, Ludwig W, Pot B, Kersters K, Schleifer K, Hammes W. Identification of lactobacilli from sourdough and description of Lactobacillus pontis sp. nov. Int J Syst Bacteriol. 1994;44:223–229. doi: 10.1099/00207713-44-2-223. [DOI] [PubMed] [Google Scholar]
- 38.Walter J, Tannock G, Tilsala-Timisjarvi A, Rodtong S, Loach D, Munro K, Alatossava T. Detection and identification of gastrointestinal Lactobacillus species by using denaturing gradient gel electrophoresis and species-specific PCR primers. Appl Environ Microbiol. 2000;66:297–303. doi: 10.1128/aem.66.1.297-303.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yu B, Tsen H. Lactobacillus cells in the rabbit digestive tract and the factors affecting their distribution. J Appl Bacteriol. 1993;75:269–275. doi: 10.1111/j.1365-2672.1993.tb02776.x. [DOI] [PubMed] [Google Scholar]
- 40.Zoetendal E, Akkermans A, deVos W. Temperature gradient gel electrophoresis analysis of 16S rRNA from human fecal samples reveals stable and host-specific communities of active bacteria. Appl Environ Microbiol. 1998;64:3854–3859. doi: 10.1128/aem.64.10.3854-3859.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]