Mechanism of transfer of NO from extracellular S-nitrosothiols into the cytosol by cell-surface protein disulfide isomerase (original) (raw)
Abstract
_N_-dansylhomocysteine (DnsHCys) is quenched on S-nitrosation. The product of this reaction,_N_-dansyl-_S_-nitrosohomocysteine, is a sensitive, direct fluorogenic substrate for the denitrosation activity of protein disulfide isomerase (PDI) with an apparent_K_M of 2 μM. _S_-nitroso-BSA (BSA-NO) competitively inhibited this reaction with an apparent_K_I of 1 μM. The oxidized form of DnsHCys,_N,N_-didansylhomocystine, rapidly accumulated in cells and was reduced to DnsHCys. The fluorescence of DnsHCys-preloaded human umbilical endothelial cells and hamster lung fibroblasts were monitored as a function of extracellular BSA-NO concentration via dynamic fluorescence microscopy. The observed quenching of the DnsHCys fluorescence was an indirect measure of cell surface PDI (csPDI) catalyzed denitrosation of extracellular _S_-nitrosothiols as decrease or increase in the csPDI levels in HT1080 fibrosarcoma cells correlated with the rate of quenching and the PDI inhibitors, 5,5′-dithio-_bis_-3-nitrobenzoate and 4-(_N_-(_S_-glutathionylacetyl) amino)phenylarsenoxide inhibited quenching. The apparent_K_M values for denitrosation of BSA-NO by csPDI ranged from 12 μM to 30 μM. Depletion of membrane N2O3 with the lipophylic antioxidant, vitamin E, inhibited csPDI-mediated quenching rates of DnsHCys fluorescence by ≈70%. The _K_M for BSA-NO increased by ≈3-fold and _V_max decreased by ≈4-fold. These findings suggest that csPDI catalyzed NO released from extracellular _S_-nitrosothiols accumulates in the membrane where it reacts with O2 to produce N2O3. Intracellular thiols may then be nitrosated by N2O3 at the membrane-cytosol interface.
Nitric oxide is a secondary messenger involved in the cGMP cascade, vasodilation, and a known inhibitor of platelet aggregation. _S_-nitrosothiols (RSNOs) are formed via nitrosation of thiols under aerobic conditions (1). Apart from being cellular sources of NO, they also prolong its half-life (2, 3). RSNOs are involved in signaling pathways, immune responses, and the actions of nitrovasodilating compounds (4–7). Therefore, a study involving the effects and transport of_S_-nitroso-BSA (BSA-NO) into live cells is of utmost physiological importance and the center of pharmacological interest (8).
Many indirect, discontinuous fluorescent, electrochemical, and colorometric assays have been developed for RSNOs (8, 9). However, none of these are conducive to measuring the transport of RSNO-bound NO into live cells, in vitro. The probe for NO influx presented here is _N_-dansylhomocysteine (DnsHCys). The fluorescence of this compound is completely quenched on its S-nitrosation, yielding_N-_dansyl-_S_-nitrosohomocysteine (DnsHCysNO). Here we show that DnsHCysNO is a direct fluorogenic substrate for protein disulfide isomerase (PDI).
PDI acts as a chaperone molecule in the endoplasmic reticulum where it catalyses protein thiol exchange reactions. Hotchkiss et al. (10) have reported that PDI is also secreted by endothelial cells as well as deposited on the cell surface. Recent studies have presented indirect evidence for the involvement of cell-surface PDI (csPDI) in the influx of RSNO-bound NO (11, 12). Stamler and coworkers (13) also have shown that the export of intracellular NO from red blood cells to be facilitated by S-nitrosation of the cysteine residues in the hemoglobin-binding cytoplasmic domain of the anion exchanger AE1. Membrane-bound PDI may catalyze the formation of AE1-SNO and the subsequent export of cytosolic NO.
Here, the extracellular RSNO-dependent quenching of the DnsHCys fluorescence was shown to be a csPDI-dependent process with the aid of antisense-mediated underexpression of PDI and the sense-mediated overexpression of PDI in HT1080 fibrosarcoma cells as well as with a cell-impermeant inhibitor that reacts with vicinal dithiols. In addition, _N_-dansylhomocystine (DnsHCys2) is shown to be a sensitive intracellular probe for the kinetic characterization of csPDI in human umbilical vein endothelial cells (HUVECs), hamster lung fibroblasts, and HT1080 fibrosarcoma cells. Based on this data, a mechanism for csPDI-meditated intracellular S-nitrosation, by extracellular RSNOs, has been proposed.
Materials and Methods
Synthesis of _S_-Nitrosoglutathione (GSNO).
Glutathione (GSH, Sigma) was dissolved in ice-cold 0.5 M HCl. Equimolar sodium nitrite was added and the reaction was carried out in the dark at 4°C for 40 min. The pH of the reaction mixture was adjusted to 7.0 and crystallized by the slow addition of cold acetone. BSA-NO was synthesized by using the above-mentioned protocol (3).
Synthesis of DnsHCysNO.
HCysNO was prepared by treating HCys (Sigma) with acidified nitrite. Dansylation was carried out in 0.1 M phosphate buffer (pH 8.5) at stochiometric amounts. The product was purified on a G-10 Sephadex column as reported by Ramachandran et al. (14).
Synthesis of N,_N_-Didansylhomocystine.
HCys was dissolved in water (80 ml) and saturated NaHCO3 (25 ml). Once the solution was clear 30 ml of acetone was added. Dansyl chloride (Sigma) (1.608 g/80 ml of acetone) was then added drop-wise over a 5-h period. The acetone was removed under reduced pressure and acidified to pH 2–3 with 4 M HCl. Solution was extracted with ethyl acetate, washed with water, dried over Na2SO4, and evaporated to dryness. The residue was subject to chromatography on a silica gel column by using the following solvent system: 1-methylene chloride, 2–50% methyl chloride/ethyl acetate, 3–80% methylene chloride/20% methanol. The purity of the product was confirmed by using NMR.
HPLC Analysis.
The DnsHCys and its derivatives were chromatographed on a C18 reverse-phase column (Varian) attached onto a Bio-Logic HPLC (Bio-Rad) with a Shimadzu RF 551 fluorescence detector. The solvents used were 0.1% trifluoroacetic acid and 60% acetonitrile with 0.1% trifluoroacetic acid. The _S_-carboxymethyl-DnsHCys standards were prepared by reducing DnsHCys2 (200 μM), with 1 mM DTT and reacting the mixture with 10 mM iodoacetate (Sigma) for 30 min (0.1 M Tris⋅HCl, pH 8.5). Appropriate amounts of this mixture were injected into the HPLC to obtain a standard curve. Hamster lung fibroblasts (1 × 106 cells) were incubated in 200 μM DnsHCys2 for 2 min. The cells were lysed by ultrasonication in buffer (0.1 M Tris⋅HCl, pH 8.5) containing 20 mM iodoacetate. The cell lysate was filtered by using a 0.45-μm syringe filter before HPLC analysis. Concentration of the lysate was interpolated from a standard curve, and the intracellular concentration was determined based on the assumption the average cell volume was ≈1 pL (15).
PDI Assay_._
PDI (1 mg, Panvera, Madison, WI) activity was determined by using DnsHCysNO as substrate. PDI inhibition studies were performed in triplicate with PDI (0.25 μM) and DnsHCysNO (0.5–20 μM) coincubated with BSA-NO (0.1 μM, 1 μM, 10 μM). The reaction was performed in PBS supplemented with 50 μM GSH, and the data were obtained over 60 sec at 100-msec intervals via a Cary Eclipse fluorometer (Varian).
Cell Culture.
Chinese hamster lung fibroblasts (V 79–4 ATCC CCL-93) were cultured in DMEM (GIBCO/BRL) supplemented with 4 mM glutamine, 1.5 g/liter sodium bicarbonate, 4.5 g/liter glucose, 1.0 mM sodium pyruvate, and 10% FBS. Cells were grown to confluence on slides (Fisher) at 37°C with 10% atmospheric CO2. HUVECs (HUVEC-CRL 1730, ATCC) were grown in GIBCO's Ham F12K (GIBCO/BRL) with 2 mMl-glutamine, 1.5 g/liter sodium carbonate, 100 μg/ml heparin (Sigma), 50 μg of endothelial cell growth supplement (Sigma), and 10% FBS. The HUVECs were grown to confluence on slides at 37°C with 10% atmospheric CO2. HT1080 cells stably underexpressing or overexpressing PDI were produced and characterized as described by Jiang et al. (12). HT1080 cells were grown in DMEM supplemented with 4 mM glutamine, 1.5 g/liter sodium bicarbonate, 4.5 g/liter glucose, 1.0 mM sodium pyruvate, 600 μg/ml G14, and 10% FBS. Cells were grown to confluence on slides at 37°C with 10% atmospheric CO2.
csPDI Inhibition Studies.
For PDI inhibition studies, HUVECs and hamster lung fibroblasts were incubated with either 100 μM 5,5′-dithio-_bis_-3-nitrobenzoate (DTNB) (Sigma) or 100 μM 4-(_N_-(_S_-glutathionylacetyl)amino)-phenylarsenoxide (GSAO) for 30 min before fluorescence studies. GSAO was synthesized according to the methodology reported by Donoghue et al. (16) and stored in 20 mM Hepes, 0.14 M NaCl, 20 mM glycine, pH 7.4. The control for inhibition by GSAO also included a 30-min incubation of HUVECs with 100 μM 4-(_N_-(_S_-glutathionylacetyl)amino)benzoic acid, where the arsenic was replaced with a carboxylic group.
Detection of Secreted PDI Protein.
Secretion of PDI by HT1080 control, HT1080s, or HT1080as cells was assessed by washing 80% confluent cultures twice and incubating in DMEM without FBS for 6 h. Conditioned media were centrifuged at 3,000 × g for 10 min to remove cell debris, and media from 3 × 104 cells were resolved on SDS/10% PAGE, transferred to poly(vinylidene difluoride) membrane, and detected by Western blot.
Detection of Reduced csPDI Protein.
HT1080 cells stably overexpressing or underexpressing PDI were produced and characterized as described by Jiang et al. (12). The active site dithiol/disulfide of PDI on the cell surface is in either the dithiol or disulfide form. Cell surface-associated PDI containing an active site dithiol was measured by labeling the transfected cells with the membrane-impermeable thiol-reactive reagent, 3-(_N_-maleimidylpropionyl)biocytin (12). The biotin-labeled proteins were collected on streptavidin-agarose, and the PDI was quantified by Western blot (12).
Fluorescence Microscopy.
Cells were incubated in PBS containing 0.2 mM DnsHCys2 for 2 min. The incubation was followed by 4 × 30-sec washes with PBS. The fluorescent cells were incubated in 1 ml of PBS; cell images were acquired at 0.5 sec for 60 sec on the addition of 100 μl of varying GSNO concentrations (1.0 mM to 10 μM). The ambient temperature was maintained at 37°C by Binomic Controller BC 100 (20/20 Technology, Wilmington, NC). Cell images were captured on the addition of GSNO by using a real-time image acquisition system (northern eclipse 6.0, Empix, Mississauga, ON). The change in fluorescence intensity was determined by computer-assisted image analysis (northern eclipse 6.0). Fluorescence of cells was observed via a Zeiss Axiovert 200 microscope by phase and fluorescence microscopy with a ×40 fluor objective. Similar protocol was used to observe BSA-NO (200–1 μM) uptake in hamster lung fibroblast and HUVECs.
Results
In a previous study, we have shown that the S-nitrosation of DnsHCys resulted in ≈90% quenching of the dansyl fluorescence (14). This effect is believed to arise from the energy transfer from the dansyl (excitation 320 nm, emission 520 nm) to the n-π* transition state of the S-NO moiety (λmax, 543 nm; ref. 14).
DnsHCysNO as a Direct Fluorogenic Assay for PDI.
To date there are no direct assays for the RSNO denitrosation activity of PDI. To test whether DnsHCysNO could be used as a direct fluorogenic substrate for PDI, 100 μM DnsHCysNO was added to a stirred cuvette in a fluorometer. The slit widths were adjusted to yield minimal photolysis of the S-NO bond. On addition of 0.25 μM PDI the fluorescence increased in a time-dependent manner (Fig.1). The increase in fluorescence in Fig.1 shows the initial linear part of the trace from which the initial rates were estimated. This enhancement arises from the conversion of the less-fluorescent DnsHCysNO to the fluorescent DnsHCys by the action of PDI. When these experiments were repeated with varying [DnsHCysNO], the initial rates of fluorescence increase were well accommodated by the Michaelis–Menten equation, yielding an apparent_K_M for DnsHCysNO of ≈2 ± 0.5 μM (Fig. 2). To estimate the affinity of PDI for the physiologically relevant RSNO, BSA-NO, the [DnsHCysNO] steady-state kinetics was performed in the presence of constant [BSA-NO]. As can be observed from Fig. 2, BSA-NO was a potent competitive inhibitor of PDI-catalyzed DnsHCysNO denitrosation with an estimated _K_I of 1 ± 0.7 μM.
Figure 1.
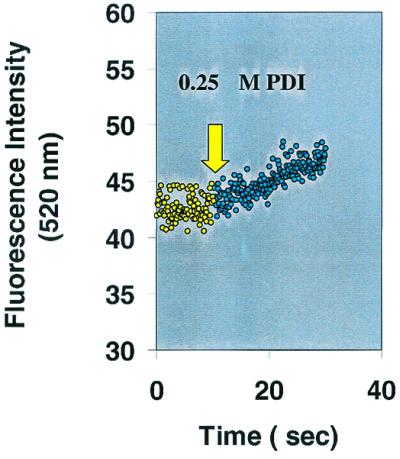
Demonstration of PDI catalyzed DnsHCysNO fluorescence increase as a function of time. The fluorescence of 25 μM DnsHCysNO before (yellow circles) and after the addition of 0.25 μM PDI (green circles). The rate of fluorescence increase on addition of PDI, 0.34 ± 0.02/sec, was significant in comparison to the blank rate, 0.02 ± 0.01/sec (n = 4).
Figure 2.
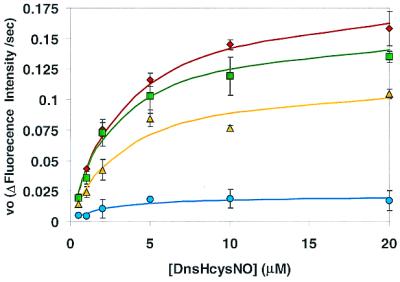
Plots of the initial rates of fluorescence increase, as a function of DnsHCysNO concentration in the presence of 0.25 μM PDI: no inhibitor (diamonds); or in the presence of 0.1 μM BSA-NO (squares), 1.0 μM BSA-NO (triangles), 10 μM BSA-NO (circles). Error bars represent SD (n = 6). The solid line represents the best fit of the data to the Michaelis–Menten equation.
DnsHCys2 as an Intracellular Probe for NO.
It is clear from the above studies that DnsHCysNO can be used to monitor PDI activity by the increase in the fluorescence signal. Therefore, if DnsHCys is placed in the cytosol, its fluorescence-quenching rate should represent the intracellular S-nitrosation flux. DnsHCys should compete with GSH as a nitrosation target and because of its hydrophobic dansyl moiety should have an advantage over GSH in the cytosol/membrane interface. To this end, the more hydrophobic, oxidized form of DnsHCys, DnsHCys2, was prepared. It was observed that DnsHCys2 rapidly accumulated in cells as the intracellular fluorescence reached a maximum within 2 min and remained constant for up to 20 min (data not shown).
Intracellular GSH concentrations are in the 1- to 10-mM range (17). Therefore, the first question was whether this concentration range (1 to 10 mM) is sufficient reducing potential for the amount of DnsHCys2 accumulated intracellularly on incubation of the cells with this compound. To test this, fibroblasts (0.5 × 106 cells) were incubated with 200 μM of DnsHCys2 for 2 min. The cells were washed and lysed in the presence of 1 μM iodoacetic acid. In this manner all of the reduced thiols would be alkylated, thus preventing their reoxidation. The cell extracts were centrifuged and chromatographed on an HPLC with a fluorescence detector. Authentic DnsHCys2 and_N_-dansyl-_S_-carboxymethylhomocysteine had retention times under the conditions used of 17.8 min and 28.9 min, respectively. The cell extract had one major fluorescent peak corresponding to the retention time of_S_-carboxymethyl-_N_-dansylhomocysteine, indicating that there is sufficient intracellular GSH to reduce the accumulated DnsHCys2. In addition, the peak area of _S_-carboxymethyl-_N_-dansylhomocysteine corresponded to ≈50 pmol/106cell. If it is assumed that the volume of a fibroblast is ≈1 pL, ≈50 μM of DnsHCys must have accumulated in the cells after 2 min (15).
DnsHCys fluorescence-quench kinetics was then performed in solution and with fibroblasts. In the solution studies, 25 μM DnsHCys2 was reduced with 100 μM GSH, and was treated with either 200 μM NO(aq), where aq is aqueous, or 200 μM GSNO. Under these conditions, [DnsHCys] in solution would be close to the value (50 μM) estimated to be accumulated in cells from the HPLC studies. Under the present experimental conditions NO(aq) when exposed to oxygenated buffer is expected to be rapidly converted to N2O3, a very efficient nitrosating agent. As expected, the fluorescence of DnsHCys was quenched on exposure to NO(aq) with_k_obs of 1.21 ± 0.067 sec−1 (Fig.3A). When GSNO was introduced to DnsHCys in solution the _k_obs was 2.14 ± 0.094 sec−1 (Fig. 3B).
Figure 3.

DnsHCys fluorescence-quench kinetics. Fluorescence decrease was monitored as a function of time. (Insets) The semiln plots from which the _k_obs was estimated. The solid line represents the best fit of the data to a first-order process (n = 3). (A) DnsHCys2 (25 μM) was reduced with 100 μM GSH and treated with 200 μM NO(aq) (triangles) in a 3.0-ml stirred fluorescence cuvette (n = 3). (B) DnsHCys2 (25 μM) was reduced with 100 μM GSH and treated with 200 μM GSNO(aq) (circles), in a 3.0-ml stirred fluorescence cuvette (n = 3). (C) The fluorescence microscope images of fibroblast cells grown on coverslips, were acquired at 0.2-msec intervals subsequent to the introduction of 100 μM GSNO to the coverslip holder compartment. (D) The quenching rates for intracellular DnsHCys by 200 μM extracellular NO(aq) (squares) were extracted with the aid ofnorthern exposure image processing software from the change in the intracellular fluorescence intensity/μm2 of microscope images of cells grown on cover slips collected at ≈250-msec intervals (n = 3). (E) The quenching rates for intracellular DnsHCys by 200 μM extracellular GSNO (diamonds) were extracted with the aid of northern exposure image processing software from the change in the intracellular fluorescence intensity/μm2 of microscope images of cells grown on cover slips collected at ≈250-msec intervals (n = 3).
The quenching rates for intracellular DnsHCys by extracellular RSNOs or NO were calculated from the change in the intracellular fluorescence intensity/μm2 of images collected at 100- to 500-msec intervals. A sample, time-dependent image series, where 200 μM GSNO was added to the culture media (at_t_ = 0), is presented in Fig. 3C. The_k_obs for extracellular NO(aq) (Fig. 3D) and GSNO (Fig. 3E) were 0.78 ± 0.022 sec−1 and 0.74 ± 0.078 sec−1, respectively. Although the_k_obs were the same within experimental error, extracellular NO (N2O3) yielded nearly 80% more intracellular nitrosation, in comparison to GSNO (≈18 μM vs. 10 μM as determined from the degree of DnsHCys quenching).
In the solution studies, the _k_obs for transnitrosation by GSNO was ≈2-fold larger than that for S-nitrosation by NO (N2O3). The fact that the rate of quenching of intracellular DnsHCys occurred with the same rate with NO or with GSNO, and that this rate was closer to that observed with NO in solution, suggests that the S-nitrosation of intracellular DnsHCys is mediated through N2O3 rather than by transnitrosation.
Up to this point it is clear that DnsHCys in the cytosol can be S-nitrosated by extracellular RSNO, but it's unknown if this process depends on csPDI. To obtain a clear answer, these experiments were repeated with HT1080 fibrosarcoma cells in which PDI was either underexpressed by antisense mediation or overexpressed. The overexpression and antisense-mediated underexpression of PDI resulted in a 70 ± 20% increase and 50 ± 25% decrease, respectively, in the surface-bound dithiol form of PDI as determined by labeling with the biotin-linked maleimide, 3-(_N_-maleimidylpropionyl)biocytin, which only reacts with free thiols. Jiang et al. (12) have established that under the conditions used here, two-thirds of the free thiols of csPDI are oxidized, thus underestimating csPDI levels by as much as 3-fold. This would appear to be the case as the total secreted PDI levels, as determined by Western blotting of the media indicated that overexpression or underexpression of PDI resulted in a 3.7-fold increase and 47% decrease, respectively, in secretion of PDI as compared with controls.
These cells were preloaded with DnsHCys and 200 μM BSA-NO was introduced to the media. In the cells in which the enzyme was underexpressed there was a small amount of intracellular fluorescence quenching (≈15% of control) (Fig. 4, triangles). On the other hand in cells where the PDI was overexpressed (Fig. 4, circles), the total intracellular fluorescence quenching was ≈3-fold larger than those of controls (Fig. 4, squares). These experiments clearly demonstrate the requirement for csPDI in the transfer of NO from extracellular RSNOs to those in the cytosol.
Figure 4.

Intracellular fluorescence intensity of DnsHCys2-treated HT1080 fibroblastoma cells on introduction of 100 μM BSA-NO (indicated by up arrow) to cells in which csPDI was underexpressed (triangles) or overexpressed (circles). Control cells transfected with vector alone are shown by the squares. Intracellular (fluorescence/μm2) was calculated from digitized images of the observation field taken every 250 msec with the aid of northern eclipse 5.0 imaging software.
In an effort to kinetically characterize csPDI in HT1080s, the cells were preincubated with 200 μM DnsHCys2 for 2 min and then exposed to varying concentrations of BSA-NO (1 μM to 200 μM). The fluorescence images were captured ≈ every 250 msec. The initial rates fluorescence quenching/μm2 were calculated with the aid of a northern eclipse image acquisition system. The plots of the initial rates as a function of [RSNO] were hyperbolic. The steady-state treatment of HT1080 cell kinetic data resulted in estimated apparent _K_M values for BSA-NO of 35 ± 2 μM and 35 ± 4 μM for control and csPDI overexpressing cells, respectively. In addition, the estimated_V_max for the csPDI overexpressing cell line was ≈2-fold larger than controls (Fig.5A).
Figure 5.

Kinetics of csPDI-catalyzed intracellular DnsHCys S-nitrosation. (A) Initial rates of intracellular DnsHCys fluorescence quenching as a function of BSA-NO concentration. Control HT1080 cells are indicated by circles, and HT1080 cells overexpressing csPDI are indicated by squares. The error bars represent SD (n = 6). The solid line represents the best fit of the data to the Michaelis–Menten equation. (B) Initial rates of intracellular DnsHCys fluorescence quenching as a function of BSA-NO concentration. HUVECs, no inhibitor (circles); 100 μM DTNB (squares). The error bars represent SD (n = 6). The solid line represents the best fit of the data to the Michaelis–Menten equation. (C) Initial rates of intracellular DnsHCys fluorescence quenching as a function of BSA-NO concentration. Hamster lung fibroblasts, no inhibitor (circles); 100 μM DTNB (squares). The error bars represent SD (n = 6). The solid line represents the best fit of the data to the Michaelis–Menten equation. (D) Dynamic fluorescence quenching of intracellular DnsHCys fluorescence on the addition of 200 μM BSA-NO (indicated by arrow). HUVECs, no inhibitor (triangles); 100 μM GSAO (circles); 100 μM 4-(_N_-(_S_-glutathionylacetyl)amino)benzoic acid (diamonds). The error bars represent SD (n = 6).
Next, kinetic studies were performed on HUVECs (Fig. 5B) and hamster lung fibroblasts (Fig. 5C) in culture, yielding apparent _K_M values for BSA-NO of 17 ± 2 μM and 12 ± 3 μM, respectively. Another piece of evidence that intracellular DnsHCys is measuring csPDI activity came from the fact that the initial rates of fluorescence quenching were inhibited by DTNB (Fig. 5 B and C, squares) and nearly totally inhibited by GSAO (Fig. 5D, circles). Time-dependent intracellular fluorescence quenching with 4-(_N_-(_S_-glutathionylacetyl)amino)benzoic acid, where the arsenic was replaced with a carboxylic group (Fig.5D, diamonds), was indistinguishable from controls (Fig.5D, triangles). DTNB is a nonspecific thiol reagent that will block free thiols on the cell surface. GSAO, on the other hand, is a trivalent arsenical with one of its sites occupied by the thiol group of GSH. The remaining two thiol sites specifically block proteins that, like PDI, contain vicinal dithiols. GSAO does not react with monothiol-containing proteins or peptides (16).
In an effort to determine the effects of membrane soluble N2O3 quenchers on the csPDI-dependent RSNO influx, HUVECs were grown in the presence of 10 μM α-tocopherol for 14 h. Jiang et al. (18) have reported that under these conditions epithelial cells were able to accumulate ≈0.9 nmol of α-tocopherol/106 cells, which translates to a membrane concentration of ≈5 mM. The rates of intracellular DnsHCys quenching by the extracellular introduction of 25 μM BSA-NO then were compared in α-tocopherol-treated and control cells (Fig.6A). As can been seen, the initial rates of intracellular nitrosation was inhibited by ≈70% in the α-tocopherol-treated cells. In addition, α-tocopherol had a very large effect on the _K_m and_V_max estimated for csPDI-catalyzed BSA-NO denitrosation as determined from the intracellular DnsHCys quenching rates (Fig. 6B). The_K_m increased from 17 μM in controls to 45 μM in α-tocopherol-treated cells, whereas the_V_max decreased by nearly 4-fold (control, 26 μM/sec; α-tocopherol-treated, 7 μM/sec).
Figure 6.
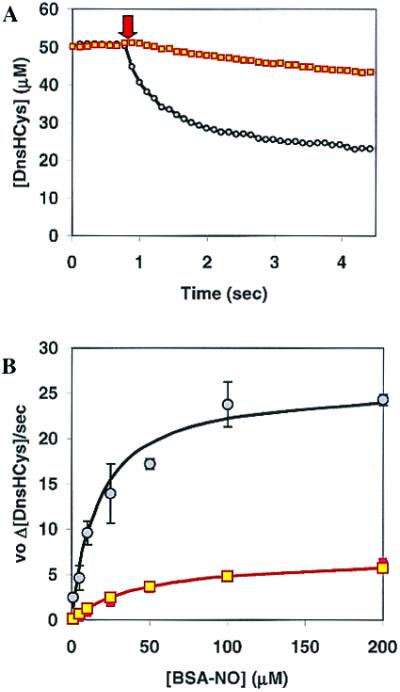
The effect of α-tocopherol on csPDI-catalyzed intracellular S-nitrosation. (A) The intracellular fluorescence of DnsHCys2-pretreated HUVECs, controls (circles), or HUVECs grown in the presence of 10 μM α-tocopherol for 14 h (squares), monitored subsequent to the extracellular addition of 25 μM BSA-NO (indicated by down arrow). (B) Initial rates of intracellular DnsHCys fluorescence quenching as a function of BSA-NO concentration. HUVECs, controls (circles), or HUVECs grown in the presence of 10 μM α-tocopherol for 14 h (squares). The error bars represent SD (n = 6). The solid line represents the best fit of the data to the Michaelis–Menten equation.
Discussion
There is increasing evidence that S-nitrosylation plays a large role in the regulation of key metabolic enzymes. And unlike NO whose effects are localized, NO derivatives of serum proteins like albumin can carry NO far from its point of synthesis to regulate vascular tone and platelet function. The other advantage of RSNOs over NO is that the delivery of RSNO-bound NO to the cytosol can be regulated (11). In addition, Mayer et al. (19) have suggested GSNO as an intermediate in the NO/cGMP signaling pathway. Transport mechanisms for RSNO-bound NO into live cells have been speculated on (11, 20). Studies of this process have been hampered by the inability to directly measure the kinetics of uptake in living cells.
If RSNOs are central players in signal transduction, there must be an efficient process by which their bound NO is transferred from the extracellular environment to the cytosol. Recent work by Zai et al. (11) suggested a central role for csPDI in this process. This membrane-bound enzyme is capable of catalyzing thiol-disulphide exchange as well as transnitrosation reactions (11, 21).
In this study, we have demonstrated that DnsHCysNO can be used as a fluorogenic substrate for PDI with an apparent_K_M of 2 μM. Using this kinetic assay, we were also able to demonstrate that BSA-NO competitively inhibited this reaction with an apparent_K_I of ≈1 μM. We also demonstrated that DnsHCys could act as an intracellular kinetic probe for csPDI in live cells in vitro. The initial rates of fluorescence quenching as a function of extracellular RSNO was hyperbolic, indicating that a saturation phenomenon was being monitored. Furthermore, this saturable RSNO-dependent process was inhibitable by compounds commonly used as inhibitors of PDI (22, 23). Perhaps the most convincing piece of evidence that we were monitoring csPDI activity was obtained in the studies where the fluorescence quenching by extracellular BSA-NO was tested with HT1080 cells underexpressing or overexpressing PDI. The results of these experiments showed that overexpression increases csPDI levels by ≈1.7 ± 0.2-fold whereas underexpression decreased csPDI levels by ≈75 ± 25%. Once these values are adjusted for the fact that 3-(_N_-maleimidylpropionyl)biocytin labeling underestimates csPDI by as much as 3-fold (12), they closely mirror the ≈3-fold increase and ≈85% decrease in intracellular nitrosation on PDI overexpression and underexpression, respectively (Fig. 4). In addition, the fact that intracellular nitrosation was totally abolished with the cell-impermeant, vicinal-dithiol-specific reagent GSAO (16) indicates the absolute requirement for csPDI and other vicinal dithiol-containing proteins in the transport of RSNO bound NO to the cytosol. We reach this conclusion because GSAO does not react with single protein thiols or with Cu- or Zn-containing proteins.
We now come to the physiological significance of the kinetic constants estimated here. The _K_I for BSA-NO was ≈1 μM (Fig. 2). The human serum albumin (HSA)-NO level in serum of healthy individuals recently has been estimated by a combination of GC and MS to be 181 ± 150 nM (n = 22) (24). This means that with the kinetic constants estimated here the enzyme would be operating at ≈17% of _V_max. However, the serum albumin-NO concentration on the cell surface may be many-fold larger as many cells contain surface receptors for HSA (25). In addition Pietraforte et al. (25) suggested that HSA-NO might concentrate on cell surface via electrostatic interactions.
The _K_M estimated from the intracellular DnsHCys quenching rates was ≈10- to 30-fold larger depending on the cell type studied here. This observation might be related to the manner and the form in which NO released by csPDI, nitrosates thiols in the cytosol. The mechanism(s) by which PDI catalyzes the release of NO (or NO+ or NO−) from extracellular RSNOs is unknown; however, Zai et al. (11) have demonstrated that OxyMyoglobin is converted to MetMyoglobin in the presence of PDI and RSNOs, indicating that NO is released during catalysis. Therefore, a proposed plausible mechanism was (11):
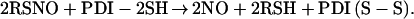
For csPDI to catalyze further denitrosation a mechanism is required to reduce the oxidized form of the protein. This may be accomplished the plasma membrane NADH-oxidoreductase system, which has been implicated in reduction of extracellular protein disulfide bonds (26). Another possibility is thioredoxin reductase (27), which is secreted by peripheral blood cells and is in plasma at a concentration of 18 ng/ml (28).
Another unknown is how the NO released by PDI nitrosates intracellular thiols. A potential clue to this comes from the work of Lancaster and coworkers (29) who demonstrated that the reaction of NO and O2, both hydrophobic molecules, is ≈300-fold more rapid in cell membranes than in aqueous media. The product of this reaction is the powerful nitrosating agent N2O3. More recently, Nudler and coworkers (30) have demonstrated that under physiological concentrations of NO, N2O3 forms inside protein-hydrophobic cores and causes autonitrosylation within the protein interior.
We propose, therefore, that csPDI-catalyzed NO released from RSNOs accumulates in the membrane where it reacts with O2 to produce N2O3. Intracellular thiols then get nitrosated by N2O3 at the membrane-cytosol interface (Fig.7). If this hypothesis is correct, depletion of membrane N2O3 with membrane-soluble quenchers such as vitamin E (α-tocopherol) should inhibit intracellular nitrosation by csPDI (31). This is indeed what was observed when HUVECs were preincubated with α-tocopherol. The rate of intracellular DnsHCys fluorescence quenching decreased by ≈70% in cells where membrane vitamin E levels were elevated. In addition, vitamin E increased the _K_M by ≈3-fold and decreased _V_max by ≈4-fold. Furthermore, this hypothesis can account for the ≈10- to 30-fold difference in the _K_I estimated for BSA-NO in the direct PDI assay and the_K_M estimated from intracellular quenching of DnsHCysNO. When the S-nitrosation probe is in the cytosol, the amount of NO produced by csPDI at low BSA-NO will likely yield smaller amounts of N2O3, which will be quenched by the normal levels of vitamin E present in the membrane. Only at high BSA-NO levels would there be enough N2O3 buildup to permit the detection of intracellular nitrosation via DnsHCys fluorescence quenching. This can explain why the apparent_K_M in the α-tocopherol-treated cells was ≈3-fold larger than in controls.
Figure 7.

Postulated mechanism for intracellular S-nitrosation by csPDI-catalyzed NO released from extracellular RSNOs.
In this study we have introduced DnsHCysNO as a direct fluorogenic substrate for PDI and using this compound have estimated the affinity of PDI for BSA-NO. We also have used DnsHCys in the cytosol as an S-nitrosation probe to demonstrate the absolute requirement for csPDI for the transfer of extracellular RSNO in to the cytosol. In addition, we have proposed and tested a mechanism by which the NO released by csPDI from RSNOs can nitrosate intracellular thiols at the membrane-cytosol interface. The fact that this process is affected by membrane-antioxidant levels raises the possibility that cells can regulate the bidirectional movement of RSNO-bound NO by csPDI activity/expression as well as by controlling the membrane redox status.
Acknowledgments
This work was supported by a research grant (to B.M.) from the Natural Sciences and Engineering Research Council of Canada and an equipment grant from the Canada Foundation for Innovation.
Abbreviations
HCys
homocysteine
DnsHCys
_N_-dansylhomocysteine
DnsHCys2
_N_-dansylhomocystine
DnsHCysNO
_S_-nitroso-_N_-dansylhomocysteine
GSNO
_S_-nitrosoglutathione
BSA-NO
_S_-nitroso-BSA
PDI
protein disulfide isomerase
csPDI
cell-surface PDI
DTNB
5,5′-dithio-_bis_-3-nitrobenzoate
RSNO
_S_-nitrosothiol
HUVEC
human umbilical vein endothelial cell
GSH
glutathione
GSAO
4-(_N_-(_S_-glutathionylacetyl)amino)-phenylarsenoxide
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Wink D A, Cook J A, Kim S Y, Vodovotz Y, Pacelli R, Krishna M C, Russo A, Mitchell J B, Jourd'heuil D, Miles A M, Grisham M B. J Biol Chem. 1997;272:11147–11151. doi: 10.1074/jbc.272.17.11147. [DOI] [PubMed] [Google Scholar]
- 2.Girard P, Potier P. FEBS Lett. 1993;320:7–8. doi: 10.1016/0014-5793(93)81645-g. [DOI] [PubMed] [Google Scholar]
- 3.Stamler J S, Simon D I, Osborne J A, Mullins M E, Jaraki O, Michel T, Singel D J, Loscalzo J. Proc Natl Acad Sci USA. 1992;89:444–448. doi: 10.1073/pnas.89.1.444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Myers P R, Minor R L, Jr, Guerra R, Jr, Bates J N, Harrison D G. Nature (London) 1990;10, 345:161–163. doi: 10.1038/345161a0. [DOI] [PubMed] [Google Scholar]
- 5.Park J K J, Kostka P. Anal Biochem. 1997;249:61–66. doi: 10.1006/abio.1997.2159. [DOI] [PubMed] [Google Scholar]
- 6.Ignarro L J, Lippton H, Edwards J C, Baricos W H, Hyman A L, Kadowitz P J, Gruetter C A. J Pharmacol Exp Ther. 1981;218:739–749. [PubMed] [Google Scholar]
- 7.Ignarro L J, Edwards J C, Gruetter D Y, Barry B K, Gruetter C A. FEBS Lett. 1980;110:275–278. doi: 10.1016/0014-5793(80)80091-3. [DOI] [PubMed] [Google Scholar]
- 8.Marzinzig M, Nussler A K, Stadler J, Marzinzig M, Barthlen W, Nussler N C, Beger H G, Morris S M, Bruckner U B. Nitric Oxide Biol Chem. 1997;1:177–189. doi: 10.1006/niox.1997.0116. [DOI] [PubMed] [Google Scholar]
- 9.Cook J A, Kim S Y, Teague D, Krishna M C, Pacelli R, Mitchell J B, Vodovotz Y, Nims R W, Christodoulou D, Miles A M, et al. Anal Biochem. 1996;238:150–158. doi: 10.1006/abio.1996.0268. [DOI] [PubMed] [Google Scholar]
- 10.Hotchkiss K A, Matthias L J, Hogg P J. Biochim Biophys Acta. 1998;1388:478–488. doi: 10.1016/s0167-4838(98)00211-8. [DOI] [PubMed] [Google Scholar]
- 11.Zai A, Rudd A, Scribner A W, Loscalzo J. J Clin Invest. 1999;103:393–399. doi: 10.1172/JCI4890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jiang X M, Fitzgerald M, Grant C M, Hogg P J. J Biol Chem. 1999;274:2416–2423. doi: 10.1074/jbc.274.4.2416. [DOI] [PubMed] [Google Scholar]
- 13.Pawloski J R, Hess T D, Stamler J S. Nature (London) 2001;409:622–626. doi: 10.1038/35054560. [DOI] [PubMed] [Google Scholar]
- 14.Ramachandran N, Jacob S, Zielinski B, Curatola G, Mazzanti L, Mutus B. Biochem Biophys Acta. 1999;1430:149–154. doi: 10.1016/s0167-4838(98)00286-6. [DOI] [PubMed] [Google Scholar]
- 15.Freshney I R. Culture of Animal Cells. New York: Wiley-Liss; 2000. [Google Scholar]
- 16.Donoghue N, Yam P T W, Jiang X, Hogg P J. Protein Sci. 2000;9:2436–2445. doi: 10.1110/ps.9.12.2436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Till U, Bosia A, Losche W, Spangenberg P, Pescarmona G P. Folia Haematol Int Mag Klin Morphol Blutforsch. 1998;115:415–419. [PubMed] [Google Scholar]
- 18.Jiang Q, Elson-Schwab I, Courtemanche C, Ames B N. Proc Natl Acad Sci USA. 2000;97:11494–11499. doi: 10.1073/pnas.200357097. . (First Published September 26, 2000; 10.1073/pnas.200357097) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mayer B, Pfeiffer S, Schrammel A, Koesling D, Schmidt K, Brunner F. J Biol Chem. 1998;273:3264–3270. doi: 10.1074/jbc.273.6.3264. [DOI] [PubMed] [Google Scholar]
- 20.Hogg N, Singh R J, Konorev E, Joseph J, Kalyanaraman B. Biochem J. 1997;323:477–481. doi: 10.1042/bj3230477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ferrari D M, Soling H D. Biochem J. 1999;339:1–10. [PMC free article] [PubMed] [Google Scholar]
- 22.Essex D W, Li M. Br J Haematol. 1999;104:448–454. doi: 10.1046/j.1365-2141.1999.01197.x. [DOI] [PubMed] [Google Scholar]
- 23.Mandel R, Ryser H J P, Ghani F, Wu M, Peak D. Proc Natl Acad Sci USA. 1993;90:4112–4119. doi: 10.1073/pnas.90.9.4112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tsikas D, Sandmann J, Gutzki F M, Stichtenoth D O, Frolich J C. J Chromatogr B. 1999;726:13–24. [PubMed] [Google Scholar]
- 25.Pietraforte D, Mallozzi C, Scorza G, Minetti M. Biochemistry. 1995;34:7177–7185. doi: 10.1021/bi00021a032. [DOI] [PubMed] [Google Scholar]
- 26.Wolvetang E J, Larm J A, Moutsoulas P, Lawen A. Cell Growth Differ. 1996;7:1315–1325. [PubMed] [Google Scholar]
- 27.Lundstrom J, Holmgren A. J Biol Chem. 1990;265:9114–9120. [PubMed] [Google Scholar]
- 28.Soderberg A, Sahaf B, Rosen A. Cancer Res. 2000;60:2281–2289. [PubMed] [Google Scholar]
- 29.Liu X, Miller M J, Joshi M S, Thomas D D, Lancaster J R. Proc Natl Acad Sci USA. 1998;95:2175–2179. doi: 10.1073/pnas.95.5.2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nedospasov A, Rafikov R, Beda N, Nudler E. Proc Natl Acad Sci USA. 2000;97:13543–13548. doi: 10.1073/pnas.250398197. . (First Published November 28, 2000; 10.1073/pnas.250398197) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hogg N, Singh J R, Goss P A S, Kalyanaraman B. Biochem Biophys Res Commun. 1996;224:696–702. doi: 10.1006/bbrc.1996.1086. [DOI] [PubMed] [Google Scholar]