Crystal structure of coproporphyrinogen III oxidase reveals cofactor geometry of Radical SAM enzymes (original) (raw)
Abstract
‘Radical SAM’ enzymes generate catalytic radicals by combining a 4Fe–4S cluster and _S_-adenosylmethionine (SAM) in close proximity. We present the first crystal structure of a Radical SAM enzyme, that of HemN, the Escherichia coli oxygen-independent coproporphyrinogen III oxidase, at 2.07 Å resolution. HemN catalyzes the essential conversion of coproporphyrinogen III to protoporphyrinogen IX during heme biosynthesis. HemN binds a 4Fe–4S cluster through three cysteine residues conserved in all Radical SAM enzymes. A juxtaposed SAM coordinates the fourth Fe ion through its amide nitrogen and carboxylate oxygen. The SAM sulfonium sulfur is near both the Fe (3.5 Å) and a neighboring sulfur of the cluster (3.6 Å), allowing single electron transfer from the 4Fe–4S cluster to the SAM sulfonium. SAM is cleaved yielding a highly oxidizing 5′-deoxyadenosyl radical. HemN, strikingly, binds a second SAM immediately adjacent to the first. It may thus successively catalyze two propionate decarboxylations. The structure of HemN reveals the cofactor geometry required for Radical SAM catalysis and sets the stage for the development of inhibitors with antibacterial function due to the uniquely bacterial occurrence of the enzyme.
Keywords: Radical enzyme mechanism/Radical SAM enzymes/_S_-adenosylmethionine configuration/tetrapyrrole biosynthesis
Introduction
Biosynthesis of heme and chlorophyll requires coproporphyrinogen III to be converted to protoporphyrinogen IX by oxidatively decarboxylating the propionate side chains of rings A and B to the corresponding vinyl groups (Figure 1A). Two unrelated enzymes catalyze this reaction: HemF, the oxygen-dependent coproporphyrinogen III oxidase; and HemN, the oxygen-independent coproporphyrinogen III oxidase (Jordan, 1981; Dailey, 2002; Friedmann and Tauer, 1992; Chadwick and Ackrill, 1994).

Fig. 1. Schematic representation of the enzymatic reaction of HemN. (A) HemN oxidatively decarboxylates coproporphyrinogen III to protoporphyrinogen IX by converting the propionate side chains of rings A and B to the corresponding vinyl groups. (B) The first reaction step common to HemN and all Radical SAM enzymes: a reduced 4Fe–4S cluster transfers an electron to the sulfonium of _S_-adenosylmethionine (SAM). The C5′–S+ bond of SAM is cleaved, producing methionine and a highly oxidizing 5′-deoxyadenosyl radical. The radical abstracts a hydrogen atom from a substrate RH (the substrate may itself be an enzyme), creating the corresponding substrate radical (R·). (C) In the reaction catalyzed by HemN, the 5′-deoxyadenosyl radical abstracts a hydrogen atom from the β-C atom of the substrate propionate side chain. CO2 is eliminated, and a single electron transfer to an electron acceptor gives rise to the vinyl group of the reaction product.
The oxygen-independent coproporphyrinogen III oxidase HemN, an iron–sulfur protein, belongs to the recently discovered ‘Radical SAM’ protein family (Sofia et al., 2001; Layer et al, 2002). All members of this protein family contain an unusual 4Fe–4S cluster coordinated through three conserved cysteine residues in a characteristic CxxxCxxC motif (Sofia et al., 2001). In its reduced state, the iron–sulfur cluster transfers a single electron to _S_-adenosylmethionine (SAM), inducing the reductive cleavage of SAM to methionine and a 5′-deoxyadenosyl radical. This highly oxidizing radical abstracts a hydrogen atom from an appropriately positioned carbon atom, creating a substrate (as in HemN, lysine-2,3-aminomutase) or a catalytic glycyl radical [as in activating enzymes of class III ribonucleotide reductase and pyruvate formate-lyase (Cheek and Broderick, 2001; Fontecave et al., 2001; Frey and Magnusson, 2003; Jarrett, 2003) (Figure 1B). Whereas SAM is consumed in some Radical SAM enzymes [HemN, biotin synthase (Uglava et al., 2003) and pyruvate formate lyase-activating enzyme (Frey et al., 1994)], it is restored and reused in others [spore photoproduct lyase (Cheek and Broderick, 2002) and lysine-2,3-aminomutase (Frey and Magnusson, 2003)]. Another feature common to all Radical SAM proteins is a glycine-rich sequence motif proposed to be the SAM-binding site. Radical SAM enzymes are found in numerous fundamental biosynthetic pathways such as vitamin, cofactor, DNA precursor or antibiotic biosyntheses. Despite catalyzing widely different reactions and having very low sequence conservation, Radical SAM enzymes are presumed to bear a common core domain (Sofia et al., 2001). Three-dimensional information has, however, been lacking so far, despite a wealth of biochemical data for many members of the family.
We present the crystal structure of HemN, the first structure determination of a member of the Radical SAM family. The iron–sulfur protein was co-crystallized with the cofactor SAM. The 2.07 Å crystal structure provides insight into two SAM-binding sites, one of these in close proximity to the iron–sulfur cluster revealing the molecular basis of the first common reaction step of all Radical SAM enzymes.
Results
Structure determination and refinement
HemN was expressed, purified and crystallized under strictly anaerobic conditions in an anaerobic chamber. This ensured the integrity of the 4Fe–4S cluster in a large proportion of the enzyme population, stabilizing the molecular structure and allowing successful protein crystallization. Diffraction data of cryo-cooled crystals were collected at the Fe K-edge and the crystal structure was solved by multiple anomalous difference (MAD) techniques (Table I). The anomalous signal extends to the resolution limit, allowing the localization of the iron–sulfur cluster by Patterson methods. AUTOSHARP (www.globalphasing.com) was used for integrated phasing (SHARP; de la Fortelle and Bricogne, 1997), solvent flattening (DM; Cowtan and Main, 1998) as well as automated model building (ARPWARP; Lamzin and Wilson, 1993) and refinement (REFMAC5; Murshudov et al., 1997), greatly enhancing the speed of structure solution. Due to severe radiation sensitivity, the 1.80 Å high-resolution data set collected after the MAD experiment proved ineffectual in phasing and of limited use in refinement, as disordered regions are significantly extended. The structure of HemN has therefore been refined against the first data set. The final _R_-factor is 15.4% (_R_free = 18.7%) with good geometry (Table I). Overall, 439 of 457 residues have been located in the electron density map. Disordered regions of the polypeptide not included in the final model include the residues 1–3, 19–21 and 446–457. Residues 4–6, 17–18 and 22–24 are partly disordered but have been included in the final model. Three residues lie outside the favored regions of the Ramachandran plot: Ser25 in the poorly defined N-terminal strand and Met287 at the interface between the catalytic domain and the N-terminus are both partly disordered and presumably adopt an ordered conformation only on substrate binding. Gln172 is involved in SAM binding. Crystal contacts exclusively involve the catalytic domain of HemN. Modeling interdomain movement during refinement (TLS refinement; Murshudov et al., 1997) indicates significant freedom of movement of the C-terminal domain and N-terminal trip-wire (see below). Correspondingly, the average temperature factor of these domains is 50.4 Å2, in contrast to 25.4 Å2 for the catalytic domain (including cofactors).
Table I. Data collection and refinement statistics.
| Data collection | |||
|---|---|---|---|
| Data set | Inflection | Peak | High energy remote |
| Wavelength (Å) | 1.742 | 1.739 | 1.542 |
| Space group | _P_63 | ||
| Unit cell lengths (Å) | a = b = 114.0, c = 76.5 | ||
| Resolution range (Å)a | 30–2.07 (2.11–2.07) | 30–2.06 (1.53–1.50) | 30–1.84 (1.86–1.84) |
| _R_merge (%)a | 6.6 (18.2) | 7.4 (14.4) | 7.7 (38.5) |
| I/σIa | 30.6 (10.2) | 29.2 (9.7) | 27.0 (3.4) |
| Completeness (%)a | 99.1 (96.8) | 97.4 (90.7) | 98.8 (84.3) |
| Redundancya | 9.4 (8.8) | 8.5 (2.7) | 8.5 (4.5) |
| Unique reflections | 34 220 | 34 613 | 48 413 |
| Wilson plot _B_-factor | 27.2 | 26.0 | 25.5 |
| Refinement | |||
| Resolutiona | 20–2.07 (2.12–2.07) | ||
| R (%)a | 15.4 (15.8) | ||
| _R_free (%)a | 18.7 (21.5) | ||
| No. of reflectionsa | |||
| Working set | 59 440 (2334) | ||
| Test set | 3170 (133) | ||
| Water molecules | 398 | ||
| Average _B_-factor (Å2)b | 34.0 (15.8) | ||
| R.m.s.d. bond lengths (Å) | 0.024 | ||
| R.m.s.d. bond angles (°) | 2.1 | ||
| Ramachandran plot (%) | |||
| Allowed | 92.3 | ||
| Additional | 7.2 | ||
| Generous | 0 | ||
| Disallowed (%) | 0.5 |
Overall structure of HemN
HemN is a monomeric protein consisting of two distinct domains (Figure 2A). The N-terminal domain, significantly larger than the C-terminal domain, comprises residues 36–364. It is characterized by a curved, 12-stranded, largely parallel β-sheet. Only three of the 12 strands, located near or at the very end of the sheet, arrange in an antiparallel fashion (Figure 2B). α-Helices predominantly decorate the outer surface of the β-sheet. The six N-terminal β-strands (residues 50–282) are part of repeated β/α motifs (or βαα variations) forming the central core of the domain. Structurally, the curved (β/α)6 repeat bears some resemblance to known (β/α)8 or TIM barrel domains, including that of the β-amylase of Bacillus cereus (PDB code: 1B9Z), human glucuronidase (1BHG) and, intriguingly, uroporphyrinogen decarboxylase (1URO) or HemE, the preceding enzyme in heme biosynthesis (Whitby et al., 1998). Compared with the TIM barrel, individual β-strands are, however, less strongly inclined relative to the barrel axis and the curvature of the β-sheet is not nearly as tight. The missing (β/α) motifs of the (β/α)6 or three-quarter barrel open the barrel laterally, resulting in a substrate-binding pocket perpendicular to the β-barrel axis rather than aligned with this axis as in many TIM barrel proteins. Similarly to TIM barrels, loops at the N-terminal ends of β-strands tend to be short while those at the C-terminal ends are often long, without much additional secondary structure. This is particularly true for the two loops following strands β1 and β6, which cover the central void of the β-barrel on the C-terminal side, while a single long loop (after β-strand 8) bearing two short β-strands connected by a hairpin loop (b1–loop–b2 in Figures 2B and 3B) plugs the void on the opposite side, leaving only the lateral opening.

Fig. 2. Structure of HemN. (A) A ribbons-type and (B) a schematic representation of the secondary structure elements. HemN consists of two distinct domains (shades of blue and red) as well as an elongated N-terminal region termed a trip-wire (green). The catalytic domain is built around a 12-stranded, largely parallel β-sheet. At its core, the N-terminal region bears a three-quarter barrel, a (βα)6 variation of the (βα)8 TIM barrel. This core binds all cofactors, a 4Fe–4S cluster and two SAM molecules. The N-terminal trip-wire and the C-terminal domain probably participate in substrate binding. A CxxxCxxC motif, conserved in all Radical SAM proteins, is located in a loop following the first β-strand of the central barrel. The three cysteines (small yellow circles) bind three of the Fe ions of the cluster.

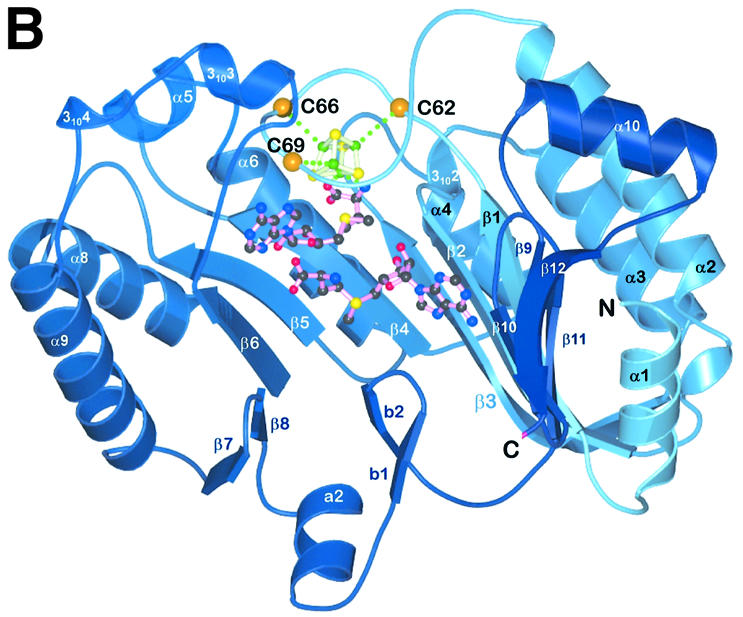

Fig. 3. Detailed views of the cofactors. (A) The electron density associated with the cofactors and the CxxxCxxC motif, conserved in all Radical SAM proteins. The 4Fe–4S cluster is rendered in green (Fe) and yellow (S), while pink-colored bonds highlight SAM1 and SAM2. Both (S)- (above) and (R)- (below) sulfonium sulfur configurations are observed for SAM1. SAM2 is rotationally disordered around the C5′–S+ bond, resulting in discontinuous electron density. (B) The cofactors occupy the central void of the catalytic domain near the C-terminal ends of the three-quarter barrel β-strands. Orange spheres mark the Cα positions of conserved cysteines. (C) A schematic depiction of inter-cofactor distances and amino acid residues involved in binding the cofactors. The (S)-sulfur is presented in yellow and the (R)-sulfur in orange. Green arcs represent hydrophobic interactions.
The β-sheet of the three-quarter barrel is extended at either end by additional β-strands deepening the substrate-binding tunnel. Though physically located in the middle of the domain, the three-quarter barrel corresponds to the N-terminal residues 50–282. At its C-terminal end, it is directly complemented by two β-strands followed by a long loop bearing the β-strand–loop–β-strand plug of the central void. The polypeptide then leads back to the N-terminal end of the three-quarter barrel, where a further four β-strands complete this domain. The combination of the 12-stranded β-sheet closed above and below the β-barrel creates a domain reminiscent of a cupped hand or crucible with a deep active site tunnel, to accommodate both the cofactors and the large coproporphyrinogen III substrate molecule (see below).
As far as we can ascertain, the N-terminal domain is unique. Initial secondary structure prediction analyses of other Radical SAM proteins indicate that the three-quarter barrel with its lateral opening may be a common feature of all Radical SAM proteins.
The C-terminal domain connected to the N-terminal domain by a short loop following the last β-strand consists of a bundle of four, roughly parallel, α-helices and a small, three-stranded antiparallel β-sheet. Significant structural homologs have not been identified. However, as this domain bears similarly highly conserved regions as the N-terminal domain, it must be functionally important to the enzyme. Most likely is a role in covering and partly filling the substrate-binding crevice of the N-terminal domain to shield the substrate from the solvent once it has bound. Sequence homology to other Radical SAM proteins could not be detected, indicating that this domain is presumably HemN specific.
Structurally, the N-terminal residues (4–35) belong to neither the catalytic nor the C-terminal domain. Instead, the first 35 residues adopt an extended conformation without pronounced secondary structure. It is loosely bound in an extended cleft between the N- and the C-terminal domains, wrapping around the latter. Although partly disordered (residues 19–21), it bears an amino acid sequence G20PRYTSYPTA29 highly conserved in all HemNs possibly involved in substrate recognition, and partly covers the entrance to the active site (see below). Structurally, it may, therefore, function akin to a ‘trip-wire’, stabilizing on substrate binding and possibly inducing the C-terminal domain to rearrange and close the active site.
The 4Fe–4S cluster and SAM cofactors
The crystal structure of HemN contains three cofactors, a 4Fe–4S cluster and two SAM molecules, henceforth denoted SAM1 and SAM2. The 4Fe–4S cluster and SAM1 are well resolved in the electron density map (Figure 3A). Rotational disorder around the C5′–S+ bond in SAM2 leads to the interruption of the electron density. All cofactors are bound in close mutual proximity within the active site pocket of the catalytic domain near the C-terminal end of the parallel β-strands of the three-quarter barrel (Figure 3B). The 4Fe–4S cluster is bound in the deepest recesses of the active site pocket, near the center of the three-quarter barrel. Three of its Fe ions are coordinated through three characteristically conserved cysteines (Cys62, Cys66 and Cys69 in HemN) of the Radical SAM CxxxCxxC motif (Sofia et al., 2001). The cysteines are located in an extended loop immediately C-terminal of the β-strand β1 (orange spheres in Figure 2). This flattened, circular loop laterally wraps around the 4Fe–4S cluster. Overall, the geometry, individual bonding distances within the cluster and distances to coordinating cysteines are typical of 4Fe–4S clusters. The environment of the cluster is, however, strongly polarized. Hydrophobic residues surround roughly one half of the cluster facing the cysteine-rich loop. Apart from the cysteines, Phe68, which precedes the third conserved cysteine and is invariantly an aromatic residue in all Radical SAM proteins (Sofia et al., 2001), as well as Leu63, Ile74, Val75 and the aliphatic part of Lys73 contribute to this hydrophobic region. Hydrophilic and charged neighbors surround the other half of the cluster. The amide nitrogen of Gly113 and the side chains of Thr114, Asp147, Arg149 and Arg184, and two water molecules are no further than 4 Å from the cluster. An elongated stretch over the fourth Fe, however, remains uncovered by protein.
Immediately adjacent to the 4Fe–4S cluster, HemN binds SAM1. The cofactor is well defined and adopts a unique bent conformation. It is held in position by numerous specific interactions (Figure 3C) extensively involving the 4Fe–4S cluster and in turn completing the coordination sphere of the latter: the amino nitrogen and one carboxylate oxygen of the methionine moiety of SAM1 coordinate the fourth iron. The respective distances to the iron are 2.6 (N) and 2.3 Å (O). Like the cysteines coordinating the 4Fe–4S cluster, most residues involved in binding SAM1 are conserved in all HemN sequences (Figure 4A). Arg184 is pivotal in binding the carboxylate group of the SAM methionine moiety, while Q172 adopts a particularly unfavorable backbone conformation to bind both O3′ and O4′ of the SAM ribose moiety. Aromatic residues Phe68, Phe240 and Tyr242 and aliphatic Ile211 surround the adenine moiety, while hydrogen bonds to backbone atoms of Phe68, Gly70 and Ala243 ensure the correct orientation. Surprisingly, the sulfonium sulfur of SAM1 occupies two alternative positions. Both are clearly defined in the anomalous difference maps and are compatible with the expected bond lengths of SAM. The implication is that HemN recognizes SAM with both an (S) and (R) configuration at the chiral sulfonium sulfur. The (S) configuration is favored, as judged by the anomalous difference and electron density maps, representing ∼60% occupancy. In the following, we will refer to (S,S)- and (R,S)-SAM as (S)- and (R)-SAM, as the second chiral center, the methionine Cα atom, invariably bears the (S) configuration.


Fig. 4. Distribution of conserved residues in HemN. (A) Amino acid sequence of E.coli HemN. Residues conserved in 34 sequences of HemN are underlaid in dark pink; residues conserved in >90, 80 and 70%, respectively, of sequences are marked by progressively lighter shades. α-Helices are represented by rectangles, β-strands by arrows. See Figure 2 for color-coding and nomenclature. Filled circles, squares and inverted triangles below individual residues mark amino acids involved in 4Fe–4S cluster, SAM1 and SAM2 binding, respectively. Filled squares denote residues in domain–domain interactions. Residues postulated to be involved in binding the external electron donor, terminal electron acceptor and the coproporphyrinogen III substrate are marked by half circles, diamonds and plus signs, respectively, Surface representation of HemN (B) front (in stereo) and (C) back view. The degree of conservation (A) is mapped onto the molecular surface of the catalytic domain. Note that the highest concentration of conserved residues is found in the active site cleft and at domain–domain interfaces. The trip-wire and C-terminal domain are represented by green and red coils. Most of the outer surface is poorly conserved (white), with the exception of the proposed entrance to the terminal electron acceptor-binding pocket and, to a lesser extent, the binding site of the external electron donor.
In addition to the nitrogen and oxygen ligands to the fourth iron, the sulfonium sulfur of the (S)-SAM stereoisomer is located a mere 3.5 Å from the same iron and 3.6 Å from a neighboring sulfur atom. Presumably, therefore, electron transfer from the reduced 4Fe–4S cluster to SAM occurs through a favorable electronic interaction between the sulfonium sulfur and an Fe–S edge of the cluster.
A second binding site for SAM
In addition to SAM1, HemN unexpectedly binds a second SAM molecule (SAM2) within the same deep cleft of the N-terminal domain that binds the 4Fe–4S cluster and SAM1. SAM2 is located adjacent to SAM1 and its adenine moiety stacks on the aromatic side chain of Tyr56, conserved in all known sequences of HemN and shown to be essential for HemN catalysis (Layer et al., 2002). A second aromatic residue (Phe310), part of a HemN- conserved 308KNFQGYTT315 motif that creates the strand–turn–strand element plugging the three-quarter barrel (see above), is located on the opposite face of the planar structure. A small change in its χ1 and χ2 torsion angles could swing the side chain to stack upon the opposite face of the adenine moiety. In addition, the ribose moiety is clearly defined in the electron density, while the anomalous difference map indicates the presence of a sulfur covalently attached to the ribose C5′ atom, clearly identifying this molecule as SAM. The electron density is discontinuous after the S, but an additional structure of density is observed adjacent to the S (Figure 3A). We have interpreted this observation as a second molecule of SAM disordered about the C5′–S+ bond. Additional continuous electron density adjacent to the sulfonium sulfur is, however, only incompletely described by the disordered methionine moiety. This may indicate that this site is also partly occupied by a 5′-deoxy-5′-(methylthio)adenosine molecule, a known degradation product of SAM (Hoffman, 1986), plus a second ligand. The identity of this ligand, however, remains unclear.
Discussion
Radical SAM enzymes
SAM is most widely associated with its function of serving as a methyl group donor to methyltransferases, a function that is vital to myriad physiological processes (Schubert et al., 2003). 4Fe–4S clusters are similarly commonly known to function as redox centers in electron transfer reactions. However, in recent years, Radical SAM enzymes have been recognized to combine these two cofactors in a novel fashion. These enzymes bind a 4Fe–4S cluster and SAM in close proximity. Under suitable conditions, reduction of the 4Fe–4S cluster induces electron transfer to the SAM sulfonium, cleaving the C5′–S+ bond to produce the strongly oxidizing 5′-deoxyadenosyl radical. This reactive radical intermediate rapidly abstracts a hydrogen atom from a suitably placed hydrogen donor (protein or substrate) to generate the corresponding radical (Sofia et al, 2001; Frey, 2003; Jarrett, 2003).
Structurally, the requirements for a Radical SAM enzyme thus deviate substantially from those of methyltransferases and 4Fe–4S-binding proteins. Methyltrans ferases need to place a methyl group acceptor near the methyl moiety of SAM to facilitate transfer of the methyl group from one to the other. 4Fe–4S clusters are often buried within proteins, allowing electron transfer to occur over fairly large distances while protecting the cluster from direct chemical interaction. Radical SAM enzymes, in contrast, need to pair SAM with a 4Fe–4S cluster in such a way as to allow electron transfer first from an external electron donor onto the 4Fe–4S cluster and then in a second electron transfer step from the reduced cluster to the sulfonium sulfur of SAM. A hydrogen atom donor must be positioned near the ribose C5′ atom preferably prior to the cleavage of the C5′–S+ bond and formation of the 5′-deoxyadenosyl radical to ensure abstraction of the correct H atom.
The structure of HemN reveals that the catalytic domain is indeed unique and unrelated to both the typical seven-stranded mixed β-sheet domain observed for SAM-dependent methyltransferases (Schluckebier et al., 1995) and the atypical SAM-binding domains of other methyltransferases (Schubert et al., 2003). It is, furthermore, also unrelated to other 4Fe–4S-binding domains. At its core, the catalytic domain of HemN contains a partial barrel related to the TIM barrel, which we refer to as a three-quarter barrel. The three-quarter barrel bears only six βα motifs opening up the domain laterally. In the case of HemN, the resulting large active site cleft allows the large substrate coproporphyrinogen III to enter the active site.
Other features clearly support its role as the functional domain of a Radical SAM protein. The inner parallel β-sheet of the three-quarter barrel covered with α-helices on its outer surface provides overall structural rigidity and stability to the enzyme. The deep lateral active site cleft allows the vital 4Fe–4S cluster to be buried within the protein. This renders it inaccessible to most cell constituents, preventing its strong reduction potential from being lost. The catalytic domain furthermore binds SAM in particular close proximity to the 4Fe–4S cluster. The distances between the cofactors (Figure 3C) are similar to those spectroscopically inferred for pyruvate formate lyase-activating enzyme (Walsby et al, 2002; Cosper et al., 2003), biotin synthase (Cosper et al., 2003) and lysine-2,3-aminomutase (Cosper et al., 2000); however, note the differences below. This further supports the notion that Radical SAM enzymes are closely related. At the same time, structural features of HemN are likely to be conserved in other members of the family. Details that are in agreement include a direct coordination of the fourth Fe ion by both amide nitrogen and one carboxylate oxygen of the SAM methionine moiety. The spectroscopic models differed in their interpretation of whether the sulfonium sulfur of SAM is located nearest the same, fourth iron or an adjacent sulfur atom (Jarrett, 2003). The structure of HemN now reveals that both are, in fact, partly correct as the sulfonium is essentially equidistant from both the fourth iron (3.5 Å) and an adjacent sulfur atom (3.6 Å; Figure 3C). An Fe–C5′ distance of 5.2 Å is, however, nearer the 4.9 Å expected for the S+–S model than the 3.6 Å predicted by the S+–Fe model.
Sequence conservation
A comparison of 34 amino acid sequences of HemN proteins (Figure 4A) indicates that HemN is remarkably well conserved. Mapping the degree of conservation onto the surface of each domain (catalytic domain shown in Figure 4B) reveals that conserved residues are concentrated in particular areas, most prominently within the active site cleft. Clearly, this correlates with the requirement to bind cofactors and substrate within this cleft. The cysteines coordinating the 4Fe–4S cluster are obviously conserved, as they are conserved in all Radical SAM enzymes (Sofia et al., 2001). Similarly, Phe68, the residue preceding the third conserved cysteine, which was found invariantly to be aromatic in Radical SAM enzymes, is a phenylalanine in ∼50% of HemN sequences and a tyrosine in all others. Located between SAM1 and the 4Fe–4S cluster, this residue contributes to the hydrophobic region of the polarized 4Fe–4S cluster environment (see above) and to binding of the adenine moiety of SAM1 both through van der Waals interactions and through a hydrogen bond from its carbonyl oxygen to the adenine amide nitrogen (Figure 3C). A residue that appears vital in correctly orientating the methionine moiety of SAM1 is Arg184, again conserved for all HemN sequences. In fact, a first analysis, based on the correlation of secondary structure elements in HemN and those predicted for biotin synthase, another well characterized Radical SAM enzyme, indicates that this arginine may correspond to a conserved arginine in this enzyme (Arg168 in the enzyme from Escherichia coli).
Similarly, a number of residues involved in binding SAM2 are conserved in all HemN sequences. These include Phe310, onto which the adenine moiety of SAM2 stacks, Ile329, which stabilizes SAM2 both through hydrophobic interactions via its side chain and through hydrogen bonds through its backbone atoms, Gly112, vital for orienting the ribose moiety, and Glu145, which hydrogen-bonds the ribose O3′ of SAM2. Highly specific interactions with so many conserved residues weigh heavily in favor of SAM2 being not merely an artifact of crystallization, but truly integral to a functional HemN enzyme.
Other conserved residues congregate at the interface between domains. The N-terminal ‘trip-wire’ bears the single longest stretch of conserved residues 21PRY TSYPTA29 (Figure 4A). Surprisingly, this motif closely corresponds to a poorly ordered region covering residues 17–25. We propose that these residues, and in particular Arg22 (see below), are crucial for substrate binding. This part of the protein could thus adopt an ordered conformation on substrate binding, possibly inducing the C-terminal domain to tip and close the active site cleft. This is supported by conserved residues of the C-terminal domain being located at the interface either with the catalytic domain or with the N-terminal ‘trip-wire’, implicating them in achieving a final, closed conformation of the protein.
Proposed electron donor-binding site
The loop bearing the cysteines of the conserved Radical SAM CxxxCxxC motif involved in coordinating three Fe ions of the 4Fe–4S cluster forms a conspicuously flattened lid-like structure that substantially covers the central void of the three-quarter barrel and separates the 4Fe–4S cluster from the surrounding medium (Figure 3B). The lid is particularly rigid as it is stabilized by three cysteinyl–Fe bonds. It covers the cluster under a single layer of residues separating the 4Fe–4S cluster by only 6–7 Å from the surrounding aqueous medium. It forms, furthermore, the bottom of a depression on the surface of HemN with a patch of hydrophobic amino acid residues in its center, some of which are conserved in HemNs (Figure 4B and C). This depression may thus function as the docking site of an external electron donor, presumed to be flavodoxin (Layer et al., 2002). Manually placing flavodoxin alongside HemN reveals a suitable surface complementarity with an estimated shortest edge–edge distance of ∼8 Å between the flavin cofactor of flavodoxin and the 4Fe–4S cluster of HemN, a distance suitable for electron transfer (not shown). Access via the active site cleft does not appear to be the route of choice, as reduction of the 4Fe–4S cluster, for steric reasons, would need to occur prior to SAM binding. Secondly, HemN catalyzes two similar oxidation reactions on a single substrate. Re-reduction of the 4Fe–4S cluster by an exterior electron donor following the first of these reactions would allow both reactions to occur without opening the active site, which would risk losing the reaction intermediate.
Residues located in this electron donor-binding site mainly involve two continuous stretches 73KIVT76 and 247TIFAAQ252. They are not as well conserved as those involved in cofactor binding. Relative to the otherwise highly variable outer surface of HemN, they are, however, invariably replaced by homologous, mostly hydrophobic residues.
Modeling the substrate implies a possible reaction mechanism
As far as we can ascertain, no protein structure has been reported previously that simultaneously binds two symmetrically unrelated SAM cofactors. Similarly, HemN has not been known previously to bind two SAM molecules. Can the observation of SAM2 thus be physiologically relevant? For SAM1, this is clearly so. Its apposition with the 4Fe–4S cluster and the strong conservation of the residues creating its binding pocket clearly confirm all criteria of a Radical SAM-bound cofactor. Residues comprising the binding pocket of SAM2 are, however, as highly conserved as those of SAM1. Also, the shape complementarity between SAM2 and its binding pocket (Figure 4B) supports its relevance. Furthermore, biotin synthase, another Radical SAM enzyme, recently has been reported to bind two SAM cofactors per dethiobiotin substrate (Uglava et al., 2003). Like HemN, biotin synthase needs independently to abstract two hydrogen atoms from unactivated carbons. The alternative would be to assume that SAM2 partly occupies the binding pocket of coproporphyrinogen III and in particular that of one of the pyrrole rings, mimicking the stacking interaction of this substrate moiety with Phe310.
To distinguish between these alternatives, we have modeled the substrate coproporphyrinogen III assuming either the absence or presence of SAM2. In contrast to tetrapyrrole cofactors such as heme and chlorophyll, coproporphyrinogen III is not rigidly planar due to its non-conjugated backbone. This restricts precise modeling of the substrate in the absence of additional structural or biochemical information. We have, therefore, as a first approximation, placed a roughly planar coproporphyrinogen III into the catalytic cleft of HemN. Thereby we may qualitatively compare the size of the substrate and the approximate distance between the propionate side chains with the size of the proposed substrate-binding site.
Placing one pyrrole ring of coproporphyrinogen III into the binding pocket of SAM2 allows the substrate to be rotated such that the propionate side chain of a neighboring ring (either A or B, Figure 1A) is placed near the C5′ atom of SAM1. This model is thus possible. It does, however, require the substrate to be placed steeply into the active site while not providing obvious interaction partners for any of the three remaining propionate side chains. In particular, the negatively charged propionate side chain of the pyrrole occupying the SAM2 site would be located in a predominantly negatively charged pocket.
Assuming that HemN does indeed bind two SAM molecules allows the substrate to be placed less steeply into the active site cleft in an orientation without obvious steric conflicts between substrate and enzyme (Figure 5). The substrate was rotated to place the propionate group of ring A near SAM1. The propionate side chains of rings C and D, both of which are not involved in the reaction and which point in roughly parallel directions (Figure 1A), could be accommodated near the N-terminal trip-wire and the C-terminal domain. Both provide suitable, conserved arginine residues without salt bridge or hydrogen bonding partners in the current structure (Arg22 and Arg430). These interactions could provide the stimulus for the N-terminal trip-wire to move into the active site cleft and adopt an ordered conformation and for the C-terminal domain to move to cover the active site cleft.

Fig. 5. The modeled substrate (cyan) is depicted in the context of the electrostatic surface potential distribution of the HemN catalytic domain. The binding pockets of SAM1 and SAM2 (pink bonds) are predominantly negatively charged (red), while the proposed substrate-binding pocket is largely positively charged (blue) to accommodate the positively charged sulfonium and the negatively charged propionate side chains, respectively, of coproporphyrinogen III. The C-terminal domain is omitted for clarity, while the N-terminal trip-wire is shown in ribbon-style representation (green). It bears a conserved positively charged residue that may help bind the substrate. This could, in turn, stabilize the trip-wire and tip the C-terminal domain to shut the active site cleft.
In this orientation, the Cβ atom of the ring B propionate side chain (BCβ) is located near C5′ of SAM2. This is crucial as the 5′-deoxyadenosyl radical is located at the C5′ atom which then abstracts a hydrogen atom from Cβ to achieve substrate oxidation and conversion to the corresponding vinyl group coupled to loss of CO2 (Seehra et al., 1983; Layer et al., 2002). Optimizing the distance between BCβ and C5′ of SAM2 indicates that the ACβ–BCβ distance is longer than that between the C5′s of SAM1 and SAM2. As a result, ACβ does not end up next to the C5′ of SAM1 but alongside the side chain of Cys71 (Figure 3C), a fourth conserved cysteine that extends the Radical SAM cysteine motif to CxxxCxxCxC in HemN and has been shown to be crucial to the catalytic mechanism of HemN (Layer et al., 2002). The sulfur of Cys71, however, is only 3.8 Å away from C5′ of SAM1, indicating that Cys71 may be involved in relaying a radical state from SAM1 C5′ to one of the propionate side chains. Such an involvement of cysteine is known to occur in pyruvate formate lyase (Becker et al., 1999) and class III ribonucleotide reductase (Logan et al., 1999), both activated by members of the Radical SAM family.
Involvement of SAM2 to produce a 5′-deoxyadenosyl radical and initiate oxidative decarboxylation of one substrate propionate side chain would require that it be the recipient of a single electron. Structurally, the sulfonium sulfur of SAM2 is located at a distance of 6 Å from the (R)-sulfur of SAM1 (Figure 3C) but positioned to produce a roughly linear arrangement between the 4Fe–4S cluster and the two sulfonium sulfurs. Direct reduction of SAM2 by the iron–sulfur cluster is thus not possible. Two water molecules between the sulfonium sulfurs and the interrupted electron density of SAM2 (Figure 3A) indicate that this moiety may not have adopted its active state conformation. The modeled position of the substrate indicates that substrate binding may induce rotation around the C5′–S+ bond, moving the methionine moiety of SAM2 and in particular its sulfonium significantly closer to that of SAM1. Simultaneously, this would bring the sulfonium sulfur of SAM2 into salt bridging distance of negatively charged Glu145, already involved in a hydrogen bond to O3′ of SAM2 (Figure 3C).
We thus propose that the first electron transferred from the 4Fe–4S cluster to the (S)-sulfonium of SAM1 is passed on to the sulfonium of SAM2, perhaps after inversion of configuration to (R)-SAM1. This would induce radical formation in SAM2, achieving decarboxylation in one propionate side chain. Re-reduction of the cluster and a second electron transfer to SAM1 would then induce radical formation in SAM1 possibly relayed to the second propionate side chain by Cys71, causing the second decarboxylation.
Each SAM may thus possibly catalyze the oxidative decarboxylation of one propionate side chain. As described, such a double reaction would eliminate the need for the active site to open following the decarboxylation of the first propionate side chain. Were both decarboxylations to be catalyzed at a single active site, the spent SAM1 would need to be replenished after the first decarboxylation. This would, in turn, require the substrate intermediate to be at least partly released to provide a route for the exchange of SAM. The substrate would, furthermore, need to be rotated to place the second propionate chain into the active site. This scenario appears altogether more cumbersome than the hypothesis of HemN achieving two related reactions at neighboring locations in the active site as suggested by the binding of both SAM1 and SAM2.
The derived model is currently not sufficiently precise to describe the stereochemistry of the reaction. A 2-fold rotational ambiguity with respect to the substrate orientation means that it is not clear which propionate side chain (ring A or B) would need to be positioned alongside which SAM. Similarly, it is not clear whether the pro-S or pro-R hydrogen at Cβ would be abstracted in either case (Seehra et al., 1983). Many details of the reaction thus await further investigation.
Configuration of SAM sulfonium sulfur
The sulfonium sulfur of SAM is chiral (De la Haba et al., 1959). Adenosylmethionine synthase, which catalyzes the formation of SAM from methionine and ATP, exclusively produces the (S) stereoisomer (Conforth et al., 1977), the stereoisomer utilized by all SAM-dependent enzymes described to date (Cannon et al., 2002). Under physiological conditions, (S)-SAM spontaneously racemizes, producing equivalent amounts of (S) and (R) stereoisomers within a few days (Wu, 1983; Hoffman, 1986). Assuming fast turnover of (S)-SAM, (R)-SAM should nevertheless slowly accumulate in living organisms. Only trace amounts of (R)-SAM have, however, been observed (Hoffman, 1986). No enzyme has been identified that can utilize (R)-SAM nor have any proteins been reported that either stabilize the (S) configuration or invert the non-physiological (R) to the useful (S) configuration.
HemN appears to be the first enzyme that binds both stereoisomers. Structurally, only the (S) configuration achieves the described distances of the sulfonium sulfur from the 4Fe–4S cluster, while the (R) configuration allows the sulfonium sulfur to be positioned significantly closer to that of SAM2. This leads us to speculate that close approach between sulfonium sulfurs of SAM1 and SAM2 and/or reduction of the 4Fe–4S cluster could induce an inversion of configuration at the sulfonium of the minority (R)-SAM1 population. A first electron transfer to (S)-SAM1 could perhaps invert the configuration to (R)-SAM1, shortening the distance to the sulfonium of SAM2 and aiding transfer of the electron to SAM2. SAM2 would thus be the first to react.
Proposed pocket of the electron acceptor
The oxidative decarboxylation of coproporphyrinogen III by a radical mechanism requires the uptake of an unpaired electron from the substrate radical intermediate (Figure 1C). The physiological electron acceptor of HemN has eluded identification to date (Layer et al., 2002). Nevertheless, the reaction depends on the presence of a small, soluble electron acceptor such as phenazine methosulfate in vitro before any product formation occurs (unpublished results). Such an electron acceptor needs to be located within a distance of a few Ångstroms of the substrate propionate side chains for electron transfer to occur. In the crystal structure of HemN, we have identified a pocket occupied by water molecules that could fit all requirements for such an electron acceptor. It is located symmetrically adjacent to both SAM1 and SAM2 sulfonium sulfurs and both propionate side chains in the substrate model. The edge–edge distance between propionate Cα and electron acceptor could be as short as 5 Å. Relative to the N-terminal domain as a whole, this pocket extends along the axis of the three-quarter barrel with an entrance at the N-terminal end of the barrel β-strands between the β-strands and the β-loop–β plug. It is predominantly lined by highly conserved, charged and polar residues (filled diamonds in Figure 4A). Two salt bridges Arg166–Glu296 and Arg236–Asp295 guarding the entrance form the only salt bridges in this channel. All other charged residues are without a direct partner and a few are observed in alternative orientations, potentially indicating a missing molecular partner. Phenazine methosulfate, an artificial electron acceptor used in biochemical investigations, structurally fits into the pocket. It appears a little small, though, and not sufficiently charged to match the charged residues in the proposed electron acceptor site.
Conclusions
Despite catalyzing widely different reactions, Radical SAM enzymes all share a common mechanism of initiating radical reactions. In combination with the conservation of a few sequence motifs, this suggests that all such proteins share a common structural core. The crystal structure of HemN fills the gap previously left by the absence of structural information on this intriguing group of proteins and provides a first view of the requirements for generating oxidizing equivalents under generally reducing conditions. Radical SAM enzymes as a group are mostly sensitive towards molecular oxygen, and many, including HemN, have oxygen-dependent or oxygen-tolerant counterparts that may have evolved to survive the accumulation of oxygen in the biosphere following the advent of oxygenic photosynthesis. Radical SAM mechanisms may thus be likened to molecular fossils from a pre-aerobic world, surviving mostly in anaerobic bacteria.
Which structural features do Radical SAM enzymes share? The three-quarter barrel of the HemN catalytic domain optimally combines a stable core with a deep active site, inside which the 4Fe–4S cluster, the SAM cofactor (or co-reactant) as well as the substrate may bind and react while shielding the aggressive radical intermediates from the surrounding medium. The conserved cysteine motif invariably will cover the central 4Fe–4S cluster and SAM-binding site in a flattened loop, shielding them while providing a route of electron transfer from an electron donor docked to the exterior of the molecule. Variations of the basic architecture will, on the other hand, allow Radical SAM enzymes to bind both large and small substrates, ranging from entire enzymes (as in ribonucleotide reductase-activating enzyme) and large organic molecules (such as coproporphyrinogen III in HemN) to much smaller substrates (such as dethiobiotin in biotin synthase, and α-l-lysine in lysine-2,3-aminomutase). Suitable models of many other members of the Radical SAM enzyme family may thus now be inferred, even in the absence of high-resolution crystal structures in every case.
Materials and methods
Protein purification and crystallization
Recombinant E.coli HemN was purified as described (Layer et al., 2002) with minor modifications. Triton X-100 was replaced by C8E4 in all buffers. After binding of HemN to the blue Sepharose column, the concentration of C8E4 was reduced from 0.1% (v/v) to 0.01% (v/v) to elute the protein. The purified protein was concentrated to 10 mg/ml and dialyzed against 10 mM HEPES pH 7.5, 3 mM dithiothreitol (DTT), 150 mM NaCl and 0.01% (v/v) C8E4. HemN was crystallized by the hanging drop vapor diffusion method at 20°C in an anaerobic chamber. HemN (10 mg/ml) was incubated with 1 mM SAM for 2 h prior to crystallization. A 5 µl aliquot of protein/SAM and 5 µl of reservoir solution (22% PEG 10000, 100 mM HEPES pH 7.0, 500 µl or 21% PEG 4000, 10% isopropanol, 100 mM HEPES pH 7.5) was mixed for crystallization drops. Crystals grew within 5–7 days and were shock-cooled in liquid N2 using reservoir solution supplemented with 20% 2-methyl-2,4-pentanediol (MPD) as cryoprotectant.
Data collection, structure determination and refinement
X-ray data were collected using synchrotron radiation and a MAR–CCD/MAR345 image plate detector on beamlines BL1 and BL2 at BESSY II, Berlin, Germany. An X-ray fluorescence scan confirmed the presence of iron in the sample. Data sets were collected at the Fe absorption edge inflection point (1.7420 Å), peak (1.7319 Å) and high-energy remote (1.5498 Å). A high-resolution data set was then collected at 0.9198 Å on BL2 using the same crystal (Table I). Data were processed using the HKL suite (Otwinowski and Minor, 1997) and TRUNCATE (CCP4, 1994). The 4Fe–4S cluster was located using Patterson methods. AUTOSHARP was used to streamline phasing, density modification and automated model building (see Results) and REFMAC5 was used for final structure refinement (Murshudov et al., 1997). O was used for manual model building, structural analysis and substrate modeling (Jones et al., 1991). The structure was validated using PROCHECK (Laskowski et al., 1993) and CHECKIT (CCP4, 1994).
Molecular depictions were prepared using MOLSCRIPT (Kraulis, 1991), surfaces by GRASP (Nicholls et al., 1993), and rendered using POV-Ray (www.povray.org).
Accession numbers
The coordinates of the structures have been deposited in the Protein Data Bank (accession No. 1OLT).
Acknowledgments
Acknowledgements
We thank Drs Uwe Müller and Martin Ehlers of the Protein Structure Factory at BESSY II (Berlin) for synchrotron beamtime and their support during data collection. This work was funded by the Deutsche Forschungsgemeinschaft (D.W.H. and D.J.).
References
- Becker A., Fritz-Wolf,K., Kabsch,W., Knappe,J., Schultz,S. and Wagner,A.F.V. (1999) Structure and mechanism of the glycyl radical enzyme pyruvate formate-lyase. Nat. Struct. Biol., 6, 969–975. [DOI] [PubMed] [Google Scholar]
- Cannon L.M., Butler,F.N., Wan,W. and Zhou,Z.S. (2002) A stereo specific colorimetric assay for (S,S)-adenosylmethionine quantification based on thiopurine methyltransferase-catalyzed, thiol methylation. Anal. Biochem., 308, 358–363. [DOI] [PubMed] [Google Scholar]
- CCP4. (1994) The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D, 50, 760–763. [DOI] [PubMed] [Google Scholar]
- Chadwick D.J. and Ackrill,K. (eds) (1994) Ciba Foundation Symposia 180: The Biosynthesis of the Tetrapyrrole Pigments. Wiley and Sons, Chichester, UK, pp. 131–155. [Google Scholar]
- Cheek J. and Broderick,J.B. (2001) Adenosylmethionine-dependent iron–sulfur enzymes: versatile clusters in a radical new role. J. Biol. Inorg. Chem., 6, 209–226. [DOI] [PubMed] [Google Scholar]
- Cheek J. and Broderick,J.B. (2002) Direct H atom abstraction from spore photoproduct C-6 initiates DNA repair in the reaction catalyzed by spore photoproduct lyase: evidence for a reversibly generated adenosyl radical intermediate. J. Am. Chem. Soc., 124, 2860–2861. [DOI] [PubMed] [Google Scholar]
- Cornforth J.W., Reichard,S.A., Talalay,P., Carrell,H.L. and Glusher,J.P. (1977) Determination of the absolute configuration at the sulfonium center of _S_-adenosylmethionine. Correlation with the absolute configuration of the diastereomeric _S_-caboxymethyl-(S)-methionine salts. J. Am. Chem. Soc. 99, 7292–7300. [DOI] [PubMed] [Google Scholar]
- Cosper N.J., Booker,S.J., Ruzicka,F., Frey,P.A. and Scott,R.A. (2000) Direct FeS cluster involvement in generation of a radical in lysine 2,3-aminomutase. Biochemistry, 39, 15668–15673. [DOI] [PubMed] [Google Scholar]
- Cosper M.M., Cosper,N.J., Hong,W., Shokes,J.E., Broderick,W.E., Broderick,J.B., Johnson,M.K and Scott,R.A. (2003) Structural studies of the interaction of _S_-adenosylmethionine with the [4Fe–4S] clusters in biotin synthase and pyruvate formate-lyase activating enzyme. Protein Sci., 12, 1573–1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowtan K. and Main,P. (1998) Miscellaneous algorithms for density modification. Acta Crystallogr. D, 54, 487–93. [DOI] [PubMed] [Google Scholar]
- Dailey H.A. (2002) Terminal steps of haem biosynthesis. Biochem. Soc. Trans, 30, 590–595. [DOI] [PubMed] [Google Scholar]
- de la Fortelle E. and Bricogne,G. (1997) Maximum-likelihood heavy-atom parameter refinement for multiple isomorphous replacement and multiwavelength anomalous diffraction methods. Methods Enzymol., 276, 472–494. [DOI] [PubMed] [Google Scholar]
- De la Haba G., Jamieson,G.A., Mudd,S.H. and Richards,H.H. (1959) _S_-Adenosylmethionine: the relation of configuration at the sulfonium center to enzymatic reactivity. J. Am. Chem. Soc., 81, 3975–3980. [Google Scholar]
- Fontecave M., Mulliez,E. and Ollagnier-de-Choudens,S. (2001) Adenosylmethionine as a source of 5′-deoxyadenosyl radicals. Curr. Opin. Chem. Biol., 5, 506–511. [DOI] [PubMed] [Google Scholar]
- Frey M., Rothe,M., Wagner,A.F.V. and Knappe,J. (1994) Adenosyl methionine-dependent synthesis of the glycyl radical in pyruvate formate-lyase by abstraction of the glycine C-2 pro-S hydrogen atom. J. Biol. Chem., 269, 12432–12437. [PubMed] [Google Scholar]
- Frey P.A. and Magnusson,O.T. (2003) _S_-Adenosylmethionine: a wolf in sheep’s clothing, or a rich man’s adenosylcobalamin? Chem. Rev., 103, 2129–2148. [DOI] [PubMed] [Google Scholar]
- Friedmann H.C. and Thauer,R.T. (1992) In Encyclopedia of Microbiology. Academic Press, New York, Vol. 3, pp. 1–19. [Google Scholar]
- Hoffman J.L. (1986) Chromatographic analysis of the chiral and covalent instability of _S_-adenosyl-l-methionine. Biochemistry, 25, 4444–4449. [DOI] [PubMed] [Google Scholar]
- Jarrett J.T. (2003) The generation of 5′-deoxyadenosyl radicals by adenosylmethionine-dependent radical enzymes. Curr. Opin. Chem. Biol., 7, 174–182. [DOI] [PubMed] [Google Scholar]
- Jones T.A., Zou,J.Y., Cowan,S.W. and Kjeldgaard,M. (1991) Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr. A, 47, 110–119. [DOI] [PubMed] [Google Scholar]
- Jordan P.M. (1981) In Neuberger,A. and van Deenen,L.M.M. (eds), New Comprehensive Biochemistry. Elsevier, Amsterdam, Vol. 19, pp. 1–66. [Google Scholar]
- Lamzin V.S. and Wilson,K.S. (1993) Automated refinement of protein models. Acta Crystallogr. D, 129–147. [DOI] [PubMed] [Google Scholar]
- Kraulis P.J. (1991) MOLSCRIPT: a program to produce both detailed and schematic plots or protein structures. J. Appl. Crystallogr., 24, 946–950. [Google Scholar]
- Laskowski R.A., MacArthur,M.W., Moss,D.S. and Thornton,J.M. (1993) PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Crystallogr., 26, 283–291. [Google Scholar]
- Layer G., Verfürth,K., Mahlitz,E. and Jahn,D. (2002) Oxygen-independent coproporphyrinogen-III oxidase HemN from Escherichia coli. J. Biol. Chem., 277, 34136–34142. [DOI] [PubMed] [Google Scholar]
- Logan D.T., Andersson,J., Sjöberg,B.-M. and Nordlund,P. (1999) A glycyl radical state in the crystal structure of a class III ribonucleotide reductase. Science, 283, 1499–1504. [DOI] [PubMed] [Google Scholar]
- Murshudov G.N., Vagin,A.A. and Dodson,E.H. (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D, 53, 240–255. [DOI] [PubMed] [Google Scholar]
- Nicholls A., Bharadwaj,R. and Honig,B. (1993) GRASP: a graphical representation and analysis of surface properties. Biophys. J., 64, A166. [Google Scholar]
- Otwinowski Z. and Minor,W. (1997) Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol., 276, 307–326. [DOI] [PubMed] [Google Scholar]
- Schluckebier G., O’Gara,M., Saenger,W. and Cheng,X. (1995) Universal catalytic domain structure of SAM-dependent methyltransferases. J. Mol. Biol., 247, 16–20. [DOI] [PubMed] [Google Scholar]
- Schubert H.L., Blumenthal,R.M. and Cheng,X. (2003) Many paths to methyltransfer: a chronically of convergence. Trends Biochem. Sci., 28, 329–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seehra J.S., Jordan,P.M. and Akhtar, (1983) Anaerobic and aerobic coproporphyrinogen III oxidases of Rhodopseudomonas sphaeroides. Biochem. J., 209, 709–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sofia H.J. Chen,G., Hetzler,B.G., Reyes-Spindola,J.F. and Miller,N.E. (2001) Radical SAM, a novel protein superfamily linking unresolved steps in familiar biosynthetic pathways with radical mechanisms: functional characterization using new analysis and information visualization methods. Nucleic Acids Res., 29, 1097–1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uglava N.B., Frederick K.K. and Jarrett,J.T. (2003) Control of adenosylmethionine-dependent radical generation in biotin synthase: a kinetic and thermodynamic analysis of substrate binding to active and inactive forms of BioB. Biochemistry, 42, 2708–2719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsby C.J., Hong,W., Broderick W.E., Cheek,J., Ortillo,D., Broderick,J.B. and Hoffman,B.M. (2002) Electron-nuclear double resonance spectroscopic evidence that _S_-adenosylmethionine binds in contact with the catalytically active [4Fe–4S]+ cluster of pyruvate formate-lyase activating enzyme. J. Am. Chem. Soc., 124, 3143–3151. [DOI] [PubMed] [Google Scholar]
- Whitby F.G., Phillips,J.D., Kushner,J.P. and Hill,C.P. (1998) Crystal structure of human uroporphyrinogen decarboxylase. EMBO J., 17, 2463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu S.-E., Huskey,W., Borchardt,R.T. and Schowen,R.L. (1983) Chiral instability at sulfur of _S_-adenosylmethionine. Biochemistry, 22, 2828–2832. [DOI] [PubMed] [Google Scholar]