JunD activates transcription of the human ferritin H gene through an antioxidant response element during oxidative stress (original) (raw)
. Author manuscript; available in PMC: 2008 May 2.
Published in final edited form as: Oncogene. 2005 Nov 17;24(51):7567–7578. doi: 10.1038/sj.onc.1208901
Abstract
Ferritin is the major intracellular iron storage protein that sequesters excess free iron to minimize generation of iron-catalysed reactive oxygen species. We previously demonstrated that expression of ferritin heavy chain (ferritin H) was induced by pro-oxidants, which is a part of cellular antioxidant response to protect cells from oxidative damage. In this study, we have identified that the antioxidant/electrophile response element (ARE) located 4.5 kb upstream to the human ferritin H transcription initiation site is responsible for the oxidant response. The human ferritin H ARE comprises two copies of bidirectional AP1 motifs. Mutations in each AP1 motif significantly impaired protein binding and the function of the ARE, indicating that both of the AP1 motifs are required for pro-oxidant-mediated activation of the ferritin H gene. We identified that JunD, an AP1 family basic-leucine zipper (bZip) transcription factor, is one of the ferritin H ARE binding proteins and activates ferritin H transcription in HepG2 hepatocarcinoma cells. Gel retardation assay demonstrated that H2O2 (hydrogen peroxide) or t-BHQ (tert-butylhydroquinone) treatment increased total protein binding as well as JunD binding to the ferritin H ARE. Chromatin immunoprecipitation assay showed that H2O2 treatment induced JunD binding to the ferritin H ARE. Both H2O2 and t-BHQ induced phosphorylation of JunD at Ser-100, an activated form of JunD. Furthermore, overexpression of JunD induced endogenous ferritin H protein synthesis. Since JunD has recently been demonstrated to protect cells from several stress stimuli including oxidative stress, these results suggest that, in addition to NFE2-related factor 2 (Nrf2) as a major ARE regulatory protein, JunD is another ARE regulatory protein for transcriptional activation of the human ferritin H gene and probably other antioxidant genes containing the conserved ARE sequences by which JunD may confer cytoprotection during oxidative stress.
Keywords: ferritin, oxidative stress, JunD, AP1, ARE, iron
Introduction
Iron plays a crucial role in a variety of activities that are essential for cell growth and viability, including enzymes involved in DNA synthesis, electron transport and oxidative metabolism (Richardson and Ponka, 1997). Iron can also be a potential toxicant at elevated levels that are implicated in the pathogenesis of a wide variety of disorders including cancer and neurodegenerative diseases (Hentze et al., 2004). Excess intracellular labile iron plays a catalytic role in the formation of highly reactive hydroxyl radical (OH•) via the Fenton reaction. Hydroxyl radical is the most damaging radical to cells because it can penetrate into lipid bilayers of membranes and it causes direct oxidation of cellular components including lipids, proteins and nucleic acids, resulting in alterations of structure and function of oxidized molecules (Meneghini, 1997). Thus the tight regulation of iron homeostasis is essential for the coordinated activities of the cells in our body.
Ferritin is a ubiquitous and highly conserved iron storage protein that plays a prominent role in maintaining intracellular iron homeostasis (Arosio and Levi, 2002; Theil, 2003). Ferritin consists of 24 subunits of the H and L types, which associate in various ratios depending on the type of tissues and the physiological state of cells (Harrison and Arosio, 1996). Ferritin is capable of sequestering up to 4500 atoms of iron and thereby detoxifying intracellular excess iron as well as storing iron in a bioavailable form. Ferritin synthesis is regulated at both transcriptional and translational levels (Torti and Torti, 2002). A preferential induction of the H subunit than the L subunit of the ferritin gene was frequently observed in response to environmental and intracellular signals during inflammation (Torti et al., 1988; Miller et al., 1991; Tsuji et al., 1991; Kwak et al., 1995), and differentiation (Chou et al., 1986; Beaumont et al., 1987, 1994) in an iron-independent manner. Alterations in the subunit composition of the ferritins have a potential to alter intracellular iron balance because there are functional differences between the H and L subunits of ferritin; the ferritin heavy chain (ferritin H) subunit has ferroxidase activity that oxidizes ferrous iron to ferric iron, while the ferritin L subunit lacks the ferroxidase center but contributes to stabilization of assembled ferritin proteins (Levi et al., 1992, 1994; Santambrogio et al., 1992). Ferritin molecules that are rich in the H subunit are therefore involved in rapid iron uptake and release, contrasting the long-term iron storage by L subunit-rich ferritin molecules (Wagstaff et al., 1978; Bomford et al., 1981; Levi et al., 1988). Thus, predominant production of ferritin H under these acute environmental changes appears to be a reasonable cellular response for adaptation. In fact, others and we demonstrated that overexpression of ferritin H is cytoprotective against oxidative stress (Epsztejn et al., 1999; Cozzi et al., 2000; Orino et al., 2001; Pham et al., 2004). Furthermore, the ferritin H gene, but not ferritin L gene, was identified as a TNF-response gene (Torti et al., 1988; Miller et al., 1991) through activation of NF-_κ_B (Kwak et al., 1995), and very recently NF-_κ_B-mediated induction of ferritin H is an essential mediator of the antioxidant and cytoprotective activities against TNF-induced apoptosis (Pham et al., 2004).
In contrast to the well-defined translational regulation of ferritin by iron through interaction of iron regulatory protein and iron response element in the 5′-untranslated region of both ferritin H and L mRNAs (Hentze and Kuhn, 1996; Rouault and Klausner, 1996; Theil, 2000), molecular mechanisms of transcriptional regulation of ferritin genes, in particular, the human ferritin H gene, have been less elucidated. It has been reported by two independent research groups that transcription of the human ferritin H gene is activated by cyclic AMP (Bevilacqua et al., 1997) and hemin (Marziali et al., 1997) via a proximal _cis_-acting element containing CCAAT motif. This element, termed B site, located within 0.1 kb upstream from transcription initiation site, is regulated by NF-Y transcription factors (Bevilacqua et al., 1997; Marziali et al., 1997) and transcriptional coactivators harboring histone acetyltransferase activity such as p300/CBP (Faniello et al., 1999) and P/CAF (Bevilacqua et al., 1998). The element also serves as a basal transcription apparatus that contributes to the tissue-specific expression of the ferritin H gene (Bevilacqua et al., 1998). However, roles of the CCAAT site and NF-Y transcription factor as well as other regulatory elements in the transcriptional activation of the human ferritin H gene in response to xenobiotics and oxidative stressors have not been elucidated.
A battery of phase II detoxification genes including glutathione _S_-transferase, NAD(P)H quinone reductase, _γ_-glutamylcysteine synthetase and heme oxygenase, are transcriptionally activated in cells exposed to a variety of xenobiotics such as oxidants, antioxidants, carcinogens and electrophilic compounds (Nguyen et al., 2003). The induction of these detoxification genes is an important cell defence mechanism from toxicity of reactive metabolites and reactive oxygen species produced during biotransformation of xenobiotics by phase I metabolic enzymes (Hayes and McMahon, 2001). The transcriptional activation of these phase II antioxidant genes is mediated by a _cis_-acting element, termed the antioxidant response element (ARE; Rushmore and Pickett, 1990) or electrophile response element (EpRE; Friling et al., 1990), identified in the 5′-flanking region of these phase II detoxification genes (Hayes and McMahon, 2001; Nguyen et al., 2003; Jaiswal, 2004). The functional ARE sequence contains a core AP1-like TGACnnnGCA motif (Rushmore et al., 1991; Wasserman and Fahl, 1997b), and basic-leucine zipper (bZip) transcription factors such as NFE2-related factor 2 (Nrf2) as well as small Maf proteins (MafK and MafG) have been demonstrated to activate or repress the ARE enhancer activity, respectively (Venugopal and Jaiswal, 1998; Dhakshinamoorthy and Jaiswal, 2000; Motohashi et al., 2002).
In this study, we cloned a 5′-regulatory region of the human ferritin H gene and demonstrated that the human ferritin H gene is subject to transcriptional activation by hydrogen peroxide and xenobiotic compounds of tert-butylhydroquinone (t-BHQ) and _β_-naphthoflavone (_β_-NF) in HepG2 human liver cells. We have identified a far-upstream ferritin H enhancer element that serves as an ARE responsible for oxidative stress-mediated transcriptional activation, while the proximal CCAAT element was not directly involved in pro-oxidant-mediated transcriptional activation of the human ferritin H gene. Furthermore, our results from gel shift and chromatin immunoprecipitation (ChIP) assays showed that JunD, a member of the AP1 family, is one of transcription factors bound to the ARE of the human ferritin H gene and serves as activator of the ferritin H transcription. JunD has been demonstrated to be involved in antioxidant defense (Gerald et al., 2004) and cytoprotection against ultraviolet irradiation and TNF-α (Weitzman et al., 2000; Lamb et al., 2003). Taken together with the cytoprotective role of ferritin H in oxidative stress (Epsztejn et al., 1999; Cozzi et al., 2000; Orino et al., 2001; Pham et al., 2004), these results indicate that the human ferritin H gene is a JunD target gene regulated through the far-upstream ARE enhancer in response to electrophilic xenobiotics and oxidative stressors.
Results
Oxidative stressors activate transcription of the human ferritin H gene
We previously demonstrated that oxidative stress induced ferritin H mRNA synthesis in several mouse cell lines (Tsuji et al., 2000). To study oxidative stress-mediated ferritin H regulation in human cells, we treated HepG2 human hepatoma cells with hydrogen peroxide (H2O2), t-BHQ or _β_-NF for 24 h and measured ferritin H mRNA expression by Northern blotting. As shown in Figure 1a, ferritin H mRNA expression was significantly induced by t-BHQ and _β_-NF, and to a lesser extent by H2O2.
Figure 1.
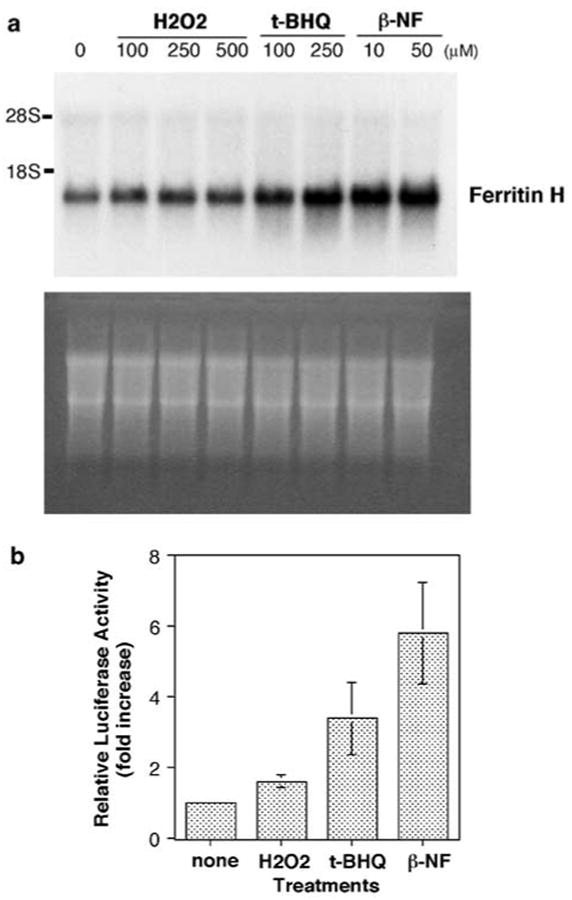
H2O2, t-BHQ and _β_-NF induce ferritin H mRNA at transcriptional level. (a) HepG2 cells were treated with indicated concentrations of H2O2, t-BHQ or _β_-NF for 24 h and 10 _μ_g of total RNA from each treatment was subjected to hybridization with a ferritin H cDNA probe (top). Equal amounts of RNA loading and transfer to nitrocellulose membrane were verified by ethidium bromide staining (bottom). (b) A measure of 1 _μ_g of −5.2 kb ferritin H-luciferase plasmid was transiently transfected into HepG2 cells by calcium phosphate method, followed by treatment with 250 _μ_m H2O2, 250 _μ_m t-BHQ or 50 _μ_m _β_-NF for 24 h. Luciferase activity with no treatment was defined as 1.0, and the results from four independent experiments and s.e.'s are shown
To date, only a proximal 0.15 kb promoter/enhancer region containing the CCAAT box (termed B site) has been elucidated in the transcriptional regulation of the human ferritin H gene (Bevilacqua et al., 1997; Marziali et al., 1997; Faniello et al., 1999). We therefore asked whether the proximal ferritin H enhancer is the sole enhancer element for transcriptional regulation of the human ferritin H gene, in particular, in response to oxidative stress. To address this question, first of all, we cloned a far-upstream 5′-enhancer/promoter region containing 5.2 kb upstream from transcription start site of the human ferritin H gene because the proximal B site containing the CCAAT box does not appear to have a canonical AP1/ARE motif (Rushmore et al., 1991; Wasserman and Fahl, 1997b). The nucleotide sequence of the 5.2 kb human ferritin H 5′-region we cloned turned out to be 99.2% identical to nucleotides 14 926–20 111 of the clone pDJ759j12 chromosome 11q13 (the NCBI Accession number AF139813). Then, we made a luciferase reporter construct driven by the 5.2 kb 5′-region of the human ferritin H gene and transfected into HepG2 cells to test whether the 5.2 kb region contains an enhancer element responsible for induction of ferritin H mRNA by these oxidative stressors. As shown in Figure 1b, H2O2, t-BHQ and _β_-NF induced luciferase expression driven by the 5′ 5.2 kb region of the ferritin H gene by 1.6-, 3.4- and 5.8-fold, respectively. These results indicate that ferritin H mRNA induction by these oxidative stressors is transcriptionally regulated by _cis_-acting element(s) in the 5.2 kb region of the human ferritin H gene.
Identification of the ARE in the human ferritin H gene
To define a _cis_-acting element responsible for transcriptional activation of the human ferritin H gene in response to these pro-oxidants, we made several deletions in the ferritin H-luciferase reporter genes, transfected them into HepG2 cells, and measured luciferase expression in response to H2O2, t-BHQ and _β_-NF. As shown in Figure 2, −4.0 kb ferritin H showed a significant decrease in basal expression of luciferase reporter compared with the −5.2 kb ferritin H construct, and there was no induction of luciferase expression by treatment with H2O2, t-BHQ or _β_-NF, whereas the −5.2 kb construct was reproducibly induced by these treatments. The −0.15 kb construct just containing the B site (CCAAT box) and TATA box of the human ferritin H gene showed similar response to the −4.0 kb construct with low basal expression and no further activation by these pro-oxidant treatments (Figure 2). However, the −0.15 kb region drove much higher luciferase expression than −0.03 kb TATA-only construct (Figure 2). These results suggest the presence of additional enhancer element(s), located between −5.2 and −4.0 kb of the human ferritin H gene, and plays roles in both basal expression and transcriptional activation by these oxidative stressors.
Figure 2.
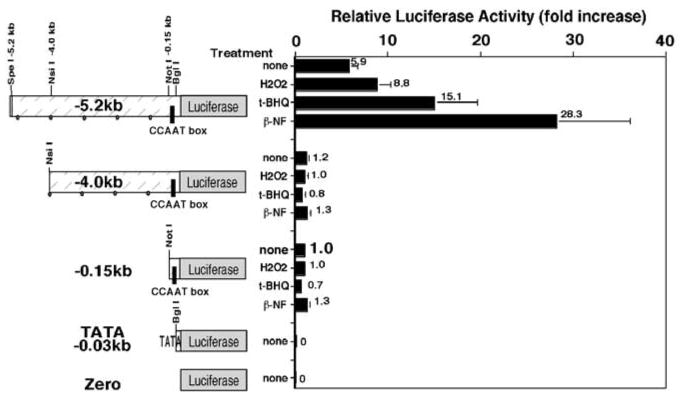
The human ferritin H gene is regulated by the proximal and a far-upstream enhancer element, and the latter is responsible for transcriptional activation by oxidative stressors. A measure of 1 _μ_g of a human ferritin H-luciferase reporter construct (−5.2, −4.0, −0.15, −0.03 kb or zero) was transiently transfected into HepG2 cells, followed by treatment with 250 _μ_m H2O2, 250 _μ_m t-BHQ or 50 _μ_m _β_-NF for 24 h and luciferase assays. Luciferase activity from HepG2 cells transfected with the −0.15 kb construct without treatment was defined as 1.0. The results from six experiments and standard errors are shown
The functional ARE enhancer elements in various phase II genes have an inverted or direct repeat of the primary core sequence of TGACnnnGCA (Rushmore et al., 1991; Jaiswal, 1994; Wasserman and Fahl, 1997b). In the −5.2 to −4.0 kb region of the human ferritin H gene, we found there is one inverted repeat of the ARE motif containing an AP1-like and AP1/NFE2 sites at −4.5 kb upstream from transcription initiation site (Figure 3a). In order to assess whether this inverted repeat of AP1 motifs is a functional ARE, we tested −4.5 and −4.4 kb ferritin H reporter genes in their response to oxidative stressors. These two reporter constructs have the only difference in the presence or absence of the 55 bp AP1-like and AP1/NFE2 sites. The −0.15 kb reporter gene containing the B site (CCAAT) was also transfected into HepG2 cells to examine its role in pro-oxidant response. Luciferase expression of the −4.5 kb ferritin H containing the inverted repeat of the ARE sequence was induced by H2O2 or t-BHQ treatment; however, the −4.4 kb construct lacking the inverted repeat of AP1 motifs or the −0.15 kb containing only the CCAAT element failed to be activated by treatment with these pro-oxidants (Figure 3a). The basal expression levels of these reporter constructs lacking the ARE motif were also lower (approximately 50%) than that of −4.5 kb reporter gene containing the ARE sequence (Figure 3a), indicating that the inverted repeat of AP1 motifs is a functional ARE.
Figure 3.
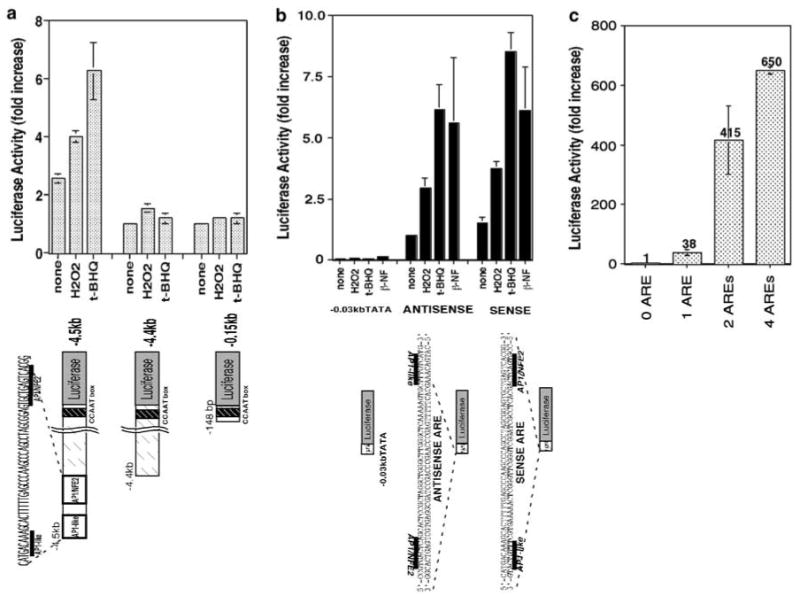
A human ferritin H ARE. (a) The 4.5 kb-upstream enhancer of the human ferritin H gene contains composite AP1 motifs. A measure of 1 _μ_g of a human ferritin H-luciferase reporter construct (−4.5, −4.4 or −0.15 kb) was transiently transfected into HepG2 cells, followed by treatment with 250 _μ_m H2O2 or 250 _μ_m t-BHQ for 24 h and luciferase assays. Luciferase activity from HepG2 cells transfected with the −4.4 kb construct without treatment was defined as 1.0. The results from four experiments and s.e.'s are shown. (b) The composite AP1 motifs serve as a basal enhancer and are sufficient to activate transcription of the ferritin H gene in response to oxidative stressors. One copy of a double-stranded oligonucleotide containing the composite AP1 motifs was inserted into −0.03 kb TATA-ferritin H luciferase construct in sense or antisense orientation, and 1 _μ_g of each luciferase construct was transiently transfected into HepG2 cells. The transfected HepG2 cells were treated with 250 _μ_m H2O2, 250 _μ_m t-BHQ or 50 _μ_m _β_-NF for 24 h followed by luciferase assays. Luciferase activity from HepG2 cells transfected with the antisense insertion construct without treatment was defined as 1.0. The results from three experiments and standard errors are shown. (c) Multiple copies of the oligonucleotides containing the composite AP1 motifs were inserted into −0.03 kb TATA-ferritin H luciferase construct in antisense orientation, and 1 _μ_g of each luciferase construct was transiently transfected into HepG2 cells and luciferase assays were carried out. Luciferase activity from HepG2 cells transfected with no insertion construct was defined as 1.0. The results from three experiments and standard errors are shown
Then we asked whether the 55 bp ferritin H ARE sequence is sufficient to induce ferritin H transcription by pro-oxidants. To address this question, we inserted one copy of the 55 bp ARE into TATA-only ferritin H-luciferase in sense or antisense orientation, then transfected these luciferase constructs into HepG2 cells and treated them with H2O2, t-BHQ or _β_-NF. Either sense or antisense insertion of the 55 bp ARE into the TATA-containing minimum ferritin H promoter increased basal expression level (no treatment) and further induced luciferase reporter expression in response to pro-oxidants (Figure 3b). Insertion of multiple copies of the 55 bp ARE further increased the basal expression level in a copy number-dependent manner (Figure 3c). Collectively, these results indicate that the 55 bp ARE located at −4.5 kb upstream from transcription initiation site of the human ferritin H gene is sufficient to pro-oxidant-mediated transcriptional activation and it also contributes to basal expression of the human ferritin H gene.
The two AP1 motifs are essential for the function of the ARE enhancer
The 55 bp ferritin H ARE sequence contains inverted repeat of AP1-like and AP1/NFE2 sites (Figure 3). We next asked whether both AP1-like and AP1/NFE2 sites are required for the function of the ARE in the human ferritin H gene. To address this question, we introduced TG to CA mutations in either AP1-like site or the AP1/NFE2 site, or in both the AP1-like and AP1/NFE2 sites in the luciferase reporter constructs and tested induction of luciferase activity in HepG2 cells following treatment with H2O2, t-BHQ or _β_-NF. In contrast to the pro-oxidant-mediated induction of luciferase expression driven by the wild-type (wt) 55 bp ARE, TG to CA mutations in either the AP1-like or AP1/NFE2 site significantly diminished both the basal luciferase expression and the response to these pro-oxidants (Figure 4). In addition, the double mutations in both the AP1-like and AP1/NFE2 sites completely destroyed the function of the ARE (Figure 4), suggesting that both the AP1-like and AP1/NFE2 elements are required for the function of the ferritin H ARE.
Figure 4.
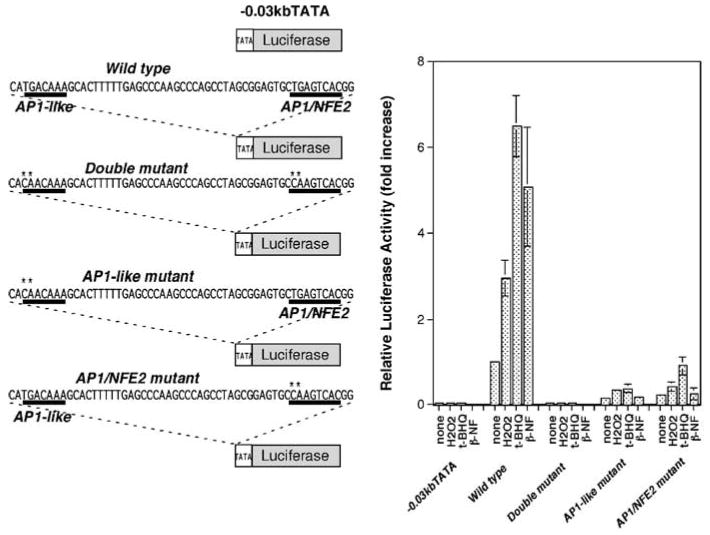
Both AP1-like and AP1/NFE2 sites are essential for the functional ARE. One copy of a double-stranded oligonucleotide containing wt AP1-like and AP1/NFE2 motifs, point mutations in either AP1-like site or AP1/NFE2 site, or mutations in both sites was inserted into a TATA-ferritin H luciferase construct, and 1 _μ_g of each luciferase construct was transiently transfected into HepG2 cells. The transfected HepG2 cells were treated with 250 _μ_m H2O2, 250 _μ_m t-BHQ or 50 _μ_m _β_-NF for 24 h followed by luciferase assays. Luciferase activity from HepG2 cells transfected with the wt insertion construct and no treatment was defined as 1.0. The results from four experiments and s.e.'s are shown
JunD is an ARE binding protein and activates ferritin H transcription
In order to study binding proteins to the ferritin H ARE and activation mechanisms, we first carried out gel retardation assays to test the effects of mutations in the AP1 motifs of the ferritin H ARE on protein binding. When the AP1/NFE2 sequence was used as a probe, protein binding was competed by the same AP1/NFE2 as well as the entire wtARE oligonucleotides, but not by the mutated AP1 oligonucleotide (Figure 5a, left). Similarly, when we used the entire ARE sequence as a probe, protein binding was competed by the wtARE or the AP1/NFE2 sequence, but not by mutated ARE or mutated AP1/NFE2 sequence (Figure 5a, right). To characterize binding proteins to the human ferritin H ARE in response to pro-oxidants, we performed gel supershift assays with HepG2 nuclear extracts using several antibodies to AP1, CREB and Maf/Nrf2 bZip family proteins (data not shown), in which JunD was reproducibly detected in the binding complex (Figure 5b). JunD binding to the human ferritin H ARE was induced by H2O2 or t-BHQ treatment in HepG2 cells in conjunction with increase in total protein binding to the ARE (Figure 5b). ChIP of HepG2 cells treated with H2O2 or t-BHQ showed that H2O2 treatment induced JunD binding to the ferritin H ARE, whereas t-BHQ had the minimum effect on JunD–ferritin H ARE interaction (Figure 5c). Nrf2, an important activator of ARE in a battery of phase II detoxification genes (Motohashi et al., 2002) including the mouse ferritin H gene (Pietsch et al., 2003), was taken as a positive control of ChIP assay, showing that Nrf2 binds to the human ferritin H ARE constitutively in HepG2 cells (Figure 5c).
Figure 5.
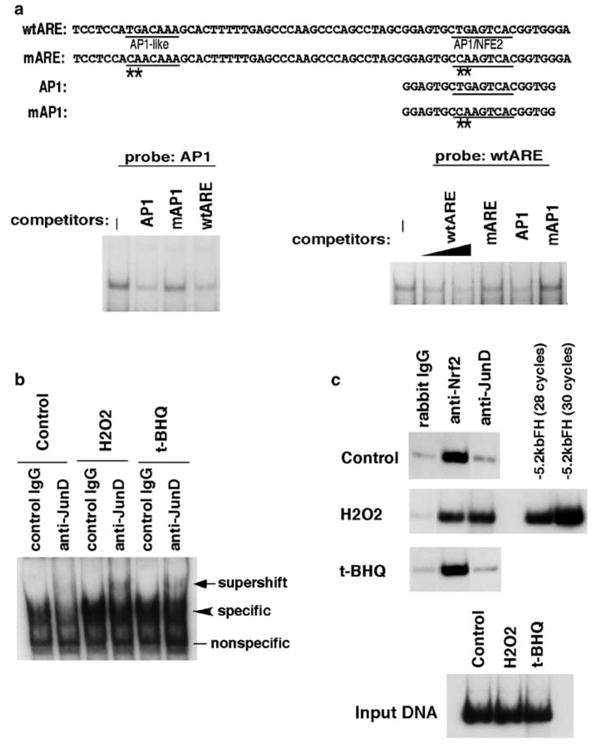
JunD is one of ferritin H ARE binding proteins. (a) Gel retardation assays were carried out by incubating 20 _μ_g of nuclear extracts isolated from growing HepG2 cells with 32P-labeled, double-stranded AP1/NFE2 probe (left) or wt ARE probe (right) in the presence of 40- to 50-fold molar excess of the indicated double-stranded competitor oligonucleotides. Nucleotide sequences of oligonucleotides used in the assays are shown on top with mutations denoted by asterisks. (b) HepG2 cells were treated with 100 _μ_m H2O2 or 100 _μ_m t-BHQ for 4 h followed by isolation of nuclear extracts. 20 _μ_g of HepG2 nuclear extracts were incubated with either rabbit IgG or anti-JunD antibody (rabbit) for 2 h at 4°C prior to the addition of a 32P-labeled AP1/NFE2 probe. A supershift band induced by anti-JunD antibody was indicated by an arrow. (c) HepG2 cells were treated with 100 _μ_m H2O2 or 100 _μ_m t-BHQ for 6 h and ChIP assay was carried out with rabbit IgG, anti-Nrf2 or anti-JunD antibody. Immunoprecipitated DNA, input DNA from each treatment (approximately 1/20 of cell lysate used for each immunoprecipitation) or 1 pg of pBluescript −5.2 kb ferritin H luciferase DNA was used for detection of 155 bp ferritin H ARE in 28 cycles of PCR reaction in addition to 30 cycles of PCR with pBluescript −5.2 kb ferritin H luciferase DNA for confirmation of a linear range of the DNA amplification
We then assessed role of JunD on the human ferritin H ARE by transient cotransfection of either wtARE- or mutant ARE-luciferase plasmid together with JunD expression plasmid. As shown in Figure 6a, JunD activated expression of luciferase driven by wtARE but not by mutant ARE. We further asked if overexpression of JunD induces endogenous ferritin H synthesis. We transfected three different JunD expression plasmids and transiently transfected cells were subjected to Western blotting for ferritin H protein expression. Cell lysates overexpressing JunD (lanes c and e) showed increased expression of ferritin protein compared with cells transfected with no DNA (lane a) or a control plasmid (lane b) or with pcDNAINeoJunD, which failed to express JunD (lane d). These results indicate that JunD is a transcriptional activator of the human ferritin H gene through the ARE.
Figure 6.
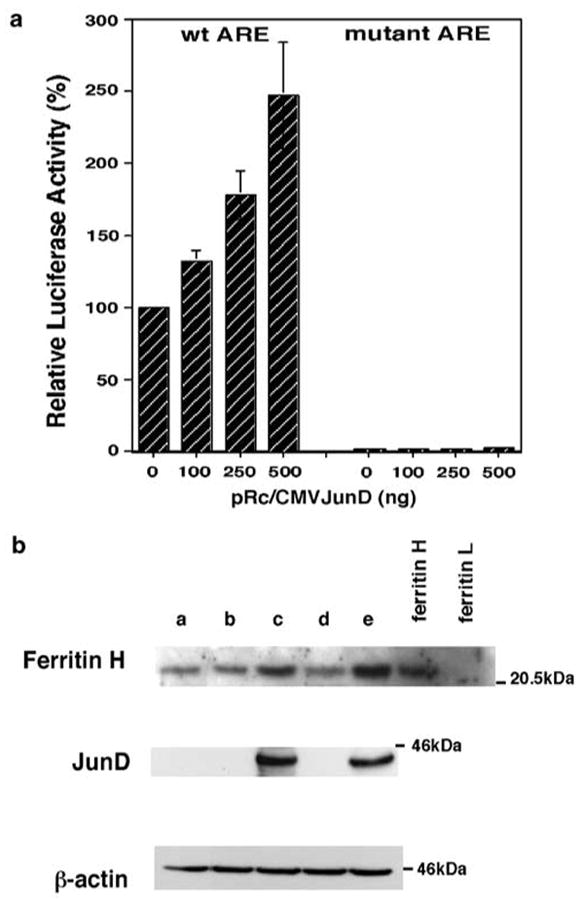
JunD activates the human ferritin H ARE. (a) A measure of 1 _μ_g of pBluescript wtARE ferritin H luciferase or mARE ferritin H luciferase (wtARE and mARE DNA sequences are shown in Figure 5a) was cotransfected with indicated amounts of pRc/CMVJunD into HepG2 cells. Total input of plasmid DNA was equalized to 2 _μ_g by adding pRc/CMV empty vector DNA and 0.1 _μ_g of pRL-CMV. Cells were harvested for dual luciferase assays 48–60 h after DNA transfection. DNA transfection was performed in duplicate, and the luciferase expression after normalization with pRL-CMV from five independent experiments are shown with s.e.'s. The luciferase activity in cell lysates cotransfected with wtARE ferritin H luciferase and pRc/CMV empty vector was defined as 100%. (b) A total of 7 × 106 K562 cells were transiently transfected with no DNA (lane a), 15 _μ_g of pRc/CMV (lane b), pRc/CMVJunD (lane c), pcDNAINeoJunD (lane d) or pCMVJunD (lane e) by electroporation and were incubated for 65 h. Cell lysates (50 _μ_g) were subjected to Western blotting for anti-ferritin H, anti-JunD and anti-_β_-actin. JunD cDNA cloned into pcDNAINeo vector was expected to express JunD protein but no expression for an uncertain reason
ChIP assay showed increased JunD binding to human ferritin H ARE by H2O2 (Figure 5). It has also been demonstrated that phosphorylation of Jun family members including JunD activate AP1 activity (Vinciguerra et al., 2004). In order to further understand how H2O2 or t-BHQ modulates JunD for the activation of the ferritin H transcription, we tested whether nuclear accumulation and/or phosphorylation of JunD was induced by these oxidative stressors. Cell lysates from HepG2 cells treated with H2O2 or t-BHQ were separated into nuclear and cytoplasmic fractions and subjected to Western blotting with anti-JunD or anti-phospho-JunD (anti-cJun phosphoserine-73, which also detects phosphorylated JunD at Ser100). In HepG2 cells, JunD was abundantly expressed and exclusively located in nucleus, and H2O2 or t-BHQ treatment had minimum effect on JunD expression but induced phosphorylation (Figure 7). Since anti-phospho-JunD antibody (Ser100) used in this experiment also detects phospho-cJun (Ser73), we attempted to confirm H2O2- or t-BHQ-mediated phosphorylation of JunD at Ser100 by transient transfection of JunD into K562 cells. Empty vector-transfected K562 cell lysates showed no bands of endogenous JunD or phosphorylated JunD or cJun (data not shown); however, in the same amount of JunD-transfected cell lysates, overexpressed JunD was detected with anti-JunD antibody, and induction of JunD phosphorylation (at Ser100) was detected by H2O2 or t-BHQ treatment (Figure 7b). Collectively, these results suggest that JunD activates transcription of the human ferritin H gene via ARE, and that H2O2 or t-BHQ may further activate the ferritin H transcription, at least in part, through phosphorylation of JunD.
Figure 7.
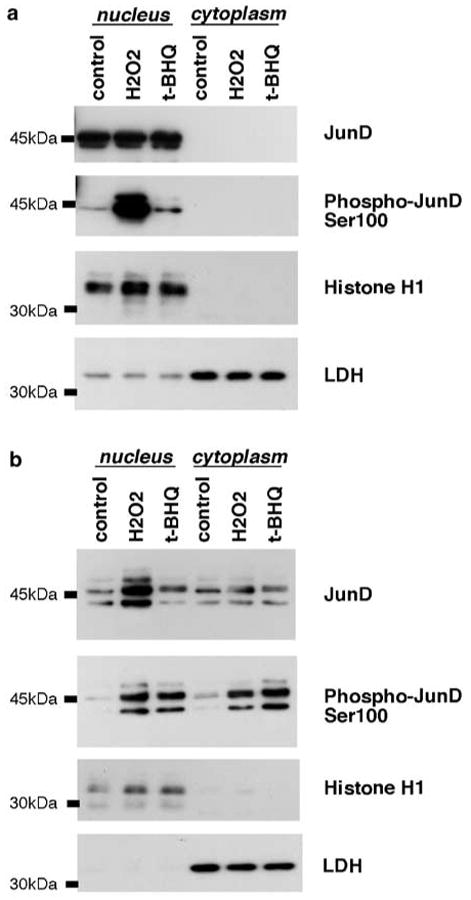
JunD expression in nucleus and phosphorylation at Ser100 during oxidative stress. (a) HepG2 cells were treated with 100 _μ_m H2O2 or 100 _μ_m t-BHQ for 1.5 h, (b) K562 cells transiently transfected with 20 _μ_g of pRc/CMVJunD were incubated for 45 h, followed by treatment with 100 _μ_m H2O2 or 100 _μ_m t-BHQ for 4 h. In both (a) and (b), cells were harvested for isolation of nuclear and cytoplasmic extracts, and 30 _μ_g of proteins were subjected to Western blotting for anti-JunD or anti-phospho-cJun (Ser73), which also detects phospho-JunD at Ser100. The purity of nuclear and cytoplasmic fractions was assessed by Western blotting with anti-histone H1 and anti-lactose dehydrogenase (LDH), respectively
Discussion
Several lines of evidence, including our previous results, indicate a role of ferritin as a cytoprotectant against damages mediated by oxidative stressors or inflammatory cytokines (Epsztejn et al., 1999; Cozzi et al., 2000; Orino et al., 2001; Pham et al., 2004). Ferritin, serving as the major iron-binding protein for storage of intracellular free iron, is subject to regulation at both transcriptional and translational levels (Torti and Torti, 2002). In this study, we attempted to shed light on pro-oxidant-mediated transcriptional regulation of the human ferritin H gene by characterization of a far-upstream 5′ regulatory region and regulatory proteins. In the human ferritin H gene, the proximal 0.15 kb promoter/enhancer region containing a GC box (recognized by Sp1) and the CCAAT box (recognized by NF-Y) were previously identified as key regulatory enhancer elements to determine tissue-specific ferritin H expression (Bevilacqua et al., 1998), by cAMP treatment (Bevilacqua et al., 1997; Faniello et al., 1999) and during monocyte to macrophage differentiation (Marziali et al., 1997). We and other groups reported previously that ferritin synthesis was regulated at transcriptional level by treatment with oxidative stressors in rat and mouse liver cells (Cairo et al., 1995; Orino et al., 1999; Tsuji et al., 2000) and in human cells (Orino et al., 2001). We therefore asked whether these proximal enhancers are the sole element for transcriptional regulation of the human ferritin H gene in basal expression as well as in response to pro-oxidative conditions.
Treatment of human hepatocarcinoma cell line HepG2 cells with H2O2, t-BHQ and _β_-NF induced ferritin H mRNA (Figure 1a). Cloning a 5.2 kb region of the 5′ human ferritin H gene and transfection of its luciferase reporter construct indicated that the ferritin H gene was transcriptionally activated in response to these pro-oxidants and that the 5.2 kb region of the ferritin H gene contains an enhancer element responsible for the transcriptional activation (Figure 1b). Further characterization indicated that the −4.0 or −0.15 kb region of the ferritin H gene containing the CCAAT box had significantly lower basal expression and failed to be activated by treatment with H2O2, t-BHQ or _β_-NF, while the −5.2 kb region exhibited higher basal expression and further activation by these oxidants (Figure 2). Therefore, we concluded that the response element to these oxidants is located between −5.2 and −4.0 kb upstream from transcription initiation site of the human ferritin H gene and it also serves as a basal enhancer. It should be noted that we confirmed previous reports by Costanzo and colleagues (Bevilacqua et al., 1997; Faniello et al., 1999) and Battistini and colleagues (Marziali et al., 1997) that the proximal region containing CCAAT box is involved in basal expression of the human ferritin H gene because we observed that the −0.15 kb region has much higher luciferase expression than that driven by −0.03 kb region containing only TATA box of the ferritin H gene (Figure 2).
Beaumont et al. (1994) and our previous mouse ferritin H studies (Tsuji et al., 1995) demonstrated that the major basal enhancer element (we initially characterized it as a target of the adenovirus E1A oncogene for transcriptional repression; (Tsuji et al., 1995, 1999)) is located approximately 4.1 kb upstream from transcription initiation site in the mouse ferritin H gene. We then identified that the E1A-targeted basal enhancer element, termed FER-1 (Tsuji et al., 1995), is a part of the ARE that activates transcription of the mouse ferritin H gene in response to hydrogen peroxide and phenolic electrophile compounds (Tsuji et al., 2000). Given the core ARE sequence TGACnnnGCA (Rushmore et al., 1991; Inamdar et al., 1996; Wasserman and Fahl, 1997b; Nioi et al., 2003), we identified an AP1-like (5′-TGACAAAGCA-3′) followed by an AP1/NFE2 (5′-TGCTGAGTCA-3′) sites located at −4.5 kb-upstream region of the human ferritin H gene (Figure 3) and these two AP1 motifs are completely conserved between the human and mouse ferritin H genes (Tsuji et al., 2000). The AP1/NFE2 site in the complementary strand (5′-TGACTCAGCA-3′) corresponds to the primary core ARE sequence, therefore, two ARE motifs are aligned in bidirectional manner. The −4.4 kb ARE(−)FH-luciferase reporter, in which both of the ARE motifs were deleted from −4.5 kb ARE(+)FH-luciferase reporter construct, resulted in decreased basal expression level similar to that driven by the CCAAT box alone, accompanied with loss of transcriptional activation by pro-oxidants (Figure 3a). Insertion of the 55 bp sequence containing bidirectional two ARE motifs into the TATA only luciferase reporter restored basal expression and also conferred induced expression by oxidant treatment (Figure 3b), indicating that the bidirectional ARE motifs are sufficient for both a basal enhancer and a functional ARE of the human ferritin H gene. Introducing mutations in either AP1-like or AP1/NFE2 site or mutations in both AP1-like and AP1/NF-E2 site abolished the ARE enhancer activity (Figure 4), suggesting that both AP1-like and AP1/NFE2 motifs are essential for the functional ARE enhancer activity.
We observed increased protein binding to the ferritin H ARE in HepG2 cells after treatment with H2O2 or t-BHQ (Figure 5). This is consistent with the results previously reported during the activation of various phase II genes by oxidative stress (Choi and Moore, 1993; Vasiliou et al., 1995; Ainbinder et al., 1997; Wild et al., 1999; Kwak et al., 2001), although no significant increase in protein binding to ARE has also been reported (Rushmore et al., 1990; Li and Jaiswal, 1992; Liu and Pickett, 1996; Wasserman and Fahl, 1997a). Among ARE binding proteins, the Nrf2 transcription factor (NFE2-related factor 2), a member of cap ‘n’ collar bZip transcription factor family, has been demonstrated to play a key role in transcriptional activation of genes encoding several phase II detoxification enzymes (Motohashi et al., 2002). Dithiolethione-mediated induction of particular phase II detoxification genes such as GSTYa and quinone reductase, but not ferritin, was impeded in Nrf2-disrupted mice (Kwak et al., 2001), implying the possibility that Nrf2 may not be a major transcription factor for transcriptional activation of ferritin genes. However, Torti and colleagues have recently demonstrated that Nrf2 is a transcriptional activator of the mouse ferritin H gene in response to chemopreventive dithiolethiones (Pietsch et al., 2003). We observed that wt Nrf2, but not mutant Nrf2 (Δ48–68, deletion of 21 amino acids in the transcription activation domain), activates the human ferritin H ARE luciferase reporter by 1.6-fold (unpublished observation). In addition, in our ChIP assays, we detected constitutive Nrf2 binding to the human ferritin H ARE, although we did not see increased binding of Nrf2 after H2O2 or t-BHQ treatment (Figure 5). Therefore, we understood that Nrf2 can be an activator of the human ferritin H ARE and maintain a basal expression of the ferritin H gene, which led us to pursue other regulatory proteins of the ferritin H ARE.
In this study, we have found that JunD is a regulatory protein that activates transcription of the human ferritin H gene via ARE. JunD has been reported to be involved in cytoprotection against apoptosis induced by UV irradiation or tumor necrosis factor-α (Weitzman et al., 2000; Lamb et al., 2003). In addition, JunD has recently been shown to regulate genes involved in antioxidant defense and protect cells from oxidative stress (Gerald et al., 2004). Regarding the role of ferritin H in oxidative stress, we and others previously demonstrated that induction of ferritin H gene expression is protective against oxidative stress-mediated cytotoxicity (Epsztejn et al., 1999; Cozzi et al., 2000; Orino et al., 2001; Pham et al., 2004). Since two AP1 motifs in the ferritin ARE are essential for the integrity of ARE (Figure 4), we asked whether JunD regulates the human ferritin H gene through ARE. In our gel shift assays, JunD binding was increased by H2O2 or t-BHQ treatment in conjunction with increased total protein binding to the ferritin H ARE (Figure 5b). Transfection of JunD activated wt ARE-luciferase, but not mutant ARE (Figure 6a). Furthermore, overexpression of JunD induced endogenous ferritin H expression (Figure 6b). ChIP assay also showed increase in JunD binding to the ferritin H ARE by H2O2 treatment (Figure 5c). In HepG2 cells, JunD was exclusively localized in nucleus but no significant increase in JunD expression after H2O2 or t-BHQ treatment (Figure 7a). Some AP1 family transcription factors including JunD are post-translationally modified, such as phosphorylation, leading to increase in transcriptional activity (Vinciguerra et al., 2004). The N-terminus of JunD has three potential MAPK phosphorylation sites (Ser90, Ser100 and Threonine 117) (Vinciguerra et al., 2004). We found that H2O2 or t-BHQ treatment induced phosphorylation of JunD in HepG2 cells and confirmed it in JunD-transfected K562 cells by Western blotting with anti-cJun-Ser73-specific antibody (Figure 7). Since this antibody detects phospho-JunD Ser100 protein due to conserved amino-acid sequences in this region between cJun and JunD, we concluded that phosphorylation of at least Ser100 of JunD was induced by H2O2 or t-BHQ treatment. JunD was shown to be activated through phosphorylation of these serines and threonine by activated ERK1/2 (Gallo et al., 2002) or JNK (Yazgan and Pfarr, 2002). We observed that H2O2 but not t-BHQ activates JNK, and both H2O2 and t-BHQ activate ERK1/2 in several cell lines including HepG2 (Y Tsuji, unpublished data), suggesting that H2O2 may utilize JNK and ERK1/2, and t-BHQ may utilize ERK1/2 for JunD phosphorylation. Phosphorylated JunD may recruit transcriptional coactivators such as p300/CBP into the ferritin H ARE for transcriptional activation (Tsuji et al., 1999).
Another post-translational activation mechanism of JunD and other AP1 family members during oxidative stress is a redox factor-mediated regulation (Nakamura et al., 1997). For instance, Ref-1 and thioredoxin were shown to reduce a conserved cysteine residue located in the DNA-binding domain of the Fos and Jun family members, including JunD (Xanthoudakis and Curran, 1996), and increase DNA-binding activity (Xanthoudakis and Curran, 1992; Hirota et al., 1997). Very recently, MBF1 was found to block oxidative modification of AP1 and ultimately increases AP1 binding to DNA (Jindra et al., 2004). It will be important to assess roles of these redox factors in JunD-mediated transcriptional regulation of ferritin H gene during oxidative stress.
Perturbations in ferritin levels and iron homeostasis are associated with malignancy, infections and immunologic insults, some of which may be due to the results of oxidative stress. The identification of an ARE enhancer element in the human ferritin H gene as well as ferritin H as one of JunD target genes through the ARE presented in this study will help our understanding of molecular mechanisms behind disorders of iron homeostasis associated with these oxidative stress conditions and diseases.
Materials and methods
Cell culture
The HepG2 human hepatocarcinoma cells and K562 human erythroleukemia cells were purchased from the American Type Culture Collection. HepG2 cells were cultured in minimum essential medium supplemented with 2 mm l-glutamine, 1 mm sodium pyruvate, 0.1 mm nonessential amino acids and 10% fetal calf serum (Mediatech). K562 cells were cultured in RPMI1640 medium containing 10% fetal calf serum. Both cell lines were incubated at 37°C in a humidified 5% CO2 atmosphere.
Cloning of the 5′-human ferritin H gene
The 5.2 kb human ferritin H promoter/enhancer region was cloned using a PCR-based cloning method, human Genome-Walker kit (Clontech). Five libraries of restriction enzyme-digested, 5′-adaptor-ligated human genomic DNA fragments were used as templates for a primary PCR with a forward primer (the adaptor sequence AP1: 5′-GTA ATA CGA CTC ACT ATA GGG C-3′) and a reverse ferritin H-specific primer (5′-TAG GAG GCG TAG AGC TCC AGG TTG ATC T-3′). The secondary PCR was carried out with the 1/2500 of the primary PCR products and a nested forward primer (AP2: 5′-ACT ATA GGG CAC GCG TGG T-3′) and a reverse ferritin H primer (5′-AAG AAC GTC TGG CCC TGC GGG TCG CTT GT-3′). As a result, the ScaI genomic DNA library gave rise to the largest DNA fragment (3.0 kb). In the similar strategy, further upstream region (additional 2.6 kb) of the human ferritin H gene was amplified by PCR. The partially overlapped three PCR fragments (2.0, 1.0 and 3.0 kb from 5′ to 3′ regions), covering approximately a 5.6 kb region 5′ to the transcription initiation site of the human ferritin H gene, were cloned into pT-Adv TA cloning vector (Invitrogen).
Since these DNA fragments have partially overlapped regions and also contain the 48 bp GenomeWalker adaptor sequence at their 5′-ends, reconstitution of the continuous 5′-region of the human ferritin H gene were carried out by PCR to remove the adaptor and overlapped sequences. pT-Adv2.0 kb was used as a template to amplify the region between −5.6 and −3.6 kb of the ferritin H gene with a set of primers (5′-AGC CGC TAG CCA AAG GTA GAG AGC CGA-3′) and (5′-GAG CAC AGG AGG AAA TGA AGG TTT CG-3′). For PCR amplification of the 0.9 kb region (between −3.7 and −2.8 kb region), pT-Adv1.0 kb was used as a template with (5′-CGA AAC CTT CAT TTC CTC CTG TGC TC-3′) and (5′-TCT TCT GAG GCT GTC AGA AGC CGT AAA-3′) primers, allowing removal of GenomeWalker adaptor sequences. In order to amplify the continuous −5.6 to −2.8 kb region, a mixture of these two PCR products with two outside primers was PCR amplified and digested with _Dra_I and _Sca_I to obtain 2.2 kb DNA (−5.2 to −3.0 kb of the human ferritin H gene). For amplification of the −3.0 kb to +17 region, pT-Adv3.0 kb was used as a template with _Sca_I (5′-ACA TAG TAC TTC ATT AAA CAT ACA AAT-3′) and _Xho_I (5′-ATA TCT CGA GAA GAA CGT CTG GCC CTG C-3′) primes. The 3.0 kb PCR fragment was digested with _Sca_l and _Xho_I, removing the GenomeWalker adaptor sequences and creating these restriction sites at 5′- and 3′-end, respectively. Then the _Dra_I-_Sca_I 2.2 kb and _Sca_I–_Xho_I 3.0 kb DNA fragments were cloned into pBluescript SK(−) vector to construct pBluescript SK(−)-5.2 kb h-ferritin H. The entire 5.2 kb 5′-ferritin H gene was verified by DNA sequencing (Qiagen Co. and SeqWright Co.) and it was 99.2% identical to nucleotides 14926–20111 in the NCBI Accession number AF139813.
Construction of luciferase reporter plasmids
pBluescript SK(−)0-luciferase was constructed by ligation of the _Xho_I–_Sal_I 2.0 kb luciferase DNA derived from pGL3 basic (Promega) to _Xho_I-digested pBluescript SK(−). pBluescript SK(−)-5.2 kb h-ferritin H-luciferase was similarly constructed by ligation of the _Xho_I–_Sal_I 2.0 kb luciferase DNA to _Xho_I-digested pBluescript SK(−)-5.2 kb h-ferritin H. The 5′ deletion mutants of h-ferritin H luciferase were constructed by digestion of pBluescript SK(−)-5.2 kb h-ferritin H-luciferase with _Spe_I and _Nsi_I (for −4.0 kb), or _Not_I (−0.15 kb), followed by treatment with the large fragment of DNA polymerase I (−0.15 kb) or T4 DNA polymerase (−4.0 kb) and the larger DNA fragment from each reaction was purified and self-ligated. The −0.03 kbTATA h-ferritin H-luciferase was constructed by digestion of pBluescript SK(−)-0.15 kb h-ferritin H-luciferase with _Bgl_I, followed by successive treatment with T4 DNA polymerase and _Xho_I, and ligation of the resultant 50 bp DNA fragment to _Eco_RV/_Xho_I sites of the pBluescript SK(−)0-luciferase. The −4.5 kb ARE(+) and −4.4 kb ARE (−) ferritin H 5′ regions were amplified by PCR using pBluescript SK(−)-5.2 kb h-ferritin H as a template in the presence of the _Xho_I primer described above and an _Spe_I primer 5′-ATA TAC TAG TCC ATG ACA AAG CAC TTT TTG A-3′ or 5′-ATA TAC TAG TGG GAC AGC AAC TCT CCA CC-3′, respectively. The 4.5 and 4.4 kb PCR fragment were cloned into pT-Adv vector, followed by digestion with _Spe_I and _Xho_I and the 4.5 and 4.4 kb _Spe_I/_Xho_I fragment were cloned into _Spe_I/_Xho_I digested pBluescript SK(−)0-luciferase vector. All plasmid constructs were characterized by digestion with several restriction enzymes, and the nucleotide sequence of deleted or mutated regions were verified by DNA sequencing (Qiagen Co. and SeqWright Co.).
DNA transfection, luciferase reporter assays and Western blotting
Transient DNA transfection into HepG2 cells was carried out by the calcium phosphate precipitation method as described previously (Tsuji et al., 1993), except the following minor modifications: cells were plated at a density of 4 × 105 cells/35 mm plate containing 2 ml of the culture medium, and a total 0.2 ml of calcium phosphate solution containing 0.5–1 _μ_g of each reporter plasmid DNA was added to the cells. As a transfection internal control, 0.1 _μ_g of pRL-CMV (CMV promoter-Renilla luciferase, Promega) or pRL-EF (elongation factor promoter) was simultaneously cotransfected. After incubation for 16–24 h, the cells were fed with fresh growth medium and incubated for 24 h, followed by treatment with H2O2, t-BHQ or _β_-NF for 8–24 h. Preparation of cell extracts and luciferase assays were performed using Dual Luciferase Assay Reagents (Promega) and the luciferase activity was measured with Luminometer (Model 20E, Turner Designs). Luciferase expression driven by the ferritin H gene in each transfected sample was normalized by Renilla luciferase activity.
Transfection of JunD expression plasmids into K562 cells was carried out by using the Bio-Rad X-Cell with a preset electroporation protocol (100 _μ_l of cell suspension in 0.2 cm cuvette at 155 V, 1000 _μ_F). The mouse JunD cDNA fragment was cut out from pBluescript SK(−) JunD (XHJ-12.4, American Type Culture Collection) and cloned into pRc/CMV (Invitrogen), pcDNAINeo (Invitrogen) or pCMV vector (Tsuji et al., 1995). Each JunD expression plasmid (15 _μ_g) was electroporated into 4–7 × 106 K562 cells and incubated for 50–72 h. As a negative control, no DNA or 15 _μ_g of pRc/CMV empty vector was similarly electroporated into K562 cells. Transfected K562 cells were harvested and subjected to Western blotting followed by ECL detection using an anti-JunD antibody, anti-ferritin H antibody (Santa Cruz Biotechnology) or anti-_β_-actin antibody (Sigma). To confirm the specificity of the ferritin H antibody, 50 ng of recombinant human ferritin H and L proteins (Calbiochem) were loaded on the same gel.
Gel retardation assay
Wt and mutant 65 bp ARE probe were labeled with 32P-dCTP by primer extension with the large fragment of DNA polymerase I, and a 22 bp AP1 double-stranded oligonucleotide was end-labeled with T4 polynucleotide kinase. HepG2 nuclear extracts were prepared by using nuclear extract isolation kit (Active Motif), and binding reaction and separation of retarded bands by polyacrylamide gel electrophoresis were described previously (Tsuji et al., 1995). Anti-JunD antibody used in gel supershift assays was purchased from Santa Cruz Biotechnology Inc.
ChIP assay
A total of 3–5 × 106 HepG2 cells/100 mm plate were treated with oxidative stressors for 4 h, followed by crosslinking chromatins with 10% formaldehyde. Preparation of cell lysates was carried out by using the ChIP assay kit (UPSTATE Biology) according to the provided protocol with minor modifications. All antibodies for immunoprecipitations were purchased from Santa Cruz Biotechnology. Quantitative PCR was performed in 50 _μ_l of reactions containing 0.5–1 _μ_Ci of [_α_-32P]dCTP and Advantage 2 PCR polymerase mix (Clontech) with 26–30 cycles at 94°C for 30 s and 68°C for 1 min. A pair of primers to amplify human ferritin H ARE-containing 0.15 kb region were 5′-CCCTCCAGGTCTTATGACTGCTC-3′ and 5′-GTTTCTGGAGGTTCAGCACGTC-3′. In this condition, 1pg or less of pBluescript −5.2 kb ferritin H-luciferase plasmid or 30–300 ng of sonicated K562 genomic DNA gave rise to proportional intensities of the 0.15 kb band. The PCR reactions were loaded and separated on 8% acrylamide gel and subjected to autoradiography.
Cell fractionation and JunD Western Blotting
HepG2 cells (2 × 106 cells/100 mm plate), or 1–2 × 107 K562 cells transfected with 20 _μ_g of pRc/CMVJunD by electroporation were treated with 100 _μ_m H2O2 or 100 _μ_m t-BHQ for 1.5–4 h. Nuclear and cytoplasmic fractions were separated using nuclear extracts isolation kit (Active Motif). Both fractions were subjected to 10% acrylamide SDS–PAGE and total JunD and phosphorylated JunD were detected by Western blotting with anti-JunD antibody (Santa Cruz Biotechnology) and anti-cJun phosphoserine-73 antibody (Cell Signaling) that crossreacts phospho-JunD at Ser100, respectively. The purity of each fraction was verified by Western blotting with anti-histone H1 antibody (Santa Cruz Biotechnology) and anti-lactate dehydrogenase antibody (Chemicon).
Acknowledgments
We are grateful to Dr Kazushi Inoue at Wake Forest University School of Medicine for helpful advice on ChIP assays. This work was supported by the National Institutes of Health research grant DK-60007 and North Carolina State University Faculty Research and Professional Development Fund SPS0064-9596 to Y Tsuji.
References
- Ainbinder E, Bergelson S, Pinkus R, Daniel V. Eur J Biochem. 1997;243:49–57. doi: 10.1111/j.1432-1033.1997.0049a.x. [DOI] [PubMed] [Google Scholar]
- Arosio P, Levi S. Free Radic‘ Biol Med. 2002;33:457–463. doi: 10.1016/s0891-5849(02)00842-0. [DOI] [PubMed] [Google Scholar]
- Beaumont C, Jain SK, Bogard M, Nordmann Y, Drysdale J. J Biol Chem. 1987;262:10619–10623. [PubMed] [Google Scholar]
- Beaumont C, Seyhan A, Yachou AK, Grandchamp B, Jones R. J Biol Chem. 1994;269:20281–20288. [PubMed] [Google Scholar]
- Bevilacqua MA, Faniello MC, Quaresima B, Tiano MT, Giuliano P, Feliciello A, Avvedimento VE, Cimino F, Costanzo F. J Biol Chem. 1997;272:20736–20741. doi: 10.1074/jbc.272.33.20736. [DOI] [PubMed] [Google Scholar]
- Bevilacqua MA, Faniello MC, Russo T, Cimino F, Costanzo F. Biochem J. 1998;335(Part 3):521–525. doi: 10.1042/bj3350521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bomford A, Conlon-Hollingshead C, Munro HN. J Biol Chem. 1981;256:948–955. [PubMed] [Google Scholar]
- Cairo G, Tacchini L, Pogliaghi G, Anzon E, Tomasi A, Bernelli-Zazzera A. J Biol Chem. 1995;270:700–703. doi: 10.1074/jbc.270.2.700. [DOI] [PubMed] [Google Scholar]
- Choi HS, Moore DD. Mol Endocrinol. 1993;7:1596–1602. doi: 10.1210/mend.7.12.8145765. [DOI] [PubMed] [Google Scholar]
- Chou CC, Gatti RA, Fuller ML, Concannon P, Wong A, Chada S, Davis RC, Salser WA. Mol Cell Biol. 1986;6:566–573. doi: 10.1128/mcb.6.2.566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cozzi A, Corsi B, Levi S, Santambrogio P, Albertini A, Arosio P. J Biol Chem. 2000;275:25122–25129. doi: 10.1074/jbc.M003797200. [DOI] [PubMed] [Google Scholar]
- Dhakshinamoorthy S, Jaiswal AK. J Biol Chem. 2000;275:40134–40141. doi: 10.1074/jbc.M003531200. [DOI] [PubMed] [Google Scholar]
- Epsztejn S, Glickstein H, Picard V, Slotki IN, Breuer W, Beaumont C, Cabantchik ZI. Blood. 1999;94:3593–3603. [PubMed] [Google Scholar]
- Faniello MC, Bevilacqua MA, Condorelli G, de Crombrugghe B, Maity SN, Avvedimento VE, Cimino F, Costanzo F. J Biol Chem. 1999;274:7623–7626. doi: 10.1074/jbc.274.12.7623. [DOI] [PubMed] [Google Scholar]
- Friling RS, Bensimon A, Tichauer Y, Daniel V. Proc Natl Acad Sci USA. 1990;87:6258–6262. doi: 10.1073/pnas.87.16.6258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallo A, Cuozzo C, Esposito I, Maggiolini M, Bonofiglio D, Vivacqua A, Garramone M, Weiss C, Bohmann D, Musti AM. Oncogene. 2002;21:6434–6445. doi: 10.1038/sj.onc.1205822. [DOI] [PubMed] [Google Scholar]
- Gerald D, Berra E, Frapart YM, Chan DA, Giaccia AJ, Mansuy D, Pouyssegur J, Yaniv M, Mechta-Grigoriou F. Cell. 2004;118:781–794. doi: 10.1016/j.cell.2004.08.025. [DOI] [PubMed] [Google Scholar]
- Harrison PM, Arosio P. Biochim Biophys Acta. 1996;1275:161–203. doi: 10.1016/0005-2728(96)00022-9. [DOI] [PubMed] [Google Scholar]
- Hayes JD, McMahon M. Cancer Lett. 2001;174:103–113. doi: 10.1016/s0304-3835(01)00695-4. [DOI] [PubMed] [Google Scholar]
- Hentze MW, Kuhn LC. Proc Natl Acad Sci USA. 1996;93:8175–8182. doi: 10.1073/pnas.93.16.8175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hentze MW, Muckenthaler MU, Andrews NC. Cell. 2004;117:285–297. doi: 10.1016/s0092-8674(04)00343-5. [DOI] [PubMed] [Google Scholar]
- Hirota K, Matsui M, Iwata S, Nishiyama A, Mori K, Yodoi J. Proc Natl Acad Sci USA. 1997;94:3633–3638. doi: 10.1073/pnas.94.8.3633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inamdar NM, Ahn YI, Alam J. Biochem Biophys Res Commun. 1996;221:570–576. doi: 10.1006/bbrc.1996.0637. [DOI] [PubMed] [Google Scholar]
- Jaiswal AK. Biochem Pharmacol. 1994;48:439–444. doi: 10.1016/0006-2952(94)90272-0. [DOI] [PubMed] [Google Scholar]
- Jaiswal AK. Methods Enzymol. 2004;378:221–238. doi: 10.1016/S0076-6879(04)78018-0. [DOI] [PubMed] [Google Scholar]
- Jindra M, Gaziova I, Uhlirova M, Okabe M, Hiromi Y, Hirose S. EMBO J. 2004;23:3538–3547. doi: 10.1038/sj.emboj.7600356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwak EL, Larochelle DA, Beaumont C, Torti SV, Torti FM. J Biol Chem. 1995;270:15285–15293. doi: 10.1074/jbc.270.25.15285. [DOI] [PubMed] [Google Scholar]
- Kwak MK, Itoh K, Yamamoto M, Sutter TR, Kensler TW. Mol Med. 2001;7:135–145. [PMC free article] [PubMed] [Google Scholar]
- Lamb JA, Ventura JJ, Hess P, Flavell RA, Davis RJ. Mol Cell. 2003;11:1479–1489. doi: 10.1016/s1097-2765(03)00203-x. [DOI] [PubMed] [Google Scholar]
- Levi S, Luzzago A, Cesareni G, Cozzi A, Franceschinelli F, Albertini A, Arosio P. J Biol Chem. 1988;263:18086–18092. [PubMed] [Google Scholar]
- Levi S, Santambrogio P, Cozzi A, Rovida E, Corsi B, Tamborini E, Spada S, Albertini A, Arosio P. J Mol Biol. 1994;238:649–654. doi: 10.1006/jmbi.1994.1325. [DOI] [PubMed] [Google Scholar]
- Levi S, Yewdall SJ, Harrison PM, Santambrogio P, Cozzi A, Rovida E, Albertini A, Arosio P. Biochem J. 1992;288(Part 2):591–596. doi: 10.1042/bj2880591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Jaiswal AK. J Biol Chem. 1992;267:15097–15104. [PubMed] [Google Scholar]
- Liu S, Pickett CB. Biochemistry. 1996;35:11517–11521. doi: 10.1021/bi960572p. [DOI] [PubMed] [Google Scholar]
- Marziali G, Perrotti E, Ilari R, Testa U, Coccia EM, Battistini A. Mol Cell Biol. 1997;17:1387–1395. doi: 10.1128/mcb.17.3.1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meneghini R. Free Radic Biol Med. 1997;23:783–792. doi: 10.1016/s0891-5849(97)00016-6. [DOI] [PubMed] [Google Scholar]
- Miller LL, Miller SC, Torti SV, Tsuji Y, Torti FM. Proc Natl Acad Sci USA. 1991;88:4946–4950. doi: 10.1073/pnas.88.11.4946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motohashi H, O'Connor T, Katsuoka F, Engel JD, Yamamoto M. Gene. 2002;294:1–12. doi: 10.1016/s0378-1119(02)00788-6. [DOI] [PubMed] [Google Scholar]
- Nakamura H, Nakamura K, Yodoi J. Annu Rev Immunol. 1997;15:351–369. doi: 10.1146/annurev.immunol.15.1.351. [DOI] [PubMed] [Google Scholar]
- Nguyen T, Sherratt PJ, Pickett CB. Annu Rev Pharmacol Toxicol. 2003;43:233–260. doi: 10.1146/annurev.pharmtox.43.100901.140229. [DOI] [PubMed] [Google Scholar]
- Nioi P, McMahon M, Itoh K, Yamamoto M, Hayes JD. Biochem J. 2003;374:337–348. doi: 10.1042/BJ20030754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orino K, Lehman L, Tsuji Y, Ayaki H, Torti SV, Torti FM. Biochem J. 2001;357:241–247. doi: 10.1042/0264-6021:3570241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orino K, Tsuji Y, Torti FM, Torti SV. FEBS Lett. 1999;461:334–338. doi: 10.1016/s0014-5793(99)01443-x. [DOI] [PubMed] [Google Scholar]
- Pham CG, Bubici C, Zazzeroni F, Papa S, Jones J, Alvarez K, Jayawardena S, De Smaele E, Cong R, Beaumont C, Torti FM, Torti SV, Franzoso G. Cell. 2004;119:529–542. doi: 10.1016/j.cell.2004.10.017. [DOI] [PubMed] [Google Scholar]
- Pietsch EC, Chan JY, Torti FM, Torti SV. J Biol Chem. 2003;278:2361–2369. doi: 10.1074/jbc.M210664200. [DOI] [PubMed] [Google Scholar]
- Richardson DR, Ponka P. Biochim Biophys Acta. 1997;1331:1–40. doi: 10.1016/s0304-4157(96)00014-7. [DOI] [PubMed] [Google Scholar]
- Rouault TA, Klausner RD. Trends Biochem Sci. 1996;21:174–177. [PubMed] [Google Scholar]
- Rushmore TH, King RG, Paulson KE, Pickett CB. Proc Natl Acad Sci USA. 1990;87:3826–3830. doi: 10.1073/pnas.87.10.3826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rushmore TH, Morton MR, Pickett CB. J Biol Chem. 1991;266:11632–11639. [PubMed] [Google Scholar]
- Rushmore TH, Pickett CB. J Biol Chem. 1990;265:14648–14653. [PubMed] [Google Scholar]
- Santambrogio P, Levi S, Arosio P, Palagi L, Vecchio G, Lawson DM, Yewdall SJ, Artymiuk PJ, Harrison PM, Jappelli R, Cesareni G. J Biol Chem. 1992;267:14077–14083. [PubMed] [Google Scholar]
- Theil EC. Biochem Pharmacol. 2000;59:87–93. doi: 10.1016/s0006-2952(99)00300-7. [DOI] [PubMed] [Google Scholar]
- Theil EC. J Nutr. 2003;133:1549S–1553S. doi: 10.1093/jn/133.5.1549S. [DOI] [PubMed] [Google Scholar]
- Torti FM, Torti SV. Blood. 2002;99:3505–3516. doi: 10.1182/blood.v99.10.3505. [DOI] [PubMed] [Google Scholar]
- Torti SV, Kwak EL, Miller SC, Miller LL, Ringold GM, Myambo KB, Young AP, Torti FM. J Biol Chem. 1988;263:12638–12644. [PubMed] [Google Scholar]
- Tsuji Y, Akebi N, Lam TK, Nakabeppu Y, Torti SV, Torti FM. Mol Cell Biol. 1995;15:5152–5164. doi: 10.1128/mcb.15.9.5152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuji Y, Ayaki H, Whitman SP, Morrow CS, Torti SV, Torti FM. Mol Cell Biol. 2000;20:5818–5827. doi: 10.1128/mcb.20.16.5818-5827.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuji Y, Miller LL, Miller SC, Torti SV, Torti FM. J Biol Chem. 1991;266:7257–7261. [PubMed] [Google Scholar]
- Tsuji Y, Moran E, Torti SV, Torti FM. J Biol Chem. 1999;274:7501–7507. doi: 10.1074/jbc.274.11.7501. [DOI] [PubMed] [Google Scholar]
- Tsuji Y, Ninomiya-Tsuji J, Torti SV, Torti FM. J Immunol. 1993;150:1897–1907. [PubMed] [Google Scholar]
- Vasiliou V, Puga A, Chang CY, Tabor MW, Nebert DW. Biochem Pharmacol. 1995;50:2057–2068. doi: 10.1016/0006-2952(95)02108-6. [DOI] [PubMed] [Google Scholar]
- Venugopal R, Jaiswal AK. Oncogene. 1998;17:3145–3156. doi: 10.1038/sj.onc.1202237. [DOI] [PubMed] [Google Scholar]
- Vinciguerra M, Vivacqua A, Fasanella G, Gallo A, Cuozzo C, Morano A, Maggiolini M, Musti AM. J Biol Chem. 2004;279:9634–9641. doi: 10.1074/jbc.M308721200. [DOI] [PubMed] [Google Scholar]
- Wagstaff M, Worwood M, Jacobs A. Biochem J. 1978;173:969–977. doi: 10.1042/bj1730969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wasserman WW, Fahl WE. Arch Biochem Biophys. 1997a;344:387–396. doi: 10.1006/abbi.1997.0215. [DOI] [PubMed] [Google Scholar]
- Wasserman WW, Fahl WE. Proc Natl Acad Sci USA. 1997b;94:5361–5366. doi: 10.1073/pnas.94.10.5361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weitzman JB, Fiette L, Matsuo K, Yaniv M. Mol Cell. 2000;6:1109–1119. doi: 10.1016/s1097-2765(00)00109-x. [DOI] [PubMed] [Google Scholar]
- Wild AC, Moinova HR, Mulcahy RT. J Biol Chem. 1999;274:33627–33636. doi: 10.1074/jbc.274.47.33627. [DOI] [PubMed] [Google Scholar]
- Xanthoudakis S, Curran T. EMBO J. 1992;11:653–665. doi: 10.1002/j.1460-2075.1992.tb05097.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xanthoudakis S, Curran T. Adv Exp Med Biol. 1996;387:69–75. [PubMed] [Google Scholar]
- Yazgan O, Pfarr CM. J Biol Chem. 2002;277:29710–29718. doi: 10.1074/jbc.M204552200. [DOI] [PubMed] [Google Scholar]