Mll fusions generated by Cre-loxP-mediated de novo translocations can induce lineage reassignment in tumorigenesis (original) (raw)
Abstract
Chromosomal translocations are primary events in tumorigenesis. Those involving the mixed lineage leukaemia (MLL) gene are found in various guises and it is unclear whether MLL fusions can affect haematopoietic differentiation. We have used a model in which chromosomal translocations are generated in mice de novo by Cre-_loxP_-mediated recombination (translocator mice) to compare the functionally relevant haematopoietic cell contexts for Mll fusions, namely pluripotent stem cells, semicommitted progenitors or committed cells. Translocations between Mll and Enl or Af9 cause myeloid neoplasias, initiating in pluripotent stem cells or multipotent myeloid progenitors. However, while Mll-Enl translocations can also cause leukaemia from T-cell progenitors, no tumours arose with Mll-Af9 translocations in the T-cell compartment. Furthermore, Mll-Enl translocations in T-cell progenitors can cause lineage reassignment into myeloid tumours. Therefore, a permissive cellular environment is required for oncogenicity of _Mll_-associated translocations and Mll fusions can influence haematopoietic lineage commitment.
Keywords: chromosomal translocations, Cre-loxP, ES cells, gene fusions, homologous recombination
Introduction
Cancer generally arises from a single cell that acquires a somatic mutation in a gene capable of eliciting a cell division property and/or immortalisation (Vogelstein and Kinzler, 2004). This initiating cell also gains the characteristic of self-renewal but may or may not be the cell that has the self-maintenance capacity of the putative cancer stem cell (Reya et al, 2001). Chromosomal translocations are primary events in cancer, and create an immortal lineage that provides cells for secondary mutations to give frank malignancy. Leukaemia and lymphomas arise within a hierarchy of progenitor, committed or terminally differentiated cells. These are exemplified, respectively, by chronic myeloid leukaemia (CML), Burkitt's lymphoma and multiple myeloma. Among the somatic mutations that characterise these tumours, recurrent chromosomal translocations are generally found at presentation of disease. Moreover, chromosomal translocations are often the sole cytogenetic chromosomal abnormality at such time (see http://cgap.nci.nih.gov/Chromosomes/Mitelman). Important studies of concordant leukaemias in monozygotic twins have shown the presence of identical MLL-AF4 (mixed lineage leukaemia) chromosomal translocations but with different immunoglobulin gene rearrangements, demonstrating the in utero origin of this leukaemic precursor cell (Mori et al, 2002; Greaves et al, 2003).
Chromosomal translocations involving the MLL gene typify gene fusion. MLL is involved in multifarious chromosomal translocations in human acute leukaemias with adverse prognosis. The MLL gene is located on human chromosome 11, band q23, and is associated with translocations, inversions and duplications of 11q23 (see Daser and Rabbitts, 2004). MLL is a homologue of the Drosophila Trx gene and Hox genes are necessary for Mll-fusion-induced leukaemia in mice (Ayton and Cleary, 2003; Kumar et al, 2004). Thus, presumably MLL-fusion proteins confer deregulation on HOX genes, in turn as a consequence of chromosomal abnormalities of chromosome 11q23. MLL protein can bind directly to HOXA9 and HOXC8 promoters (Milne et al, 2002; Nakamura et al, 2002) and can act as a histone methyltransferase (methylating Lys4 of histone H3) (Sobulo et al, 1997; Birke et al, 2002; Milne et al, 2002; Xia et al, 2003; Hess, 2004; Yokoyama et al, 2004).
MLL-associated translocations represent a cross-section of possible associations found in acute leukaemias. There are more than 30 different known chromosome partners involving MLL, each appearing to result in a fusion of MLL with a gene from a different chromosomal region (see Daser and Rabbitts, 2004). A notable feature of MLL translocation leukaemias is that the main fusion partners (i.e. AF4, AF9, ENL, ELL) are found in tumours of either lymphoid lineage (AF4) or myeloid lineage (AF9 or ELL) or of both lineages (ENL). It is not known if MLL fusions are simply oncogenic in any haematopoietic cell, either progenitor or committed cells. Furthermore, it is still unclear if an MLL-fusion protein has an instructive property on a cell, driving differentiation into specific haematopoietic lineages. Mouse models have begun to address these issues. Studies of retroviral transduction of bone marrow stem cells with viruses expressing MLL fusions (Ayton and Cleary, 2001; So et al, 2003; Zeisig et al, 2003), gene targeting knock-in models expressing Mll-AF9 fusion (Corral et al, 1996; Dobson et al, 1999) or a translocator mouse model, in which de novo translocations of Mll-Enl occur (Forster et al, 2003), show that haematopoietic stem cells (HSC) are targets for Mll fusions. In addition, retroviral transduction of semicommitted, common myeloid progenitors (CMP) (Miyamoto et al, 2002) has shown that similar leukaemias result from these progenitors as from transduced bone marrow HSC (Cozzio et al, 2003). Recently, an Mll-CBP conditional knock-in model indicated targeting of granulocyte–macrophage progenitors (Wang et al, 2005).
In the translocator mouse model (Buchholz et al, 2000; Collins et al, 2000), chromosomal translocations are made de novo in a cell-specific manner, using Cre-loxP recombination in vivo (Smith et al, 1995; van Deursen et al, 1995). Chromosomal translocations can thus be generated as primary genetic changes, initially in the absence of any other changes and at predetermined stages of haematopoiesis. Using Lmo2-Cre to express Cre recombinase, chromosomal translocations were made in pluripotent stem cells, and Mll-Enl fusion resulted in myeloid leukaemia (Forster et al, 2003). We have now applied the translocator approach using an Lck-Cre allele that specifically causes expression of Cre recombinase in T cells and their progenitors (McCormack et al, 2003; Codrington et al, 2005). We find that Mll-Enl fusions can be oncogenic in the lymphoid lineage as in human leukaemias. However, when Mll-Af9 translocations are induced in similar T-cell populations via Lck-Cre expression, no leukaemias arose, although Mll-Af9 translocations in uncommitted progenitors do induce myeloid leukaemias. Therefore, Mll fusions are not necessarily oncogenic in any haematopoietic cell. Furthermore, the Mll-associated leukaemic precursor cell expressing the Mll-Enl fusion can undergo lineage reassignment from the lymphoid lineage into myeloid lineage.
Results
Mll-Enl and Mll-Af9 fusions cause myeloid neoplasias in translocator mice
Mice have been described (Collins et al, 2000; Forster et al, 2003) carrying Mll, Enl or Af9 genes with loxP sites within the appropriate introns for Cre-mediated chromosomal translocations equivalent to those found in human leukaemias (the loxP sites were introduced by homologous recombination in embryonic stem (ES) cells and these targeted ES cells were used to generate mice). These mice were designated translocator mice (Forster et al, 2003). The Mll, Enl or Af9 alleles are diagrammatically shown in Figure 1A, indicating the intronic location of the loxP sites. Cre recombinase can recognise these sites and cause interchromosomal translocations resulting in translocation t(9;4) (Mll-Af9) or t(9;17) (Mll-Enl) (Figure 1A).
Figure 1.
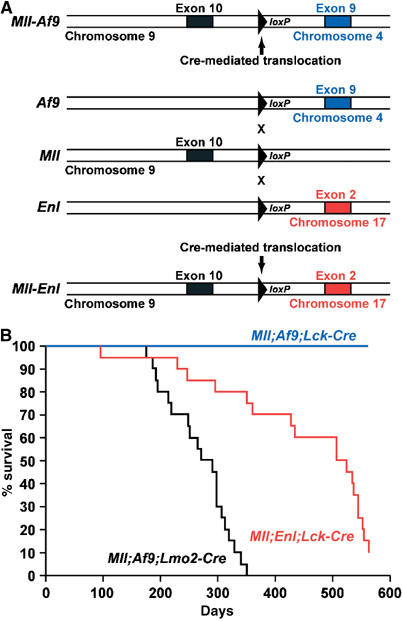
Incidence of leukaemias in Mll translocator mice. (A) Diagram of Mll, Enl and Af9 targeted alleles in translocator mice (Collins et al, 2000; Forster et al, 2003). LoxP sites were introduced, by homologous recombination in ES cells, into introns of mouse Mll, Enl or Af9, corresponding to the human introns where chromosomal translocations are typically found. Specific cells in mice carrying loxP sites in both Mll and Enl or Mll and Af9 genes can undergo chromosomal translocations, mediated by Cre recombinase, to create fusion genes analogous to those of human leukaemias, as indicated. (B) Survival curves for translocator mice. Cohorts of 20 translocator mice expressing Cre under the control of Lck or Lmo2 promoters (McCormack et al, 2003) were analysed over a period of about 1.5 years.
The effects of targeting the Mll chromosomal translocations to haematopoietic cells of differing maturities, and comparative assessment of chromosomal translocations between Mll and different fusion partners such as Enl or Af9 genes, were analysed with these Mll; Enl or Mll; Af9 translocators. We used a strain of mice with Cre recombinase expression under the control of the Lck promoter (Wildin et al, 1995), which restricts Cre expression to the T-cell lineage (McCormack et al, 2003; Codrington et al, 2005), or the Lmo2 promoter, which works in HSC (Yamada et al, 1998; McCormack et al, 2003; Schlaeger et al, 2004). Cohorts of 20 mice were established with Mll plus Enl or Mll plus Af9 loxP alleles and carrying the Lck-Cre transgene (designated, respectively, Mll; Enl; Lck-Cre or Mll; Af9; Lck-Cre mice). In addition, a cohort of Mll; Af9; Lmo2-Cre mice was studied. Comparable control groups with only the Lck-Cre allele, the Mll plus Enl loxP or Mll plus Af9 loxP alleles were also studied. All mice were monitored for possible adverse effects. Out of the 20 Mll; Enl; Lck-Cre mice, 18 developed haematopoietic neoplasms within 18 months (Figure 1B) and all the mice in the Mll; Af9; Lmo2-Cre cohort developed haematological malignancies. No haematological malignancies were observed in the mice of the Mll; Af9; Lck-Cre cohort.
Mll; Enl; Lck-Cre translocators develop either lymphoid or myeloid tumours
Haematopoietic neoplasias in the Mll-Enl translocators were dependent on Lck-Cre and in Mll-Af9 translocators on Lmo2-Cre, supporting a mandatory role for chromosomal translocations in neoplasia in these mice. The presence of chromosomal translocations in the cells characterising the haematological malignancies was verified by either genomic DNA filter hybridisation to detect the chromosomal fusion point or by fluorescence in situ hybridisation (FISH) (Figure 2). Exemplification is presented in Figure 2, showing filter hybridisation of Mll; Enl; Lck-Cre or Mll; Af9; Lmo2-Cre translocator DNA (tumour, thymus and liver biopsy) with 5′ Mll, 3′ Mll (Figure 2A and B), 3′ Enl probes (Figure 2C) or a 3′ Af9 probe (Figure 2D). The translocated alleles were observed in all three tissues examined, even in the liver where some sections have high numbers of cells with the chromosomal translocation. An independent assessment of the presence of chromosomal translocations in the tumour cells was obtained by FISH. Metaphase spreads from spleen cells of tumour mice were hybridised with whole chromosome paints for chromosome 9 (Mll, green) and chromosome 17 (Enl, red) (Figure 2C) or chromosome 4 (Af9, red; Figure 2D). In addition, expression of the _Mll_-fusion genes was confirmed by RT–PCR analysis of RNA prepared from cell lines established from tumours (see below). We could detect the relevant _Mll_-fusion mRNA in RNA prepared from Mll-AF9 knock-in, Mll; Enl; Lmo2-Cre and Mll; Af9; Lmo2-Cre translocator mice (Figure 2E). The cells present in the tumours, therefore, express _Mll_-fusion mRNA, carry reciprocal chromosomal translocations and the malignancies are caused by the Mll-fusion protein since they are dependent on the presence of Mll-loxP, Enl-loxP or Af9-loxP plus the _Cre_-expressing alleles.
Figure 2.
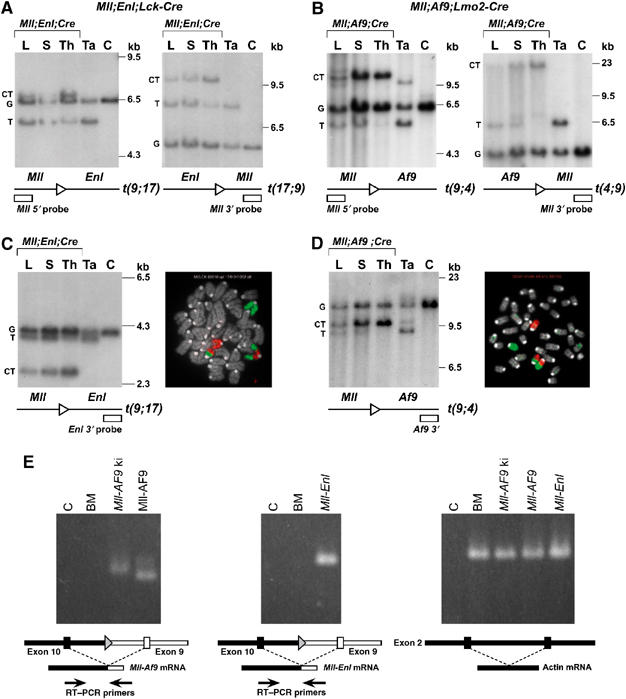
Haematological malignancies in Mll translocators have chromosomal translocations. Mll; Enl; Lck-Cre (A, C) or Mll; Af9; Lmo2-Cre (B, D) tumours were examined for the presence of chromosomal translocations by filter hybridisation and by FISH. For the former, DNA samples are designated as follows: C: control C57BL/6 tail or CCB ES DNA; Ta: tail; Th: thymus; S: spleen; L: liver. For each panel, the spleen, liver and thymus DNAs were prepared from Mll; Enl; Lck-Cre or Mll; Af9; Lmo2-Cre mice (overlined samples). Hybridising bands correspond to targeted (T), germline (G) or chromosomal translocation (CT) alleles. Hybridisation probes from either side of the translocation junction, namely 5′ and 3′ Mll probes and a 3′ Enl probe (A and C, respectively) or 5′ and 3′ Mll probes and a 3′ Af9 probe (B and D, respectively), were used to detect rearrangements. For FISH analysis (C, D), metaphase spreads were made with cells from the spleen and painted with whole chromosome paints for chromosome 9 (green) and chromosome 4 (Af9) or 17 (Enl) (red). Painted chromosomes from an Mll; Enl; Lck-Cre (C) or Mll; Af9; Lmo2-Cre spleen (D) are shown. (E) RT–PCR analysis of _Mll_-fusion mRNA. Cell lines were established from Mll-AF9 knock-in mice (Corral et al, 1996; Dobson et al, 1999), Mll; Enl; Lmo2-Cre translocators (Forster et al, 2003) and Mll; Af9; Lmo2-Cre translocators (this paper). RNA was made and converted to cDNA for RT–PCR amplification with primers spanning junctions of Mll and Enl or Af9 RNA sequences as indicated. Total bone marrow RNA from a wild-type mouse was used as a negative control for cDNA lacking Mll-fusion sequences and RT–PCR with actin primers was used for quality control of cDNA. C: no template (H2O) control; BM: bone marrow; ki: knock-in.
Post-mortem analysis revealed splenomegaly typifying the haematological malignancy in both the Mll; Enl; Lck-Cre and Mll; Af9; Lmo2-Cre translocators, due to an almost total repopulation of the spleen with malignant cells (Figure 3) represented by loss of distinct white and red pulp architecture. In Mll; Af9; Lmo2-Cre translocators, perivascular deposits of these same cells are evident with associated infiltration of large areas of the liver (a similar picture was found with kidney and lung, data not shown) and involvement of lymph nodes. These features, together with myeloid surface marker expression on the neoplastic cells (Figure 4 and Supplementary Table 1B), suggest that these mice consistently develop myeloid lineage malignancies. Blood smears show circulating leukaemic cells of various maturities, including a large proportion of well-differentiated cells. By the Bethesda classification of nonlymphoid tumours (Kogan et al, 2002), these mice have myeloproliferative (MPD)-like myeloid leukaemia. Blood smears of the Mll; Enl; Lck-Cre translocator mice showed little evidence of circulating leukaemic cells (Figure 3), unlike the Mll; Enl; Lmo2-Cre translocators previously described (Forster et al, 2003) and the Mll; Af9; Lmo2-Cre translocators described here (Figures 3 and 4), both of which have a similar MPD-like myeloid leukaemia. This distinction may reflect differences in the leukaemic precursors in the translocator models since the chromosomal translocations are caused by Cre expression controlled by different Cre alleles.
Figure 3.
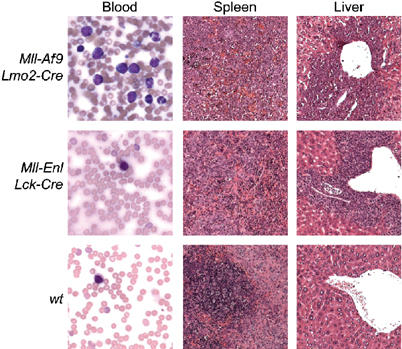
Histopathology of tumours from Mll-Enl and Mll-Af9 translocator mice. Tissues were fixed in 10% formalin and wax embedded prior to generation of histological sections. The sections were stained with H&E and photographed at × 40 magnification. May–Grünwald–Giemsa (MGG)-stained blood films and sections shown are from an Mll; Af9; Lmo2-Cre translocator mouse (top row), from an Mll; Enl; Lck-Cre translocator mouse (middle row) or from a C57BL/6 wild-type mouse (bottom row).
Figure 4.
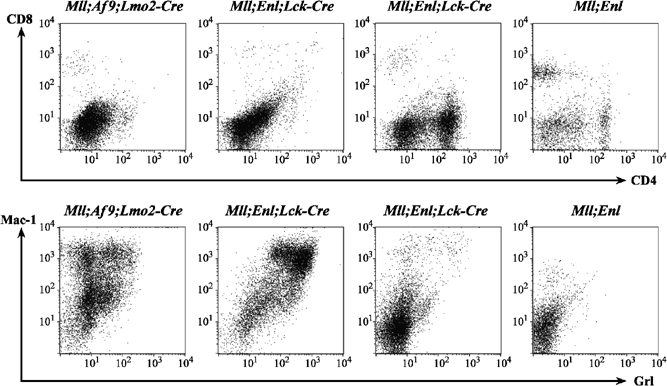
Cell surface profiles of leukaemias vary in different translocator mice. Single-cell suspensions from splenic tumours of Mll; Enl; Lck-Cre or Mll; Af9; Lmo2-Cre translocator mice were compared by flow cytometry with splenic leucocytes from a control (Mll; Enl) mouse lacking the Cre allele. Surface proteins were labelled with fluorescent antibodies detecting CD8 plus CD4 (T-cell markers) or Mac-1 plus Gr1 (myeloid markers).
A total of 90% of the Mll; Enl; Lck-Cre translocator mice developed haematological malignancy in the 18-month period of the experimental analysis. The haematological malignancy in these translocators was analysed by surface protein expression profile using FACS analysis to determine the major cell types from spleens of mice with splenomegaly. Figure 4 and Supplementary Table 1 embody findings with the Mll; Enl; Lck-Cre translocators compared with Mll; Af9; Lmo2-Cre translocator mice. Unlike the uniform finding of MPD-like myeloid leukaemia in Mll; Af9; Lmo2-Cre translocator mice described here and Mll; Enl; Lmo2-Cre mice (Forster et al, 2003), the neoplasias found in Mll; Enl; Lck-Cre mice were either of the T-cell or the myeloid lineage. Examples are shown in Figure 4, where a Mac-1/Gr-1 double positive myeloid tumour is compared with a CD4-positive, CD8-negative T-cell tumour. Data for 18 tumours in the Mll; Enl; Lck-Cre cohort are shown in Supplementary Table 1A. In addition to the cell surface marker phenotype, examination of histopathology defined the tumours as T-cell lineage. The characteristics of these T-cell tumours (including T-cell receptor Tcrb gene rearrangements; see Figure 5) were splenomegaly, lymphadenopathy without enlargement of thymus and expression of T-cell receptor-associated CD4 and/or CD8 molecules. This classifies the tumours as mature T-cell neoplasms, specifically small T-cell lymphoma (STL) according to the Bethesda classification (Morse et al, 2002).
Figure 5.
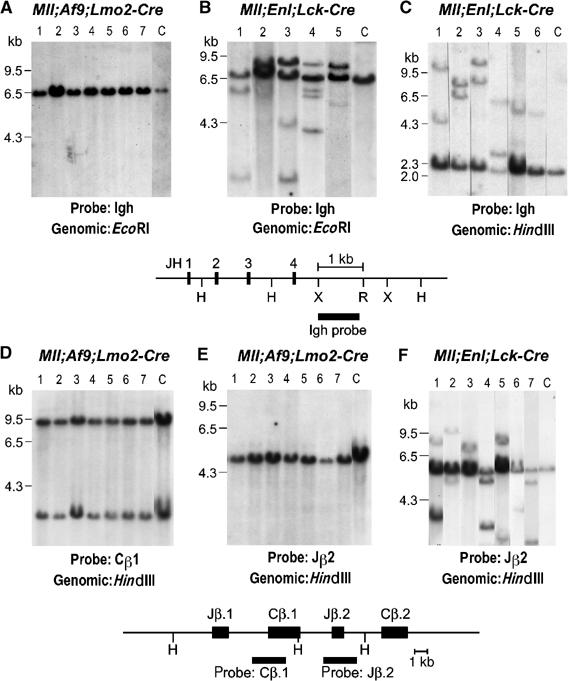
Rearranged antigen receptor genes in tumours from Mll; Enl; Lck-Cre translocator mice. Genomic DNA from splenic tumours of Mll; Af9; Lmo2-Cre (A, D, E) or Mll; Enl; Lck-Cre (B, C, F) translocator mice were analysed by filter hybridisation using an Igh, Tcr C_β_1 or Tcr J_β_2 probe. For hybridisation of the DNAs with the Igh probes, digestions were either with _Eco_RI (A, B), which will detect all joining events to the whole Ig JH region, or with _Hin_dIII (C). Hybridisation of TCR probes was with _Hin_dIII-digested DNA. V-D-J or D-J joins were detected with C_β_1 or J_β_2 probes. Partial restriction maps of the JH or C_β_1–J_β_2 genomic regions are shown, indicating the location of the probe fragments. H: _Hin_dIII; X: _Xba_I; R: _Eco_RI.
While some Mll; Enl; Lck-Cre mice developed STL, others developed tumours expressing myeloid markers (Figure 4 and Supplementary Table 1), which showed few, poorly differentiated cells in blood, myeloid cell histology in spleen and had infiltrates of neoplastic cells in the liver and kidneys. These criteria demonstrated the presence of myeloid leukaemia without maturation in these mice, by the Bethesda classification of nonlymphoid neoplasms (Kogan et al, 2002).
All Mll; Enl; Lck-Cre tumours have a footprint of Rag V-D-J recombinase activity
The finding of T-cell or myeloid tumours in Mll; Enl; Lck-Cre mice is intriguing because of the specific expression of the Lck-Cre transgene in T cells (McCormack et al, 2003). This implies that both the STL and the myeloid neoplasias in the Mll; Enl; Lck-Cre translocator mice have their origin within cells of the lymphoid lineage. The precursors of the STLs were presumably committed to T-cell differentiation, whereas the myeloid leukaemias could have arisen from early lymphoid progenitor cells retaining various differentiation options, allowing reassignment of cellular commitment, as suggested by early studies of human acute myeloid leukaemia (AML) (Boehm et al, 1987a, 1987b; Adriaansen et al, 1991). The corollary of Lck-Cre expression, which initiates in immature thymocytes, is Mll; Enl; Lck-Cre translocators should undergo chromosomal translocation around the time of V-D-J recombinase gene expression (Rag1 and Rag2). Thus, Rag proteins may have caused antigen receptor gene rearrangement leaving a ‘footprint' at the DNA level reflected by new ‘rearranged' restriction fragments for T-cell receptor (Tcr) or immunoglobulin H chain (Igh) genes.
We examined the genomic DNA of the Mll; Enl; Lck-Cre translocator splenic tumours with Tcr β chain and Igh probes. All except one (tumour 11, which showed a myeloid surface marker pattern of splenic tumour cells) showed rearrangement of the Tcr β chain and/or Igh probes (Figure 5 and Supplementary Table 1A). Most exhibited bi-allelic rearrangements, and some showed four rearranged bands implying that the tumour is either not clonal or comprises two distinct populations of cells (e.g. Figure 5B, lane 4). This is not a random rearrangement phenomenon as neither the myeloid lineage tumours from Mll; Enl; Lmo2-Cre mice (F Cano and TH Rabbitts, unpublished) nor splenic tumours from the Mll; Af9; Lmo2-Cre translocators described here showed Tcr β chain or Igh gene alterations (14 Mll; Af9; Lmo2-Cre tumours were compared and seven are shown for reference in Figure 5A, D and E). Thus, the cells that constitute all the Mll; Enl; Lck-Cre translocator splenic tumours have passed through a differentiation stage during which the Rag recombinase genes were expressed and the Rag proteins were actively performing their function of V-D-J recombination in these precursors. This suggests an intermediate stage of T-lymphoid differentiation in all these tumours, even those that present with a myeloid lineage phenotype.
Cell-specific effects of Mll-Af9 chromosomal translocations
Our observations with Mll-Enl translocator mice showed that haematopoietic malignancies ensue if de novo chromosomal translocations are induced by Cre expression from the Lmo2-Cre allele (Forster et al, 2003) or from the _Lck-Cr_e allele (this paper). We previously found that mice expressing an Mll-AF9 fusion knock-in allele developed haematological malignancies with a myeloid phenotype (Corral et al, 1996; Dobson et al, 1999). We have compared the effects of Mll; Af9 translocator mice made with Lck-Cre or Lmo2–Cre alleles. A cohort of Mll; Af9; Lck-Cre translocators was studied for an 18-month period. Surprisingly, no signs of ill health or post-mortem abnormalities were found in these mice at the 18-month point. This contrasts with malignancies in the cohort of Mll; Af9; Lmo2-Cre translocators, which all succumbed to leukaemia within about 1 year (Figure 1B). These latter mice have a similar pathology to the Mll; Enl; Lmo2-Cre translocators, showing MPD-like myeloid leukaemia. The tumour cells carry reciprocal chromosomal translocations as judged by filter hybridisation with 5′ or 3′ Mll DNA probes (Figure 2B) or with a 3′ Af9 probe (Figure 2D). In addition, FISH analysis of spleen cells from the tumour-bearing mice showed the reciprocal chromosome (Figure 2D) and the typically diploid state of the tumours.
Thus, while Mll; Af9; Lmo2-Cre translocators can develop leukaemias, Mll; Af9; Lck-Cre do not. It is possible that chromosome territory within mouse lymphoid cells specifically precludes chromosomal translocations between the Mll and Af9 genes. We investigated the presence of Mll and Af9 interchromosomal translocations in asymptomatic mice using a genomic PCR assay dependent on the ability to amplify a product if Mll and Af9 sequences were abutted following translocation (Figure 6). Pups were identified from 8-day postnatal litters of Mll; Enl; Lck-Cre, Mll; Af9; Lmo2-Cre or Mll; Af9; Lck-Cre genotype. Thymus and bone marrow DNA was prepared and used in nested PCR analysis for the presence of chromosomal translocations. In each case, five pups of appropriate genotypes were used. Examination of DNA from Mll; Enl; Lck-Cre pups showed that one of the five had detectable Mll-Enl translocations in both thymus and bone marrow DNA (Figure 6A). By contrast, all five Mll; Af9; Lck-Cre pups showed chromosomal translocations in thymic DNA but none had any detectable in bone marrow (Figure 6C). This striking difference is despite the fact that Mll; Af9; Lck-Cre translocators do not develop leukaemias whereas essentially all Mll; Enl; Lck-Cre do. Finally, all the Mll; Af9; Lmo2-Cre pups had translocations in both thymus and bone marrow (Figure 6B) consistent with the rapid leukaemogenesis in these mice. Therefore, despite the absence of tumorigenesis, Mll-Af9 chromosomal translocations do occur in _Lck-Cre_-expressing cells and furthermore, they occur in T cells very early in the life of the Mll; Af9; Lck-Cre mice.
Figure 6.
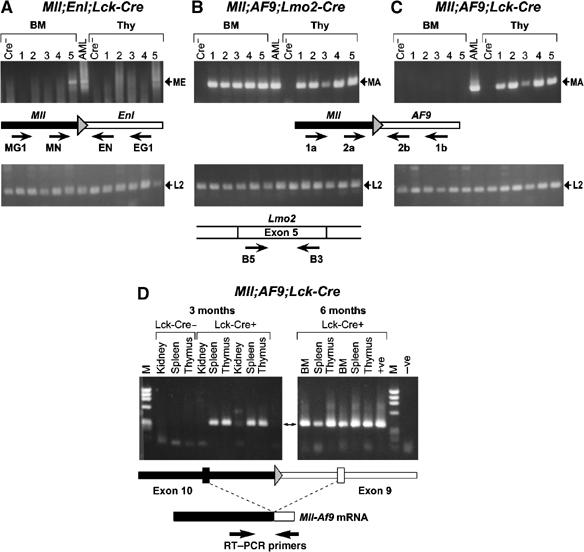
Chromosomal translocations are present in Mll; Af9; Lck-Cre translocators by postnatal day 8 and Mll-Af9 expressed at 6 months. (A–C) Bone marrow (BM) and thymus (Thy) genomic DNA was prepared from 8-day-old pups (five littermates for each genotype) from Mll; Af9; Lck-Cre or Mll; Enl; Lck-Cre translocators. PCR was performed for 70 cycles using nested primers to amplify either (A) Mll plus Enl (MG1+EG1, followed by MN+EN; Forster et al, 2003) or (B, C) Mll plus Af9 primers (1a+1b, followed by 2a+2b; Collins et al, 2000). Positive controls were performed on DNA from Mll; Af9 or Mll; Enl translocator splenic tumours (indicated as AML) using 35 cycles of amplification, and negative controls were performed, using 70 cycles of amplification, on DNA prepared from spleens of P8 litter mates of Mll; Af9 or Mll; Enl mice without the Cre gene (indicated as Cre−). Quality control of DNA was determined by PCR amplification of the Lmo2 gene with primers B5 and B3 (McCormack et al, 2003) using 35 cycles of amplification. Primer locations are diagrammatically shown. Bands corresponding to amplified products are indicated as follows: ME: Mll-Enl junction; MA: Mll-Af9 junction; L2: Lmo2 gene product. (D) Litters of Mll; Af9; Lck-Cre mice, including Mll; Af9 controls lacking Cre gene, were established and at 3 or 6 months, RNA was prepared from the spleen, thymus, kidney and bone marrow (latter at 6 months only). RT–PCR was carried out with primers from exon 10 of Mll or exon 9 of Af9 to detect RNA products of the Mll-Af9 translocated fusion allele. The double-headed arrow indicates the gel band corresponding to the product of this translocated allele. Mll-Af9 fusion mRNA transcript was present in the spleen and thymus at 3 months and in addition detected in these tissues and in bone marrow at 6 months of translocator mice. M: φX174 _Hae_III-digested DNA markers; +ve: RT–PCR product from an Mll-Af9 leukaemia produced in Mll; Af9; Lmo2-Cre mice; −ve: RT–PCR in the absence of template; BM: bone marrow.
The expression of Mll-Af9 fusion mRNA was demonstrated by RT–PCR with RNA-cDNA made from Mll; Af9; Lck-Cre mice at 3 or 6 months of age (Figure 6D). We found that RNA from the spleen, thymus and bone marrow, even at 6 months, contained Mll-Af9 transcripts, whereas other tissues such as the kidney did not. Thus, sustained expression of the chromosomal translocation occurs in Mll; Af9; Lck-Cre mice. These results suggest that Mll-Af9 is inert in committed T cells.
Mll-Af9 and Mll-Enl translocator myeloid tumours are transplantable
The ability of the Mll-fusion-dependent tumours to undergo independent growth was assessed by establishment of tissue culture lines from Mll-AF9 knock-in mice (Corral et al, 1996; Dobson et al, 1999) and Mll; Enl; Lmo2-Cre translocators (Collins et al, 2000; Forster et al, 2003). We have been unable to make lines from Mll; Enl; Lck-Cre translocators, irrespective of the lymphoid or myeloid phenotype. The tumorigenicity of the other lines was tested in a nude mouse assay in which cells were inoculated subcutaneously and tumour growth was assessed. All the lines developed into tumours in a high proportion of recipients, with latencies less than 3 weeks (Table I). In addition, the retention of either the Mll-AF9 knock-in allele or the Mll-Af9 or Mll-Enl chromosomal translocation was confirmed by filter hybridisation (data not shown).
Table 1.
Translocator tumours are transplantable
| Genotype | Cell line | Cell surface phenotype | No. of tumours | Average time for growth (days) |
|---|---|---|---|---|
| Mll-AF9 knock-in | #1 | Mac+; Gr1− | 8/10 | 18 |
| #2 | Mac+; Gr1− | 9/10 | 17 | |
| #3 | Mac+; Gr1− | 9/10 | 14 | |
| Mll; Enl; Lmo2-Cre | #1 | Mac+; Gr1low | 11/14 | 18 |
| #2 | Mac+; Gr1low | 11/14 | 18 | |
| #3 | Mac+; Gr1low | 11/14 | 13 | |
| Cell lines from either Mll-AF9 knock-in or Mll-Enl translocator mice were cultured in the presence of growth factors (see Materials and methods) and 1–5 × 106 cells were injected into the flanks of 6- to 8-week-old female MF1 nude mice. Tumours were resected at approximately 1.5 cm size and post-tumour growth evaluation was conducted for genotype, presence of the relevant translocation alleles and surface marker expression. Number (No.) of tumours is expressed as fraction of tumours/number of injection sites. | ||||
| All lines were negative with antibodies recognising Sca1, C-kit, Ter119, CD4, CD8, Thy1, CD25 and B220. |
Discussion
The translocator model recapitulates analogous events in human cancers
The translocator mouse model (Smith et al, 1995; van Deursen et al, 1995; Buchholz et al, 2000; Collins et al, 2000) can recapitulate chromosomal translocations found in human tumours by allowing cell-specific de novo chromosomal translocations to take place. This means that reciprocal translocations can be engineered in vivo for the first time. We have shown that _Mll_-associated translocations can be induced by Cre recombinase between two pairs of chromosomes (i.e. between chromosomes 9 and 4 or between chromosomes 9 and 17) in similar cell types, as well as in distinct lineages. Chromosomes 4 and 17 therefore seem equally accessible to translocate with chromosome 9, indicating that chromosome territory might not impinge too heavily on the ability of translocation between different Mll partners. In addition to mimicking the ability of MLL to fuse with various partners and cause leukaemia, the outcome of the mouse Mll-Enl and Mll-Af9 chromosomal translocations parallels the tumour phenotypes found in humans, where the former is seen in T-cell tumours or in myeloid tumours whereas MLL-AF9 is almost exclusively seen in myeloid tumours.
Leukaemic precursor cells in Mll-associated leukaemias
Chromosomal translocations are primary events in haematopoietic tumours (Rowley, 1999) and the putative self-renewing leukaemic stem cell (LSC) must carry the chromosomal translocation. The oncogenicity of _Mll_-fusion proteins in different cells of haematopoiesis has been evaluated using knock-in alleles (Corral et al, 1996; Dobson et al, 1999, 2000; Wang et al, 2005), or retroviral delivery of MLL fusions to HSC (Slany et al, 1998; Lavau et al, 2000; Deguchi et al, 2003; So et al, 2003; Zeisig et al, 2003) or translocator mice (Forster et al, 2003). These demonstrate that tumours develop with a differentiated phenotype. Furthermore, transduction of _Mll-Enl_-expressing retroviruses into purified GMP progenitor cells showed that leukaemias could also develop from these (Cozzio et al, 2003).
In studies of MLL-AF4 chromosomal translocations in monozygotic twins, leukaemias were found that arose with clonal origin in utero but with different immunoglobulin rearrangements in the leukaemias at presentation (Greaves and Wiemels, 2003). This provides strong evidence that the MLL-AF4 chromosomal translocations were primary in the LSC and that MLL-AF4 can exert an effect on pluripotent HSC. However, the fusion protein may also have molecular consequences for later cells, which are no longer pluripotent. We show in our translocator model that the primary effects of Mll-Enl fusion can occur in noncommitted cells or cells that are partially or fully committed to the T-lymphocyte lineage. Thus, Mll fusions do not need the environment of noncommitted cell to be oncogenic, suggesting that MLL translocation-associated leukaemia in man is not a mandatory stem cell leukaemia.
Mll-fusion proteins require a permissive environment for oncogenicity
The Mll-Af9 fusion appears to be unable to support tumorigenesis in T-lymphopoiesis whereas it is a potent inducer of myeloid leukaemias. The absence of Mll; Af9; Lck-Cre-induced leukaemias is not due to the lack of translocations or expression of Mll-Af9 fusion mRNA in Mll; Af9; Lck-Cre nonleukaemic mice (Figure 6). Therefore, Mll-Af9 is inert in the multipotent progenitors destined to produce the T-cell lineage. This ‘inertia' (depicted in Figure 7) may be due to the nonresponsiveness of target genes to lineage activation by Mll-Af9, or secondary mutations necessary for Mll-Af9 complementation do not typically occur in T cells or cofactors needed for Mll-Af9 function do not exist in T cells. The first possibility raises intriguing questions about the nature of MLL-fusion protein target genes, namely the possibility of common and specific target genes for MLL fusions in haematopoietic cells. If all MLL fusions have common effects on the transcriptome (e.g. control of HOX gene expression), we should anticipate the same consequence of having Mll-Enl and Mll-Af9 translocations in T cells. Our data therefore suggest separation of MLL-fusion protein function into distinct parts, one being related to cell phenotype control (instructive function and controlling putative Mll-fusion-specific target genes) and another to development of oncogenic phenotype.
Figure 7.
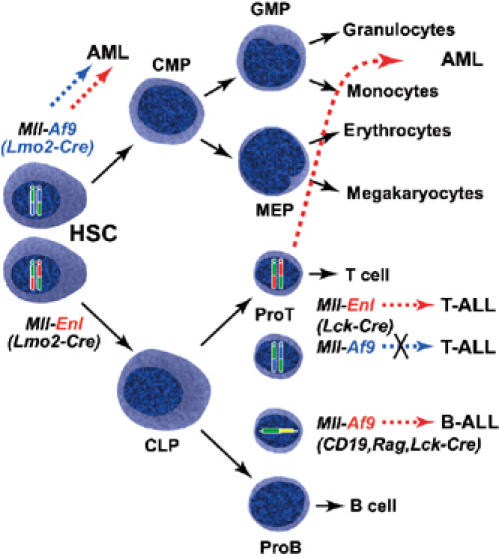
Summary of Mll fusions in haematopoiesis and tumorigenesis. When chromosomal translocations are generated de novo in Mll-Af9 or Mll-Enl translocator mice, haematopoietic malignancies arise if translocations are mediated via Cre expression with either Lmo2-Cre (Mll-Af9 or Mll-Enl) or Lck-Cre (Mll-Enl only) alleles. No tumours arose when Mll-Af9 chromosomal translocations were induced by Lck-Cre expression, even though both chromosomal translocations and Mll-Af9 fusion mRNA could be detected. Thus, when Mll translocations are induced in the pluripotent stem cells or in the multipotent myeloid progenitors using Lmo2-Cre, myeloid malignancies arise (this paper and Forster et al, 2003). A more complex situation was observed when the chromosomal translocations between Mll and Enl genes were induced by _Lck-Cre_-controlled expression of Cre recombinase. In this case, either T-cell malignancies or myeloid leukaemias appeared; however, in both types, the leukaemia progenitors had arisen from cells that had expressed functional Rag V-D-J recombinase. Thus, the myeloid malignancies of the Mll; Enl; Lck-Cre translocator mice appear to arise from cells that have undergone a lineage reassignment from a semicommitted, Rag+ lymphoid cell to a myeloid cell. This effect may occur by reprogramming the transcriptome of cells with the chromosomal translocations to realigning differentiation into myeloid from lymphoid status. HSC: haematopoietic stem cells; CMP: common myeloid progenitors; GMP: granulocyte–monocyte precursors; MEP: megakaryocyte–erythrocyte precursors; CLP: common lymphoid progenitors.
The Lck-Cre expression causes Mll-Af9 translocations but not leukaemia
We have shown that Mll-Enl is an effective oncogene when chromosomal translocations are caused by Cre recombinase made from an Lck-Cre allele. Virtually all the Mll; Enl; Lck-Cre mice developed leukaemia (Figure 1) but the phenotypes of these tumours were disparate, only a proportion being T-cell neoplasias and the majority myeloid tumours. Crucially, irrespective of the cell lineage, the tumours had detectable rearrangements of immunoglobulin and/or T-cell receptor genes (95% of tumours). This footprint of Rag gene expression means that the precursors of all these tumours have been through a stage during which Rag recombination takes place, which is a feature of lymphoid development (Gellert, 2002). This critical finding places the initiating chromosomal translocation event for the myeloid tumours of Mll; Enl; Lck-Cre mice within cells after the common lymphocyte precursor stage, implying that these are noncommitted lymphoid progenitors (see below).
An additional fundamental issue is the specificity of the Lck-Cre allele for Cre expression in the T-cell lineage as misexpression in progenitors might explain the myeloid tumours in Mll; Enl; Lck-Cre translocators. Several lines of evidence demonstrate that Lck-Cre is T-cell specific. Only those cells designated as being within the T-cell pathway (from DN1 cells onwards, which is just a stage before endogenous Lck activity is apparent; Wolfer et al, 2001, 2002) showed Cre-dependent deletion of a floxed chromosomal segment of the Lmo2 gene as a reporter (McCormack et al, 2003) or activation of the lacZ gene in the ROSA locus with the _Lck-Cre_-controlled Cre (Codrington et al, 2005). There was no evidence of Lck-Cre activity in progenitors or in purified Mac-1- or Gr-1-positive cells (using a radiolabelled hybridisation assay for sensitive detection of deletion of a _loxP_-flanked reporter gene (McCormack et al, 2003) and the other direct detection of β-gal-positive cells (Codrington et al, 2005)). The analysis has been extended by the use of the ROSA-YFP reporter line (Srinivas et al, 2001) in which no progenitor or myeloid cells were found coexpressing relevant surface markers and YFP (markers were CD34, Sca1, Ter119, Mac-1 or Gr-1) (LF Drynan & TH Rabbitts, unpublished). Further, no Cre activity could be detected after selection of cells expressing B-cell markers (CD19, B220, IgM, IgD or IgG).
A critical biological control that provides a very powerful assessment of the specific association of Cre activity with T-cell lineage precursors or mature T cells is our observation that no tumours arose in translocator mice with Mll-Af9 translocations induced by Lck-Cre. Our previous work with Mll-AF9 knock-in mice (Corral et al, 1996; Dobson et al, 1999), and with Mll-Af9 translocators in this paper, shows that this fusion creates a very potent myeloid oncogenic protein. In addition, we show that Mll-Af9 chromosomal translocations occur in all thymuses of Mll;Af9; Lck-Cre mice tested as early as 8 days after birth and the fusion gene is expressed at least to 6 months of age (Figure 6). This shows that these translocator T cells can undergo translocation and there is no obvious clonal deletion of the cells carrying the expressed Mll-Af9 fusion allele. If the myeloid tumours in the Mll; Enl; Lck-Cre arose because the Lck-Cre allele was expressing at some level in myeloid progenitors or in some occasional progenitor cell, we would expect myeloid tumours in the Mll; Af9; Lck-Cre translocators. A final piece of evidence of a T-cell progenitor origin for the myeloid tumours in Mll; Enl; Lck-Cre translocators is that they have Tcr/Igh gene rearrangements, unlike the myeloid tumours arising in progenitors such as those made in Mll; Af9; Lmo2-Cre or Mll; Enl; Lmo2-Cre translocator mice. We conclude that the Lck-Cre allele is specific for the T-cell lineage and that the myeloid tumours originate in cells after the CLP stage in the Mll; Enl; Lck-Cre translocator mice.
Mll-Enl can cause lineage reassignment
We have found that either T-cell or myeloid type leukaemias arise from Mll-Enl chromosomal translocations mediated by the T-cell-specific Cre recombinase made from our Lck-Cre transgenic mouse (McCormack et al, 2003). These findings suggest that expression of Mll-Enl in immature T-cell precursors elicits reprogramming of the cells into the myeloid lineage. A process of lineage reassignment in human leukaemias has already been indicated by previous observations that AML often have rearranged antigen receptor genes (Boehm et al, 1987a, 1987b; Adriaansen et al, 1991). Direct experimental support for reprogramming of lymphoid precursors into myeloid cells has been provided by ectopic expression of v_-raf_ (Klinken et al, 1988) or CEB/P (Xie et al, 2004), indicating that aberrant gene expression can force cells to respond to different developmental programmes. These data indicate that the boundaries in haematopoietic lineage assignment may be blurred, as strict development along individual pathways is subvertable (Klinken et al, 1988; Xie et al, 2004). The ability of Mll-Enl to reassign lymphoid cells to myeloid lineage reflects a pathogenic subversion of lineage commitment and supports an instructive function to this (and possibly other) MLL fusion, in accordance with the master gene model for the effects of oncogene activation following chromosomal translocations in acute leukaemias (Rabbitts, 1991). The model that emerges is of a noncommitted T-cell progenitor that would normally have only one fate, namely to develop into a mature thymocyte (i.e. after Rag recombinase expression). If an Mll-Enl translocation occurs in this early, noncommitted T-cell progenitor, the influence of the Mll-Enl fusion is reassignment of lineage into the myeloid pathway. Conversely, if the translocations occur after the cell has progressed sufficiently far along the T-cell differentiation pathway, the neoplastic cell can only present as a T-cell leukaemia. The molecular events by which this lineage reassignment occurs will be interesting, as it should identify molecular targets of instructive MLL-fusion protein function distinct from other putative common functions.
Materials and methods
Analysis of translocator mouse strains
Mouse lines carrying loxP sites in either an Mll intron, an Af9 intron or an Enl intron have been described (Collins et al, 2000; Forster et al, 2003). The general structure of these alleles is summarised in Figure 1A. Mice expressing Cre recombinase either under the control of the Lmo2 or the Lck promoter (McCormack et al, 2003) were bred with those with Mll, Enl or Af9 loxP alleles to generate cohorts of 20 mice for each genotype. Mice carrying Mll and Enl or Af9 alleles with loxP sites with or without a Cre allele are designated respectively (Mll; Enl; Lmo2-Cre), (Mll; Enl; Lck-Cre) or (Mll; Enl) or (Mll; Af9; Lmo2-Cre), (Mll; Af9; Lck-Cre) or (Mll; Af9). At the signs of ill health, post mortem was carried out and tissues were removed as needed. Histological sections (4 μm in thickness) were made from fixed tissue using wax embedding and were stained with haematoxylin and eosin (H&E). Flow cytometry was conducted using a FACScaliber and surface proteins were detected with fluorescent antibodies (BD-Pharmingen).
Detection of chromosomal translocations was carried out using Southern filter hybridisation or FISH as described (LeFranc et al, 1986; Forster et al, 2003). Filters were incubated for 24 h with 32P-labelled probes at 65°C in a 3 × SSC buffer and washed at 65°C with 0.1 × SCC and 0.1% SDS. Hybridisation signal was detected with pre-fogged X-ray films at −70°C with intensifying screens. FISH analysis was carried out with metaphase spreads of spleen cells and metaphase spreads were painted with whole chromosome paints for chromosome 9 (green, Mll chromosome), 17 (red, Enl chromosome) or 4 (red, Af9 chromosome) (Cambio).
Antigen receptor gene rearrangements were detected by Southern filter hybridisation. Splenic tumour genomic DNA or CCB ES cell DNA was restriction digested, fractionated and transferred to nylon membranes followed by hybridisation to either a mouse Igh probe (Neuberger and Williams, 1986) or T-cell receptor probes (C_β_1 or J_β_2) (Malissen et al, 1984). DNA was cleaved with either _Hin_dIII or _Eco_RI for Igh gene status or _Hin_dIII for Tcr gene status.
Detection of Mll chromosomal translocations and mRNA in nonleukaemic mice
Genomic PCR was carried out using nested primers and 0.5 μg genomic DNA per initial 25 μl PCR reactions. A 1 μl portion of this reaction was used with nested primers, again in 25 μl reaction. PCR reactions were initially denatured at 95°C for 2 min followed by 35 cycles comprising 95°C for 1 min, 65°C for 1 min and 72°C for 1 min with a final 72°C step for 10 min. The PCR primers flanking the Mll-Enl translocation junction were MG1+EG1 followed by nested primers MN+EN (Forster et al, 2003). Primers flanking the Mll-Af9 translocation junction were 1a+1b followed by nested primers 2a+2b (Collins et al, 2000). As an internal control for DNA quality, the Lmo2 primers B5+B3 were used to amplify an Lmo2 gene segment (McCormack et al, 2003). For inter-exon RT–PCR on cDNA, MEX7F+F9B2L were used corresponding to sequences present in exon 10 of Mll and exon 9 of Af9. RT–PCR primers for amplification of Mll-Enl mRNA were as described (Forster et al, 2003).
Cell lines
Bone marrows were cultured in DMEM media in 10% FCS plus growth factors (1 μl/ml IL7 (Roche), 2 μl/ml IL2 (Roche), 0.5 μl/ml IL6 (Roche), 5% IL3 (WEHI supernatant) and 1 μl/ml GMSCF (Boehringer) at 37°C under 5% CO2. For tumorigenicity assays, 1–5 × 106 cells were injected subcutaneously into flanks of 6- to 8-week-old female MF1 nude mice. Tumour growth was terminated at ∼1.5 cm diameter. Tumours were resected, single-cell suspensions were made, followed by flow cytometry and genomic DNA filter hybridisation analysis to confirm concordance between the cultured cell lines and the tumours.
Supplementary Material
Supplementary Table 1
Acknowledgments
This work was funded by the Medical Research Council. N Chan was the recipient of a Croucher Foundation studentship and F Cano of a Darwin Milstein studentship. We thank Tina Hamilton for expert technical assistance, Shankar Srinivas for ROSA-YFP mice, and Angela Middleton, Gareth King, Claire Peace, Charlotte Rickett and Richard Berks for animal husbandry.
References
- Adriaansen HJ, Soeting PW, Wolvers-Tettero IL, van Dongen JJ (1991) Immunoglobulin and T-cell receptor gene rearrangements in acute non-lymphocytic leukemias. Analysis of 54 cases and a review of the literature. Leukemia 5: 744–751 [PubMed] [Google Scholar]
- Ayton PM, Cleary ML (2001) Molecular mechanisms of leukemogenesis mediated by MLL fusion proteins. Oncogene Rev 20: 5695–5707 [DOI] [PubMed] [Google Scholar]
- Ayton PM, Cleary ML (2003) Transformation of myeloid progenitors by MLL oncoproteins is dependent on Hoxa7 and Hoxa9. Genes Dev 17: 2298–2307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birke M, Schreiner S, Garcia-Cuellar MP, Mahr K, Titgemeyer F, Slany RK (2002) The MT domain of the proto-oncoprotein MLL binds to CpG-containing DNA and discriminates against methylation. Nucleic Acids Res 30: 958–965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boehm T, Werle A, Drahousky D (1987a) Immunoglobulin heavy chain and T-cell receptor α and β chain gene rearrangements in acute myeloid leukaemias. Mol Biol Med 4: 51–62 [PubMed] [Google Scholar]
- Boehm TL, Werle A, Ganser A, Kornhuber B, Drahovsky D (1987b) T cell receptor gamma chain variable gene rearrangements in acute lymphoblastic leukemias of T and B lineage. Eur J Immunol 17: 1593–1597 [DOI] [PubMed] [Google Scholar]
- Buchholz F, Refaeli Y, Trumpp A, Bishop JM (2000) Inducible chromosomal translocation of AML1 and ETO genes through Cre/loxP-mediated recombination in the mouse. EMBO Rep 1: 133–139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Codrington R, Pannell R, Forster A, Drynan LF, Daser A, Lobato MN, Metzler M, Rabbitts TH (2005) The Ews-ERG fusion protein can initiate neoplasia from lineage committed haematopoietic cells. Publ Lib Sci Biol 3: e242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins EC, Pannell R, Simpson EM, Forster A, Rabbitts TH (2000) Inter-chromosomal recombination of Mll and Af9 genes mediated by cre-_lox_P in mouse development. EMBO Rep 1: 127–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corral J, Lavenir I, Impey H, Warren AJ, Forster A, Larson TA, Bell S, McKenzie ANJ, King G, Rabbitts TH. (1996) An Mll-Af9 fusion gene made by homologous recombination causes acute leukemia in chimeric mice: a method to create fusion oncogenes. Cell 85: 853–861 [DOI] [PubMed] [Google Scholar]
- Cozzio A, Passegue E, Ayton PM, Karsunky H, Cleary ML, Weissman IL (2003) Similar MLL-associated leukemias arising from self-renewing stem cells and short-lived myeloid progenitors. Genes Dev 17: 3029–3035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daser A, Rabbitts TH (2004) Extending the repertoire of the mixed lineage leukemia gene MLL in leukemogenesis. Genes Dev 18: 965–974 [DOI] [PubMed] [Google Scholar]
- Deguchi K, Ayton PM, Carapeti M, Kutok JL, Snyder CS, Williams IR, Cross NC, Glass CK, Cleary ML, Gilliland DG (2003) MOZ-TIF2-induced acute myeloid leukemia requires the MOZ nucleosome binding motif and TIF2-mediated recruitment of CBP. Cancer Cell 3: 259–271 [DOI] [PubMed] [Google Scholar]
- Dobson CL, Warren AJ, Pannell R, Forster A, Lavenir I, Corral J, Smith AJH, Rabbitts TH (1999) The Mll-AF9 gene fusion in mice controls myeloproliferation and specifies acute myeloid leukaemogenesis. EMBO J 18: 3564–3574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobson CL, Warren AJ, Pannell R, Forster A, Rabbitts TH (2000) Tumorigenesis in mice with a fusion of the leukaemia oncogene Mll and the bacterial lacZ gene. EMBO J 19: 843–851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forster A, Pannell R, Drynan LF, McCormack M, Collins EC, Daser A, Rabbitts TH (2003) Engineering de novo reciprocal chromosomal translocations associated with Mll to replicate primary events of human cancer. Cancer Cell 3: 449–458 [DOI] [PubMed] [Google Scholar]
- Gellert M (2002) V(D)J recombination: RAG proteins, repair factors, and regulation. Annu Rev Biochem 71: 101–132 [DOI] [PubMed] [Google Scholar]
- Greaves MF, Maia AT, Wiemels JL, Ford AM (2003) Leukemia in twins: lessons in natural history. Blood 102: 2321–2333 [DOI] [PubMed] [Google Scholar]
- Greaves MF, Wiemels J (2003) Origins of chromosome translocations in childhood leukaemia. Nat Rev Cancer 3: 639–649 [DOI] [PubMed] [Google Scholar]
- Hess JL (2004) MLL: a histone methyltransferase disrupted in leukemia. Trends Mol Med 10: 500–507 [DOI] [PubMed] [Google Scholar]
- Klinken SP, Alexander WS, Adams JM (1988) Hemopoietic lineage switch: v-raf oncogene converts Emu-myc transgenic B cells into macrophages. Cell 53: 857–867 [DOI] [PubMed] [Google Scholar]
- Kogan SC, Ward JM, Anver MR, Berman JJ, Brayton C, Cardiff RD, Carter JS, de Coronado S, Downing JR, Fredrickson TN, Haines DC, Harris AW, Harris NL, Hiai H, Jaffe ES, MacLennan IC, Pandolfi PP, Pattengale PK, Perkins AS, Simpson RM, Tuttle MS, Wong JF, Morse HC (2002) Bethesda proposals for classification of nonlymphoid hematopoietic neoplasms in mice. Blood 100: 238–245 [DOI] [PubMed] [Google Scholar]
- Kumar AR, Hudson WA, Chen W, Nishiuchi R, Yao Q, Kersey JH (2004) Hoxa9 influences the phenotype but not the incidence of Mll-AF9 fusion gene leukemia. Blood 103: 1823–1828 [DOI] [PubMed] [Google Scholar]
- Lavau C, Luo RT, Du C, Thirman MJ (2000) Retrovirus-mediated gene transfer of MLL-ELL transforms primary myeloid progenitors and causes acute myeloid leukemias in mice. Proc Natl Acad Sci USA 97: 10984–10989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeFranc M-P, Forster A, Baer R, Stinson MA, Rabbitts TH (1986) Diversity and rearrangement of the human T cell rearranging γ genes: nine germ-line variable genes belonging to two subgroups. Cell 45: 237–246 [DOI] [PubMed] [Google Scholar]
- Malissen M, Minard K, Mjolsness S, Kronenberg M, Goverman J, Hunkapillar T, Prystowsky MB, Yoshikai Y, Fitch F, Mak TW, Hood L (1984) Mouse T cell antigen receptor: structure and organization of constant and joining gene segments encoding the β polypeptide. Cell 37: 1101–1110 [DOI] [PubMed] [Google Scholar]
- McCormack MP, Forster A, Drynan LF, Pannell R, Rabbitts TH (2003) The LMO2 T-cell oncogene is activated via chromosomal translocations or retroviral insertion during gene therapy but has no mandatory role in normal T-cell development. Mol Cell Biol 23: 9003–9013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milne TA, Briggs SD, Brock HW, Martin ME, Gibbs D, Allis CD, Hess JL (2002) MLL targets SET domain methyltransferase activity to Hox gene promoters. Mol Cell 10: 1107–1117 [DOI] [PubMed] [Google Scholar]
- Miyamoto T, Iwasaki H, Reizis B, Ye M, Graf T, Weissman IL, Akashi K (2002) Myeloid or lymphoid promiscuity as a critical step in hematopoietic lineage commitment. Dev Cell 3: 137–147 [DOI] [PubMed] [Google Scholar]
- Mori H, Colman SM, Xiao Z, Ford AM, Healy LE, Donaldson C, Hows JM, Navarrete C, Greaves M (2002) Chromosome translocations and covert leukemic clones are generated during normal fetal development. Proc Natl Acad Sci USA 99: 8242–8247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morse HC III, Anver MR, Fredrickson TN, Haines DC, Harris AW, Harris NL, Jaffe ES, Kogan SC, MacLennan IC, Pattengale PK, Ward JM (2002) Bethesda proposals for classification of lymphoid neoplasms in mice. Blood 100: 246–258 [DOI] [PubMed] [Google Scholar]
- Nakamura T, Mori T, Tada S, Krajewski W, Rozovskaia T, Wassell R, Dubois G, Mazo A, Croce CM, Canaani E (2002) ALL-1 is a histone methyltransferase that assembles a supercomplex of proteins involved in transcriptional regulation. Mol Cell 10: 1119–1128 [DOI] [PubMed] [Google Scholar]
- Neuberger MS, Williams GT (1986) Construction of novel antibodies by use of DNA transfection: design of plasmid vectors. Philos Trans R Soc London 317: 425–432 [Google Scholar]
- Rabbitts TH (1991) Translocations, master genes, and differences between the origins of acute and chronic leukemias. Cell 67: 641–644 [DOI] [PubMed] [Google Scholar]
- Reya T, Morrison SJ, Clarke MF, Weissman IL (2001) Stem cells, cancer, and cancer stem cells. Nature 414: 105–111 [DOI] [PubMed] [Google Scholar]
- Rowley JD (1999) The role of chromosome translocations in leukemogenesis. Semin Hematol 36: 59–72 [PubMed] [Google Scholar]
- Schlaeger TM, Schuh A, Flitter S, Fisher A, Mikkola H, Orkin SH, Vyas P, Porcher C (2004) Decoding hematopoietic specificity in the helix–loop–helix domain of the transcription factor SCL/Tal-1. Mol Cell Biol 24: 7491–7502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slany RK, Lavau C, Cleary ML (1998) The oncogenic capacity of HRX-ENL requires the transcriptional transactivation activity of ENL and the DNA binding motifs of HRX. Mol Cell Biol 18: 122–129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith AJH, De Sousa MA, Kwabi-Addo B, Heppell-Parton A, Impey H, Rabbitts PH (1995) A site-directed chromosomal translocation induced in embryonic stem cells by Cre-loxP recombination. Nat Genet 9: 376–384 [DOI] [PubMed] [Google Scholar]
- So CW, Karsunky H, Passegue E, Cozzio A, Weissman IL, Cleary ML (2003) MLL-GAS7 transforms multipotent hematopoietic progenitors and induces mixed lineage leukemias in mice. Cancer Cell 3: 161–171 [DOI] [PubMed] [Google Scholar]
- Sobulo OM, Borrow J, Tomek R, Reshmi S, Harden A, Schlegelberger B, Housman D, Doggett NA, Rowley JD, Zeleznik-Le NJ (1997) MLL is fused to CBP, a histone acetyltransferase, in therapy-related acute myeloid leukemia with a t(11;16)(q23;p13). Proc Natl Acad Sci USA 94: 8732–8737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivas S, Watanabe T, Lin CS, William CM, Tanabe Y, Jessell TM, Costantini F (2001) Cre reporter strains produced by targeted insertion of EYFP and ECFP into the ROSA26 locus. BMC Dev Biol 1: 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Deursen J, Fornerod M, van Rees B, Grosveld G (1995) Cre-mediated site-specific translocation between nonhomologous mouse chromosomes. Proc Natl Acad Sci USA 92: 7376–7380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogelstein B, Kinzler KW (2004) Cancer genes and the pathways they control. Nat Med 10: 789–799 [DOI] [PubMed] [Google Scholar]
- Wang J, Iwasaki H, Krivtsov A, Febbo PG, Thorner AR, Ernst P, Anastasiadou E, Kutok JL, Kogan SC, Zinkel SS, Fisher JK, Hess JL, Golub TR, Armstrong SA, Akashi K, Korsmeyer SJ (2005) Conditional MLL-CBP targets GMP and models therapy-related myeloproliferative disease. EMBO J 24: 368–381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wildin RS, Wang HU, Forbush KA, Perlmutter RM (1995) Functional dissection of the murine lck distal promoter. J Immunol 155: 1286–1295 [PubMed] [Google Scholar]
- Wolfer A, Bakker T, Wilson A, Nicolas M, Ioannidis V, Littman DR, Lee PP, Wilson CB, Held W, MacDonald HR, Radtke F (2001) Inactivation of Notch 1 in immature thymocytes does not perturb CD4 or CD8T cell development. Nat Immunol 2: 235–241 [DOI] [PubMed] [Google Scholar]
- Wolfer A, Wilson A, Nemir M, MacDonald HR, Radtke F (2002) Inactivation of Notch1 impairs VDJbeta rearrangement and allows pre-TCR-independent survival of early alpha beta lineage thymocytes. Immunity 16: 869–879 [DOI] [PubMed] [Google Scholar]
- Xia ZB, Anderson M, Diaz MO, Zeleznik-Le NJ (2003) MLL repression domain interacts with histone deacetylases, the polycomb group proteins HPC2 and BMI-1, and the corepressor C-terminal-binding protein. Proc Natl Acad Sci USA 100: 8342–8347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie H, Ye M, Feng R, Graf T (2004) Stepwise reprogramming of B cells into macrophages. Cell 117: 663–676 [DOI] [PubMed] [Google Scholar]
- Yamada Y, Warren AW, Dobson C, Forster A, Pannell R, Rabbitts TH (1998) The T cell leukaemia LIM protein Lmo2 is necessary for adult mouse haematopoiesis. Proc Natl Acad Sci USA 95: 3890–3895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoyama A, Wang Z, Wysocka J, Sanyal M, Aufiero DJ, Kitabayashi I, Herr W, Cleary ML (2004) Leukemia proto-oncoprotein MLL forms a SET1-like histone methyltransferase complex with menin to regulate Hox gene expression. Mol Cell Biol 24: 5639–5649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeisig BB, Garcia-Cuellar MP, Winkler TH, Slany RK (2003) The oncoprotein MLL-ENL disturbs hematopoietic lineage determination and transforms a biphenotypic lymphoid/myeloid cell. Oncogene 22: 1629–1637 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table 1