Two internal ribosome entry sites mediate the translation of p53 isoforms (original) (raw)
Abstract
The p53 tumour suppressor protein has a crucial role in cell-cycle arrest and apoptosis. Previous reports show that the p53 messenger RNA is translated to produce an amino-terminal-deleted isoform (ΔN-p53) from an internal initiation codon, which acts as a dominant-negative inhibitor of full-length p53. Here, we show that two internal ribosome entry sites (IRESs) mediate the translation of both full-length and ΔN-p53 isoforms. The IRES directing the translation of full-length p53 is in the 5′-untranslated region of the mRNA, whereas the IRES mediating the translation of ΔN-p53 extends into the protein-coding region. The two IRESs show distinct cell-cycle phase-dependent activity, with the IRES for full-length p53 being active at the G2–M transition and the IRES for ΔN-p53 showing highest activity at the G1–S transition. These results indicate a novel translational control of p53 gene expression and activity.
Keywords: IRES, p53, translation, cell cycle
Introduction
The tumour suppressor protein p53 has a key role in maintaining genomic integrity by controlling cell-cycle progression and cell survival (Levine, 1997). Consistent with this view, about 50% of human tumours carry mutations in the p53 gene (Hollstein et al, 1991). On exposure to stress stimuli, p53 is activated through post-translational modifications, resulting in increased protein stability and activity (Somasundaram & El-Deiry, 2000). The increased levels of p53 protein may also result from the enhancement of messenger RNA translation (Ko & Prives, 1996). Previous studies have reported that a 40/47 kDa isoform of p53 (ΔN-p53), lacking the amino-terminal transactivation domain, is generated by alternative translation initiation from an AUG codon at position 40 of the p53 mRNA (Courtois et al, 2002; Yin et al, 2002). ΔN-p53, the expression of which was regulated in a cell-cycle phase-dependent manner, inhibited the transcriptional and growth-suppressive activities of full-length p53 in a dominant-negative manner (Courtois et al, 2002). However, the precise mechanism regulating the expression of the p53 isoforms remains unknown. Internal ribosome entry site (IRES)-mediated translation, where the ribosome is directly recruited to a site within the 5′-untranslated region (5′UTR) of the mRNA, is the most important mode of alternative translation initiation of eukaryotic mRNAs (Vagner et al, 2001). IRES-mediated translation has been demonstrated in the picornavirus RNAs and in several cellular mRNAs (IRES database, http://www.rangueil.inserm.fr/iresdatabase). Many of these mRNAs encode proteins that are associated with the control of cell proliferation and cell death (Holcik & Sonenberg, 2005). A common feature of many of the IRESs is their utilization of non-canonical translation initiation factors referred to as IRES-interacting _trans_-acting factors (ITAFs; Stoneley & Willis, 2004). The spatial and temporal distribution of the ITAFs may be an important mode of regulation of IRES activity and thereby of gene expression.
In this study, we show that the translation of full-length and ΔN-p53 proteins is directed by two distinct IRESs that show differential cell-cycle phase-dependent activity. These results indicate a novel mode of regulation of p53 gene expression at the level of translation initiation.
Results
p53 5′UTR mediates cap-independent translation
The MFOLD algorithm (Zuker, 2003) predicted the presence of strong secondary structures in the 134 nt 5′UTR and the 5′ 251 nt region of the p53 mRNA (Fig 1A), with Δ_G_ values of −57 and −92 kcal, respectively. The presence of these sequences upstream of a green fluorescent protein (GFP) reporter gene reduced translation by about 50% compared with a capped GFP RNA (compare black bars in Fig 1B), but still allowed translation of the reporter gene irrespective of the presence or absence of the cap. Interestingly, addition of the cap analogue failed to inhibit the translation from p53(+39)GFP RNA and only partially inhibited the translation from the p53(−1)GFP RNA, whereas it inhibited the translation from capped GFP RNA (compare grey with black bars in Fig 1B). Similar observations were made with a hepatitis C virus (HCV) 5′UTR-GFP RNA, which is known to be translated in a cap-independent manner (Fig 1B). These results indicated that the p53(+39) and (−1) 5′UTRs allowed cap-independent translation initiation.
Figure 1.
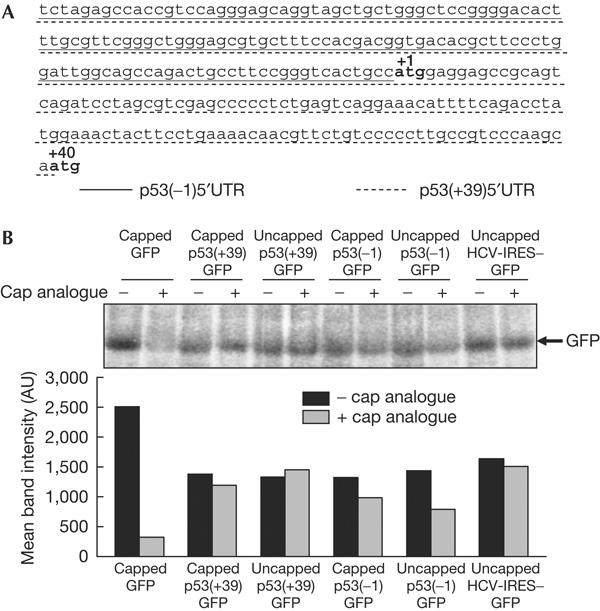
p53 5′-untranslated regions mediate cap-independent translation. (A) Nucleotide sequence of p53 5′-untranslated region (5′UTR) and part of the protein-coding region. The 134 nt canonical 5′UTR is underlined by a solid line, whereas the 251 nt 5′UTR for ΔN-p53 is underlined by a dotted line. (B) In vitro translation of capped and uncapped monocistronic RNAs in the presence or absence of exogenously added cap analogue. The band intensities were quantified and are represented graphically.
p53 5′UTR sequences contain IRESs
To investigate whether the translation initiation by the p53 5′UTR sequences was by the internal entry of ribosomes, transient transfections were carried out using bicistronic constructs, where the upstream cistron Renilla luciferase (Rluc) is translated in a cap-dependent manner, whereas the downstream firefly luciferase (Fluc) will be translated if the intercistronic sequence contains an IRES (Fig 2A). Bicistronic plasmids pRp53(+39)F and pRp53(−1)F, transfected into HeLa cells, showed around 11- and 5-fold higher Fluc activity, respectively, than that of the control bicistronic DNA pRnullF, which had an unrelated sequence in the intercistronic space (Fig 2B(i)). These results indicated the presence of IRES elements in both p53(+39) and (−1) sequences. To rule out the ribosomal read-through of the intercistronic sequences, they were cloned in the plasmid pRΔEF, downstream of a stable RNA structure derived from the encephalomyocarditis virus (EMCV) IRES that inhibits ribosomal read-through (Johannes et al, 1999). These bicistronic DNAs, pRΔEp53(+39)F and pRΔEp53(−1)F, produced 13- and 12-fold higher Fluc activity, respectively, than that from the control vector pRΔEF (Fig 2B(ii)). Similar results were obtained from the p53-null human lung carcinoma cell line H1299 (Fig 2C(i,ii)). Ribosomal read-through of the second cistron was further ruled out by inserting the 440 nt EMCV sequence upstream of the Rluc gene in the bicistronic constructs. This resulted in a strong reduction in the translation of Rluc (Fig 2D, black bars), but no significant change in Fluc expression (Fig 2D, grey bars). Together, these results indicated that the translation from the p53(+39) and (−1) 5′UTRs was IRES mediated and not due to ribosomal read-through.
Figure 2.
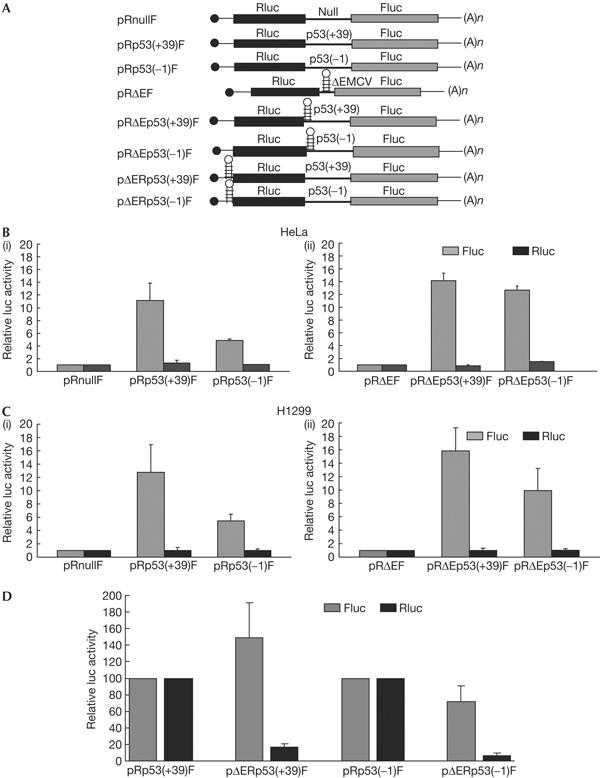
p53 5′-untranslated region sequences mediate internal ribosome entry site-dependent translation. (A) Schematic representation of bicistronic plasmids used in transient transfections. (B(i)) Transfection of bicistronic plasmids pRnullF, pRp53(+39)F and pRp53(−1)F containing a 264 nt unrelated sequence, p53 5′UTR (+39) and (−1) sequences, respectively, in the intercistronic space into HeLa cells. (ii) Transfection of bicistronic plasmids containing p53 5′UTR (+39) and (−1) downstream of the ΔEMCV sequence. The control bicistronic plasmid pRΔEF contained only the ΔEMCV sequence. Transfection efficiencies were normalized by co-transfecting with a β-galactosidase plasmid. The luciferase activities for Fluc and Rluc are shown separately as fold increase compared with that from control, taken as 1. (C(i,ii)) Transfection of the same set of bicistronic plasmids into H1299 cells. The data are represented as in (B). (D) Transfection of bicistronic plasmids containing p53 5′UTR (+39) or (−1) sequences in the intercistronic place and the ΔEMCV sequence upstream of Rluc into HeLa cells. The Fluc and Rluc activities from the ΔEMCV-containing plasmids are shown as fold increase or decrease with respect to the corresponding controls, taken as 100.
The efficiency of the p53(+39) IRES in HeLa cells was similar to that of the heavy chain immunoglobulin binding protein (BiP) IRES, a known cellular IRES. The efficiency of the (−1) IRES was similar to that of the hepatitis A virus IRES (supplementary Fig 1A online). We also investigated whether the translation mediated by the p53(+39) IRES was independent of that mediated by the p53(−1) IRES. Specifically, we wanted to rule out the possibility that the translation mediated by the p53(+39) sequence was due to ribosome binding to the (−1) IRES and initiation from the +1 initiation codon. This was necessitated by the fact that the ATG of the Fluc gene was in-frame with the +1 ATG of the p53(+39) sequence in the bicistronic construct. However, transfection of a p53(+39) bicistronic construct with the +1 ATG mutated to AAG did not show any significant change in Fluc activity compared with the wild-type p53(+39) bicistronic DNA (supplementary Fig 1B online). This indicated that the translation mediated by p53(+39) IRES was independent of that mediated by the (−1) IRES, and supported earlier observations that the mutation of the +1 ATG did not abrogate the translation of ΔN-p53.
p53 5′UTR does not have splice sites or cryptic promoters
To rule out the possibility that the Fluc activity from the p53(+39) bicistronic DNA was due to the presence of splice sites or cryptic promoters, which would generate monocistronic Fluc RNAs in vivo, RNA analyses were carried out with the RNA extracted from HeLa cells transfected with p53 bicistronic DNAs. Reverse transcription–PCR (RT–PCR) and northern blot analysis showed the presence of full-length bicistronic RNA and no smaller RNA species, demonstrating that the Rluc and Fluc cistrons were part of a single transcript (Fig 3A,B).
Figure 3.
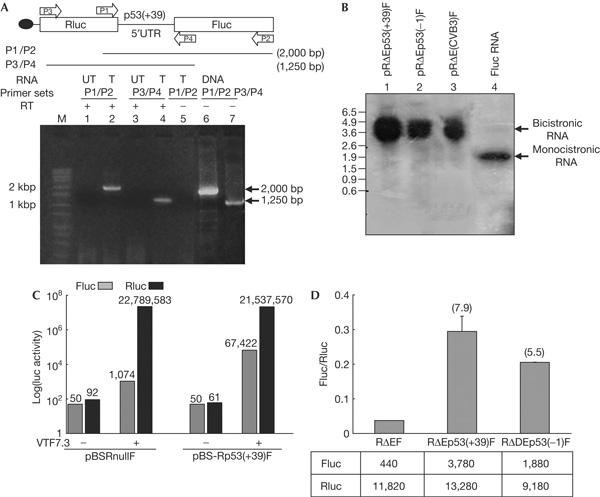
p53 bicistronic plasmid does not show splicing or cryptic promoter activity. (A) Reverse transcription–PCR analysis, using two sets of primers P1/P2 and P3/P4, of RNA extracted from pRp53(+39)F bicistronic plasmid-transfected (T) and untransfected (UT) HeLa cells. Lane 5 shows reverse transcriptase-negative control, whereas lanes 6 and 7 show PCR products amplified from the bicistronic DNA. (B) Northern blot of total RNA extracted from HeLa cells transfected with pRp53(+39)F (lane 1), pRp53(−1)F (lane 2), pR(coxsackievirusB3-IRES)F (lane 3) DNAs and _in vitro_-synthesized Fluc RNA (lane 4) using a 32P-labelled riboprobe corresponding to Fluc. (C) Transfection of bicistronic plasmids pBS-Rp53(+39)F and pBSRnullF lacking eukaryotic promoters into HeLa cells in the absence and presence of infection by VTF7-3. The luciferase activity values are indicated above the respective bars. (D) Transfection of HeLa cells with capped bicistronic RNAs containing the p53(+39) and (−1) 5′UTRs. Fluc/Rluc ratios are shown as fold increase compared with that from a control bicistronic RNA lacking p53 sequences.
However, there is a possibility that small amounts of monocistronic transcripts of the second cistron may be generated, which are not detectable by the RNA analyses but may contribute to Fluc activity. To rule out this possibility, a bicistronic plasmid pBS-Rp53(+39)F, lacking a eukaryotic promoter but containing a T7 promoter, was transfected into HeLa cells. This plasmid did not produce any Fluc or Rluc activity (Fig 3C), demonstrating the absence of a cryptic promoter within the p53 5′UTR sequence that could drive the transcription of Fluc RNA. Only when the cells were infected with a vaccinia virus expressing T7 RNA polymerase (VTF7-3; Fuerst et al, 1986) was high expression of both Fluc and Rluc observed (Fig 3C). The negative control pBSRnullF did not show any luciferase activity when transfected alone, but showed only high Rluc activity when the cells were infected with VTF7-3. Finally, transfection of HeLa cells with the capped bicistronic RNAs showed a Fluc/Rluc ratio significantly higher than that from an empty bicistronic RNA (Fig 3D). Together, these results demonstrated that the expression of the downstream cistron from p53 bicistronic constructs is not due to splicing or cryptic promoter activity.
Cell-cycle phase-dependent regulation of p53 IRES
The expression of ΔN-p53 was shown to vary during cell-cycle progression, with the maximum expression being observed during the G1–S transition, which was accompanied by a transient decrease of full-length p53. We investigated the activities of the IRESs mediating translation of ΔN-p53 and full-length p53 at different phases of the cell cycle. HeLa cells were transfected with pRp53(+39)F or pRp53(−1)F bicistronic plasmids and then blocked at G2–M transition by nocodazole treatment. On releasing the cells and assaying for luciferase activity at different time points, the p53(+39) IRES showed maximum activity at the 24 h time point (Fig 4B). Flow cytometric analysis showed that this coincided with the highest number of cells being in the S phase (Fig 4A). This suggested that the p53(+39) IRES activity was maximum during the progression into S phase, and is supported by the earlier observation that the level of ΔN-p53 protein, which is translated by this IRES, is maximum at this phase.
Figure 4.
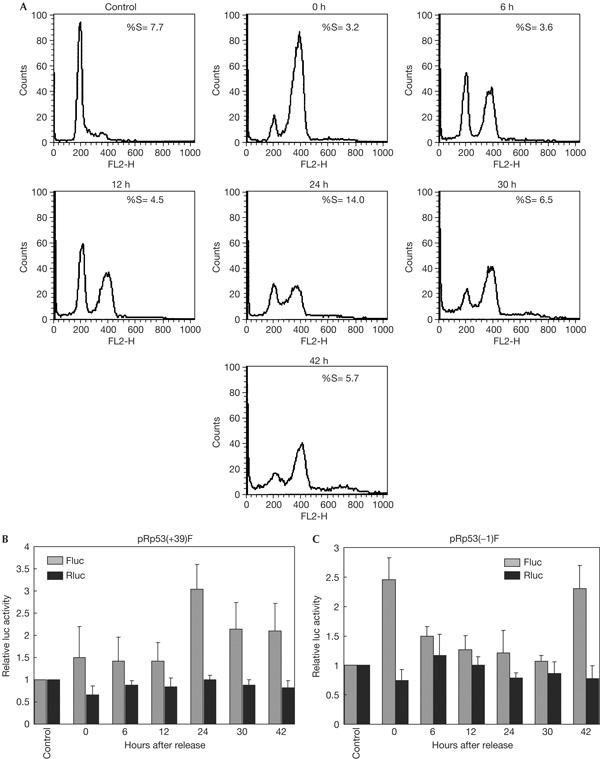
Cell cycle-dependent p53 internal ribosome entry site activity in G2/M synchronized cells. (A) Flow cytometric analysis of HeLa cells collected at different time points after being synchronized at G2/M phase by nocodazole treatment. The percentage of cells in S phase at each time point is indicated. (B) Luciferase assay of cells transfected with p53(+39) and (−1) bicistronic constructs and synchronized at G2/M phase at various time points after release. Fluc and Rluc activities at each time point are expressed as fold of the activity obtained from non-synchronized, transfected cells taken as control. The data mean±s.d. from three independent experiments.
On the other hand, the maximum activity of the p53(−1) IRES was found to be at the G2–M transition (0 h) and decreased subsequently (Fig 4C). Interestingly, it again increased at the 42 h time point, which coincided with the increase in number of cells in the G2–M phase. These observations suggested that the p53(−1) IRES, which evidently controls the translation of full-length p53, is most active during the G2–M transition. In support of these observations, when we blocked the transfected cells at the end of the G1 phase by double thymidine treatment and carried out luciferase assays at subsequent time points, the p53(+39) IRES and the p53(−1) IRES were again found to be maximally active at the G1–S and G2–M transitions, respectively (supplementary Fig 2 online).
Discussion
IRES-mediated translation is an important mode of alternative translation initiation of cellular mRNAs and has been implicated in the regulation of gene expression under different physiological conditions (Hellen & Sarnow, 2001). p53 has a crucial role in cell-cycle progression and has both G1/S and G2/M checkpoint functions. Earlier results have suggested that ΔN-p53, which is highly expressed at the onset of S phase, might facilitate G1–S transition by inhibiting wild-type p53 activity (Courtois et al, 2002). Our study showed that the IRES, mediating the translation of ΔN-p53, is most active at the G1–S transition. The increase in IRES activity at this stage would result in a higher level of expression of ΔN-p53, which is expected to exert a significant negative effect on full-length p53 activity, especially because the ΔN-p53 isoform is resistant to Mdm2-mediated degradation (Yin et al, 2002). This would counteract the cell-cycle checkpoint activity of p53 in normal cells and allow the progression into the S phase (supplementary Fig 3 online).
On the other hand, the IRES mediating the translation of full-length p53 showed the highest activity at the G2–M transition. Cellular cap-dependent translation is known to be downregulated during the G2–M transition. IRESs mediating the translation of several mRNAs show high activity during this phase of the cell cycle (Stoneley & Willis, 2004), suggesting that IRES-mediated translation provides an alternative mode of protein synthesis at this stage. As p53 activity is required for the G2/M checkpoint (Bunz et al, 1998), the increased activity of this IRES at the G2–M transition may be responsible for maintaining the synthesis of basal levels of p53 protein. As the efficiency of the IRES mediating the translation of full-length p53 is low, the small amounts of full-length p53 protein would be efficiently degraded by the Mdm-2-mediated pathway, allowing the progression of cells into the next phase of the cell cycle. Our observations suggest a very intricate regulation of p53 gene expression by the differential activities of the two IRESs (supplementary Fig 3 online). Other cell-cycle-regulated IRESs, such as that of the PITSLRE protein kinase, have been reported previously (Cornelis et al, 2000). The presence of two independent IRESs, differentially regulating vascular endothelial growth factor expression, has been reported before in the vascular endothelial growth factor mRNA (Huez et al, 1998). Also, IRESs present within coding sequences and mediating the translation of truncated versions of proteins have been reported in the case of p58PITSLRE (Cornelis et al, 2000) and oestrogen receptor (Barraille et al, 1999) mRNAs. This study, for the first time, suggests the presence of two IRESs mediating the translation of two different isoforms of the same protein. However, we cannot rule out the possibility that only one IRES is present in the p53 mRNA, which alternatively allows translation initiation from two different downstream initiation codons, and the use of either of these initiation codons is regulated in a cell-cycle-dependent manner.
The interaction of different ITAFs with the cellular IRESs is an important mechanism of regulation of translation initiation. Several proteins, including p53 itself, have been reported to interact with the 5′UTR of the p53 mRNA. p53 interaction with the 5′UTR was found to inhibit the translation of the p53 mRNA, thereby exerting a negative autoregulatory control (Mosner et al, 1995). Interestingly, it was also shown that although p53 mRNA production peaks at the G1–S phase of the cell cycle, the protein level is maximal at mid-S. This suggests that the relative ratios of p53 and ΔN-p53 may be crucial in regulating the translation of p53. High levels of ΔN-p53, produced at the G1–S transition by the activity of the (+39) IRES, might prevent the binding of full-length p53 to the 5′UTR and thereby allow the production of full-length p53 at mid-S phase. Following synthesis of a sufficient level of full-length p53, it would interact with the 5′UTR of its own mRNA and inhibit its translation, creating a negative feedback loop and allowing the cells to enter the G2–M phase of the cell cycle (supplementary Fig 3 online). This model suggests an intricate regulation of p53 expression by RNA–protein interactions, which is required for allowing the normal progression of the cell cycle and for eliciting cell-cycle checkpoint controls under conditions such as DNA damage. Recently, two other proteins, ribosomal protein L26a and nucleolin, have been shown to interact with the p53 5′UTR and modulate its translation (Takagi et al, 2005). Interestingly, nucleolin and other ribosomal proteins have been shown to interact with viral IRESs (Fukushi et al, 2001; Izumi et al, 2001). Therefore, the interaction of nucleolin and RPL26a with the p53 mRNA 5′UTR might regulate the activities of the p53 IRESs. Future studies on the role of these and other p53 IRES-interacting proteins would provide further insights into the regulation of p53 gene expression under various physiological and stress conditions.
Methods
Plasmid constructs. The complementary DNAs corresponding to the 5′ 251 and 134 nt region of p53 mRNA were isolated from the blood of a healthy individual and cloned and sequenced (GenBank accession number AY627884). The 251 and 134 nt DNAs were cloned into pCDNA3 vector (Invitrogen, Carlsbad, CA, USA) upstream of GFP to generate monocistronic constructs. The HCV-IRES–GFP construct had the HCV IRES upstream of GFP. All bicistronic constructs had the respective 5′UTR sequences cloned between Rluc and Fluc genes, in pCDNA3.1, which does not contain a chimeric intron. p53 5′UTR sequences were also cloned downstream of the inactive ΔEMCV IRES sequence in the intercistronic region of the plasmid pRΔDEF (Johannes et al, 1999). The ΔEMCV sequence was also cloned upstream of Rluc gene in the bicistronic plasmids. The ATG codon at position +1 in p53 5′UTR (+39) was mutated to AAG and cloned into a bicistronic construct. The eukaryotic promoter-less bicistronic construct pBS-Rp53(+39)F had the bicistronic cassette containing the p53(+39) 5′UTR cloned into the vector pBluescript-KS (Stratagene, La Jolla, CA, USA) under the T7 promoter.
In vitro transcription. Monocistronic plasmids were linearized downstream of GFP and transcribed using T7 RNA polymerase in the presence/absence of RNA Cap Analog (Promega, Madison, WI, USA). Bicistronic plasmids were linearized downstream of Fluc and transcribed to generate capped RNAs.
In vitro translation. In vitro translation was carried out in rabbit reticulocyte lysate medium (Promega) using [35S]methionine (Perkin-Elmer Life Sciences, Singapore) and the products were resolved on SDS–polyacrylamide gel electrophoresis (as described previously, Ray & Das, 2004). A 2 mM RNA Cap Analog (Promega) or an equivalent amount of GTP was added to reactions, as indicated.
Reverse transcription–PCR analysis. Total RNA isolated by Trizol reagent (Sigma) from untransfected and transfected HeLa cells was pretreated with DNase I and analysed by RT–PCR using two sets of primers: P1(forward)/P2(reverse), corresponding to the 3′ region of Rluc gene and 3′ region of Fluc gene, respectively, and P3(forward)/P4(reverse), corresponding to the 5′ region of Rluc and 5′ region of Fluc, respectively.
Northern blotting. Total RNAs from HeLa cells, transfected with p53(+39), p53(−1) and CVB3-IRES bicistronic plasmids, and Control Fluc RNA (Promega) were resolved on 1% agarose gel, blotted on nylon membrane and hybridized with a 32P-labelled riboprobe corresponding to the Fluc gene.
Cell transfection and synchronization. HeLa S3 and human lung carcinoma H1299 cells were maintained in DMEM (Invitrogen) with 10% fetal bovine serum. Cells were transfected with various bicistronic plasmids or capped bicistronic RNAs using Tfx 20 reagent (Promega). Luciferase assay was carried out in DNA-transfected cells 24 h after transfection and in RNA-transfected cells 6 h after transfection. In experiments using eukaryotic promoter-less bicistronic constructs, cells were infected with a vaccinia virus expressing T7 RNA polymerase (VTF7.3) before transfection with bicistronic plasmids.
At 24 h after transfection with bicistronic plasmids, cells were synchronized either at G2/M phase by nocodazole (500 ng/ml) or at S phase by double thymidine treatment (2 mM). Subsequently, the blocked cells were released by washing with PBS and collected at various time points for either the luciferase assay or flow cytometry. Flow cytometry was carried out after staining DNA with propidium iodide (Sigma-Aldrich, St Louis, MO, USA).
Supplementary information is available at EMBO reports online (http://www.nature.com/embor/journal/vaop/ncurrent/extref/7400623-s1.pdf).
Supplementary Material
Supplementary Information
Acknowledgments
We thank Dr P. Sarnow (Stanford), Dr R. Elliot (Glasgow) and Dr B. Moss (National Institutes of Health) for plasmid constructs and virus. We thank Dr K. Somasundaram (Indian Institute of Science (IISc.), Bangalore), Dr U. Varshney (IISc., Bangalore) and Dr P. Fox (Cleveland Clinic Foundation, Cleveland) for critical reading of the manuscript and members of our laboratory for help and suggestions. This work was supported by a research grant to S.D. from the Department of Biotechnology-Genomics Initiative at IISc. R.G. is supported by a pre-doctoral fellowship from the Council of Scientific and Industrial Research, India.
References
- Barraille P, Chinestra P, Bayard F, Faye JC (1999) Alternative initiation of translation accounts for a 67/45 kDa dimorphism of the human estrogen receptor ERα. Biochem Biophys Res Commun 257: 84–88 [DOI] [PubMed] [Google Scholar]
- Bunz F, Dutriaux A, Lengauer C, Waldman T, Zhou S, Brown JP, Sedivy JM, Kinzler KW, Vogelstein B (1998) Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science 282: 1497–1501 [DOI] [PubMed] [Google Scholar]
- Cornelis S, Bruynooghe Y, Denecker G, Van Huffel S, Tinton S, Beyaert R (2000) Identification and characterization of a novel cell cycle-regulated internal ribosome entry site. Mol Cell 5: 597–605 [DOI] [PubMed] [Google Scholar]
- Courtois S, Verhaegh G, North S, Luciani MG, Lassus P, Hibner U, Oren M, Hainaut P (2002) N-p53, a natural isoform of p53 lacking the first transactivation domain, counteracts growth suppression by wild-type p53. Oncogene 21: 6722–6728 [DOI] [PubMed] [Google Scholar]
- Fuerst TR, Niles EG, Studie FW, Moss B (1986) Eukaryotic transient-expression system based on recombinant vaccinia virus that synthesizes bacteriophage T7 RNA polymerase. Proc Natl Acad Sci USA 83: 8122–8126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukushi S, Okada M, Stahl J, Kageyama T, Hoshino FB, Katayama K (2001) Ribosomal protein S5 interacts with the internal ribosome entry site of hepatitis C virus. J Biol Chem 276: 20824–20826 [DOI] [PubMed] [Google Scholar]
- Hellen CUT, Sarnow P (2001) Internal ribosome entry sites in eukaryotic mRNA molecules. Genes Dev 15: 1593–1612 [DOI] [PubMed] [Google Scholar]
- Holcik M, Sonenberg N (2005) Translational control in stress and apoptosis. Nat Rev Mol Cell Biol 6: 318–327 [DOI] [PubMed] [Google Scholar]
- Hollstein M, Sidransky D, Vogelstein B, Harris CC (1991) p53 mutations in human cancers. Science 253: 49–53 [DOI] [PubMed] [Google Scholar]
- Huez I, Creancier L, Audigier S, Gensac MC, Prats AC, Prats H (1998) Two independent internal ribosome entry sites are involved in translation initiation of vascular endothelial growth factor mRNA. Mol Cell Biol 18: 6178–6190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izumi RE, Valdez B, Banerjee R, Srivastava M, Dasgupta A (2001) Nucleolin stimulates viral internal ribosome entry site-mediated translation. Virus Res 76: 17–29 [DOI] [PubMed] [Google Scholar]
- Johannes G, Carter MS, Eisen MB, Brown PO, Sarnow P (1999) Identification of eukaryotic mRNAs that are translated at reduced cap binding complex eIF4F concentrations using a cDNA microarray. Proc Natl Acad Sci USA 96: 13118–13123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko LJ, Prives C (1996) p53: puzzle and paradigm. Genes Dev 10: 1054–1072 [DOI] [PubMed] [Google Scholar]
- Levine AJ (1997) p53, the cellular gatekeeper for growth and division. Cell 88: 323–331 [DOI] [PubMed] [Google Scholar]
- Mosner J, Mummenbrauer T, Bauer C, Sczakiel G, Grosse F, Deppert W (1995) Negative feedback regulation of wild-type p53 biosynthesis. EMBO J 14: 4442–4449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray PS, Das S (2004) Inhibition of hepatitis C virus IRES-mediated translation by small RNAs analogous to stem–loop structures of the 5′-untranslated region. Nucleic Acids Res 32: 1678–1687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somasundaram K, El-Deiry WS (2000) Tumor suppressor p53: regulation and function. Front Biosci 5: 424–437 [DOI] [PubMed] [Google Scholar]
- Stoneley M, Willis AE (2004) Cellular internal ribosome entry segments: structures, _trans_-acting factors and regulation of gene expression. Oncogene 23: 3200–3207 [DOI] [PubMed] [Google Scholar]
- Takagi M, Absalon MJ, McLure KG, Kastan MB (2005) Regulation of p53 translation and induction after DNA damage by ribosomal protein L26 and nucleolin. Cell 123: 40–63 [DOI] [PubMed] [Google Scholar]
- Vagner S, Galy B, Pyronnet S (2001) Irresistible IRES. Attracting the translation machinery to internal ribosome entry sites. EMBO Rep 10: 893–898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin Y, Luciani MG, Fahraeus R (2002) p53 stability and activity is regulated by Mdm2-mediated induction of alternative p53 translation products. Nat Cell Biol 4: 462–467 [DOI] [PubMed] [Google Scholar]
- Zuker M (2003) Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res 31: 3406–3415 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information