Bladder Cancer Predisposition: A Multigenic Approach to DNA-Repair and Cell-Cycle–Control Genes (original) (raw)
Abstract
The candidate-gene approach in association studies of polygenic diseases has often yielded conflicting results. In this hospital-based case-control study with 696 white patients newly diagnosed with bladder cancer and 629 unaffected white controls, we applied a multigenic approach to examine the associations with bladder cancer risk of a comprehensive panel of 44 selected polymorphisms in two pathways, DNA repair and cell-cycle control, and to evaluate higher-order gene-gene interactions, using classification and regression tree (CART) analysis. Individually, only XPD Asp312Asn, RAG1 Lys820Arg, and a p53 intronic SNP exhibited statistically significant main effects. However, we found a significant gene-dosage effect for increasing numbers of potential high-risk alleles in DNA-repair and cell-cycle pathways separately and combined. For the nucleotide-excision repair pathway, compared with the referent group (fewer than four adverse alleles), individuals with four (odds ratio [OR] = 1.52, 95% CI 1.05–2.20), five to six (OR = 1.81, 95% CI 1.31–2.50), and seven or more adverse alleles (OR = 2.50, 95% CI 1.69–3.70) had increasingly elevated risks of bladder cancer (P for trend <.001). Each additional adverse allele was associated with a 1.21-fold increase in risk (95% CI 1.12–1.29). For the combined analysis of DNA-repair and cell-cycle SNPs, compared with the referent group (<13 adverse alleles), the ORs for individuals with 13–15, 16–17, and ⩾18 adverse alleles were 1.22 (95% CI 0.84–1.76), 1.57 (95% CI 1.05–2.35), and 1.77 (95% CI 1.19–2.63), respectively (P for trend = .002). Each additional high-risk allele was associated with a 1.07-fold significant increase in risk. In addition, we found that smoking had a significant multiplicative interaction with SNPs in the combined DNA-repair and cell-cycle–control pathways (P<.01). All genetic effects were evident only in “ever smokers” (persons who had smoked ⩾100 cigarettes) and not in “never smokers.” A cross-validation statistical method developed in this study confirmed the above observations. CART analysis revealed potential higher-order gene-gene and gene-smoking interactions and categorized a few higher-risk subgroups for bladder cancer. Moreover, subgroups identified with higher cancer risk also exhibited higher levels of induced genetic damage than did subgroups with lower risk. There was a significant trend of higher numbers of bleomycin- and benzo[_a_]pyrene diol-epoxide (BPDE)–induced chromatid breaks (by mutagen-sensitivity assay) and DNA damage (by comet assay) for individuals in higher-risk subgroups among cases of bladder cancer in smokers. The P for the trend was .0348 for bleomycin-induced chromosome breaks, .0036 for BPDE-induced chromosome breaks, and .0397 for BPDE-induced DNA damage, indicating that these higher-order gene-gene and gene-smoking interactions included SNPs that modulated repair and resulted in diminished DNA-repair capacity. Thus, genotype/phenotype analyses support findings from CART analyses. This is the first comprehensive study to use a multigenic analysis for bladder cancer, and the data suggest that individuals with a higher number of genetic variations in DNA-repair and cell-cycle–control genes are at an increased risk for bladder cancer, confirming the importance of taking a multigenic pathway-based approach to risk assessment.
The candidate-gene approach is hypothesis driven, uses a priori knowledge of SNP and gene functions, and has yielded sometimes informative but often conflicting data in cancer association studies. In many studies where a significant association is reported, the odds ratios (ORs) for individual variants are <2 (Goode et al. 2002; Neumann et al. 2005). There are innumerable instances in which association studies have been unable to replicate an initial positive candidate-gene finding. Among the reasons for this lack of replication are small sample size, inadequate statistical methods, and failure to evaluate the effect of multiple pathophysiologically related genes (Horne et al. 2005). The low risk conferred by an individual polymorphism is not surprising, given that carcinogenesis is usually a multistep, multigenic process, and it is unlikely that any one single genetic polymorphism would have a dramatic effect on cancer risk. Therefore, single-gene studies are likely to provide limited value in predicting risk. A pathway-based genotyping approach, which assesses the combined effects of a panel of polymorphisms that interact in the same pathway, may amplify the effects of individual polymorphisms and enhance the predictive power. Several recent small-scale multigenic studies provide evidence of the promising potential of applying such a pathway-based multigenic approach in association studies (Han et al. 2004; Popanda et al. 2004; Cheng et al. 2005; Gu et al. 2005). In this article, we use bladder cancer as the cancer prototype and focus on two relevant physiologic pathways, DNA repair and cell-cycle control, to illustrate our theme.
Bladder cancer is the malignancy with the fifth highest incidence in the United States, with ∼63,210 new cases in 2005 (Jemal et al. 2005). Cigarette smoking is the predominant risk factor and is estimated to be responsible for half the cases in men and for a third in women. Occupational exposure to carcinogens is the second major risk factor. Despite the overwhelming evidence that most bladder cancers are attributable in part to environmental carcinogenic exposures, only a fraction of exposed individuals actually develop bladder cancer, and the working hypothesis is that there are also predisposing genetic factors (Shields and Harris 2000; Wu et al. 2004).
DNA damage repair and cell-cycle checkpoints are two primary defense mechanisms against mutagenic exposures. There are four major DNA-repair pathways in human cells: mismatch repair, nucleotide-excision repair (NER), base-excision repair (BER), and double-strand–break (DSB) repair (Christmann et al. 2003). The NER pathway mainly removes bulky DNA adducts typically generated from exposure to polycyclic aromatic hydrocarbons in tobacco smoke. The BER pathway is responsible for removal of oxidized DNA bases that may arise endogenously or from exogenous agents. The DSB pathway is responsible for repairing double-strand breaks caused by a variety of exposures, including ionizing radiation, free radicals, and telomere dysfunction. There are two distinct and complementary pathways for DSB repair—namely, homologous recombination (HR) and nonhomologous end joining (NHEJ). Cell-cycle checkpoints are regulatory pathways that control the order and timing of cell-cycle transitions to ensure the fidelity of critical events such as DNA replication and chromosome segregation (Elledge 1996). Cells may be arrested at any of the checkpoints, temporarily halting the cell cycle and allowing DNA repair to be completed. Checkpoint loss and perturbation of cell-cycle control results in genomic instability and is a hallmark of cancer, as evidenced by the frequent inactivation of cell-cycle–control genes, including p53, p16, and Rb, in various cancers (Hanahan and Weinberg 2000).
There is considerable interindividual variation in DNA-repair capacity (DRC) and strong evidence that reduced DRC is associated with increased cancer risk (Berwick and Vineis 2000; Spitz et al. 2003; Wu et al. 2004). Polymorphisms in DNA-repair genes are hypothesized to be a contributor to this individual DRC variation (Mohrenweiser et al. 2003). There have been numerous studies, often with conflicting results, assessing the associations of polymorphisms in DNA-repair and cell-cycle genes with cancer risk on the basis of the hypothesis that individuals with “adverse” genotypes that result in reduced DRC or perturbed cell-cycle control are at a higher risk of developing cancer than the general population (Goode et al. 2002; Wu et al. 2004; Neumann et al. 2005).
In this study, we applied a pathway-based multigenic approach to examine the associations of a comprehensive panel of polymorphisms in DNA-repair and cell-cycle genes with bladder cancer risk. We selected 13 SNPs from the NER pathway, 8 SNPs from the BER pathway, 8 SNPs from the HR pathway, 5 SNPs from the NHEJ pathway, and 10 SNPs from the cell-cycle–control pathway. The majority of these SNPs were selected from published association studies, and a few were chosen from dbSNP on the basis of their location (promoter or coding regions) and minor-allele frequencies (>5%). To our knowledge, this is the largest multigenic cancer association study reported. We examined the combined effects of the minor alleles and evaluated higher-order gene-gene interactions, using several statistical models. In addition, we used two functional assays assessing genetic instability to determine genotype-phenotype correlations, in an attempt to validate our analytic approach. This pathway-based multigenic approach may provide a refinement of epidemiologic profiles associated with risk.
Material and Methods
Study Subjects
This study included patients with newly diagnosed bladder cancer and age-, gender-, and ethnicity-matched control subjects. The cases were enrolled at The University of Texas M. D. Anderson Cancer Center and the Scott Department of Urology at Baylor College of Medicine between 1999 and 2003. All patients had histopathologically confirmed bladder cancer, and none had received chemotherapy or radiation before enrollment. The control subjects were healthy individuals with no prior history of cancer (except nonmelanoma skin cancer) who were recruited from Kelsey Seybold, the largest multispecialty, managed-care physician group in the Houston metropolitan area. We also excluded control subjects with chronic urinary tract disease, obstructive airway disease, and diabetes. Control subjects were matched to the case patients by age (±5 years), sex, and ethnicity. The potential control subjects were first surveyed with a short questionnaire to elicit willingness to participate in the study and to provide preliminary demographic data for matching. A Kelsey-Seybold staff member provided the questionnaire to each potential control subject during clinical registration. The potential control subjects were contacted by telephone at a later date to confirm their willingness to participate and to schedule an interview appointment at a Kelsey-Seybold clinic convenient to the participant. Informed consent was obtained from all study participants before the collection of epidemiological data and blood samples by trained M. D. Anderson staff interviewers. The response rates were 75% for the controls and 92% for the cases.
Epidemiology Data Collection
In 45-min interviews, trained M. D. Anderson staff interviewers collected data on demographics, smoking history, and family history of cancer. At the end of the interviews, 40-mL blood samples were drawn into coded heparinized tubes. Individuals who had smoked at least 100 cigarettes in their lifetimes were defined as “ever smokers.” Participants who had quit smoking at least 1 year before the study were categorized as former smokers. All participants signed informed consent agreements, and the institutional review boards of M. D. Anderson, Baylor College of Medicine, and Kelsey-Seybold Clinic, in accordance with the U.S. Department of Health and Human Services, approved the study.
Genotyping
Genomic DNA was extracted from peripheral blood lymphocytes by proteinase K digestion, followed by isopropanol extraction and ethanol precipitation; DNA samples were stored at −80°C. Genotyping was performed using the Taqman method with a 7900 HT sequence detector system (Applied Biosynthesis), except for XPA, XPD Asp312Asn, and p53 intron 3 polymorphism, which were genotyped using PCR-RFLP (Spitz et al. 2001; Wu et al. 2002, 2003). The primer and probe sequences for each SNP are available upon request. Amplification mixes (5 μl) contained sample DNA (5 ng), 1× TaqMan buffer A, deoxynucleotide triphosphate (200 μM), MgCl2 (5 mM), AmpliTaq Gold (0.65 units), each primer (900 nM), and each probe (200 nM). The thermal cycling conditions consisted of 1 cycle for 10 min at 95°C, 40 cycles for 15 s at 95°C, and 40 cycles for 1 min at 60°C. SDS version 2.1 software (Applied Biosystems) was used to analyze end-point fluorescence, in accordance with the allelic discrimination technique. Water control, ample internal controls, and previously genotyped samples were included in each plate to ensure accuracy of the genotyping. Positive and negative controls were used in each genotyping assay, and 5% of the samples were randomly selected and run in duplicates with 100% concordance.
Genetic Instability Index
Genetic instability was measured by two in vitro assays. Mutagen sensitivity was assessed in vitro in lymphocyte cultures by counting the number of chromatid breaks induced by exposure to either bleomycin or benzo[_a_]pyrene diol-epoxide (BPDE), as described elsewhere (Hsu et al. 1989; Wu et al. 1998_a_). In brief, blood cultures were incubated for 3 d and then exposed to either bleomycin (0.03 U/mL) for 5 h or BPDE (2 μM) for 24 h. Cells were harvested, and chromatid breaks were counted in 50 metaphases per sample and were recorded as the mean number of breaks per cell. Laboratory personnel read the slides without knowledge of the individual’s cancer status. The comet assay under alkaline conditions was performed also as described elsewhere (Schabath et al. 2003). In brief, blood cultures were challenged with 2 μM BPDE. Then, the cell culture was mixed with agarose gel and was adhered to a microscope slide. The cells were lysed by submersing the slides in freshly prepared lysis buffer for 1 h at 4°C to remove all the cellular proteins. The slides were next placed in alkali buffer (pH >12.0) to denature and unwind the DNA and to expose the alkali-labile sites. The slides were subjected to electrophoresis and were stained with ethidium bromide, then were neutralized, were fixed in 100% methanol, and were stored in the dark at room temperature until ready for analysis. Consecutive comet cells (_n_=50 [25 cells from each end of the slide]) were manually selected and were automatically quantified using the Komet 4.0.2 (Kinetic Imaging) imaging software attached to a fluorescent microscope. The Olive tail moment was used as the parameter for DNA damage computed by the imaging system software. The averages of the Olive tail moment for each subject were calculated for the baseline and mutagen-induced comets. All assays were performed in a blinded manner, and a single lab technician performed the entire procedure for each assay to minimize interindividual variation. Detailed scanning criteria for the comet assay have also been well established to ensure consistency. There were 877 samples with mutagen-sensitivity data and 957 samples with comet assay data. The functional assays started during the second year of recruitment, and all the samples were measured consecutively. There were no significant differences in characteristics between subjects with functional data and those without functional data.
Statistical Analysis
The χ2 and Fisher’s exact tests were used to assess patient characteristics by genotype and to compare cases with controls. Student’s t test was used to test for differences between the cases and controls for continuous variables. ORs were calculated as estimates of relative risk. Unconditional multivariable logistic regression was performed to control for possible confounding by age, sex, ethnicity, and smoking status, where appropriate. Since we found no definitive prior associations in our review of the literature between any of the variants studied and bladder cancer risk, our primary analysis treated the minor-variant allele at each locus as the “risk” allele. However, the assumption that the minor variant is associated with increased risk may not be accurate. We, therefore, also evaluated other analytic approaches. In particular, we developed a method for cross-validation. First, we evaluated the main effect associated with each minor allele in half of the data. We then reassigned the minor-allele risk status if the main effect of the minor variant was found to show a negative association with risk (i.e., OR <1), and we then re-evaluated the risk in the validation set. To increase the efficiency of this testing approach, we cross-validated 10 randomly selected samples from the original data set. The average risk of each SNP in the validation sets was used to obtain pooled estimates of risk for each allele and for sums of alleles. The SE (δ2) for the allele effect of each marker is expected to be identical and to show a covariance of 1/4 across randomly selected subsets. The mean estimate, 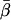 , of the allele effect across n replicate samples is normally distributed with mean μ (the per-replicate estimate of the allele effect) and variance
, of the allele effect across n replicate samples is normally distributed with mean μ (the per-replicate estimate of the allele effect) and variance 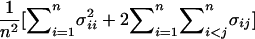 , where σ2_ii_ is the variance of the allele effect and σ_ij_ is the covariance, which is 1/4σ2_ii_. After simplification, the SE of the average-risk estimate across cross-validation sets is (1/4+3/[4_n_])δ2, where n is the number of cross-validation replicates.
, where σ2_ii_ is the variance of the allele effect and σ_ij_ is the covariance, which is 1/4σ2_ii_. After simplification, the SE of the average-risk estimate across cross-validation sets is (1/4+3/[4_n_])δ2, where n is the number of cross-validation replicates.
For higher-order gene-gene interactions, classification and regression tree (CART) analysis was performed using the HelixTree Genetics Analysis Software (version 4.1.0, Golden Helix). CART is a binary recursive-partitioning method that produces a decision tree to identify subgroups of subjects at higher risk. Specifically, the recursive-partitioning algorithm in HelixTree starts at the first node (with the entire data set) and uses a statistical hypothesis-testing method—formal inference-based recursive modeling—to determine the first locally optimal split and each subsequent split of the data set, with multiplicity-adjusted P values to control tree growth. This process continues until the terminal nodes have no subsequent statistically significant splits or the terminal nodes reach a prespecified minimum size (at least 10 subjects for each terminal node in our analysis). The data were divided randomly into a learning set (90% of the data) and a testing set (10% of the data). The learning set was used to construct the tree model, and the testing set was used to internally validate the resulting tree model.
Results
Characteristics of Subjects
There were a total of 1,484 study subjects recruited for this study. The population consisted of 1,325 whites (89.3%), 77 Mexican Americans (5.2%), 67 African Americans (4.5%), and 15 others (1.0%). Among whites, there were 696 patients with newly diagnosed, histologically confirmed bladder cancer and 629 unaffected controls. There was no significant age difference between the cases (63.91 ± 11.17 years) and the controls (62.77 ± 10.50 years) (_P_=.06). There were more males (78.45%) among the cases than among the controls (72.66%) (_P_=.014). As would be predicted, the cases had a significantly higher percentage of current (25.29%) and ever smokers (73.55%) than the controls (8.21% and 53.74%, respectively) (P<.0001). Among ever smokers, cases reported significantly higher levels of consumption of cigarettes than controls, as assessed by computing mean pack-years (42.95 vs. 28.28, P<.0001).
Risk Associated with Individual SNPs
Since ∼90% of the study subjects were white, we limited all our analyses to this population. The distributions of the selected panel of SNPs in the major genes involved in DNA-repair and cell-cycle–control pathways are summarized in tables 1–4. The allele frequencies of each SNP in the cases and controls and the corresponding ORs for the heterozygous and homozygous variant genotypes and for the combined heterozygote and rare homozygote genotypes are listed. For main effects of the DNA-repair genes, individually, only carriers of the variant Asn allele of XPD, Asp312Asn (_OR_=1.28, 95% CI 1.01–1.62), and the variant Arg allele of the RAG1, Lys820Arg (_OR_=1.32, 95% CI 1.00–1.73), exhibited significantly increased risks for bladder cancer (tables 1–4). For the cell-cycle–control genes, a protective effect was found for carriers of at least one rare allele of the p53 intron 3 SNP (_OR_=0.72, 95% CI 0.55–0.94) (table 4). The other selected SNPs in the cell-cycle–control pathway did not show significant main effects for bladder cancer risk (table 4). With consideration of the borderline CIs and multiple comparisons performed, the impact of any individual SNP on bladder cancer risk, if it exists, would be minimal.
Table 1.
NER Genotype Frequencies and Risks for Bladder Cancer
| Finding for SNP | ||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ERCC1 3′ UTRa (rs3212986) | XPD K751Qb (rs1052559) | XPD D312Nc (rs1799793) | XPA 5′ UTRd (rs1800975) | XPF S662Pe (rs2020955) | XPG H1104Df (rs17655) | XPC Intron 9 (AT)ng | XPC K939Qh (rs2228001) | XPC A499Vi (rs2228000) | RAD23B A249Vj (rs1805329) | CCCNH V270Ak (rs2266690) | ERCC6 M1097Vl (rs2228526) | ERCC6 R1230Pm (rs4253211) | ||||||||||||||||||||||||||||||||||||||||
| Parameter | Case | Control | OR | 95% CI | Case | Control | OR | 95% CI | Case | Control | OR | 95% CI | Case | Control | OR | 95% CI | Case | Control | OR | 95% CI | Case | Control | OR | 95% CI | Case | Control | OR | 95% CI | Case | Control | OR | 95% CI | Case | Control | OR | 95% CI | Case | Control | OR | 95% CI | Case | Control | OR | 95% CI | Case | Control | OR | 95% CI | Case | Control | OR | 95% CI |
| Rare-allele frequency | .252 | .252 | .371 | .344 | .351 | .316 | .598 | .583 | .006 | .003 | .225 | .206 | .353 | .335 | .397 | .408 | .232 | .239 | .204 | .192 | .217 | .199 | .218 | .188 | .094 | .106 | ||||||||||||||||||||||||||
| Common homozygote (n) | 336 | 334 | 1.0 | 245 | 256 | 1.0 | 264 | 283 | 1.0 | 99 | 108 | 1.0 | 600 | 594 | 1.0 | 364 | 371 | 1.0 | 278 | 292 | 1.0 | 219 | 211 | 1.0 | 355 | 333 | 1.0 | 387 | 393 | 1.0 | 371 | 385 | 1.0 | 371 | 392 | 1.0 | 500 | 477 | 1.0 | |||||||||||||
| Heterozygote (n) | 232 | 224 | 1.06 | .83–1.36 | 282 | 274 | 1.07 | .83–1.38 | 283 | 243 | 1.27 | .98–1.63 | 272 | 265 | 1.17 | .83–1.64 | 7 | 3 | 2.19 | .53–9.12 | 225 | 211 | 1.06 | .83–1.36 | 242 | 219 | 1.21 | .94–1.56 | 293 | 284 | 1.03 | .79–1.34 | 216 | 232 | .88 | .69–1.13 | 193 | 176 | 1.10 | .85–1.43 | 206 | 183 | 1.25 | .97–1.62 | 204 | 182 | 1.14 | .88–1.47 | 98 | 112 | .87 | .64–1.19 |
| Rare homozygote (n) | 36 | 38 | .88 | .54–1.45 | 87 | 69 | 1.38 | .95–2.02 | 78 | 65 | 1.31 | .89–1.92 | 214 | 204 | 1.16 | .82–1.65 | 0 | 0 | 26 | 18 | 1.58 | .83–2.99 | 97 | 93 | 1.16 | .82–1.65 | 94 | 101 | .91 | .64–1.30 | 32 | 25 | 1.09 | .61–1.93 | 27 | 26 | .94 | .53–1.68 | 28 | 27 | 1.03 | .58–1.83 | 30 | 21 | 1.53 | .84–2.79 | 8 | 7 | .96 | .33–2.75 | ||
| Heterozygote and rare homozygote | 1.03 | .82–1.31 | 1.03 | .82–1.31 | 1.28 | 1.01–1.62 | 1.17 | .86–1.61 | 1.10 | .87–1.40 | 1.19 | .94–1.51 | .99 | .78–1.27 | .90 | .71–1.15 | 1.08 | .84–1.38 | 1.23 | .96–1.58 | 1.17 | .92–1.50 | .88 | .65–1.19 | ||||||||||||||||||||||||||||
| Genotyped (n) | 604 | 596 | 614 | 599 | 625 | 591 | 585 | 577 | 607 | 597 | 615 | 600 | 617 | 604 | 606 | 596 | 603 | 590 | 607 | 595 | 605 | 595 | 605 | 595 | 606 | 595 |
Table 2.
BER Genotype Frequencies and Risks for Bladder Cancer
| Finding for SNP | ||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| MBD4 E346Ka (rs140693) | MUTYH Q335Hb (rs3219489) | OGG1 S326Cc (rs1052133) | APEX1 D148Ed (rs3136820) | XRCC1 R194We (rs1799782) | XRCC1 R399Qf (rs25487) | ADPRT V762Ag (rs1136410) | POLD1 R119Hh (rs1726801) | |||||||||||||||||||||||||
| Parameter | Case | Control | OR | 95% CI | Case | Control | OR | 95% CI | Case | Control | OR | 95% CI | Case | Control | OR | 95% CI | Case | Control | OR | 95% CI | Case | Control | OR | 95% CI | Case | Control | OR | 95% CI | Case | Control | OR | 95% CI |
| Rare-allele frequency | .250 | .226 | .218 | .240 | .467 | .482 | .064 | .064 | .340 | .337 | .151 | .162 | .073 | .067 | ||||||||||||||||||
| Common homozygote (n) | 597 | 591 | 1.0 | 334 | 340 | 1.0 | 375 | 348 | 1.0 | 176 | 166 | 1.0 | 539 | 524 | 1.0 | 266 | 267 | 1.0 | 437 | 416 | 1.0 | 501 | 495 | 1.0 | ||||||||
| Heterozygote (n) | 0 | 0 | 240 | 224 | 1.08 | .84–1.38 | 209 | 216 | .91 | .71–1.17 | 283 | 279 | .94 | .71–1.23 | 71 | 75 | .9 | .63–1.29 | 277 | 256 | 1.08 | .84–1.39 | 155 | 165 | .88 | .67–1.15 | 70 | 75 | .90 | .63–1.30 | ||
| Rare homozygote (n) | 8 | 5 | 1.74 | .54–5.63 | 31 | 30 | 1.08 | .62–1.87 | 29 | 36 | .73 | .43–1.24 | 137 | 145 | .85 | .61–1.19 | 4 | 1 | 4.17 | .42–41.24 | 70 | 73 | .92 | .63–1.36 | 14 | 14 | .96 | .44–2.10 | 7 | 1 | 4.93 | .58–41.81 |
| Heterozygote and rare homozygote | 1.08 | .85–1.37 | .89 | .70–1.12 | .90 | .70–1.18 | .94 | .66–1.34 | 1.05 | .83–1.33 | .89 | .68–1.15 | .97 | .68–1.38 | ||||||||||||||||||
| Genotyped (n) | 605 | 596 | 605 | 594 | 613 | 600 | 596 | 590 | 614 | 600 | 613 | 596 | 606 | 595 | 578 | 571 |
Table 3.
HR and NEHEJ Genotype Frequencies and Risks for Bladder Cancer
| Finding for SNP | ||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| HR | NHEJ | |||||||||||||||||||||||||||||||||||||||||||||||||||
| XRCC3 T241Ma (rs861539) | XRCC3 5′ UTRb (rs1799794) | XRCC3 A17893Gc (rs1799796) | XRCC2 R188Hd (rs3218536) | XRCC2 3′ UTR C/T atNucleotide 41657e | NBS1 E185Qf (rs1805794) | BRCA2 N372Hg (rs144814) | RAG1 K820Rh (rs2227973) | KU70 G593Gi (rs132788) | KU80 3′ UTRj (rs1051685) | LIG4 T9Ik (rs1805388) | XRCC4 Splice Sitel (rs1805377) | ATM D1853Nm (rs1801516) | ||||||||||||||||||||||||||||||||||||||||
| Parameter | Case | Control | OR | 95% CI | Case | Control | OR | 95% CI | Case | Control | OR | 95% CI | Case | Control | OR | 95% CI | Case | Control | OR | 95% CI | Case | Control | OR | 95% CI | Case | Control | OR | 95% CI | Case | Control | OR | 95% CI | Case | Control | OR | 95% CI | Case | Control | OR | 95% CI | Case | Control | OR | 95% CI | Case | Control | OR | 95% CI | Case | Control | OR | 95% CI |
| Rare-allele frequency | .387 | .362 | .175 | .176 | .320 | .347 | .080 | .083 | .044 | .056 | .345 | .308 | .290 | .255 | .150 | .121 | .359 | .353 | .100 | .109 | .178 | .189 | .120 | .117 | .158 | .144 | ||||||||||||||||||||||||||
| Common homozygote (n) | 230 | 250 | 1.0 | 402 | 398 | 1.0 | 279 | 256 | 1.0 | 512 | 498 | 1.0 | 546 | 524 | 1.0 | 254 | 282 | 1.0 | 306 | 332 | 1.0 | 434 | 457 | 1.0 | 251 | 244 | 1.0 | 494 | 474 | 1.0 | 414 | 384 | 1.0 | 463 | 471 | 1.0 | 434 | 439 | 1.0 | |||||||||||||
| Heterozygote (n) | 290 | 261 | 1.21 | .93–1.56 | 185 | 185 | .96 | .74–1.24 | 258 | 261 | .90 | .70–1.16 | 89 | 95 | .89 | .64–1.24 | 51 | 62 | .84 | .56–1.27 | 283 | 259 | 1.13 | .88–1.45 | 246 | 223 | 1.12 | .87–1.43 | 160 | 132 | 1.30 | .99–1.72 | 263 | 268 | .96 | .74–1.23 | 106 | 112 | .96 | .71–1.31 | 168 | 194 | .81 | .62–1.05 | 133 | 112 | 1.20 | .89–1.61 | 156 | 136 | 1.12 | .85–1.48 |
| Rare homozygote (n) | 92 | 85 | 1.19 | .84–1.71 | 12 | 12 | 1.13 | .49–2.61 | 63 | 75 | .82 | .55–1.22 | 4 | 2 | 2.87 | .51–16.07 | 1 | 2 | .25 | .02–3.21 | 67 | 54 | 1.28 | .84–1.93 | 52 | 40 | 1.32 | .83–2.10 | 11 | 6 | 1.66 | .58–4.74 | 83 | 72 | 1.13 | .79–1.67 | 8 | 9 | .75 | .27–2.03 | 24 | 15 | 1.42 | .71–2.84 | 6 | 14 | .42 | .15–1.16 | 18 | 17 | 1.02 | .51–2.07 |
| Heterozygote and rare homozygote | 1.20 | .95–1.53 | .97 | .75–1.25 | .89 | .70–1.12 | .93 | .67–1.28 | .82 | .55–1.22 | 1.15 | .90–1.46 | 1.15 | .91–1.46 | 1.32 | 1.00–1.73 | 1.00 | .79–1.27 | .95 | .70–1.27 | .85 | .66–1.10 | 1.11 | .84–1.48 | 1.11 | .85–1.45 | ||||||||||||||||||||||||||
| Genotyped (n) | 612 | 596 | 599 | 595 | 600 | 592 | 605 | 595 | 598 | 588 | 604 | 595 | 604 | 595 | 605 | 595 | 597 | 584 | 608 | 595 | 606 | 593 | 605 | 595 | 608 | 592 |
Table 4.
Cell-Cycle SNPs Genotype Frequencies and Risks for Bladder Cancer in Whites
| Finding for SNP | ||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CCND1 P241Pa (rs603965) | P53 Intron 3 16 bp Duplicationb | P53 Intron 6c(rs1625895) | P53 R72Pd (rs1042522) | CDK4 Promotere (rs2072052) | P27 5′ UTRf (rs34330) | P21 3′ UTRg (rs1059234) | CDK6 3′ UTRh (rs42309) | STK15 F31Ii (rs2273535) | STK15 I57Vj (rs1047972) | |||||||||||||||||||||||||||||||
| Parameter | Case | Control | OR | 95% CI | Case | Control | OR | 95% CI | Case | Control | OR | 95% CI | Case | Control | OR | 95% CI | Case | Control | OR | 95% CI | Case | Control | OR | 95% CI | Case | Control | OR | 95% CI | Case | Control | OR | 95% CI | Case | Control | OR | 95% CI | Case | Control | OR | 95% CI |
| Rare-allele frequency | .413 | .438 | .124 | .154 | .126 | .149 | .215 | .217 | .301 | .292 | .240 | .239 | .076 | .075 | .250 | .258 | .182 | .197 | .171 | .147 | ||||||||||||||||||||
| Common homozygote (n) | 213 | 186 | 1.0 | 492 | 446 | 1.0 | 472 | 437 | 1.0 | 390 | 390 | 1.0 | 304 | 296 | 1.0 | 348 | 342 | 1.0 | 513 | 506 | 1.0 | 339 | 318 | 1.0 | 403 | 373 | 1.0 | 420 | 432 | 1.0 | ||||||||||
| Heterozygote (n) | 293 | 300 | .87 | .67–1.14 | 130 | 154 | .72 | .55–.96 | 128 | 142 | .85 | .64–1.12 | 186 | 156 | 1.13 | .86–1.47 | 237 | 246 | .98 | .76–1.26 | 219 | 216 | .99 | .79–1.27 | 86 | 82 | 1.04 | .74–1.47 | 223 | 240 | .82 | .64–1.05 | 180 | 213 | .81 | .63–1.04 | 162 | 148 | 1.15 | .88-1.51 |
| Rare homozygote (n) | 106 | 113 | .83 | .58–1.17 | 14 | 18 | .72 | .35–1.51 | 13 | 18 | .72 | .34–1.53 | 39 | 52 | .77 | .48–1.22 | 63 | 50 | 1.25 | .82–1.89 | 35 | 33 | 1.09 | .65–1.83 | 3 | 3 | 1.35 | .27–6.82 | 39 | 32 | 1.23 | .74–2.06 | 20 | 11 | 1.6 | .73–3.50 | 22 | 13 | 1.78 | .86–3.72 |
| Heterozygote and rare homozygote | .86 | .67–1.11 | .72 | .55–.94 | .83 | .63–1.09 | 1.04 | .81–1.33 | 1.02 | .80–1.29 | 1.00 | .79–1.27 | 1.05 | .75–1.47 | .87 | .68–1.10 | .85 | .66–1.08 | 1.20 | .92–1.56 | ||||||||||||||||||||
| Genotyped (n) | 612 | 599 | 636 | 618 | 613 | 597 | 615 | 598 | 604 | 592 | 602 | 591 | 602 | 591 | 601 | 590 | 603 | 597 | 604 | 593 |
Combined Analysis of Multiple SNPs
To test our hypothesis that multiple SNPs in the same pathway may have an additive effect on bladder cancer risk, we estimated the combined effect of these SNPs (table 5). For those genes with multiple SNPs assayed, only a single SNP was included in this combined analysis and others were excluded because of linkage disequilibrium. The excluded polymorphisms were XPD (K751Q), XPC (intron 9 and K939Q), ERCC6 (R1230P), XRCC1 (R194W), XRCC2 (R188H), XRCC3 (T241M and 3′ UTR), p53 (intron 6 and R72P), and STK15 (I57V). For the primary analysis, we treated the minor allele at each locus as the “adverse” allele and tallied the total number of adverse alleles for each individual. For the NER pathway, the number of adverse alleles for each individual ranged from 0 to 10. We categorized individuals on the basis of the quartile distribution of the number of adverse alleles in controls, and we set individuals with fewer than four adverse alleles as the reference group. Compared with the reference group, individuals with four (_OR_=1.52, 95% CI 1.05–2.20), five to six (_OR_=1.81, 95% CI 1.31–2.50), or seven or more adverse alleles (_OR_=2.50, 95% CI 1.69–3.70) exhibited significantly higher risks of bladder cancer, with a significant trend of increasing risk with increasing numbers of high-risk alleles (P for trend <.001). Each additional high-risk allele of the NER pathway gene was associated with a 21% increase in risk (95% CI 1.12–1.29) (table 5).
Table 5.
Combined Effects of Minor Alleles in the NER, BER, HR, NHEJ, and Cell-Cycle Pathways
| Summation of Minor Alleles | |||
|---|---|---|---|
| Pathway and No.of Minor Allelesa | Case/Control | OR (95% CI)b | Validationc |
| NER: | |||
| <4 | 122/186 | Reference | Reference |
| 4 | 108/105 | 1.52 (1.05–2.20) | .79 (.54–1.17) |
| 5–6 | 195/167 | 1.81 (1.31–2.50) | 1.32 (.95–1.84) |
| ⩾7 | 117/69 | 2.50 (1.69–3.70) | 2.29 (1.52–3.46) |
| Per allele | 1.21 (1.12–1.29) | 1.43 (1.27–1.60) | |
| P for trend | <.001 | ||
| HR: | |||
| <2 | 151/171 | Reference | Reference |
| 2 | 157/190 | .88 (.64–1.20) | .97 (.71–1.33) |
| 3 | 147/140 | 1.10 (.79–1.54) | 1.15 (.81–1.64) |
| ⩾4 | 147/89 | 1.70 (1.19–2.43) | 1.26 (.85–1.88) |
| Per allele | 1.13 (1.03–1.24) | 1.14 (.98–1.32) | |
| P for trend | .002 | ||
| NHEJ: | |||
| 0 | 100/101 | Reference | Reference |
| 1 | 220/196 | 1.22 (.86–1.73) | 1.29 (.95–1.76) |
| 2 | 158/186 | .91 (.63–1.31) | .71 (.51–.98) |
| ⩾3 | 109/98 | 1.16 (.77–1.75) | 1.08 (.74–1.59) |
| Per allele | .99 (.89–1.11) | .94 (.79–1.13) | |
| P for trend | .907 | ||
| BER: | |||
| <2 | 212/199 | Reference | Reference |
| 3 | 151/162 | .82 (.61–1.12) | .60 (.43–.85) |
| 4 | 113/116 | .91 (.65–1.28) | .95 (.65–1.39) |
| ⩾5 | 93/83 | 1.01 (.70–1.45) | 1.45 (.96–2.19) |
| Per allele | .98 (.90–1.07) | 1.00 (.87–1.14) | |
| P for trend | .924 | ||
| Cell cycle: | |||
| <3 | 206/174 | Reference | Reference |
| 3 | 146/145 | .86 (.62–1.18) | 1.10 (.78–1.54) |
| 4 | 120/130 | .78 (.56–1.09) | .69 (.48–.98) |
| ⩾5 | 119/131 | .79 (.56–1.10) | .91 (.64–1.29) |
| Per allele | .94 (.87–1.01) | .87 (.77–.98) | |
| P for trend | .109 | ||
| All repair: | |||
| <10 | 74/136 | Reference | Reference |
| 10–11 | 109/115 | 1.59 (1.07–2.37) | .64 (.43–.94) |
| 12–14 | 194/149 | 2.10 (1.46–3.04) | 1.55 (1.10–2.17) |
| ⩾15 | 111/81 | 2.31 (1.53–3.51) | 2.30 (1.50–3.55) |
| Per allele | 1.11 (1.06–1.16) | 1.21 (1.12–1.30) | |
| P for trend | <.001 | ||
| Repair and cell cycle: | |||
| <13 | 86/122 | Reference | Reference |
| 13–15 | 152/162 | 1.22 (.84–1.76) | .82 (.58–1.16) |
| 16–17 | 115/93 | 1.57 (1.05–2.35) | 1.12 (.74–1.68) |
| ⩾18 | 128/97 | 1.77 (1.19–2.63) | 1.88 (1.27–2.77) |
| Per allele | 1.07 (1.03–1.12) | 1.13 (1.05–1.20) | |
| P for trend | .002 |
For the HR pathway, using individuals with fewer than two adverse alleles as the referent group, we noted a significantly elevated association with bladder cancer risk only for individuals with four or more adverse alleles with a similar gene-dosage trend (_OR_=1.70, 95% CI 1.19–2.43; P for trend .002) (table 5). Each additional HR adverse allele was associated with a 1.13-fold increase in risk (95% CI 1.03–1.24). No similar patterns were observed for the NHEJ, BER, or cell-cycle pathway polymorphisms (table 5). However, a significant gene-dosage trend was evident when all DNA-repair pathway genes were combined. Compared with the reference group (<10 adverse alleles), individuals with 10–11, 12–14, and ⩾15 adverse alleles had ORs that increased to 1.59 (95% CI 1.07–2.37), 2.10 (95% CI 1.46–3.04), and 2.31 (95% CI 1.53–3.51), respectively (P for trend <.001) (table 5). Each additional high-risk allele carried a 1.11-fold increase in risk (95% CI 1.06–1.16). When the DNA-repair genes and cell-cycle–control genes were combined, compared with the reference group of <13 adverse alleles, individuals with 13–15 adverse alleles had a 1.22-fold increased risk (95% CI 0.84–1.76), individuals with 16–17 adverse alleles had a 1.57-fold increased risk (95% CI 1.05–2.35), and individuals with ⩾18 adverse alleles had a 1.77-fold increased risk (95% CI 1.19–2.63). Each additional high-risk allele conferred a 1.07-fold increase in risk (95% CI 1.03–1.12) (table 5). In comparing the relative contributions of each pathway, we found the risk conferred by each additional adverse allele to be the highest for the NER pathway (_OR_=1.2, 95% CI 1.12–1.29), intermediate for the combined DNA-repair pathways (_OR_=1.11, 95% CI 1.06–1.16), and lowest for DNA-repair and cell-cycle pathways (_OR_=1.07, 95% CI 1.03–1.12) (table 5). These data reaffirm that the NER pathway appears to be the the most significant modulator of bladder cancer risk, with other pathways playing less-prominent roles.
We also developed a statistical method for cross-validation. First, we evaluated the main effect associated with each minor allele in half of the data set. We then reassigned the minor-allele risk status if the main effect of the minor allele was found to show a negative association with risk (i.e., OR <1) and then re-evaluated the risk in the validation set. To increase the efficiency of this testing approach, we used 10 randomly selected half-sample sets from the original data set to perform the cross-validation 10 times. The average risk of each SNP in the validation sets was used to obtain pooled estimates of risk for each allele and for sums of alleles. This cross-validation scheme confirmed the significant trend of increasing risk with increasing numbers of high-risk alleles in the NER pathway, in the entire DNA-repair pathways, and in DNA-repair plus cell-cycle pathway genes (table 5, “Validation” column).
Interaction Between Smoking and Genetic Factors
Since smoking is the predominant risk factor for bladder cancer, we also stratified our analyses by smoking status (table 6). For the NER pathway, individuals with four, five to six, and seven or more adverse alleles were compared with the referent group (fewer than four adverse alleles). There was no evidence of statistically significantly increased risks for never smokers (_OR_=1.45, 95% CI 0.77–2.72; _OR_=1.27, 95% CI 0.75–2.13; and _OR_=1.40, 95% CI 0.72–2.73, respectively, for the three strata). However, for ever smokers, significantly increased risks were found (_OR_=1.53, 95% CI 0.99–2.37; _OR_=2.23, 95% CI 1.50–3.33; and _OR_=3.37, 95% CI 2.08–5.48, respectively) (P for trend <.0001). When we combined the DNA-repair and cell-cycle–control pathways, there was again evidence of gene-smoking interaction, with elevated risks evident only among ever smokers. When all DNA-repair pathways were combined, compared with individuals with <10 adverse alleles, those with 10–11, 12–14, and ⩾15 adverse alleles all exhibited significantly increased risks (_OR_=2.03, 95% CI 1.23–3.36; _OR_=2.90, 95% CI 1.84–4.59; and _OR_=3.53, 95% CI 2.08–5.99, respectively), whereas the risk was not significant in any strata for never smokers (P for trend = .45). Similarly, when we combined the DNA-repair and cell-cycle genes, no significant association was found in never smokers, but a higher number of adverse alleles conferred progressively increased risks in ever smokers (P for trend <.0001). In the logistic model, the interaction between ever smoking and the combined variant alleles was statistically significant (P<.01). Cross-validation verified that the risks conferred by the adverse alleles were evident only among ever smokers (table 6, “Validation” column). A note of caution for the lack of associations in never smokers: we had approximately twice the number of smokers as nonsmokers, which limited our statistical power to detect significant associations in never smokers. Further studies with more never smokers are needed to confirm these observations.
Table 6.
Combined Effects of Minor Alleles in the NER, DNA-Repair, and Cell-Cycle Pathways, Stratified by Smoking Status
| Summation of Minor Alleles | |||
|---|---|---|---|
| Pathway, Smoking Status, and No. of Minor Allelesa | Case/Control | OR (95% CI)b | Validation |
| NER: | |||
| Never: | |||
| <4 | 38/83 | Reference | Reference |
| 4 | 26/39 | 1.45 (.77–2.72) | 1.44 (.73–2.84) |
| 5–6 | 50/87 | 1.27 (.75–2.13) | 1.13 (.66–1.94) |
| ⩾7 | 22/34 | 1.40 (.72–2.73) | 1.04 (.51–2.14) |
| P for trend | .324 | ||
| Ever: | |||
| <4 | 56/84 | Reference | Reference |
| 4 | 54/60 | 1.53 (.99–2.37) | .51 (.30–.86) |
| 5–6 | 95/66 | 2.23 (1.50–3.33) | 1.46 (.90–2.35) |
| ⩾7 | 66/29 | 3.37 (2.08–5.48) | 3.91 (2.16–7.10) |
| P for trend | <.0001 | ||
| DNA-repair: | |||
| Never: | |||
| <10 | 29/62 | Reference | Reference |
| 10–11 | 32/53 | 1.30 (.69–2.42) | .90 (.47–1.72) |
| 12–14 | 43/64 | 1.43 (.79–2.57) | .93 (.52–1.66) |
| ⩾15 | 23/41 | 1.20 (.61–2.36) | 1.26 (.64–2.51) |
| P for trend | .45 | ||
| Ever: | |||
| <10 | 45/74 | Reference | Reference |
| 10–11 | 77/62 | 2.03 (1.23–3.36) | .52 (.32–.85) |
| 12–14 | 151/85 | 2.90 (1.84–4.59) | 1.97 (1.28–3.05) |
| ⩾15 | 88/40 | 3.53 (2.08–5.99) | 3.60 (2.00–6.48) |
| P for trend | <.0001 | ||
| DNA-repair and cell-cycle: | |||
| Never: | |||
| <13 | 35/54 | Reference | Reference |
| 13–15 | 35/72 | .76 (.42–1.37) | .76 (.42–1.37) |
| 16–17 | 30/37 | 1.25 (.65–2.38) | 1.40 (.70–2.77) |
| ⩾18 | 26/54 | .75 (.40–1.41) | .90 (.48–1.68) |
| P for trend | .702 | ||
| Ever: | |||
| <13 | 51/68 | Reference | Reference |
| 13–15 | 117/90 | 1.73 (1.09–2.73) | .81 (.53–1.25) |
| 16–17 | 85/56 | 2.03 (1.24–3.35) | .97 (.58–1.61) |
| ⩾18 | 102/43 | 3.12 (1.87–5.20) | 3.18 (1.87–5.40) |
| P for trend | <.0001 |
CART Analysis
CART uses a binary recursive-partitioning method that identifies subgroups of high-risk subjects and detects higher-order interactions among a large number of variables. Initial CART analyses for each specific pathway identified the XPD Asp312Asn, RAG1 Lys820Arg, and p53 intron 3 polymorphisms as the initial splits in their respective pathways (NER, HR, and cell cycle, respectively) (data not shown), consistent with their main effects observed by logistic-regression analyses. To further explore gene-gene and gene-environment interactions, we performed CART analysis incorporating both the genetic and smoking-status variables. Figure 1 depicts the resultant tree structure generated. There was an initial split on smoking status, confirming that smoking is the most important risk factor for bladder cancer. Further inspection of the CART structure suggested distinct patterns for ever smokers and never smokers. As documented in previous analyses, the NER gene polymorphisms were relevant only in smokers. This was especially significant since four NER SNPs (CCNH V270A, XPD D312N, ERCC6 M1097V, and RAD23B A249V) all appeared at early splits during the recursive-partitioning process, indicating a biologically meaningful interaction between NER and smoking. The subgroups with the highest bladder cancer risk were those smokers with variant genotypes of CCNH V270A, ERCC6 M1097V, and RAD23B A249V SNPs (node 14 and node 16). In nonsmokers, the three important SNPs were ADPRT V762A, ATM D1853N, and RAG1 K820R. These three genes are involved in DSB and BER and play critical roles in maintaining genomic stability. Table 7 summarizes the risk estimates of all the terminal subgroups compared with the subgroups with the least percentage of cases (node 1). There was a clear separation of individuals with varying bladder cancer risks. However, the estimated ORs should be interpreted with caution, since they were derived from groups identified through a data-mining technique applied to the same data set, and, thus, the level of uncertainty was underestimated.
Figure 1.

Classification and regression tree analysis of all the DNA-repair and cell-cycle gene polymorphisms. High-risk groups are identified by red-colored boxes, medium-risk groups by brown-colored boxes, and low-risk groups by blue-colored boxes.
Table 7.
Risk Estimates of CART Terminal Nodes
| Node | Genotype of Individuals in Each Node | Case | Control | CaseRatea(%) | Adjusted ORb(95% CI) |
|---|---|---|---|---|---|
| Nonsmokers: | |||||
| Node 1 | ADPRT(M)-ATM(W) | 19 | 68 | 22 | Reference |
| Node 2 | ADPRT(M)-ATM(M)-XRCC3UTR(W) | 8 | 19 | 30 | 1.43 (.54–3.79) |
| Node 4 | ADPRT(W)-RAG1(W) | 82 | 146 | 36 | 1.97 (1.10–3.50) |
| Node 8 | ADPRT(W)-RAG1(M) | 39 | 38 | 51 | 3.68 (1.87–7.25) |
| Node 12 | ADPRT(M)-ATM(M)-XRCC3UTR(M) | 8 | 3 | 73 | 8.87 (2.13–36.90) |
| Smokers: | |||||
| Node 3 | CCNH(W)-XPD312(W)-RAD23(M) | 22 | 43 | 34 | 1.72 (.83–3.58) |
| Node 5 | CCNH(M)-ERCC6-1097(M)-RAD23(W)-KU80(M) | 5 | 8 | 39 | 2.24 (.65– 7.67) |
| Node 6 | CCNH(M)-ERCC6-1097(W)-NBS1(W) | 28 | 39 | 42 | 2.47 (1.22–5.00) |
| Node 7 | CCNH(W)-XPD312(W)-RAD23(W)-RAG1(W) | 53 | 57 | 48 | 3.16 (1.67–5.99) |
| Node 9 | CCNH(W)-XPD312(W)-RAD23(W)-RAG1(M)-XRCC3-241(W) | 9 | 8 | 53 | 3.88 (1.31–11.45) |
| Node 10 | CCNH(W)-XPD312(M) | 158 | 104 | 60 | 5.12 (2.88–9.09) |
| Node 11 | CCNH(M)-ERCC6-1097(W)-NBS1(M) | 56 | 32 | 64 | 6.03 (3.08–11.83) |
| Node 13 | CCNH(M)-ERCC6-1097(M)-RAD23(W)-KU80(W) | 36 | 12 | 75 | 10.2 (4.43– 23.54) |
| Node 14 | CCNH(M)-ERCC6-1097(M)-RAD23(M)-MUTYH(M) | 20 | 4 | 83 | 16.78 (5.10–55.26) |
| Node 15 | CCNH(W)-XPD312(W)-RAD23(W)-RAG1(M)-XRCC3-241(M) | 17 | 2 | 89 | 27.84 (5.87–132.13) |
| Node 16 | CCNH(M)-ERCC6-1097(M)-RAD23(M)-MUTYH(W) | 35 | 0 | 100 | N/A |
To gain insight into the potential mechanism for the increased cancer risk conferred by these different genotype combinations and to attempt to validate our analytic approach, we next determined genotype-phenotype correlations in subjects in all the terminal nodes (table 8). We applied two widely used methods of detecting latent genomic instability—the mutagen-sensitivity assay and the comet assay (Hsu et al.1989; Wu et al. 1998_a_, 1998_b_; Schabath et al. 2003). We categorized the terminal nodes into three groups based on the case percentage in each node: low-risk group (case percentage <55%), medium-risk group (case percentage 55%–75%), and high-risk group (case percentage >75%). In smokers, there was a significant trend of higher numbers of bleomycin- and BPDE-induced chromosome breaks (by mutagen-sensitivity assay) and DNA damage (by comet assay) for individuals in higher-risk subgroups among cases. The P for the trend was .0348 for bleomycin-induced chromatid breaks, .0036 for BPDE-induced chromatid breaks, and .0397 for BPDE-induced DNA damage, as measured by the comet assay. There were no similar patterns in the controls among these subgroups, suggesting that suboptimal DRC in cases contributes to their increased cancer risk. Interestingly, among cases, never smokers exhibited higher bleomycin- and BPDE-induced chromatid breaks (_P_=.039 and .031, respectively) than ever smokers (data not shown), suggesting that genetic instability plays a more prominent role in predisposing never smokers to bladder cancer, whereas, in ever smokers, gene-smoking interaction plays a central role in bladder cancer etiology.
Table 8.
Chromosome Breaks and DNA Damage in Terminal Nodes at Smoking Side
| Bleomycin Sensitivity | BPDE Sensitivity | BPDE Comet | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Risk | N | Mean(SD) | P Value | N | Mean(SD) | P Value | N | Mean(SD) | P Value |
| Cases: | |||||||||
| Low riska | 86 | .75 (.33) | Reference | 95 | .61 (.28) | Reference | 74 | 1.48 (.98) | Reference |
| Medium riskb | 168 | .84 (.34) | .0448 | 180 | .69 (.30) | .0323 | 134 | 1.77 (1.01) | .0347 |
| High riskc | 80 | .89 (.36) | .0101 | 87 | .72 (.31) | .0128 | 73 | 1.83 (.95) | .0295 |
| P for trend | .0348 | .0036 | .0397 | ||||||
| Controls: | |||||||||
| Low risk | 107 | .82 (.31) | Reference | 127 | .57 (.27) | Reference | 108 | 1.77 (1.78) | Reference |
| Medium risk | 82 | .84 (.33) | .6696 | 118 | .51 (.22) | .0688 | 107 | 2.04 (2.08) | .3075 |
| High risk | 11 | .83 (.40) | .9212 | 13 | .64 (.27) | .3748 | 15 | 1.73 (1.86) | .9355 |
| P for trend | .9200 | .3339 | .9253 |
Discussion
In this study, we have used a multigenic approach to systematically examine the associations between a panel of polymorphisms in DNA-repair and cell-cycle genes and bladder cancer risk. To the best of our knowledge, this is the most comprehensive multigenic study evaluating the largest number of SNPs. The most significant finding in this study is that combined analyses of multiple SNPs in the same or relevant pathways may reveal otherwise undetectable associations between individual SNPs with cancer risk. Furthermore, we have shown that high-risk subgroups selected by the analysis exhibited high levels of latent genetic instability. Our results support the concept that genetic polymorphisms can be used as cancer risk predictors; however, a single polymorphism may have little predictive value in risk assessment, but a more comprehensive pathway-based multigenic approach combining multiple polymorphisms gives more-precise delineation of risk groups and may suggest the future direction of association studies.
There have been many studies reporting associations between genetic polymorphisms and susceptibility to common, complex diseases, including cancer. Most of these studies have used a candidate-gene approach, investigating one or only a few selected polymorphisms at a time. Since a complex disease like cancer typically occurs through a multifactorial interplay between many genetic and environmental factors, the effect of each individual SNP is unlikely to be substantial. For example, two of the most intensively studied DNA-repair gene SNPs, XPD Asp312Asn and Lys751Gln, were hypothesized to modify lung cancer risk, since several phenotypic studies suggested that individuals with the variant Asn or Gln alleles exhibit lower DRC (Spitz et al. 2001; Stern et al. 2002). Yet, two recent meta-analyses of XPD SNPs in lung cancer from nine case-control studies found that the variant alleles confer only an ∼20% increased lung cancer risk for either SNP (Hu et al. 2004; Benhamou and Sarasin 2005). Given that the cancer risk is, at most, modestly altered by individual variants and that most single studies have used relatively small sample sizes, it has been estimated that only about a quarter of previously reported associations were real positive associations with common diseases (Ioannidis et al. 2001; Lohmueller et al. 2003).
In our main-effect analysis, the XPD Asp312Asn polymorphism was one of only two DNA-repair SNPs that exhibited a statistically significant association with bladder cancer risk (_OR_=1.28, 95% CI 1.01–1.62). This association is biologically plausible, since the Asn allele is associated with reduced DRC (Ioannidis et al. 2001), and a significant association of the Asn allele with lung cancer risk has been observed in a meta-analysis of lung cancer (Hu et al. 2004; Benhamou and Sarasin 2005). However, considering the borderline CI (95% CI 1.01–1.62) and the multiple comparison issue, we cannot rule out that this individual association was due to type I error. Moreover, the modest effects of individual variants would have limited practical value in predicting cancer risk, highlighting the need for a more comprehensive approach for association studies.
Our data provide strong evidence for the improvement of a multigenic approach over the single candidate-gene approach. We found a significant trend of increased risk with increasing numbers of adverse alleles in the NER pathway, in the entire DNA-repair pathways, and in the DNA-repair and cell-cycle pathways combined. The most significant additive effect of multiple polymorphisms in a single pathway was observed in the NER pathway. Individuals with the highest number of adverse alleles exhibited a 2.50-fold increased bladder cancer risk (95% CI 1.69–3.70) compared with individuals carrying fewer than four high-risk alleles. The difference in the combined effects of multiple polymorphisms among each individual pathway may reflect the differences in the contribution of each DNA-repair pathway to bladder cancer risk. For instance, the NER pathway mainly removes bulky DNA adducts, which are typically generated from exposure to polycyclic aromatic hydrocarbons in tobacco smoke. Therefore, the NER pathway would be expected to play a more significant role in repairing tobacco carcinogen–induced DNA damage, whereas the other DNA-repair pathways play a less prominent role. Our CART analysis confirmed and strengthened this conclusion by showing that NER gene polymorphisms were selected only in smokers, whereas a few general maintenance-gene polymorphisms were relevant in nonsmokers. More interestingly, subsets of individuals with higher cancer risks were identified through CART analysis based on simple combinations of smoking and genotypes. The simple, intuitive nature of the CART algorithm may allow rapid identification of potential genetic and environmental interactions when dealing with large numbers of variables in complex diseases. The number of possible interactions grows exponentially as each additional variable is added. Another strength of this study is the ability to evaluate changes in levels of induced genetic damage assessed by three in vitro assays among subjects in terminal nodes. However, the CART analysis is a postdata-mining tool and the results should be interpreted with some degree of caution. External validation in an independent epidemiology study is warranted to confirm the potential high-risk groups.
Although our primary analysis treated the minor-variant allele at each locus as the risk allele, this assumption may not be accurate in the absence of knowledge of the functional consequences of the variants under study. We therefore evaluated other approaches to the designation of “at-risk” alleles. For example, if the main effects for the minor-variant allele exhibited an OR <1 in the initial multivariate analyses, we assigned the major allele as the adverse allele. We observed similarly significant trends of increasing risk with increasing numbers of high-risk alleles, and all associations were stronger (data not shown), which was not surprising just by the way the variable was defined. We also performed the data analysis by reversing the minor allele when OR <1 and P<.05 or P<.1, and the results were similar to what we presented in tables 5 and 6, which was also expected since these criteria affected only a single polymorphism (p53 intron 3) (data not shown). Choosing the at-risk allele on the basis of the data set under analysis may lead to data-driven findings. There is currently not a standard way to assign at-risk alleles, and more studies are needed to develop an optimal way to assign at-risk alleles.
Matullo et al. (2003) previously found a dose-response relationship between the number of adverse alleles in DNA-repair gene SNPs and levels of DNA adducts in peripheral blood cells, suggesting a stepwise decrease in DRC as the number of adverse alleles increases and lending biological support for analyzing combined effects of multiple-variant alleles rather than investigating single SNPs in modulating cancer risk. Our phenotypic assays also demonstrated that higher-risk subgroups, as identified from the CART analysis, exhibited higher chromosome breaks and DNA damage (table 8), thereby providing additional biological plausibility and validity to our approach.
Although we tried to be as inclusive as possible in our selection of genes and SNPs, the selection criteria was based on potential functional SNPs in genes with higher possibilities of being related to cancer risk. A more comprehensive pathway-based approach—for example, selecting tagging SNPs in most key genes in a defined pathway—would offer more convincing evidence for the relevance of the evaluated pathway in cancer risk. Nevertheless, this is the most comprehensive study to date to use a multigenic approach to analyze multiple genetic polymorphisms in DNA-repair and cell-cycle genes in bladder cancer risk. Our results suggest that individuals with higher numbers of variants in DNA-repair genes are at an increased risk for bladder cancer, verifying the importance of taking a pathway-based approach to improve the resolution of the risk-assessment process. This study not only is important for bladder cancer risk assessment but is also applicable to risk assessment of many complex diseases incorporating low-penetrance genes. These data confirm the notion that the future of risk assessment for multigenic complex diseases needs to move beyond analysis of single polymorphisms.
Acknowledgments
The work reported here was supported by National Institutes of Health grants CA 74880 and CA 91846.
Web Resources
The URL for data presented herein is as follows:
References
- Benhamou S, Sarasin A (2005) ERCC2 /XPD gene polymorphisms and lung cancer: a HuGE review. Am J Epidemiol 161:1–14 10.1093/aje/kwi018 [DOI] [PubMed] [Google Scholar]
- Berwick M, Vineis P (2000) Markers of DNA repair and susceptibility to cancer in humans: an epidemiologic review. J Natl Cancer Inst 92:874–897 10.1093/jnci/92.11.874 [DOI] [PubMed] [Google Scholar]
- Cheng TC, Chen ST, Huang CS, Fu YP, Yu JC, Cheng CW, Wu PE, Shen CY (2005) Breast cancer risk associated with genotype polymorphism of the catechol estrogen-metabolizing genes: a multigenic study on cancer susceptibility. Int J Cancer 113:345–353 10.1002/ijc.20630 [DOI] [PubMed] [Google Scholar]
- Christmann M, Tomicic MT, Roos WP, Kaina B (2003) Mechanisms of human DNA repair: an update. Toxicology 193:3–34 10.1016/S0300-483X(03)00287-7 [DOI] [PubMed] [Google Scholar]
- Elledge SJ (1996) Cell cycle checkpoints: preventing an identity crisis. Science 274:1664–1672 10.1126/science.274.5293.1664 [DOI] [PubMed] [Google Scholar]
- Goode EL, Ulrich CM, Potter JD (2002) Polymorphisms in DNA repair genes and associations with cancer risk. Cancer Epidemiol Biomarkers Prev 11:1513–1530 [PubMed] [Google Scholar]
- Gu J, Zhao H, Dinney CP, Zhu Y, Leibovici D, Bermejo CE, Grossman HB, Wu X (2005) Nucleotide excision repair gene polymorphisms and recurrence after treatment for superficial bladder cancer. Clin Cancer Res 11:1408–1415 10.1158/1078-0432.CCR-04-1101 [DOI] [PubMed] [Google Scholar]
- Han J, Colditz GA, Samson LD, Hunter DJ (2004) Polymorphisms in DNA double-strand break repair genes and skin cancer risk. Cancer Res 64:3009–3013 10.1158/0008-5472.CAN-04-0246 [DOI] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA (2000) The hallmarks of cancer. Cell 100:57–70 10.1016/S0092-8674(00)81683-9 [DOI] [PubMed] [Google Scholar]
- Horne BD, Anderson JL, Carlquist JF, Muhlestein JB, Renlund DG, Bair TL, Pearson RR, Camp NJ (2005) Generating genetic risk scores from intermediate phenotypes for use in association studies of clinically significant endpoints. Ann Hum Genet 69:176–186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu TC, Johnston DA, Cherry LM, Ramkissoon D, Schantz SP, Jessup JM, Winn RJ, Shirley L, Furlong C (1989) Sensitivity to genotoxic effects of bleomycin in humans: possible relationship to environmental carcinogenesis. Int J Cancer 43:403–409 [DOI] [PubMed] [Google Scholar]
- Hu Z, Wei Q, Wang X, Shen H (2004) DNA repair gene XPD polymorphism and lung cancer risk: a meta-analysis. Lung Cancer 46:1–10 10.1016/j.lungcan.2004.03.016 [DOI] [PubMed] [Google Scholar]
- Ioannidis JP, Ntzani EE, Trikalinos TA, Contopoulos-Ioannidis DG (2001) Replication validity of genetic association studies. Nat Genet 29:306–309 10.1038/ng749 [DOI] [PubMed] [Google Scholar]
- Jemal A, Murray T, Ward E, Samuels A, Tiwari RC, Ghafoor A, Feuer EJ, Thun MJ (2005) Cancer statistics, 2005. CA Cancer J Clin 55:10–30 [DOI] [PubMed] [Google Scholar]
- Kuschel B, Auranen A, McBride S, Novik KL, Antoniou A, Lipscombe JM, Day NE, Easton DF, Ponder BA, Pharoah PD, Dunning A (2002) Variants in DNA double-strand break repair genes and breast cancer susceptibility. Hum Mol Genet 11:1399–1407 10.1093/hmg/11.12.1399 [DOI] [PubMed] [Google Scholar]
- Lohmueller KE, Pearce CL, Pike M, Lander ES, Hirschhorn JN (2003) Meta-analysis of genetic association studies supports a contribution of common variants to susceptibility to common disease. Nat Genet 33:177–182 10.1038/ng1071 [DOI] [PubMed] [Google Scholar]
- Matullo G, Peluso M, Polidoro S, Guarrera S, Munnia A, Krogh V, Masala G, Berrino F, Panico S, Tumino R, Vineis P, Palli D (2003) Combination of DNA repair gene single nucleotide polymorphisms and increased levels of DNA adducts in a population-based study. Cancer Epidemiol Biomarkers Prev 12:674–677 [PubMed] [Google Scholar]
- Mohrenweiser HW, Wilson DM 3rd, Jones IM (2003) Challenges and complexities in estimating both the functional impact and the disease risk associated with the extensive genetic variation in human DNA repair genes. Mutat Res 526:93–125 [DOI] [PubMed] [Google Scholar]
- Neumann AS, Sturgis EM, Wei Q (2005) Nucleotide excision repair as a marker for susceptibility to tobacco-related cancers: a review of molecular epidemiological studies. Mol Carcinog 42:65–92 10.1002/mc.20069 [DOI] [PubMed] [Google Scholar]
- Popanda O, Schattenberg T, Phong CT, Butkiewicz D, Risch A, Edler L, Kayser K, Dienemann H, Schulz V, Drings P, Bartsch H, Schmezer P (2004) Specific combinations of DNA repair gene variants and increased risk for non-small cell lung cancer. Carcinogenesis 25:2433–2441 10.1093/carcin/bgh264 [DOI] [PubMed] [Google Scholar]
- Schabath MB, Spitz MR, Grossman, HB, Zhang K, Dinney CP, Zheng PJ, Wu X (2003) Genetic instability in bladder cancer assessed by the comet assay. J Natl Cancer Inst 95:540–547 [DOI] [PubMed] [Google Scholar]
- Shields PG, Harris CC (2000) Cancer risk and low-penetrance susceptibility genes in gene-environment interactions. J Clin Oncol 18:2309–2315 [DOI] [PubMed] [Google Scholar]
- Spitz MR, Wei Q, Dong Q, Amos CI, Wu X (2003) Genetic susceptibility to lung cancer: the role of DNA damage and repair. Cancer Epidemiol Biomarkers Prev 12:689–698 [PubMed] [Google Scholar]
- Spitz MR, Wu X, Wang Y, Wang LE, Shete S, Amos CI, Guo Z, Lei L, Mohrenweiser H, Wei Q (2001) Modulation of nucleotide excision repair capacity by XPD polymorphisms in lung cancer patients. Cancer Res 61:1354–1357 [PubMed] [Google Scholar]
- Stern MC, Johnson LR, Bell DA, Taylor JA (2002) XPD codon 751 polymorphism, metabolism genes, smoking, and bladder cancer risk. Cancer Epidemiol Biomarkers Prev 11:1004–1011 [PubMed] [Google Scholar]
- Wu X, Gu J, Amos CI, Jiang H, Hong WK, Spitz MR (1998_a_) A parallel study of in vitro sensitivity to benzo[a]pyrene diol epoxide and bleomycin in lung cancer cases and controls. Cancer 83:1118–1127 [DOI] [PubMed] [Google Scholar]
- Wu X, Gu J, Hong WK, Lee JJ, Amos CI, Jiang H, Winn RJ, Fu KK, Cooper J, Spitz MR (1998_b_) Benzo[a]pyrene diol epoxide and bleomycin sensitivity and susceptibility to cancer of upper aerodigestive tract. J Natl Cancer Inst 90:1393–1399 10.1093/jnci/90.18.1393 [DOI] [PubMed] [Google Scholar]
- Wu X, Zhao H, Amos CI, Shete S, Makan N, Hong WK, Kadlubar FF, Spitz MR (2002) p53 genotypes and haplotypes associated with lung cancer susceptibility and ethnicity. J Natl Cancer Inst 94:681–690 [DOI] [PubMed] [Google Scholar]
- Wu X, Zhao H, Suk R, Christiani DC (2004) Genetic susceptibility to tobacco-related cancer. Oncogene 23:6500–6523 10.1038/sj.onc.1207811 [DOI] [PubMed] [Google Scholar]
- Wu X, Zhao H, Wei Q, Amos CI, Zhang K, Guo Z, Qiao Y, Hong WK, Spitz MR (2003) XPA polymorphism associated with reduced lung cancer risk and a modulating effect on nucleotide excision repair capacity. Carcinogenesis 24:505–509 10.1093/carcin/24.3.505 [DOI] [PubMed] [Google Scholar]