Calbindin-D28K dynamically controls TRPV5-mediated Ca2+ transport (original) (raw)
Abstract
In Ca2+-transporting epithelia, calbindin-D28K (CaBP28K) facilitates Ca2+ diffusion from the luminal Ca2+ entry side of the cell to the basolateral side, where Ca2+ is extruded into the extracellular compartment. Simultaneously, CaBP28K provides protection against toxic high Ca2+ levels by buffering the cytosolic Ca2+ concentration ([Ca2+]i) during high Ca2+ influx. CaBP28K consistently colocalizes with the epithelial Ca2+ channel TRPV5, which constitutes the apical entry step in renal Ca2+-transporting epithelial cells. Here, we demonstrate using protein-binding analysis, subcellular fractionation and evanescent-field microscopy that CaBP28K translocates towards the plasma membrane and directly associates with TRPV5 at a low [Ca2+]i. 45Ca2+ uptake measurements, electrophysiological recordings and transcellular Ca2+ transport assays of lentivirus-infected primary rabbit connecting tubule/distal convolute tubule cells revealed that associated CaBP28K tightly buffers the flux of Ca2+ entering the cell via TRPV5, facilitating high Ca2+ transport rates by preventing channel inactivation. In summary, CaBP28K acts in Ca2+-transporting epithelia as a dynamic Ca2+ buffer, regulating [Ca2+] in close vicinity to the TRPV5 pore by direct association with the channel.
Keywords: Ca2+ homeostasis, kidney, TIRF, transient receptor potential, vitamin D
Introduction
The vitamin D-dependent Ca2+-binding proteins, named calbindins (CaBPs), are expressed in cells that are challenged by a high Ca2+ influx such as in brain, bone, teeth, inner ear, placenta, mammary gland, kidney and intestine. In these tissues, CaBPs (i.e., CaBP9K and CaBP28K) are widely regarded as a key component in cellular Ca2+ handling. In Ca2+-transporting epithelial cells, CaBPs display the potential to facilitate multiple steps in the process of transcellular Ca2+ transport. First, Ca2+ influx in these cells is mediated by the epithelial Ca2+ channels TRPV5 and TRPV6, which are distinct members of the transient receptor potential (TRP) family (Montell et al, 2002) acting as gatekeepers facilitating cellular Ca2+ entry due to a steep inward electrochemical gradient across the luminal membrane of epithelial cells (Hoenderop et al, 2005). The activity of these highly Ca2+-selective channels is tightly regulated by the Ca2+ concentration in close vicinity to the channel mouth (Hoenderop et al, 1999b). Thus, adequate buffering of Ca2+ is essential for a continuous influx of Ca2+ through these channels. Second, during high rates of transcellular Ca2+ transport, strict regulation of the intracellular Ca2+ concentration ([Ca2+]i) is crucial in protecting the cell against the cytotoxic high levels of Ca2+ (Tymianski, 1996). Buffering of Ca2+ by specialized Ca2+-binding proteins is, therefore, required (Lukas and Jones, 1994; Pauls et al, 1996; Schwaller et al, 2002). Third, Ca2+ entering at the luminal side of the cell has to diffuse, without affecting other intracellular processes, to the basolateral side, where the Na+/Ca2+ exchanger (NCX1) and/or the plasma membrane ATPase (PMCA1b) extrude Ca2+ into the extracellular compartment. In transepithelial Ca2+ transport, CaBPs have been implicated in facilitated diffusion of Ca2+ from the luminal membrane to the basolateral surface by increasing the diffusional range of Ca2+ (Bronner and Stein, 1988; Bronner, 1989). Moreover, the importance of CaBPs in Ca2+-transporting epithelial cells is underlined by the consistent coexpression with the Ca2+ transport proteins including TRPV5, TRPV6, NCX1 and PMCA1b (Lambers et al, 2006).
Negative feedback regulation of channel activity by an increased [Ca2+]i is not restricted to the epithelial Ca2+ channels. A broad range of both voltage- and non-voltage-operated ion channels are regulated by [Ca2+]i (Kits and Mansvelder, 1996; Jones et al, 1999). Ion channels that are negatively regulated by Ca2+ inactivate upon a local rise in [Ca2+] in close proximity to the channel pore. Previous studies indicated that the Ca2+ sensor calmodulin is responsible for the Ca2+-dependent regulation of particularly voltage-operated Ca2+ channels (Zuhlke et al, 1999; DeMaria et al, 2001). However, how [Ca2+] near a channel pore is regulated is not well defined. Several studies implicate CaBPs in facilitated diffusion of Ca2+. However, other than mathematical models (Feher et al, 1992) and coordinated regulation of renal Ca2+ transport proteins (van Abel et al, 2005), limited experimental data are available to substantiate these hypotheses.
In addition, previous studies indicated that CaBP28K acts as a cytosolic Ca2+ buffer to protect neurons against large fluctuations in [Ca2+]i (Iacopino and Christakos, 1990; Lukas and Jones, 1994; Guo et al, 1998). Here, the buffer capacity of CaBP28K affects the shape of the postsynaptic Ca2+ signals and may underlie paired pulse facilitation of synapses (Airaksinen et al, 1997; Blatow et al, 2003; Schmidt et al, 2003). Recently, it was shown in cerebellar Purkinje neurons that CaBP28K directly interacts with membrane-targeted inositol-1,4,5-trisphosphate and acts as a Ca2+ sensor to regulate the degradation of inositol messengers in an activity-dependent manner (Schmidt et al, 2005). Thus, CaBP28K displays several properties that imply an important role in Ca2+-induced signal transmission and hence may function not only as a Ca2+ buffer, but also as a Ca2+ sensor affecting downstream processes in the cell.
A key biochemical property specifying a Ca2+ sensor is the presence of EF-hand motifs that undergo a conformational change upon Ca2+ binding. CaBP28K is equipped with six EF-hand motifs that bind Ca2+ in a highly cooperative fashion. First, Ca2+ binds to the high-affinity EF-hand site 1, followed by EF-hands 4 and 5 and finally EF-hand 3 is loaded with Ca2+, in contrast to EF-hands 2 and 6, which do not bind Ca2+ (Venters et al, 2003). Although the crystal structure of CaBP28K has not been determined, structural analysis measured by nuclear magnetic resonance indicated that conformational changes occur in CaBP28K upon Ca2+ binding (Berggard et al, 2002; Venyaminov et al, 2004).
The aim of the present study was to determine how CaBPs contribute to the regulation of TRPV5 channel activity, intracellular Ca2+ handling and transcellular Ca2+ transport in epithelial cells. By a comprehensive approach using protein-binding analysis, life cell imaging and functional assays, this study reveals a crucial role of CaBP28K in TRPV5-mediated transepithelial Ca2+ transport in kidney. Here, we show that CaBP28K acts as a dynamic Ca2+ buffer regulating the local [Ca2+] in close vicinity to the TRPV5 pore by direct association with the channel.
Results
Concomitant regulation of calbindin-D28K and TRPV5 in kidney
In kidney, CaBP28K and TRPV5 colocalized in the distal convoluted tubule (DCT) and the connecting tubule (CNT) (Figure 1A). These cells transport Ca2+ transcellularly from the pro-urine to the blood compartment (Hoenderop et al, 2005). To investigate whether there is a correlation between the expression of TRPV5 and CaBP28K, the abundance of these proteins was analyzed in different animal models that were treated with calciotropic hormones, exposed to various dietary Ca2+ levels or ablated for genes encoding Ca2+ transport proteins (Figure 1B). Importantly, a highly significant correlation between the TRPV5 expression and CaBP28K abundance was consistently observed. CaBP28K is predominantly localized along the apical side of TRPV5-expressing renal cells (Figure 1A). Therefore, the apical localization of CaBP28K was investigated in kidneys of TRPV5−/− compared to wild-type mice. To this end, the immuno-positive staining of CaBP28K was semiquantified in five subcellular regions of DCT and CNT cells (Figure 1C). In TRPV5−/− kidney, CaBP28K was equally distributed throughout the tubular cells with no apparent apical localization, whereas CaBP28K was significantly more abundant in the apical region in wild-type kidney cells. This finding further substantiates that the apical localization of CaBP28K is dependent on the presence of TRPV5.
Figure 1.
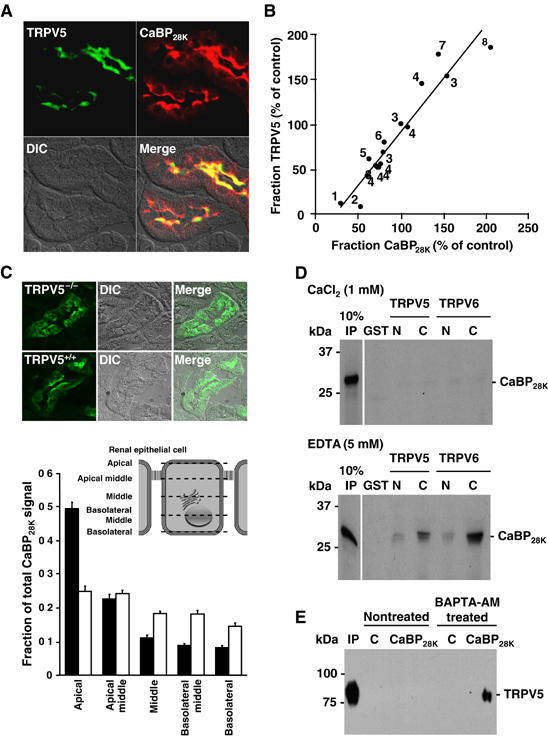
Coordinated expression and direct association of TRPV5 and CaBP28K. (A) Colocalization of TRPV5 (green) and CaBP28K (red) in the DCT and CNT. (B) Correlation in TRPV5 and CaBP28K expression after treatment (1parathyroidectomized (van Abel et al, 2005); 2tacrolimus (Nijenhuis et al, 2005); 3acidose/alkalose (Nijenhuis et al, 2006); 4thiazide (Nijenhuis et al, 2004, 2005); 5calcimemetics (van Abel et al, 2005); 6ovariectomized (van Abel et al, 2005); 7dexamethasone (Nijenhuis et al, 2004); 8ovariectomized+vitamine D3 (van Abel et al, 2002)). _R_2=0.9068. (C) Localization of CaBP28K in kidney sections of TRPV5−/− and wild-type mice. The epithelial cells were divided in different regions including apical, apical-middle, middle, basolateral-middle and basolateral as indicated. The immuno-positive CaBP28K staining of these different cellular regions in 30 cells of six different tubules was calculated as described in Materials and methods. Significant differences in CaBP28K intensities within the group are indicated by an asterisk. Open bars represent TRPV5%, closed bars represent wild-type. (D) [35S]Methionine-labeled, _in vitro_-translated CaBP28K was incubated either in the presence (1 mM CaCl2) or absence (5 mM EDTA) of Ca2+, with GST or GST fused to the N- or C-terminus of TRPV5 and TRPV6 immobilized on glutathione-Sepharose 4B beads. Input control (IP) represents 10% of the total pull-down input. (E) Cells were cotransfected with pEBG-CaBP28K (GST-CaBP28K) and pCINeo-TRPV5-IRES-EGFP or pEBG (GST) and pCINeo-TRPV5-IRES-EGFP (control, C). To decrease the [Ca2+]i, cells were treated with BAPTA-AM. Lysates were loaded on glutathione-Sepharose 4B beads, and after extensive washing co-precipitation was investigated by immunoblotting using the guinea-pig anti-TRPV5 antibody (IP=input). The two TRPV5 immuno-positive bands correspond to the core (lower) and glycosylated forms of the protein.
Calbindin-D28K interacts with TRPV5 in a Ca2+-dependent manner
Ion channels in epithelial cells are frequently regulated by associated intracellular protein complexes. A potential interaction between CaBP28K and TRPV5 was investigated using GST pull-down assays. In addition, the interaction of CaBP28K with other TRPV members, including the highly homologous TRPV6 and closely related TRPV4 channels, was studied. CaBP28K bound to both the N- and C-termini of TRPV5 and TRPV6 in the absence of Ca2+ (5 mM EDTA), whereas the binding was virtually abolished in the presence of Ca2+ (1 mM CaCl2) (Figure 1D). However, binding between CaBP28K and TRPV4 could not be detected either in the presence or absence of Ca2+ (data not shown). Subsequently, the interaction of TRPV5 and CaBP28K was evaluated by co-precipitation from GST-CaBP28K- and TRPV5-expressing human embryonic kidney (HEK293) cells. The Ca2+ dependency of this association was investigated by incubation of the cells with BAPTA-AM to decrease [Ca2+]i. To this end, cell lysates were incubated with glutathione-coupled Sepharose beads to precipitate GST-CaBP28K complexes and immunoblots containing the precipitated proteins were analyzed for the presence of TRPV5. TRPV5 co-precipitated with GST-CaBP28K in cells that were treated with BAPTA-AM, whereas the channel was not precipitated from non-treated cells (Figure 1E). In addition, co-precipitation of TRPV5 could not be detected in GST- and TRPV5-expressing cells, demonstrating the specificity of the CaBP28K association.
CaBP28K translocates towards the plasma membrane in a Ca2+- and TRPV5-dependent manner
The physiological characteristics of the interaction of CaBP28K with TRPV5 suggests that this Ca2+-binding protein translocates towards the plasma membrane at a low [Ca2+]i. This hypothesis was investigated by the isolation of plasma membrane-enriched fractions of TRPV5- and CaBP28K-transfected HeLa cells. As compared to the endogenously expressed Na,K-ATPase, CaBP28K was more abundant (260±105%, _n_=3, P<0.05) in the plasma membrane-enriched fraction of cells that were treated with BAPTA-AM compared to non-treated cells (Figure 2A, left panel). Importantly, cells lacking TRPV5 did not reveal an elevated expression of CaBP28K in the plasma membrane-enriched fraction after BAPTA-AM treatment (Figure 2A, right panel). Subsequently, CaBP28K expression in plasma membrane-enriched fractions of primary rabbit CNT/cortical collecting duct (CNT/CCD) cultures endogenously expressing TRPV5 and CaBP28K (Hoenderop et al, 2005) was evaluated. In line with the experiments using transfected cells, CaBP28K was more abundant (146±6%, _n_=3, P<0.05) in plasma membrane-enriched fractions of BAPTA-AM-treated primary CNT/CCD cultures as compared to non-treated cultures (Figure 2B).
Figure 2.
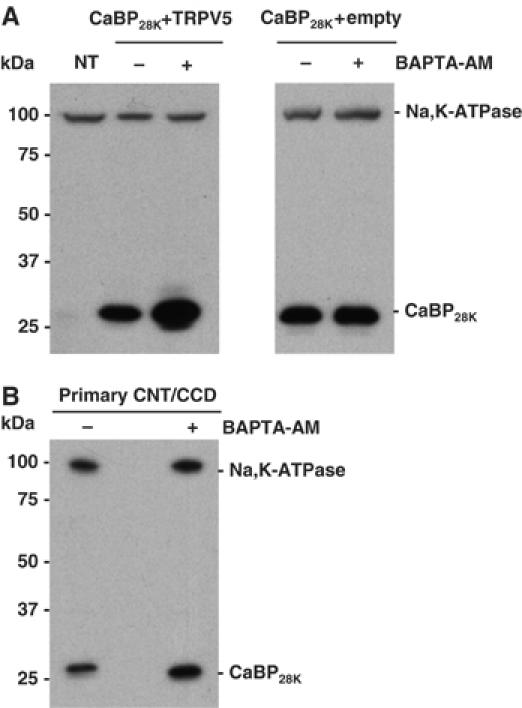
Subcellular localization of CaBP28K at low intracellular Ca2+ concentrations. (A) Cells were transfected with CaBP28K and TRPV5 (left panel) or CaBP28K and empty vector (right panel) and treated with or without BAPTA-AM. Plasma membrane-enriched fractions were probed for the presence of endogenously expressed Na,K-ATPase and exogenously expressed CaBP28K using anti-Na,K-ATPase and anti-CaBP28K antibodies, respectively. NT=non-transfected. (B) Plasma membrane-enriched fractions of primary CNT/CCD cultures, either treated with or without BAPTA-AM, were isolated and probed for the presence of endogenously expressed CaBP28K and Na,K-ATPase. Representative blots of three independent experiments are shown.
Next, the Ca2+-dependent translocation of CaBP28K towards the plasma membrane in TRPV5-expressing HEK293T cells was investigated by total internal reflection fluorescence (TIRF) or evanescent-field microscopy. TIRF microscopy illuminates fluorophores within 100–200 nm of the plasma membrane–glass cover slip interface, with excitation intensity depending on the relative indices of refraction and the angle of incidence. Light intensity falls exponentially in the axial (z) direction, such that fluorescence output is inversely proportional to the distance from the plasma membrane. TIRF therefore enabled us to study the dynamic process of CaBP28K translocation towards the plasma membrane in a direction vertical to the plasma membrane, without interference of cytosolic CaBP28K fluorescence. The presence of EYFP-CaBP28K in the TIRF signal of cells cotransfected with EYFP-CaBP28K and TRPV5-ECFP was followed in the presence and absence of BAPTA-AM, respectively. After BAPTA-AM treatment, the EYFP-CaBP28K TIRF signal increased in time as compared to non-treated cells (Figure 3A and B). In line with the cell fractionation experiments, this increase of EYFP-CaBP28K TIRF signal was not observed in cells lacking TRPV5 (Figure 3C and D). Furthermore, the BAPTA-dependent changes in TIRF signal were not detected in cells expressing TRPV5-IRES-EGFP (bicistronic expression of TRPV5 and EGFP) (Figure 3E and F), demonstrating that the observed alterations in EYFP-CaBP28K fluorescence represent a cellular redistribution of CaBP28K.
Figure 3.
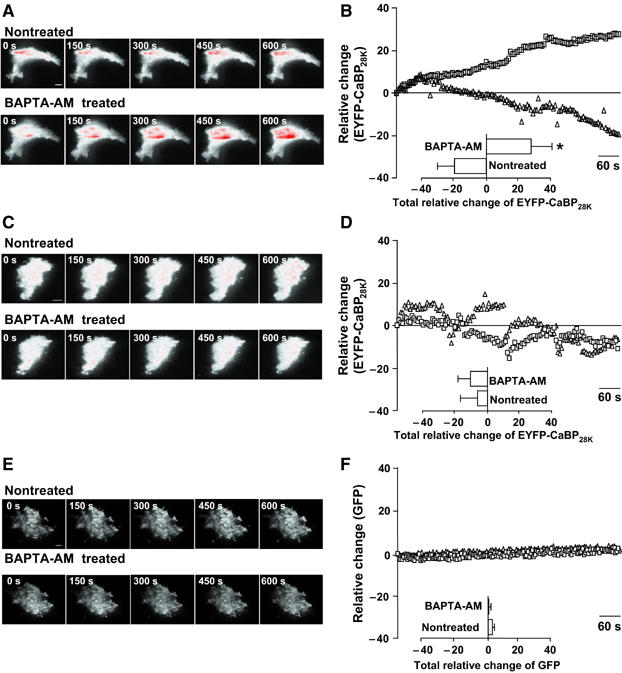
CaBP28K translocation at low intracellular Ca2+ concentrations. (A) TIRF images of a single cell expressing EYFP-CaBP28K and TRPV5-ECFP that was treated with (lower panel) or without (upper panel) BAPTA-AM. Scale bar=5 μm. (B) Average time courses of the TIRF signal of EYFP-CaBP28K in EYFP-CaBP28K- and TRPV5-ECFP-expressing cells that were treated either with (▪) or without (Δ) BAPTA-AM (_n_=7). Significant differences in total EYFP changes after BAPTA-AM treatment (inset) are indicated by an asterisk (P<0.05). (C) TIRF images of a single cell expressing EYFP-CaBP28K that was treated with (lower panel) or without (upper panel) BAPTA-AM. Scale bar=5 μm. (D) Average time courses of the TIRF signal of EYFP-CaBP28K-expressing cells that were treated either with (▪) or without (Δ) BAPTA-AM (_n_=5). (E) TIRF images of single cells expressing TRPV5-IRES-EGFP treated with (lower panel) or without (upper panel) BAPTA-AM. Scale bar=5 μm. (F) Average time courses of the TIRF signal in TRPV5-IRES-EGFP-expressing cells that were treated either with (▪) or without (Δ) BAPTA-AM (_n_=5). In all images, a gradient filter was applied such that saturation of TIRF fluorescence turns red and the intensities were measured between 5 and 10% of the visible ‘footprint' of the cell.
Coexpression of CaBP28K elevates 45Ca2+ uptake in TRPV5-expressing MDCK cells
In order to investigate the functional role of the Ca2+-binding EF-hand motifs in CaBP28K, a Ca2+-insensitive mutant (CaBP28KΔEF) was constructed and characterized by 45Ca2+ overlay and pull-down experiments. As compared to the negative control (GST), disruption of the EF-hand motifs in GST-CaBP28K resulted in a Ca2+-insensitive CaBP28K mutant, whereas wild-type GST-CaBP28K displayed normal 45Ca2+ binding (Figure 4A). In addition, pull-down analysis demonstrated that CaBP28KΔEF binds in the absence (5 mM EDTA) as well as presence (1 mM CaCl2) of Ca2+ to the N- and C-terminus of both TRPV5 and TRPV6 (Figure 4B). To investigate whether CaBP28KΔEF competes with wild-type CaBP28K for TRPV5 binding, increasing amounts of non-radioactive _in vitro_-translated CaBP28KΔEF were added during the pull-down assay. This resulted in a dose-dependent reduction of CaBP28K binding, indicating that CaBP28KΔEF competed with wild-type CaBP28K for TRPV5 association (Figure 4C).
Figure 4.
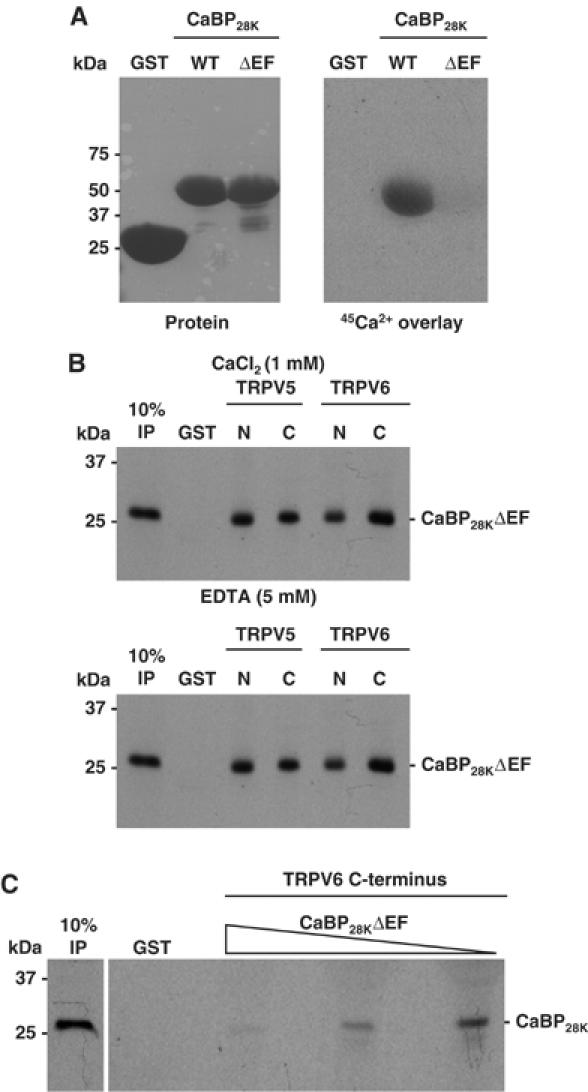
Characterization of a Ca2+-insensitive CaBP28K mutant. (A) Wild-type CaBP28K and CaBP28KΔEF were fused to GST (left panel) and 45Ca2+ binding was determined (right panel). (B) [35S]Methionine-labeled, _in vitro_-translated CaBP28KΔEF was incubated either in the presence (1 mM CaCl2) or absence (5 mM EDTA) of Ca2+, with GST or GST fused to the N- and C-termini of TRPV5 and TRPV6 immobilized on glutathione-Sepharose 4B beads. Input control (IP) represents 10% of the total pull-down input. (C) [35S]Methionine-labeled _in vitro_-translated CaBP28K was incubated in the presence of increasing amounts of non-radioactive _in vitro_-translated CaBP28KΔEF with GST or GST fused to the C-termini of TRPV5 immobilized on glutathione-Sepharose 4B beads. This pull-down experiment was performed in the absence of Ca2+ (5 mM EDTA). Input control (IP) represents 10% of the input.
The role of CaBP28K and the Ca2+-binding EF-hand motifs in CaBP28K in TRPV5-mediated 45Ca2+ uptake was assessed. To this end, stably transfected Madin–Darby Canine Kidney type-I epithelial (MDCK) cell lines were generated that express TRPV5 and CaBP28K or CaBP28KΔEF. Total expression of EGFP-TRPV5 remained constant in the MDCK-TRPV5, MDCK-TRPV5-CaBP28K and MDCK-TRPV5-CaBP28KΔEF cell lines (Figure 5A). Furthermore, immunoblot analysis demonstrated the integrity of the EGFP-TRPV5 fusion protein by the protein band of ∼100 kDa. In addition, flow cytometry analysis of the EGFP signal was employed to investigate the percentage of EGFP-TRPV5-positive cells. Importantly, >98% of the cells expressed EGFP-TRPV5, indicating that virtually all cells contribute to 45Ca2+ uptake in the MDCK-TRPV5, MDCK-TRPV5-CaBP28K and MDCK-TRPV5-CaBP28KΔEF cell lines (Figure 5B). These results demonstrated that differences in 45Ca2+ uptake are not owing to alterations in TRPV5 expression. Stable expression of EGFP-TRPV5 in MDCK cells (MDCK-TRPV5) resulted in an ∼6-fold increase of ruthenium red-sensitive 45Ca2+ uptake compared to empty vector-transfected cells (mock). Stable expression of CaBP28K in these cells (MDCK-TRPV5-CaBP28K) (Figure 5C) further increased the ruthenium red-sensitive 45Ca2+ uptake by ∼2-fold as compared to EGFP-TRPV5- and empty vector-expressing (MDCK-TRPV5-mock) cells (Figure 5D). Stable expression of CaBP28KΔEF in EGFP-TRPV5-expressing cells (MDCK-TRPV5-CaBP28KΔEF) did not result in an increase of ruthenium red-sensitive 45Ca2+ uptake, indicating the importance of the Ca2+-binding EF-hand motifs in CaBP28K.
Figure 5.
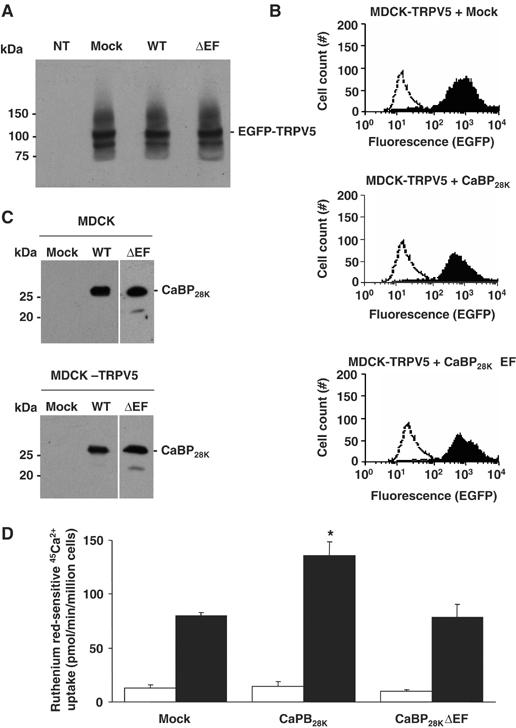
Role of CaBP28K in TRPV5-mediated 45Ca2+ uptake. EGFP-TRPV5 and CaBP28K, CaBP28KΔEF or empty vector were stably expressed in MDCK cells. The expression level of EGFP-tagged TRPV5 as determined using rabbit anti-GFP antibody (A) or flow cytometry analysis (B; black peaks) reveals that the expression of TRPV5 in empty vector- and CaBP28K-expressing cells is equal. The open peak in the three panels indicates background fluorescence in non-transfected cells. The two TRPV5 immuno-positive bands correspond to the core (lower) and glycosylated forms of EGFP-TRPV5. (C) The stable expression of CaBP28K and CaBP28KΔEF in MDCK cells or MDCK cells expressing EGFP-TRPV5 was verified using anti-CaBP28K antibodies. (D) Ruthenium red-sensitive 45Ca2+ uptake of MDCK cells stably expressing both (black bars) TRPV5 and CaBP28K, CaBP28KΔEF or empty vector and MDCK cells stably expressing (open bars) CaBP28K, CaBP28KΔEF or empty vector. Significant differences in 45Ca2+ uptake are indicated by an asterisk (P<0.05).
CaBP28K has no effect on the current characteristics of TRPV5
As CaBP28K binds to TRPV5 and possesses the ability to regulate downstream cellular processes, CaBP28K could directly affect channel activity. The effect of CaBP28K on the electrophysiological characteristics of TRPV5 was investigated by controlling [Ca2+]i in a spatially uniform manner using uncaging of Ca2+ from the photolyzable Ca2+ chelator DMNP-EDTA. TRPV5 activity in the presence and absence of CaBP28K and CaBP28KΔEF was correlated to [Ca2+]i (see Supplementary Figure S1 for additional information). Regulating [Ca2+]i enabled us to study the effect of CaBP28K on the characteristics of TRPV5 without the influence of Ca2+ buffering by CaBP28K. Neither CaBP28K nor CaBP28KΔEF modulated the Ca2+ sensitivity of TRPV5 when [Ca2+]i was gradually increased from 100 to 600 nM (Figure 6A and B).
Figure 6.
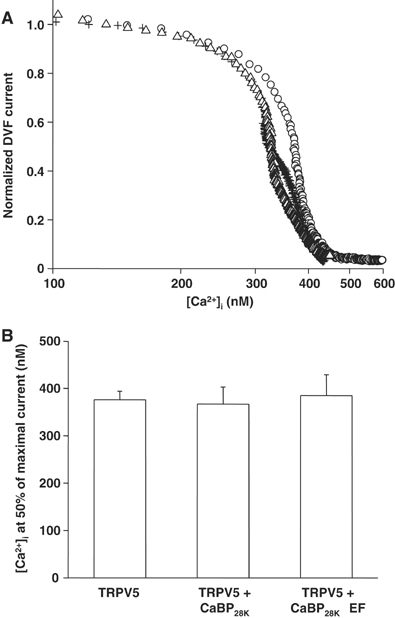
Direct influence of CaBP28K on the characteristics of TRPV5. (A) Dose–response curves showing the effect of an increasing [Ca2+]i on the normalized Na+ inward current in divalent-free extracellular solution (DVF) of cells expressing TRPV5 (O, _n_=9), TRPV5 and CaBP28K (+, _n_=11) or TRPV5 and CaBP28KΔEF (Δ, _n_=12). (B) Averaged IC50 for each of the transfections and recordings shown in panel A.
CaBP28K_Δ_EF dominant-negatively inhibits transcellular Ca2+ transport in primary rabbit CNT/CCD cultures
If the translocation of CaBP28K towards and subsequent interaction with TRPV5 is of any physiological relevance, disruption of the CaBP28K–TRPV5 association will result in a decreased Ca2+ buffering at the entry gate and coherent impairment of transcellular Ca2+ transport. This hypothesis was experimentally evaluated by using the lentiviral expression system to express EGFP-CaBP28KΔEF in primary rabbit CNT/CCD cultures and subsequent Ca2+ transport measurements across these infected confluent cell monolayers. Viral expression of EGFP-CaBP28KΔEF resulted in a dominant-negative effect on transcellular Ca2+ transport across primary CNT/CCD cultures (Figure 7A). As compared to non-infected cultures, viral expression of only EGFP did not influence transcellular Ca2+ transport. Viral infection with efficiencies of ∼50% (Supplementary Figure S2) did not affect the transepithelial resistance in any of the conditions tested, confirming the integrity of the CNT/CCD monolayer (data not shown). Importantly, only half of the CNT/CCD cells expressed the CaBP28KΔEF mutant, indicating that the observed inhibition is significantly underestimated. To investigate whether CaBP28KΔEF inhibited TRPV5-mediated transcellular Ca2+ transport in primary CNT/CCD cultures, the potent TRPV5 channel blocker ruthenium red was included during the Ca2+ transport assay. Addition of 10 μM ruthenium red to the apical side of the cell monolayer abolished transcellular Ca2+ transport in the absence or presence of exogenous CaBP28KΔEF (Figure 7A).
Figure 7.
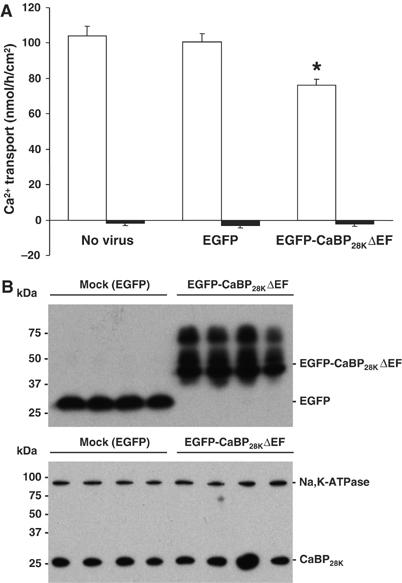
Effect of CaBP28KΔEF on transcellular Ca2+ transport. (A) Effect of lentivirus-mediated overexpression of GFP and GFP-CaBP28KΔEF on transcellular Ca2+ transport in primary rabbit CNT/CCD cells. Averaged transcellular Ca2+ transport for each infection is expressed as mean±s.e.m. A 10 μM portion of ruthenium red was added to the apical side of the cell monolayer during the transport assay to estimate the TRPV5-mediated Ca2+ transport. Significant differences as compared to mock-infected cells are indicated by an asterisk (P<0.05). (B) The expression of GFP and GFP-CaBP28KΔEF (upper panel) was assessed with immunoblotting using rabbit anti-GFP antibody and the expression of endogenous CaBP28K was checked with monoclonal anti-CaBP28K antibody that does not recognize GFP-CaBP28KΔEF (lower panel). To check for equal loading, the expression of the endogenously expressed Na,K-ATPase was measured (lower panel).
Next, we measured expression of wild-type CaBP28K and EGFP-CaBP28KΔEF in these primary renal cell cultures (Figure 7B). No differences in wild-type CaBP28K expression were detected between EGFP- and EGFP-CaBP28KΔEF-infected cells as compared to the endogenously expressed Na,K-ATPase, indicating that the observed effects were not due to downregulation of endogenous CaBP28K.
Discussion
The present study identified CaBP28K as a dynamic Ca2+ buffer facilitating TRPV5-mediated Ca2+ transport by buffering [Ca2+] in close vicinity to the channel mouth. This conclusion is based on the following observations. First, CaBP28K translocates towards TRPV5-containing plasma membranes upon a decrease in [Ca2+]i. Second, CaBP28K directly associates with TRPV5 at a low [Ca2+]i. Third, expression of CaBP28K in TRPV5-expressing cells increases TRPV5-mediated Ca2+ influx, whereas inactivation of the EF-hand motifs in CaBP28K abolishes this stimulatory effect. Fourth, the Ca2+-insensitive CaBP28K mutant (CaBP28KΔEF) competes with wild-type CaBP28K for TRPV5 association, resulting in a dominant-negative inhibition of transepithelial Ca2+ transport in primary rabbit CNT/CCD cultures.
In the past, CaBP28K was identified as a high-capacity Ca2+ buffer with Ca2+ affinities fitting the classical properties of a Ca2+ buffer. However, sequential Ca2+ binding and conformational changes suggested that CaBP28K can act as a Ca2+ sensor controlling downstream cellular processes (Gross et al, 1987; Winsky and Kuznicki, 1995; Berggard et al, 2002; Venyaminov et al, 2004). CaBP28K is highly expressed in Ca2+-transporting epithelia where it colocalizes with TRPV5. This study reveals that the expressions of CaBP28K and TRPV5 are positively correlated, suggesting a fundamental role of CaBP28K in the regulation of TRPV5 activity. In various studies exploring the regulatory role of the calciotropic hormones including vitamin D, estrogens, parathyroid hormone and dietary Ca2+, we observed the concomitant regulation of TRPV5 and CaBP28K in kidney (Lambers et al, 2006). Likewise, genetic ablation of TRPV5 in mice resulted in a decreased expression of CaBP28K (Hoenderop et al, 2003). Blockage of TRPV5 by ruthenium red eliminated parathyroid hormone-stimulated transepithelial Ca2+ transport in primary CNT/CCD cultures and simultaneously decreased the expression of CaBP28K. The magnitude of the Ca2+ influx through TRPV5 predominantly controlled the expression of CaBP28K (van Abel et al, 2005). Interestingly, Arnold and Heintz (1997) identified a 40-bp element in the CaBP28K promoter that forms a cell-specific and Ca2+-sensitive transcriptional regulatory mechanism that may play a key role in setting the Ca2+ buffering capacity of Purkinje cells (Arnold and Heintz, 1997). Together, these data assure adequate CaBP28K expression to facilitate sufficient buffering of the TRPV5-mediated Ca2+ influx.
Here, we demonstrated that in Ca2+-transporting epithelia, CaBP28K also directly associates with TRPV5 in a Ca2+-dependent fashion. In line with this interaction, in kidney cells CaBP28K is found in the cytosol as well as along the apical membrane, where it colocalizes with TRPV5. This apical localization of CaBP28K is significantly disturbed in kidney cells of TRPV5−/− mice where CaBP28K is distributed evenly throughout the cytosol. In addition, both static and dynamic measurements using cells coexpressing CaBP28K and TRPV5 revealed that CaBP28K accumulates at the plasma membrane when [Ca2+]i is low. Importantly, this translocation only occurs in the presence of TRPV5. Taken together, these data suggest that CaBP28K possesses the ability to specifically target to TRPV5-expressing plasma membranes at a low [Ca2+]i. CaBP28K translocation is supported by previous findings showing that a fraction of CaBP28K specifically associates with particular subcellular domains (Hubbard and McHugh, 1995; Winsky and Kuznicki, 1995) and that CaBP28K redistributes after Ca2+ sensing (Nemere et al, 1991). Further evidence for targeted CaBP28K mobility has been provided by Schmidt et al (2005), who demonstrated a specific association of CaBP28K with membrane-associated inositol-1,4,5-triphosphate, but not with cytosolic inositol-1,4,5-triphosphate.
In addition to CaBP28K, other Ca2+-binding proteins were found to associate with and affect the activity of TRPV5 via distinct mechanisms. Functional expression of TRPV5 requires binding of the S100A10–annexin 2 complex, whereas 80K-H acts as a Ca2+ sensor regulating TRPV5 activity (van de Graaf et al, 2003; Gkika et al, 2004). Regulation by the ubiquitous calmodulin, however, seems to be restricted to the closely related family member TRPV6 (Lambers et al, 2004). This multifaceted regulation of TRPV5 enables a strict control of transcellular Ca2+ transport at the apical entry gate. It is, however, not clear yet how these Ca2+-binding proteins integrate in this complex regulatory network to balance TRPV5-mediated Ca2+ influx.
Coexpression of CaBP28K in TRPV5-expressing HEK293 cells increased the Ca2+ uptake, whereas inactivation of the EF-hand motifs in CaBP28K blocked this stimulatory effect. CaBP28KΔEF was still able to bind TRPV5, although the Ca2+ dependency of the association was abolished. This implies that binding of CaBP28K to TRPV5 does not directly affect the physiological properties of TRPV5, otherwise the presence of Ca2+-insensitive CaBP28K would also lead to an increase in TRPV5-mediated Ca2+ uptake. Furthermore, this emphasizes the importance of the EF-hand motifs in CaBP28K. The activity of TRPV5 is tightly regulated by [Ca2+]i in such a way that Ca2+ entering through TRPV5 exerts a negative feedback on channel activity (Hoenderop et al, 1999b). The present data showing Ca2+-dependent association of TRPV5 and CaBP28K and an elevated 45Ca2+ uptake in TRPV5 and CaBP28K coexpressing cells suggests that the increase in 45Ca2+ uptake is due to CaBP28K Ca2+ buffering in close vicinity of the channel mouth. The activity of TRPV5 in the presence of CaBP28K or CaBP28KΔEF was measured at various, experimentally controlled, [Ca2+]i to further investigate whether CaBP28K directly affects TRPV5 current characteristics. In response to a gradual increase of [Ca2+]i by UV-induced uncaging of Ca2+, the activity of TRPV5 decreased, revealing the negative feedback of Ca2+ on channel activity. Coexpression of CaBP28K or CaBP28KΔEF did not change these observations, indicating that CaBP28K does not directly affect channel inactivation characteristics at controlled [Ca2+]i. Thus, CaBP28K associates with TRPV5 and stimulates TRPV5-mediated Ca2+ influx by increasing the buffering of [Ca2+] in close proximity to the channel mouth. This dynamic buffering and association is a unique process in comparison to channel regulation by other Ca2+-binding proteins like calmodulin. Channel-associated calmodulin senses [Ca2+]i and regulates channel activity by directly affecting channel (in)activation (Zuhlke et al, 1999; DeMaria et al, 2001).
Association of CaBP28K with TRPV5 and the consequent local buffering of Ca2+ is essential to facilitate the process of transcellular Ca2+ transport, which is evident from the dominant-negative effect of CaBP28KΔEF on transcellular Ca2+ transport in primary CNT/CCD cultures. CaBP28KΔEF lacks Ca2+ buffering capacity, but competes with endogenous CaBP28K for binding sites on TRPV5, as revealed by the reduction of CaBP28K and TRPV5 association in the presence of CaBP28KΔEF. This will reduce the local buffering of Ca2+ entering the epithelial cell via TRPV5, which subsequently results in inactivation of the channel and reduced transcellular transport rates. Note that addition of ruthenium red abolished transcellular Ca2+ transport in the absence or presence of exogenous CaBP28KΔEF. Thus, these findings demonstrate that CaBP28K regulates TRPV5-mediated Ca2+ transport.
Our previous findings that during transcellular Ca2+ transport in renal epithelial cells no apparent changes in the overall [Ca2+]i could be detected (Koster et al, 1995) indicate that overall Ca2+ buffering is efficiently controlled in these cells. These measurements were, however, not performed on the subcellular level or in close proximity to the channel mouth where [Ca2+] fluctuates during transcellular Ca2+ transport. Opening of epithelial Ca2+ channels likely elicits Ca2+ influx of such a large magnitude that spatial distribution of Ca2+ is complicated by insufficient local Ca2+ buffers as suggested for voltage-operated Ca2+ channels (Neher, 1998). The dominant-negative effect of CaBP28KΔEF on transcellular Ca2+ transport in primary CNT/CCD cultures, however, revealed that a channel-associated Ca2+ buffer is of utmost importance for buffering of the Ca2+ influx through TRPV5.
Together, these findings constitute the first direct evidence that CaBP28K is essential for transepithelial Ca2+ transport and elucidate the molecular role of CaBP28K as a dynamic Ca2+ buffer. At a low [Ca2+]i, CaBP28K translocates towards the plasma membrane and associates with TRPV5. Here, it buffers Ca2+ that enters the cell via TRPV5, thereby counteracting local accumulation of cytosolic free Ca2+ and coherent inactivation of the channel. Upon Ca2+ binding, CaBP28K diffuses from TRPV5 and subsequently facilitates transport of Ca2+ to the basolateral membrane. Furthermore, this illustrates an intrinsic mechanism of targeted Ca2+ buffering by spatial interactions to subcellular domains where local Ca2+ levels become detrimental. Probably, similar mechanisms occur in brain, bone, teeth, inner ear, placenta and intestine where CaBPs are abundantly expressed and where cells tolerate large fluctuations in [Ca2+]i.
Materials and methods
Molecular biology
TRPV4, 5 and 6 N- and C-termini were cloned into the pGEX6p-2 vector (Amersham Pharmacia Biotech, Roosendaal, The Netherlands). TRPV5 was cloned into the pCINeo/IRES-EGFP expression vector as described (Nilius et al, 2002) and fused to the C-terminus of EGFP or ECFP by subcloning into pEGFP-C1 and pECFP (Clontech, Palo Alto, CA). CaBP28K was cloned into the Xenopus laevis oocyte expression vector pT7Ts and pEBG (Tanaka et al, 1995), pGEX6p-2 and pCINeo/IRES-EGFP expression vectors using PCR. An EYFP-CaBP28K fusion construct was generated by cloning CaBP28K into pEYFP-C1 (Clontech, Palo Alto, CA) and a Ca2+-insensitive CaBP28K mutant (CaBP28KΔEF) was constructed by mutating the EF-hand motifs (EF1: D24A, D26A; EF2: D69A, D70A; EF3: D111A, D113A, E119A, E121A, E122A; EF4: D155A, D159A, E163A, E166A; EF5: D212A, D217A, E220A; EF6: D251A). All constructs were verified by sequence analysis and the integrity of the fusion constructs was investigated by Western blot analysis using a rabbit anti-GFP antibody.
Cell culture
HEK293, HEK293T, HeLa and MDCK cells were grown and transfected as described (Lambers et al, 2004). MDCK cells stably expressing both EGFP-TRPV5 and CaBP28K were constructed as previously described (van de Graaf et al, 2006) and maintained in medium containing 800 μg/ml G418/700 μg/ml hygromycin (GIBCO Europe, Breda, The Netherlands). Expression was verified using mouse or rabbit anti-CaBP28K (Sigma-Aldrich, Zwijndrecht, The Netherlands) and rabbit anti-GFP antibodies, respectively. Five cell line clones were pooled to eliminate differences between independent clones. For flow cytometry analysis of EGFP-TRPV5 expression, relative fluorescence intensity was measured on a FACSCalibur™ (BD Biosciences, Amsterdam, The Netherlands). Primary cultures of rabbit CNT/CCD were isolated and transepithelial Ca2+ transport was measured as described previously (Hoenderop et al, 1999a).
Protein-binding analysis
GST pull-down assays with [35S]methionine-labeled proteins in TBS–HCl pH 7.4 containing 0.5% (v/v) NP-40 and 1 mM Ca2+ or 5 mM EDTA were performed as described (Lambers et al, 2004). For binding competition assays, increasing amounts of non-radioactive _in vitro_-translated CaBP28KΔEF were included during the pull-down experiment of CaBP28K and TRPV5.
HEK293 cells were transiently cotransfected with pEBG-CaBP28K and pCINeo-TRPV5- IRES-EGFP. Cells were loaded with or without 50 μM BAPTA-AM for 30 min at 37°C and lysed by incubation for 1 h on ice in TBS pH 7.4 containing 0.5% (v/v) NP-40, 5 mM EDTA and the protease inhibitors leupeptin (0.01 mg/ml), pepstatin (0.05 mg/ml), phenylmethylsulfonyl fluoride (1 mM) and aprotinin (5 mg/ml). The lysates were centrifuged for 30 min at 16 000 g and supernatants were incubated with glutathione Sepharose beads (Amersham Bioscience, Piscataway, NJ) for 16 h. After extensive washing in lysis buffer, co-precipitation was performed by immunoblot analysis using guinea-pig anti-TRPV5 (Hoenderop et al, 1999b).
Cell fractionation assay
Primary renal CNT/CCD cultures and transiently CaBP28K- and TRPV5-transfected HeLa cells were homogenized in fractionation buffer (300 mM sucrose, 25 mM imidazole–HCl pH 7.4, 5 mM EDTA and protease inhibitors leupeptin (0.01 mg/ml), pepstatin (0.05 mg/ml), phenylmethylsulfonyl fluoride (1 mM) and aprotinin (5 mg/ml)). After centrifugation for 5 min at 4000 g 4°C to remove intact cells, nuclei and mitochondria, the supernatant was centrifuged at 16 000 g 4°C. The plasma membrane-enriched pellet fraction was subjected to SDS–PAGE and immunoblots were analyzed with rabbit anti-CaBP28K and rabbit anti-Na,K-ATPase (Hoenderop et al, 1999b) antibodies. The intensity of immuno-positive bands was measured and changes in the CaBP28K signal were expressed as a ratio of the internal control Na,K-ATPase.
Confocal, TIRF microscopy and image analysis
For TRPV5 and CaBP28K colocalization, a Zeiss LSM510meta (Carl Zeiss GmbH, Jena, Germany) confocal laser scanning microscope was used. Images were taken with a PlanApochromatic 63 × 1.4 oil immersion DIC lens (Carl Zeiss GmbH, Jena, Germany). The cytosolic localization of CaBP28K was quantified using Image J (NIH) software. Renal epithelial cells were divided in five regions (apical, apical-middle, middle, basolateral-middle and basolateral) and the intensities of the individual regions in 30 renal cells of six different tubules were calculated according to the following equation:
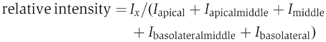
where I x is the intensity of domain X, _I_apical is the apical intensity, _I_apical-middle is the apical-middle intensity, _I_middle is the middle intensity, _I_basolateral-middle is the basolateral-middle intensity and _I_basolateral is the basolateral intensity.
To image EYFP-CaBP28K in EYFP-CaBP28K- and ECFP-TRPV5-transfected HEK293T cells, a custom-built objective-based TIRF microscope was used (Bezzerides et al, 2004). Briefly, a 488 nm solid-state diode laser (Coherent, Santa Clara, CA) was focused onto a single-mode optical fiber (Newport, Irvine, CA) with a 5-axis fiber-coupler (New Focus, San Jose, CA) and guided through the rear illumination port of an Olympus IX70 fluorescence microscope. The laser light reflected from a specially coated dichroic mirror (Z488RDC; Chroma Technology, Rockingham, VT) passes through a high-numerical aperture objective (NA 1.45, × 60, Olympus, Melville, NY) and was totally internally reflected by the glass–water interface (_n_=1.37). Fluorescence emitted from tagged proteins passed through an HQ515-30 m (Chroma) filter for EGFP/EYFP before being collected by a cooled CCD (ORCA ER II; Hamamatsu, Bridgewater, NJ). All imaging acquisition was performed with MetaMorph (Universal Imaging, West Chester, PA). Cells grown on glass coverslips were placed in a custom chamber with standard external solution (135 mM NaCl, 5 mM KCl, 1.5 mM MgCl2, 1.5 mM CaCl2, 20 mM HEPES, 10 mM D-glucose, pH 7.4 with HCl) and imaged at room temperature. Cells were screened for the expression of ECFP-TRPV5 and EYFP-CaBP28K using the normal light fluorescence setup before switching to TIRF for EYFP imaging. Cells were analyzed for 10 min (1 frame/5 s) to establish a baseline in whole-cell TIRF measurements before 50 μM BAPTA-AM was added. After 15 min, to allow BAPTA loading of the cells, cells were again followed for 10 min (1 frame/5 s). To quantify changes in TIRF, intensities of the regions of interests of several (_n_=5–7) cells, with areas between 5 and 10% of the visible ‘footprint' of the cell, were averaged and normalized according to the following equation:
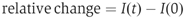
where I(0) is the intensity at the beginning of the time series and I(t) is the intensity at time point t.
45Ca2+ binding and uptake assay
GST-CaBP28K and GST were expressed and purified according to the manufacturers' protocol (Amersham Biosciences, Piscataway, NJ). Blots were washed in overlay-buffer (10 mM imidazole–HCl pH 7.4, 60 mM KCl, 0.5 mM MgCl2) before incubation with 45CaCl2 (1 μCi/ml) in overlay-buffer. After extensive washing in 50% (v/v) ethanol and drying of the blots, bound 45Ca2+ was determined by autoradiography. 45Ca2+ uptake was determined using confluent layers of MDCK cells as described (den Dekker et al, 2005). To block TRPV5-mediated 45Ca2+ uptake, cells were incubated with 10 μM ruthenium red.
Electrophysiology
Whole-cell currents in HEK293 cells transiently transfected with TRPV5 and CaPB28K were measured using an EPC-9 patch-clamp amplifier and Pulse software (HEKA Elektronik, Lambrecht, Germany). Monovalent cation currents were measured in divalent-free extracellular solution containing 150 mM NaCl, 10 mM EDTA and 10 mM HEPES–NaOH, pH 7.4. The internal (pipette) solution contained 120 mM NaCl, 1 mM CaCl2, 20 mM HEPES–NaOH and 1 mM Fura-2, supplemented with 5 mM 1-(4,5-metoxy-2-nitrophenyl)1,2-aminoethane-_N,N,N_′,_N_′-tetraacetic acid (DMNP-EDTA). DMNP-EDTA is a Ca2+ chelator that exhibits a high affinity for Ca2+ ions with an increase in its _K_d for Ca2+ upon photolysis (_K_d rises from 5 nM to 3 mM). All experiments were performed at room temperature. Current–voltage relationships were measured from linear 400-ms voltage ramps, from −100 to +100 mV. Ramps were applied every 5 s from a holding potential of +20 mV with a sampling interval of 0.8 ms. All current amplitudes in the dose–response curves were normalized to values of the inward current at −80 mV and a intracellular Ca2+ concentration ([Ca2+]i) of 135 nM and subsequently fitted with a Hill function (Origin 7.0 software, OriginLab Corporation, Northampton, MA). For photolytic release of Ca2+ and measurement of [Ca2+]i during patch-clamp, Fura-2 was excited with light alternated between 350 and 380 nm using a monochromator (Polychrome IV, TILL Photonics, Planegg, Germany), and the resulting fluorescent signal was measured using a photodiode. The ratio of the fluorescent signal was converted into [Ca2+ ]i values according to the Grynkiewicz equation (Grynkiewicz et al, 1985). Increases in [Ca2+]i were achieved by increasing the duration of 350/380 nm illumination from 15 to 150 ms each.
Lentiviral infection of primary rabbit CNT/CCD cultures
Third-generation lentiviruses were produced by cotransfection of the packaging vectors pRSV-Rev, pMDL g/p RRE and pMD2G (from Tronolab, Lausanne, Switzerland) into HEK293T cells as described (Dull et al, 1998). The virus titer was determined by p24 HIV ELISA (Murex Diagnostics, Dartford, UK). Primary rabbit CNT/CCD cultures were infected with lentiviruses containing EGFP or EGFP-CaBP28KΔEF immediately before plating in the presence of polybrene (8 μg/ml) using 20 virus particles per cell (20 MOI). Virus was removed after 24 h and subsequently transepithelial Ca2+ transport was measured 6 days after infection in the presence of 10 μM forskolin as described previously (Hoenderop et al, 1999a).
Statistical analysis
In all experiments, the data are expressed as mean±s.e.m. Overall statistical significance was determined by analysis of variance followed by Bonferroni to investigate individual significance. _P_-values below 0.05 were considered significant.
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Acknowledgments
We thank Mr J Israel and Mrs S van Gessel for excellent experimental assistance, and Dr J Hoeben (LUMC Leiden, The Netherlands) and Dr D Trono (Lausanne, Switzerland) for providing lentiviral vectors. This work was supported by the Dutch Organization of Scientific Research (Zon-Mw 016.006.001, NWO-ALW 805.09.042), Human Frontiers Science Program (RGP32/2004), the Dutch Kidney Foundation (C03.6017) and the Onderzoeksraad KU Leuven (GOA 2004/07, FWO G.0214.99, FWO G.0136.00, FWO G.0172.03, Interuniversity Poles of Attraction Program, Prime Ministers Office IUAP). A work visit of Mr TT Lambers to the lab of Dr D Clapham was further supported by a grant of the van Walree Fund from the Royal Dutch Academy of Sciences.
References
- Airaksinen MS, Eilers J, Garaschuk O, Thoenen H, Konnerth A, Meyer M (1997) Ataxia and altered dendritic calcium signaling in mice carrying a targeted null mutation of the calbindin D28K gene. Proc Natl Acad Sci USA 94: 1488–1493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold DB, Heintz N (1997) A calcium responsive element that regulates expression of two calcium binding proteins in Purkinje cells. Proc Natl Acad Sci USA 94: 8842–8847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berggard T, Miron S, Onnerfjord P, Thulin E, Akerfeldt KS, Enghild JJ, Akke M, Linse S (2002) Calbindin D28K exhibits properties characteristic of a Ca2+ sensor. J Biol Chem 277: 16662–16672 [DOI] [PubMed] [Google Scholar]
- Bezzerides VJ, Ramsey IS, Kotecha S, Greka A, Clapham DE (2004) Rapid vesicular translocation and insertion of TRP channels. Nat Cell Biol 6: 709–720 [DOI] [PubMed] [Google Scholar]
- Blatow M, Caputi A, Burnashev N, Monyer H, Rozov A (2003) Ca2+ buffer saturation underlies paired pulse facilitation in calbindin-D28K-containing terminals. Neuron 38: 79–88 [DOI] [PubMed] [Google Scholar]
- Bronner F (1989) Renal calcium transport: mechanisms and regulation—an overview. Am J Physiol 257: F707–F711 [DOI] [PubMed] [Google Scholar]
- Bronner F, Stein WD (1988) CaBPr facilitates intracellular diffusion for Ca2+ pumping in distal convoluted tubule. Am J Physiol 255: F558–F562 [DOI] [PubMed] [Google Scholar]
- DeMaria CD, Soong TW, Alseikhan BA, Alvania RS, Yue DT (2001) Calmodulin bifurcates the local Ca2+ signal that modulates P/Q-type Ca2+ channels. Nature 411: 484–489 [DOI] [PubMed] [Google Scholar]
- den Dekker E, Schoeber J, Topala CN, van de Graaf SF, Hoenderop JG, Bindels RJ (2005) Characterization of a Madin–Darby canine kidney cell line stably expressing TRPV5. Pflugers Arch 450: 236–244 [DOI] [PubMed] [Google Scholar]
- Dull T, Zufferey R, Kelly M, Mandel RJ, Nguyen M, Trono D, Naldini L (1998) A third-generation lentivirus vector with a conditional packaging system. J Virol 72: 8463–8471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feher JJ, Fullmer CS, Wasserman RH (1992) Role of facilitated diffusion of calcium by calbindin in intestinal calcium absorption. Am J Physiol 262: C517–C526 [DOI] [PubMed] [Google Scholar]
- Gkika D, Mahieu F, Nilius B, Hoenderop JG, Bindels RJ (2004) 80K-H as a new Ca2+ sensor regulating the activity of the epithelial Ca2+ channel transient receptor potential cation channel V5 (TRPV5). J Biol Chem 279: 26351–26357 [DOI] [PubMed] [Google Scholar]
- Gross MD, Nelsestuen GL, Kumar R (1987) Observations on the binding of lanthanides and calcium to vitamin D-dependent chick intestinal calcium-binding protein. Implications regarding calcium-binding protein function. J Biol Chem 262: 6539–6545 [PubMed] [Google Scholar]
- Grynkiewicz G, Poenie M, Tsien RY (1985) A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem 260: 3440–3450 [PubMed] [Google Scholar]
- Guo Q, Christakos S, Robinson N, Mattson MP (1998) Calbindin-D28K blocks the proapoptotic actions of mutant presenilin 1: reduced oxidative stress and preserved mitochondrial function. Proc Natl Acad Sci USA 95: 3227–3232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoenderop JG, Nilius B, Bindels RJ (2005) Calcium absorption across epithelia. Physiol Rev 85: 373–422 [DOI] [PubMed] [Google Scholar]
- Hoenderop JG, Vaandrager AB, Dijkink L, Smolenski A, Gambaryan S, Lohmann SM, de Jonge HR, Willems PH, Bindels RJ (1999a) Atrial natriuretic peptide-stimulated Ca2+ reabsorption in rabbit kidney requires membrane-targeted, cGMP-dependent protein kinase type II. Proc Natl Acad Sci USA 96: 6084–6089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoenderop JG, van der Kemp AW, Hartog A, van de Graaf SF, van Os CH, Willems PH, Bindels RJ (1999b) Molecular identification of the apical Ca2+ channel in 1,25-dihydroxyvitamin D3-responsive epithelia. J Biol Chem 274: 8375–8378 [DOI] [PubMed] [Google Scholar]
- Hoenderop JG, van Leeuwen JP, van der Eerden BC, Kersten FF, van der Kemp AW, Merillat AM, Waarsing JH, Rossier BC, Vallon V, Hummler E, Bindels RJ (2003) Renal Ca2+ wasting, hyperabsorption, and reduced bone thickness in mice lacking TRPV5. J Clin Invest 112: 1906–1914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubbard MJ, McHugh NJ (1995) Calbindin-D28K and calbindin-D30K (calretinin) are substantially localised in the particulate fraction of rat brain. FEBS Lett 374: 333–337 [DOI] [PubMed] [Google Scholar]
- Iacopino AM, Christakos S (1990) Corticosterone regulates calbindin-D28k mRNA and protein levels in rat hippocampus. J Biol Chem 265: 10177–10180 [PubMed] [Google Scholar]
- Jones LP, DeMaria CD, Yue DT (1999) N-type calcium channel inactivation probed by gating-current analysis. Biophys J 76: 2530–2552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kits KS, Mansvelder HD (1996) Voltage gated calcium channels in molluscs: classification, Ca2+ dependent inactivation, modulation and functional roles. Invert Neurosci 2: 9–34 [DOI] [PubMed] [Google Scholar]
- Koster HP, Hartog A, Van Os CH, Bindels RJ (1995) Calbindin-D28K facilitates cytosolic calcium diffusion without interfering with calcium signaling. Cell Calcium 18: 187–196 [DOI] [PubMed] [Google Scholar]
- Lambers TT, Bindels RJ, Hoenderop JG (2006) Coordinated control of renal Ca2+ handling. Kidney Int 69: 650–654 [DOI] [PubMed] [Google Scholar]
- Lambers TT, Weidema AF, Nilius B, Hoenderop JG, Bindels RJ (2004) Regulation of the mouse epithelial Ca2+ channel TRPV6 by the Ca2+-sensor calmodulin. J Biol Chem 279: 28855–28861 [DOI] [PubMed] [Google Scholar]
- Lukas W, Jones KA (1994) Cortical neurons containing calretinin are selectively resistant to calcium overload and excitotoxicity in vitro. Neuroscience 61: 307–316 [DOI] [PubMed] [Google Scholar]
- Montell C, Birnbaumer L, Flockerzi V, Bindels RJ, Bruford EA, Caterina MJ, Clapham DE, Harteneck C, Heller S, Julius D, Kojima I, Mori Y, Penner R, Prawitt D, Scharenberg AM, Schultz G, Shimizu N, Zhu MX (2002) A Unified Nomenclature for the Superfamily of TRP Cation Channels. Mol Cell 9: 229–231 [DOI] [PubMed] [Google Scholar]
- Neher E (1998) Vesicle pools and Ca2+ microdomains: new tools for understanding their roles in neurotransmitter release. Neuron 20: 389–399 [DOI] [PubMed] [Google Scholar]
- Nemere I, Leathers VL, Thompson BS, Luben RA, Norman AW (1991) Redistribution of calbindin-D28k in chick intestine in response to calcium transport. Endocrinology 129: 2972–2984 [DOI] [PubMed] [Google Scholar]
- Nijenhuis T, Hoenderop JG, Bindels RJ (2004) Downregulation of Ca2+ and Mg2+ transport proteins in the kidney explains tacrolimus (FK506)-induced hypercalciuria and hypomagnesemia. J Am Soc Nephrol 15: 549–557 [DOI] [PubMed] [Google Scholar]
- Nijenhuis T, Renkema KY, Hoenderop JG, Bindels RJ (2006) Acid–base status determines the renal expression of Ca2+ and Mg2+ transport proteins. J Am Soc Nephrol 17: 617–626 [DOI] [PubMed] [Google Scholar]
- Nijenhuis T, Vallon V, van der Kemp AW, Loffing J, Hoenderop JG, Bindels RJ (2005) Enhanced passive Ca2+ reabsorption and reduced Mg2+ channel abundance explains thiazide-induced hypocalciuria and hypomagnesemia. J Clin Invest 115: 1651–1658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilius B, Prenen J, Hoenderop JG, Vennekens R, Hoefs S, Weidema AF, Droogmans G, Bindels RJ (2002) Fast and slow inactivation kinetics of the Ca2+ channels ECaC1 and ECaC2 (TRPV5 and 6): role of the intracellular loop located between transmembrane segment 2 and 3. J Biol Chem 277: 30852–30858 [DOI] [PubMed] [Google Scholar]
- Pauls TL, Cox JA, Berchtold MW (1996) The Ca2+-binding proteins parvalbumin and oncomodulin and their genes: new structural and functional findings. Biochim Biophys Acta 1306: 39–54 [DOI] [PubMed] [Google Scholar]
- Schmidt H, Schwaller B, Eilers J (2005) Calbindin-D28K targets myo-inositol monophosphatase in spines and dendrites of cerebellar Purkinje neurons. Proc Natl Acad Sci USA 102: 5850–5855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt H, Stiefel KM, Racay P, Schwaller B, Eilers J (2003) Mutational analysis of dendritic Ca2+ kinetics in rodent Purkinje cells: role of parvalbumin and calbindin-D28K. J Physiol 551: 13–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwaller B, Meyer M, Schiffmann S (2002) ‘New' functions for ‘old' proteins: the role of the calcium-binding proteins calbindin-D28K, calretinin and parvalbumin, in cerebellar physiology. Studies with knockout mice. Cerebellum 1: 241–258 [DOI] [PubMed] [Google Scholar]
- Tanaka M, Gupta R, Mayer BJ (1995) Differential inhibition of signaling pathways by dominant-negative SH2/SH3 adapter proteins. Mol Cell Biol 15: 6829–6837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tymianski M (1996) Cytosolic calcium concentrations and cell death in vitro. Adv Neurol 71: 85–105 [PubMed] [Google Scholar]
- van Abel M, Hoenderop JG, Dardenne O, St-Arnaud R, van Os C, Van Leeuwen JP, Bindels RJ (2002) 1,25(OH)2D3-independent stimulatory effect of estrogen on the expression of ECaC1 in kidney. J Am Soc Nephrol 13: 2102–2109 [DOI] [PubMed] [Google Scholar]
- van Abel M, Hoenderop JG, van der Kemp AW, Friedlaender MM, van Leeuwen JP, Bindels RJ (2005) Coordinated control of renal Ca2+ transport proteins by parathyroid hormone. Kidney Int 68: 1708–1721 [DOI] [PubMed] [Google Scholar]
- van de Graaf SF, Chang Q, Mensenkamp AR, Hoenderop JG, Bindels RJ (2006) Direct interaction with Rab11a targets the epithelial Ca2+ channels TRPV5 and TRPV6 to the plasma membrane. Mol Cell Biol 26: 303–312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van de Graaf SF, Hoenderop JG, Gkika D, Lamers D, Prenen J, Rescher U, Gerke V, Staub O, Nilius B, Bindels RJ (2003) Functional expression of the epithelial Ca2+ channels (TRPV5 and TRPV6) requires association of the S100A10–annexin 2 complex. EMBO J 22: 1478–1487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venters RA, Benson LM, Craig TA, Bagu J, Paul KH, Kordys DR, Thompson R, Naylor S, Kumar R, Cavanagh J (2003) The effects of Ca2+ binding on the conformation of calbindin D28K: a nuclear magnetic resonance and microelectrospray mass spectrometry study. Anal Biochem 317: 59–66 [DOI] [PubMed] [Google Scholar]
- Venyaminov SY, Klimtchuk ES, Bajzer Z, Craig TA (2004) Changes in structure and stability of calbindin-D28K upon calcium binding. Anal Biochem 334: 97–105 [DOI] [PubMed] [Google Scholar]
- Winsky L, Kuznicki J (1995) Distribution of calretinin, calbindin-D28K, and parvalbumin in subcellular fractions of rat cerebellum: effects of calcium. J Neurochem 65: 381–388 [DOI] [PubMed] [Google Scholar]
- Zuhlke RD, Pitt GS, Deisseroth K, Tsien RW, Reuter H (1999) Calmodulin supports both inactivation and facilitation of L-type calcium channels. Nature 399: 159–162 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure S1
Supplementary Figure S2